

Background: Metalloproteinase-9 (MMP-9) is detected at the early but not late stages of osteoarthritis.
Results: Application of high fluid shear to chondrocytes recapitulates the temporal regulation of MMP-9 expression.
Conclusion: The antagonistic actions of shear-induced IL-1β and 15d-PGJ2 regulate the temporal pattern of MMP-9 synthesis.
Significance: Reconstructing the signaling network of shear-mediated MMP-9 synthesis in chondrocytes is crucial for developing strategies to treat osteoarthritis.
Keywords: Interleukin, Matrix Metalloproteinase (MMP), Osteoarthritis, Prostaglandins, Shear Stress
Abstract
Mechanical overloading of articular cartilage producing hydrostatic stress, tensile strain, and fluid flow results in irreversible cartilage erosion and osteoarthritis (OA). Application of high fluid shear to chondrocytes recapitulates the earmarks of OA as evidenced by the induction of proinflammatory cytokines and prostaglandins, which are capable of inducing the expression of matrix-degrading enzymes. Matrix metalloproteinase-9 (MMP-9) synthesis is detected at early but not late stages of OA. However, the underlying mechanism(s) of the MMP-9 temporal regulation remains unknown. Using the T/C-28a2 chondrocyte cell line as a model system, we demonstrated that high fluid shear induces a marked increase in MMP-9 expression at short shear exposure times (3–6 h), which falls below basal levels after prolonged shear exposure (12–48 h). High fluid shear stress induced the rapid and sustained synthesis of IL-1β, activating PI3K, ERK1/2, and JNK, which are in turn responsible for MMP-9 expression. Prolonged shear exposure (>12 h) induced 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) synthesis, which exerted an antagonistic effect on IL-1β-mediated PI3K-, ERK1/2-, and JNK-dependent NF-κB activation, thereby suppressing MMP-9 expression in human chondrocytes. Reconstructing the signaling network that regulates shear-mediated MMP-9 expression in human chondrocytes may provide insights for developing strategies to treat arthritic disorders.
Introduction
Osteoarthritis (OA)2 is a musculoskeletal disorder characterized by the irreversible erosion of the articular cartilage tissue that covers the moving joints. Excessive chronic or repetitive mechanical loading of articular cartilage producing hydrostatic stress, tensile strain, and fluid flow (1) has been implicated in the development and progression of OA (2). Indeed, prolonged application of high fluid shear to human chondrocytes in vitro recapitulates gene expression profiles associated with OA in vivo (3). Although OA is classified as a noninflammatory joint disease, the progressive erosion of cartilage involves the action of cytokines and prostaglandins (PGs), which are capable of inducing the expression of matrix-degrading enzymes such as matrix metalloproteinases (MMPs) (4). For instance, IL-1β is a key mediator in the pathogenesis of OA (5). Interestingly, chondrocytes from OA patients express higher levels of IL-1 receptor type I (IL-1R1) than normal controls (5). The enhanced expression of IL-1R1 in osteoarthritic chondrocytes renders these cells highly sensitive to the effects of IL-1β, thereby leading to alterations in gene expression of cartilage-degrading proteinases such as MMPs (5). Treatment of human chondrocytes with exogenous IL-1β stimulates MMP expression in vitro (6).
Previous studies have reported contradictory results regarding the relative expression/activity levels of MMP-9 in OA and healthy chondrocytes. For example, some studies have shown that MMP-9 is markedly up-regulated in OA versus normal chondrocytes (7, 8). However, Tetlow et al. (9) reported that MMP-9 is not prominently expressed in OA cartilage compared with normal, age-matched articular cartilage. Moreover, Naito et al. (10) disclosed that the plasma levels of MMP-9 are lower in OA patients than in normal subjects. These conflicting results might be reconciled by observations showing that MMP-9 expression in chondrocytes varies not only with the zone (depth) of the chondrocyte in the cartilage but also with the grade of OA (11). It is noteworthy that MMP-9 is detected at early but not late stages of OA (11). Nevertheless, the underlying mechanisms of MMP-9 synthesis at the early and late disease stages have yet to be delineated.
We have shown that exposure of the human T/C-28a2 chondrocytic cell line to fluid shear recapitulates the earmarks of OA, as evidenced by the release of proinflammatory cytokines such as IL-6, detected at the early stages of OA (or short shear exposure times in vitro), and chondrocyte senescence observed in advanced stages of OA in vivo (12) (or prolonged shear exposure times in vitro) (3, 13–15). In agreement with in vivo results (11), we demonstrate herein the temporal regulation of MMP-9 expression in shear-activated T/C-28a2 chondrocytes, which reaches maximal levels at short shear exposure times and declines below basal levels after prolonged shear exposure. Moreover, we show that the antagonistic actions of endogenous shear-induced IL-1β and 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) regulate the temporal synthesis of MMP-9 in human chondrocytes. We further demonstrate the critical roles of the extracellular signal-regulated kinase (ERK1/2), phosphatidylinositol 3-kinases (PI3K), and c-Jun NH2-terminal kinase (JNK) signaling pathways, which are downstream of IL-1β and 15d-PGJ2, in the regulation of MMP-9 expression via the NF-κB p65 subunit at short and long shear exposure times.
EXPERIMENTAL PROCEDURES
Reagents
LY294002, Wortmannin, U0126, PD98059, SB203580, SP600125, GW1929, GW9662, human recombinant IL-1β, and antibodies specific for mPGES-1 and L-PGD synthase (L-PGDS) were obtained from Sigma-Aldrich. AH6809, sulprostone, and 15d-PGJ2 were obtained from Enzo Life International Inc. (Plymouth Meeting, PA). Akt, ERK1/2, p38, c-Jun, scramble siRNAs, and antibodies specific for β-actin, Akt, p-Akt (Ser-473), ERK1/2, p-ERK1/2 (Thr-202/Tyr-204), c-Jun, and p-c-Jun (Ser-63) were purchased from Cell Signaling Technology (Danvers, MA). The IL-1β enzyme immunoassay kit and the anti-PPARγ polyclonal antibody were from Cayman Chemical (Ann Arbor, MI), whereas the 15d-PGJ2 enzyme immunoassay kit was obtained from Assay Designs (Ann Arbor, MI). The mAb against p-PPARγ (Ser-82) was from Millipore (Bedford, MA). The monoclonal neutralizing antibody specific for IL-1β was obtained from Antibodies-online (Atlanta, GA). mPGES-1, L-PGDS, and scramble siRNAs were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The L-PGDS cDNA plasmids were obtained from OriGene Technologies (Rockville, MD) and subcloned to pCMV6-XL vector. The mmp-9 promoter constructs, pMMP-9-luc1723 (−1723/−6), pMMP-9-luc1017 (−1017/−6), pMMP-9-luc604 (−604/−6), pMMP-9-luc553 (−553/−6), pMMP-9-luc324 (−324/−6), and pMMP-9-luc1017ΔNF-κB (NF-κB site mutation −332/−323), were gifts from Dr. Richard Leigh (Health Research Innovation Centre, University of Calgary, Canada) (16). The pRL-SV40 vector encoding the Renilla luciferase gene and the Dual-Luciferase reporter assay kit were purchased from Promega (Madison, WI). The chromatin immunoprecipitation (ChIP) EZ-ChIP kit was purchased from Upstate Biotechnology. All reagents for qRT-PCR and SDS-PAGE experiments were purchased from Bio-Rad Laboratories. All other reagents were from Invitrogen unless otherwise specified.
Cell Culture and Shear Stress Exposure
Human primary articular chondrocytes (Cell Applications, San Diego) or T/C-28a2 chondrocytic cells were grown (at 37 °C in 5% CO2) on glass slides in F12/DMEM supplemented with 10% FBS as described previously (3, 13–15, 17–19). Before shear exposure, cells were incubated for 18 h in serum-free medium supplemented with 1% Nutridoma-SP (Roche Applied Science), a low serum replacement that maintains chondrocyte phenotype and establishes quiescence in the monolayer (3, 13–15, 17–19). Cells were then subjected to a shear stress level of 20 dynes/cm2 for up to 48 h in medium containing 1% Nutridoma-SP, using a streamer gold flow device (Flexcell International Corp., Hillsborough, NC). In select experiments, the pharmacological agents were added to the medium at the indicated concentrations just before the onset of shear exposure. In separate experiments, T/C-28a2 chondrocytes were seeded on 6-cm tissue culture dishes (106 cells/dish) in DMEM/F12 supplemented with 10% FBS (20, 21). 24 h later, T/C-28a2 cells were grown in serum-free medium for another 24 h before being incubated with specific pharmacological inhibitors in the presence or absence of exogenously added IL-1β (20, 21).
Transient Transfection
T/C-28a2 chondrocytes were transfected with a siRNA (100 nm) oligonucleotide sequence specific for L-PGDS, mPGES-1, Akt, ERK1/2, p38, or c-Jun. In control experiments, cells were transfected with 100 nm scramble siRNA. For ectopic expression of L-PGDS, T/C-28a2 cells were transfected with 1.6 μg/slide of plasmid containing the L-PGDS cDNA by using Lipofectamine 2000. In control experiments, cells were transfected with 1.6 μg/slide of the empty pCMV6-XL vector (OriGene Technologies). In promoter assays, T/C-28a2 cells were transfected with 1.6 μg/slide of the mmp-9 promoter-reporter construct together with the pRL-SV40 vector. Transfected cells were allowed to recover for at least 12 h in growth medium and then incubated overnight in medium containing 1% Nutridoma-SP before exposure to shear stress.
Quantitative Real-time PCR (qRT-PCR)
qRT-PCR assays were performed on the iCycler iQ detection system (Bio-Rad) using total RNA, the iScript one-step RT-PCR kit with SYBR Green (Bio-Rad), and primers. The GenBankTM accession numbers and forward (F) and reverse (R) primers are as follows: IL-1β (NM_000576), F-ACTACAGCAAGGGCTTCAGG and R-TCTTTCAACACGCAGGACAG; MMP-9 (NM_004994), F-GGATGGGAAGTACTGGCGATTC and R-CACTTGGTCCACCTGGTTCAAC.
The GenBankTM accession numbers and forward (F-) and reverse (R-) primers for GAPDH are provided in our previous publications (13, 20). GAPDH was used as internal control. Reaction mixtures were incubated at 50 °C for 15 min followed by 95 °C for 5 min, and then 35 PCR cycles were performed with the following temperature profile: 95 °C for 15 s, 58 °C for 30 s, 68 °C for 1 min, and 77 °C for 20 s. Data were collected at the 77 °C 20 s step to remove possible fluorescent contribution from dimer primers (13, 20). Gene expression values were normalized to GAPDH.
Western Blot Analysis
T/C-28a2 chondrocytes, from static and sheared specimens, were lysed in radioimmune precipitation assay buffer (25 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, and 0.1% SDS) containing a mixture of proteinase inhibitors (Pierce). The protein content of the cell lysates was determined using bicinchoninic acid (BCA) protein assay reagent (Pierce). Total cell lysates (4 μg) were subjected to SDS-PAGE, transferred to a membrane, and probed with a panel of specific antibodies. Each membrane was only probed using one antibody. β-Actin was used as the loading control. All Western hybridizations were performed at least in triplicate with a different cell preparation used each time.
Measurement of IL-1β and 15d-PGJ2 Concentration in Medium
IL-1β and 15d-PGJ2 levels in both static and sheared medium were determined using an enzyme immunoassay kit following the manufacturer's instructions. The concentration of total protein in the medium was used as the loading control, and the results were expressed as picograms of IL-1β or 15d-PGJ2/microgram of total protein.
Zymography
Gelatinase activity was determined according to a previously described method (22). In brief, the conditioned media of cells were collected and mixed with non-reducing sample buffer containing 0.5 m Tris, pH 6.8, 5% SDS, 20% glycerol, and 1% bromphenol blue in a 1:1 ratio and electrophoresed directly on 10% SDS-polyacrylamide gels (SDS-PAGE). After electrophoresis, gels were washed for 1 h at room temperature in a 2.5% (v/v) Triton X-100 solution to remove SDS, transferred to zymogram development solution (10 mm CaCl2, 50 mm Tris-HCl, pH 7.4, and 0.02% NaN3), and incubated for 16 h at 37 °C. Gels were stained for 30 min with 0.1% (w/v) Coomassie Brilliant Blue in 50% (v/v) methanol/10% (v/v) acetic acid and destained in 20% (v/v) methanol/10% (v/v) acetic acid. Areas of lysis were observed as white bands against a dark background.
Promoter Activity Assays
Firefly and Renilla luciferase activities were measured by use of the Dual-Luciferase Reporter Assay kit (Promega). Firefly luciferase activities were normalized to the Renilla luciferase controls. Data are expressed as ratios of shear to static normalized firefly luciferase activity unless otherwise stated.
ChIP Assay
This assay was performed using the EZ ChIP kit following the manufacturer's instructions (Upstate Biotechnology) as described previously (14, 20, 21). Forward (F) and reverse (R) primers for mmp-9 promoter amplification by qPCR are as follows: F-CTTGGCTGACCACTGGAGGC and R-GACAGGCAAGTGCTGACTCA.
Statistics
Data represent the mean ± S.E. of at least three independent experiments. Statistical significance of differences between means was determined by Student's t test or by one-way analysis of variance wherever appropriate. If means were shown to be significantly different, multiple comparisons by pairs were performed using the Tukey test (23).
RESULTS
Shear Stress Induces IL-1β and 15d-PGJ2 Synthesis, Which in Turn Antagonistically Regulate MMP-9 Expression in Human T/C-28a2 Chondrocytes
Matrix-degrading enzymes such as MMPs are thought to be critical mediators of cartilage erosion during OA. Although some studies reveal that MMP-9 expression is markedly elevated in OA relative to healthy chondrocytes (7, 8), others have shown the exact opposite (10). Interestingly, MMP-9 expression in chondrocytes has been reported to be up-regulated at the early but not the late stages of OA (11). In view of these observations (11) and given that application of high shear stress to T/C-28a2 chondrocytes for short versus long exposure times recapitulates the earmarks of OA at early and late disease stages, respectively (3, 13–15), we hypothesized that MMP-9 expression is temporally regulated by fluid shear. The human T/C-28a2 chondrocytic cell line was chosen as a model system because T/C-28a2 cells have been shown to behave much like primary human chondrocytes when cultured under appropriate conditions (3, 13–15, 17–20). Interestingly, continuous exposure of human T/C-28a2 chondrocytes to high shear stress (20 dynes/cm2) induces maximal MMP-9 expression and activity after 6 h of cell stimulation (Fig. 1A). However, MMP-9 mRNA and activity levels decline below basal levels at 12 h and remain suppressed after 48 h of shear exposure (Fig. 1A). To validate previously published observations showing that T/C-28a2 cells represent an appropriate model for the study of chondrocyte function in vitro (3, 13–15, 17–21), we examined the responses of human primary articular chondrocytes to high fluid shear. Our data revealing significant similarities between primary articular chondrocytes and T/C-28a2 cells in the regulation of MMP-9 mRNA or activity in response to shear stress (Fig. 1, A and B) reinforce the aforementioned notion. Nevertheless, the signaling pathway of temporal regulation of MMP-9 expression has yet to be delineated in human chondrocytes.
FIGURE 1.

Shear stress mediates the temporal regulation of MMP-9 and IL-1β mRNA and protein expression in human chondrocytes. T/C-28a2 chondrocytes (A, C, and D) or human primary articular chondrocytes (B) were subjected to fluid shear stress (20 dynes/cm2) or static conditions (0 dyne/cm2) for the indicated time intervals. A and B, MMP-9 mRNA and activity levels were determined by qRT-PCR and zymography, respectively. GAPDH and MMP-2 served as internal controls in qRT-PCR and zymography assays, respectively. C and D, IL-1β mRNA expression and secretion were determined using qRT-PCR and an IL-1β enzyme immunoassay kit, respectively. GAPDH and total protein in the medium served as internal controls for the qRT-PCR and IL-1β enzyme immunoassay, respectively. Data represent the mean ± S.E. of three independent experiments. *, p < 0.05 with respect to static control.
Prior work has shown that proinflammatory cytokines such as IL-1β direct MMP-mediated digestion of cartilage matrix in OA (24). On the other hand, 15d-PGJ2 has been reported to repress MMP-9 activity in different cell types such as human smooth muscle cells and pancreatic cancer cells (25, 26). Moreover, we have recently reported that 15d-PGJ2 is synthesized by human chondrocytes after prolonged shear exposure (14, 15). We thus evaluated the potential roles of IL-1β and 15d-PGJ2 in the regulation of MMP-9 activity in shear-activated human chondrocytes. Time course experiments revealed that endogenous IL-1β mRNA expression (Fig. 1C) reached maximal levels after 1 h of T/C-28a2 chondrocyte stimulation with high fluid shear. Shear-induced IL-1β mRNA levels remained up-regulated (>5-fold) relative to static controls after 48 h of shear exposure. Continuous application of high fluid shear to T/C-28a2 chondrocytes for 48 h induced the progressive accumulation of secreted IL-1β in the medium (Fig. 1D). In view of these findings, we next examined the effects of exogenous IL-1β on MMP-9 synthesis. Our data reveal that IL-1β induces MMP-9 mRNA expression in a dose- and time-dependent manner (Fig. 2, A and B). Maximal MMP-9 mRNA levels were achieved after stimulation of T/C-28a2 chondrocytes with 100 ng/ml exogenous IL-1β (Fig. 2Α). This dose (100 ng/ml) induced the sustained synthesis of MMP-9, which reached a plateau after 1 h of cell stimulation (Fig. 2B). Of note, marked up-regulation of MMP-9 gelatinolytic activity (Fig. 2E) was detected after prolonged (48 h) incubation of T/C-28a2 chondrocytes with exogenous IL-1β.
FIGURE 2.

Effects of exogenously added IL-1β or 15d-PGJ2 on MMP-9 synthesis in human T/C-28a2 chondrocytes. T/C-28a2 chondrocytes were treated with the indicated concentrations of either exogenous IL-1β (A) or 15d-PGJ2 (C) for 48 h. In select experiments, T/C-28a2 chondrocytes were treated with either IL-1β (100 ng/ml) (B and E) or 15d-PGJ2 (2 μm) (D and F) for the indicated time intervals. In separate experiments, T/C-28a2 cells were treated with IL-1β (100 ng/ml), 15d-PGJ2 (2 μm), or a combination of them (G) for 48 h. MMP-9 mRNA (A–D and G (lower panel)) and activity (E, F, and G (upper panel)) levels were determined by qRT-PCR and zymography, respectively. GAPDH and MMP-2 served as internal controls in qRT-PCR and zymography assays, respectively. Data represent the mean ± S.E. of three independent experiments. *, p < 0.05 with respect to vehicle control; ▴, p < 0.05 with respect to IL-1β treatment.
High fluid shear suppresses 15d-PGJ2 production by human chondrocytes at short exposure times (2–6 h), whereas it stimulates 15d-PGJ2 release at long (48 h) exposure times (14, 15). Exogenous 15d-PGJ2 markedly represses MMP-9 transcript levels in a dose- and time-dependent manner (Fig. 2, C and D). 15d-PGJ2 also suppresses MMP-9 activity in T/C-28a2 chondrocytes (Fig. 2F). Concurrent treatment of T/C-28a2 cells with IL-1β (100 ng/ml) and 15d-PGJ2 (2 μm) partially reverses the MMP-9 inhibition noted in 15d-PGJ2-activated chondrocytes (Fig. 2G). Of note, neither IL-1β nor 15d-PGJ2 affects MMP-2 levels in human chondrocytes. Taken together, these data illustrate that exogenous IL-1β and 15d-PGJ2 exert opposing effects on MMP-9 synthesis in human chondrocytes. In view of these observations and the distinct temporal regulation of endogenous IL-1β (Fig. 1, C and D) and 15d-PGJ2 (14, 15) release by human chondrocytes, we hypothesized that the balance between endogenous IL-1β and 15d-PGJ2 regulates MMP-9 expression in shear-activated chondrocytes.
As a next step, we wished to examine the potential involvement of endogenously secreted 15d-PGJ2 in the regulation of MMP-9 expression in shear-activated chondrocytes. L-PGDS is responsible for the biosynthesis of PGD2 and its dehydration end product, 15d-PGJ2. Because prolonged (48 h) application of high fluid shear stimulates 15d-PGJ2 release (14, 15), we wished to determine the effects of L-PGDS knockdown on shear-induced MMP-9 expression. To this end, experiments were carried out using T/C-28a2 chondrocytes transfected with a siRNA oligonucleotide sequence specific for L-PGDS or scramble control before exposure to shear stress for 48 h. This genetic intervention suppresses L-PGDS mRNA and protein expression (Fig. 3A) and 15d-PGJ-2 release (14) in sheared T/C-28a2 chondrocytes relative to cells transfected with a scramble siRNA. Our data reveal that L-PGDS depletion reverses the suppression of MMP-9 expression and activity in T/C-28a2 chondrocytes after prolonged (48 h) shear exposure (Fig. 3B). Specifically, L-PDGS knockdown resulted in a ∼2-fold up-regulation of MMP-9 transcript levels in shear-activated chondrocytes relative to static controls (Fig. 3B). Most importantly, incubation of L-PGDS-depleted chondrocytes with a monoclonal neutralizing anti-IL-1β antibody reduced MMP-9 expression and activity levels down to base-line static controls (Fig. 3B).
FIGURE 3.

Shear-induced IL-1β and 15d-PGJ2 antagonistically regulate the MMP-9 synthesis in human T/C-28a2 chondrocytes. T/C-28a2 cells were subjected to fluid shear stress (20 dynes/cm2) or static conditions (0 dyne/cm2) for 48 h (A and B) or 6 h (C–H). In select experiments, T/C-28a2 chondrocytes were transfected with a siRNA oligonucleotide sequence specific for L-PGDS before being exposed to high fluid shear in the absence or presence of an IL-1β-neutralizing antibody (A and B). In separate experiments, T/C-28a2 chondrocytes were transfected with a plasmid containing the cDNA of L-PGDS or the empty vector prior to shear exposure in the absence or presence of IL-1β (100 ng/ml) (C and D). In distinct experiments, T/C-28a2 chondrocytes were transfected with an siRNA oligonucleotide sequence specific for mPGES-1 (E and G) before being exposed to high fluid shear or treated with the EP2 receptor antagonist AH6809 (3 μm) or the EP3 receptor agonist sulprostone (1 μm) in high fluid shear exposure (F and H). MMP-9 mRNA and activity levels were determined by qRT-PCR and zymography, respectively (B, D, E (right panel), and F). GAPDH and MMP-2 served as internal controls. L-PGDS (A and C) and mPGES-1 (E, left panel) protein expression is shown by immunoblotting using specific Abs. β-Actin was probed as the loading control. These gels are representative of three independent experiments, all revealing similar results. The intensity of bands was quantified relative to β-actin for each treatment using the Bio-Rad gel image system. G and H, IL-1β secretion was determined using an IL-1β enzyme immunoassay kit. Data represent the mean ± S.E. of three independent experiments. *, p < 0.05 as compared with static control; ▴, p < 0.05 with respect to shear stress treatment; §, p < 0.05 with respect to L-PGDS siRNA knockdown or L-PGDS ectopic overexpression samples.
Because high shear stress represses 15d-PGJ2 release at short exposure times, experiments were performed using T/C-28a2 cells transfected with a plasmid containing the cDNA of L-PGDS prior to their shear exposure for 6 h. As shown in Fig. 3, C and D, this genetic intervention not only markedly induced L-PGDS mRNA and protein levels but also blocked shear-induced MMP-9 up-regulation. Moreover, incubation of T/C-28a2 cells overexpressing L-PGDS with exogenous IL-1β restored MMP-9 synthesis to the levels of sheared controls (Fig. 3D). Collectively, these data illustrate the antagonistic actions of IL-1β and 15d-PGJ2 in the regulation of MMP-9 expression.
Shear-induced MMP-9 Expression Proceeds via a PGE2/EP2/EP3-dependent IL-1β Induction Mechanism
In view of our recent observations showing that shear stress induces the sequential induction of mPGES-1 and L-PGDS in human chondrocytes (14), we evaluated the potential role of mPGES-1 in MMP-9 regulation in human T/C-28a2 chondrocytes. Given the up-regulation of mPGES-1 expression in sheared chondrocytes at short shear exposure times (14), experiments were performed using T/C-28a2 cells transfected with an siRNA oligonucleotide sequence specific for mPGES-1 before their exposure to high fluid shear for 6 h. The efficacy of this genetic intervention is verified at the mRNA and protein levels (Fig. 3E, left panel). mPGES-1 knockdown inhibited the shear-induced MMP-9 up-regulation at 6 h without affecting the MMP-2 basal expression levels (Fig. 3E, right panel). Because mPGES-1 is responsible for the biosynthesis of PGE2 in chondrocytes, we next evaluated the effects of PGE2 receptors on shear-induced MMP-9 regulation in T/C-28a2 chondrocytes. We have previously reported that: (a) T/C-28a2 chondrocytes express EP2, EP3, and very low levels of EP4 but lack EP1 receptors; and (b) the application of high fluid shear concurrently up-regulates EP2 and down-regulates EP3 expression at both the mRNA and protein levels in T/C-28a2 chondrocytes (13). In light of these observations, we investigated the effects of an EP2 receptor antagonist, AH6809, and an EP3 receptor agonist, sulprostone, on shear-induced MMP-9 expression. Incubation of T/C-28a2 chondrocytes with AH6809 (3 μm) or sulprostone (1 μm) inhibits shear-induced MMP-9 mRNA and activity levels in T/C-28a2 chondrocytes (Fig. 3F). Furthermore, selective knockdown of mPGES-1 or treatment with the EP2 receptor antagonist AH6809 or the EP3 receptor agonist sulprostone abrogates the shear-induced IL-1β secretion by T/C-28a2 chondrocytes (Fig. 3, G and H). However, these interventions failed to affect 15d-PGJ2 production at long shear exposure times (supplemental Fig. S1). Therefore, we concluded that shear-induced MMP-9 expression proceeds via a PGE2-/EP2-/EP3-dependent IL-1β induction mechanism.
IL-1β Regulates MMP-9 Expression in Human T/C-28a2 Chondrocytes via PI3K-, ERK1/2-, and JNK-dependent Pathways
We next aimed to elucidate the signaling cascade of MMP-9 synthesis in sheared chondrocytes. In view of the critical roles of IL-1β and 15d-PGJ2 in shear-induced MMP-9 regulation, we first determined the signaling pathways modulated by exogenous IL-1β and 15d-PGJ2. In light of our recent observations showing that the PI3K, PKA, and ERK1/2 pathways are suppressed by 15d-PGJ2 in human chondrocytes (14), we evaluated their potential regulation in response to exogenously added IL-1β. IL-1β (100 ng/ml) rapidly stimulates the phosphorylation of Akt (Ser-473) without affecting total Akt levels in human T/C-28a2 chondrocytes (Fig. 4A). Elevated p-Akt levels were detected within 10 min of IL-1β stimulation, and these levels remained elevated for 48 h (Fig. 4A), thereby suggesting that IL-1β induces pronounced and sustained PI3K activity. Treatment of T/C-28a2 chondrocytes with the specific PI3K inhibitor LY294002 (10 μm) or wortmannin (1 μm) abolished IL-1β-induced Akt phosphorylation (Fig. 4B) and MMP-9 mRNA and activity levels (Fig. 4C). On the other hand, inhibition of the PKA pathway using H89 (10 μm) (13, 20) failed to modulate MMP-9 expression in IL-1β-activated T/C-28a2 chondrocytes (data not shown).
FIGURE 4.

IL-1β induces MMP-9 synthesis in human T/C-28a2 chondrocytes via PI3K-, ERK1/2-, and JNK- dependent pathways. T/C-28a2 cells were treated with IL-1β (100 ng/ml) for the indicated time intervals (A). In selected experiments, T/C-28a2 cells were incubated with the ERK1/2 inhibitor PD98059 (20 μm) or U0126 (10 μm), the PI3K/Akt inhibitor LY294002 (10 μm) or wortmannin (1 μm), the JNK inhibitor SP600125 (10 μm or 20 μm), or the p38 inhibitor SB203580 (5 μm or 10 μm) in the absence or presence of IL-1β (100 ng/ml) for 48 h (B–D). In distinct experiments, cells were transfected with a siRNA oligonucleotide sequence specific for Akt, ERK1/2, p38, or c-Jun in the absence or presence of IL-1β (100 ng/ml) for 48 h (E and F). Phosphorylated Akt (Ser-473), ERK1/2 (Thr-202/Tyr-204), and c-Jun (Ser-63) and total Akt, ERK1/2, and c-Jun protein expression are shown by immunoblotting using specific Abs. Equal loading in each lane is ensured by the similar intensities of β-actin (A, B, and E). The intensity of bands was quantified relative to β-actin for each treatment using the Bio-Rad gel image system. MMP-9 mRNA expression and activity levels were determined by qRT-PCR and zymography, respectively (C, D, and F). GAPDH and MMP-2 served as internal controls in qRT-PCR and zymography assays, respectively. These gels are representative of three independent experiments, all revealing similar results. Data represent the mean ± S.E. of three independent experiments. *, p < 0.05 as compared with the control; ▴, p < 0.05 with respect to IL-1β treatment.
The MAPK family consists of three members: ERK, JNK, and p38-MAPK (27). We thus evaluated the potential contributions of these pathways to IL-1β-induced MMP-9 expression. Exogenous IL-1β induces ERK1/2 phosphorylation (Thr-202/Tyr-204) in T/C-28a2 chondrocytes, which is maximal at the 2 h time point and remains elevated at 48 h (Fig. 4A). Incubation of T/C-28a2 cells, with either the specific ERK1 inhibitor PD98058 (20 μm) or the cell-permeable ERK1/2 inhibitor U0126 (10 μm) (14, 21), markedly represses p-ERK1/2 phosphorylation (Fig. 4B) and the induction of MMP-9 mRNA and activity levels in IL-1β-treated cells (Fig. 4C). Similarly, exogenously added IL-1β induces a rapid and sustained phosphorylation of c-Jun (Ser-63) without affecting total c-Jun expression levels in human chondrocytes (Fig. 4A). The JNK inhibitor SP600125 (10 or 20 μm) attenuates c-Jun phosphorylation (Fig. 4B) and abrogates MMP-9 synthesis in IL-1β-treated chondrocytes (Fig. 4D). In distinct contrast, the p38 inhibitor SB203580 (5 μm or 10 μm) fails to alter MMP-9 expression (Fig. 4D). Given the rather non-selective nature of the aforementioned pharmacological inhibitors, experiments were carried out to verify the involvement of the PI3K, ERK1/2, and JNK pathways in shear-induced MMP-9 expression by transfecting human T/C-28a2 chondrocytes with an siRNA oligonucleotide sequence specific for Akt, ERK1/2, or c-Jun prior to IL-1β treatment for 48 h. The efficacy of these genetic interventions was verified at the protein level (Fig. 4E). Akt, ERK1/2, or c-Jun, but not p38, knockdown inhibited IL-1β-induced MMP-9 mRNA and activity levels (Fig. 4F), thereby confirming the critical roles of PI3K, ERK1/2 and JNK (but not p38) pathways in this process.
In contrast to the effects of exogenous IL-1β, we recently reported that 15d-PGJ2 inhibits shear-induced phosphorylation of Akt and ERK1/2 (14, 21) in human T/C-28a2 chondrocytes. We demonstrate herein that exogenous 15d-PGJ2 (2 μm) markedly suppresses c-Jun expression in both shear-activated and static control human chondrocytes (Fig. 5, A, B, D, and E) and further diminishes MMP-9 mRNA and activity levels (Fig. 5, C and F) after 6 and 48 h of shear activation.
FIGURE 5.

15d-PGJ2 treatment reverses shear-induced c-Jun and MMP-9 expression in human T/C-28a2 chondrocytes. T/C-28a2 chondrocytes were subjected to shear stress (20 dynes/cm2) or static conditions (0 dyne/cm2) in the absence or presence of 15d-PGJ2 (2 μm) for 6 h (A–C) or 48 h (D–F). c-Jun protein expression is shown by immunoblotting using specific Abs (A, B, D, and E). β-Actin served as the loading control. The intensity of bands was quantified relative to β-actin for each treatment using the Bio-Rad gel image system. MMP-9 mRNA expression and activity levels were determined by qRT-PCR and zymography, respectively (C and F). GAPDH and MMP-2 served as internal controls in qRT-PCR and zymography assays, respectively. These gels are representative of three independent experiments, all revealing similar results. Data represent the mean ± S.E. of three independent experiments. *, p < 0.05 as compared with the control; ▴, p < 0.05 with respect to shear stress treatment.
We next examined the potential contributions of the PI3K, PKA, ERK1/2, JNK and p38 pathways to shear-induced MMP-9 expression. Continuous application of high fluid shear stress to T/C-28a2 chondrocytes resulted in a pronounced increase in the phosphorylation levels of Akt at Ser-473 and ERK1/2 at Thr-202/Tyr-204 at 6 h (Fig. 6A), which was negated at 48 h (14). Of note, the total Akt and ERK1/2 levels remain unaffected by shear stress (Fig. 6A). Treatment of T/C-28a2 cells with the selective PI3K inhibitors LY294002 (10 μm) and wortmannin (1 μm) repressed Akt phosphorylation (Fig. 6A) and the induction of MMP-9 mRNA and activity levels in shear-activated cells (Fig. 6B). Similarly, the specific ERK1 inhibitor PD98058 (20 μm) or the cell-permeable ERK1/2 inhibitor U0126 (10 μm) inhibited ERK1/2 phosphorylation (Fig. 6A) and MMP-9 up-regulation in sheared chondrocytes (Fig. 6B). Exposure of T/C-28a2 chondrocytes to high shear stress for 6 h also induced c-Jun activity (Fig. 6A). Incubation of T/C-28a2 chondrocytes with the JNK inhibitor SP600125 (10 μm or 20 μm) abolished MMP-9 induction in response to high fluid shear stress stimulation (Fig. 6B). This pharmacological intervention suppressed c-Jun phosphorylation at Ser-63 while also tending to modestly suppress Akt and ERK1/2 phosphorylation (Fig. 6A). On the other hand, the p38 inhibitor SB203580 (5 or 10 μm) and the PKA inhibitor H89 (10 μm) failed to modulate MMP-9 synthesis in shear-activated chondrocytes (Fig. 6B and data not shown). To exclude any potential nonspecific effects of the pharmacological inhibitors, experiments were carried out by transfecting T/C-28a2 cells with an siRNA oligonucleotide sequence specific for Akt, ERK1/2, p38, or c-Jun before exposing cells to high fluid shear for 6 h. The efficacy of these genetic interventions was verified at the protein level (Fig. 6C). Akt, ERK1/2, or c-Jun, but not p38, knockdown inhibited shear-induced MMP-9 mRNA and activity levels (Fig. 6D), thereby confirming the critical roles of PI3K, ERK1/2, and JNK (but not p38) pathways in this process.
FIGURE 6.

Involvement of PI3K, ERK1/2, and JNK signaling pathways in MMP-9 synthesis in shear-activated human chondrocytes. T/C-28a2 cells were subjected to fluid shear stress (20 dynes/cm2) or static conditions (0 dyne/cm2) in the absence or presence of the ERK1/2 inhibitor PD98059 (20 μm) or U0126 (10 μm), the PI3K/Akt inhibitor LY294002 (10 μm) or wortmannin (1 μm), the JNK inhibitor SP600125 (10 μm or 20 μm), or the p38 inhibitor SB203580 (5 or 10 μm) for 6 h (A and B). In distinct experiments, cells were transfected with an siRNA oligonucleotide sequence specific for Akt, ERK1/2, p38, or c-Jun before being exposed to high fluid shear for 6 h (C and D). Phosphorylated Akt (Ser-473), ERK1/2 (Thr-202/Tyr-204), and c-Jun (Ser-63) and total Akt, ERK1/2, and c-Jun protein expression are shown by immunoblotting using specific Abs. Equal loading in each lane is ensured by the similar intensities of β-actin (A and C). The intensity of bands was quantified relative to β-actin for each treatment using the Bio-Rad gel image system. MMP-9 mRNA expression and activity levels were determined by qRT-PCR and zymography, respectively (B and D). GAPDH and MMP-2 served as internal controls in qRT-PCR and zymography assays, respectively. These gels are representative of three independent experiments, all revealing similar results. Data represent the mean ± S.E. of three independent experiments. *, p < 0.05 as compared with the control; ▴, p < 0.05 with respect to shear stress treatment.
To further document that PI3K/Akt, ERK1/2, and JNK constitute downstream targets of endogenous IL-1β and 15d-PGJ2 in sheared chondrocytes, experiments were carried out to alter L-PGDS expression in the presence or absence of a monoclonal neutralizing antibody specific for IL-1β. Our data reveal that L-PGDS depletion stimulates Akt, ERK1/2, and c-Jun phosphorylation (Fig. 7A) and induces MMP-9 mRNA and activity levels (Fig. 3B) in T/C-28a2 chondrocytes subjected to high shear stress for 48 h. The addition of an IL-1β-neutralizing antibody just before the onset of shear application diminishes the phosphorylation of Akt, ERK1/2, and c-Jun (Fig. 7A) and markedly reduces MMP-9 expression (Fig. 3B) in L-PGDS-depleted T/C-28a2 chondrocytes. On the other hand, ectopic expression of L-PGDS suppresses Akt, ERK1/2, and c-Jun phosphorylation (Fig. 7B) and diminishes MMP-9 mRNA and activity levels (Fig. 3D) in T/C-28a2 chondrocytes exposed to high fluid shear for 6 h. The addition of exogenous IL-1β (100 ng/ml) just before the onset of shear application reverses the inhibition of phosphorylation of Akt, ERK1/2, and c-Jun (Fig. 7B) and markedly induces MMP-9 expression (Fig. 3D) in L-PGDS-overexpressed T/C-28a2 chondrocytes. In view of our observations showing that shear-induced IL-1β synthesis precedes MMP-9 expression in a PGE2/EP2/EP3-dependent manner (Fig. 3, E–H), we further examined the potential role of the PGE2/EP2/EP3 signaling cascade in regulating PI3K, ERK1/2, and JNK pathways. Our data reveal that shear-induced Akt, ERK1/2, and c-Jun phosphorylation are significantly suppressed in mPGES-1-depleted (Fig. 7C) and AH6809 (EP2 receptor antagonist)- or sulprostone (EP3 receptor agonist)-treated (Fig. 7D) T/C-28a2 chondrocytes. Collectively, our data suggest that shear-induced MMP-9 expression is regulated via the antagonistic actions of endogenously released IL-1β and 15d-PGJ2 and their downstream effectors, the PI3K/Akt, ERK1/2, and JNK-dependent signaling pathways.
FIGURE 7.

Shear-induced IL-1β and 15d-PGJ2 antagonistically regulate the activity of PI3K, ERK1/2, and JNK pathways in human T/C-28a2 chondrocytes. T/C-28a2 cells were subjected to fluid shear stress (20 dynes/cm2) or static conditions (0 dyne/cm2) for 48 h (A) or 6 h (B–D). In select experiments, T/C-28a2 chondrocytes were transfected with an siRNA oligonucleotide sequence specific for L-PGDS before exposure to high fluid shear in the absence or presence of an IL-1β-neutralizing antibody (A). In separate experiments, T/C-28a2 chondrocytes were transfected with a plasmid containing the cDNA of L-PGDS or the empty vector prior to shear exposure in the absence or presence of IL-1β (100 ng/ml) (B). In distinct experiments, T/C-28a2 chondrocytes were transfected with an siRNA oligonucleotide sequence specific for mPGES-1 (C) before being exposed to high fluid shear or treated with the EP2 receptor antagonist AH6809 (3 μm) or the EP3 receptor agonist sulprostone (1 μm) under high fluid shear exposure (D). Phosphorylated Akt (Ser-473), ERK1/2 (Thr-202/Tyr-204), and c-Jun (Ser-63) and total Akt, ERK1/2, and c-Jun protein expression are shown by immunoblotting using specific Abs. Equal loading in each lane is ensured by the similar intensities of β-actin. These gels are representative of three independent experiments, all revealing similar results. The intensity of bands was quantified relative to β-actin for each treatment using the Bio-Rad gel image system. Data represent the mean ± S.E. of three independent experiments. *, p < 0.05 as compared with static control; ▴, p < 0.05 as compared with shear stress treatment; §, p < 0.05 with respect to L-PGDS siRNA knockdown or L-PGDS ectopic overexpression samples.
PPARγ Is Involved in MMP-9 Regulation in Human Chondrocytes
In view of our data showing that 15d-PGJ2 suppresses MMP-9 expression, we next examined whether 15d-PGJ2 exerts its effects via its primary receptor PPARγ (28). As shown in Fig. 8A, exogenous IL-1β stimulates the phosphorylation of PPARγ without affecting total PPARγ protein levels in human T/C-28a2 chondrocytes. PPARγ phosphorylation in IL-1β-activated chondrocytes is abolished by inhibiting the ERK1/2 pathway using the pharmacological inhibitors U0126 and PD98059 (Fig. 8A). However, inhibition of the PI3K, JNK, or p38 pathway failed to alter the extent of PPARγ phosphorylation in IL-1β-stimulated T/C-28a2 cells (Fig. 8A). Interestingly, treatment of human T/C-28a2 cells with the PPARγ-specific agonist GW1929 (10 μm) significantly attenuated MMP-9 mRNA and activity levels and decreased p65 phosphorylation at both Ser-536 and Ser-276 (Fig. 8, C and D) without affecting the phosphorylation of Akt, ERK1/2, or c-Jun (Fig. 8B). On the other hand, treatment of human T/C-28a2 cells with the PPARγ-specific antagonist GW9662 (1 μm) significantly augmented MMP-9 mRNA and activity levels and increased p65 phosphorylation at both sites (Fig. 8, C and D) without affecting the phosphorylation of Akt, ERK1/2, or c-Jun (Fig. 8B). Of note, neither GW9662 nor GW1929 altered the total levels of p65 (Fig. 8D). Taken together, these data suggest that PPARγ is downstream of ERK1/2 and is partially involved in the modulation of MMP-9 expression in human chondrocytes.
FIGURE 8.

Involvement of PPARγ in MMP-9 synthesis in shear-activated human T/C-28a2 chondrocytes. T/C-28a2 chondrocytes were treated with IL-1β (100 ng/ml) in the absence or presence of PI3K/Akt, ERK1/2, p38, or JNK inhibitors for 48 h (A). Phosphorylated PPARγ (Ser-82) is shown by immunoblotting using a specific antibody. Equal loading in each lane is ensured by the similar intensities of total PPARγ and β-actin. In select experiments, T/C-28a2 chondrocytes were treated with the PPARγ agonist GW1929 (10 μm) or the PPARγ antagonist GW9662 (1 μm) (B–D). Phosphorylated Akt (Ser-473), ERK1/2 (Thr-202/Tyr-204), c-Jun (Ser-63), p65 (Ser-536), and p65 (Ser-276) are shown by immunoblotting using specific Abs. Equal loading in each lane is ensured by the similar intensities of total Akt, ERK1/2, c-Jun, p65, and β-actin (B and D). The intensity of bands was quantified relative to β-actin for each treatment using the Bio-Rad gel image system. MMP-9 mRNA expression and activity levels were determined by qRT-PCR and zymography, respectively (C). GAPDH and MMP-2 served as internal controls in qRT-PCR and zymography assays, respectively. These gels are representative of three independent experiments, all revealing similar results. Data represent the mean ± S.E. of three independent experiments. *, p < 0.05 as compared with the control.
NF-κB p65 Subunit Is Involved in Shear-regulated MMP-9 Expression
We recently showed that exposure of human chondrocytes to high fluid shear transactivates the NF-κB p65 subunit via phosphorylation at Ser-276 and Ser-536 (13, 14). In light of prior work showing that MMP-9 expression is regulated by NF-κB in airway epithelial cells (16), we sought to determine whether shear-induced MMP-9 synthesis is also dependent on NF-κB. To identify the critical NF-κB binding sites on the mmp-9 promoter that regulate shear-induced MMP-9 synthesis, promoter analysis experiments were performed. First, T/C-28a2 chondrocytes were transiently transfected with a construct encompassing the 5′-flanking region of the human mmp-9 gene from −1723 to −6 bp (−1723/−6) (Fig. 9A) prior to their exposure to high fluid shear. Application of high shear stress for 6 h markedly increased (∼10-fold) the mmp-9 promoter activity in T/C-28a2 chondrocytes (Fig. 9A). A similar 10-fold up-regulation was noted upon transfection of cells with plasmids deleted from −1723 to −1017 bp (−1017/−6) or from −1723 to −604 bp (−604/6) or from −1723 to −553 bp (−553/−6), thereby suggesting that the DNA region between −1723 and −553 bp upstream of the transcriptional start site is not critical to the induction of shear-mediated mmp-9 promoter activity (Fig. 9A). However, subsequent deletion from −553 to −324 bp (−324/−6 bp) markedly suppressed the luciferase activity (Fig. 9A). Bioinformatics analysis of the consensus sequence in (−553/−324) region revealed the presence of an NF-κB site situated at −332/−323 bp, which may be responsible for shear-induced MMP-9 synthesis. Indeed, introduction of a point mutation into the aforementioned NF-κB site resulted in a marked decrease in shear-induced luciferase activity relative to the reported wild type (Fig. 9B).
FIGURE 9.

Fluid shear induces the binding of the NF-κB p65 subunit to the mmp-9 promoter in human chondrocytes. T/C-28a2 cells were subjected to high fluid shear (20 dynes/cm2) or static conditions (0 dyne/cm2) for 6 h (A, B, C (left two lanes), and D) or 48 h (C (right two lanes) and E). In select experiments, T/C-28a2 cells were subjected to fluid shear stress (20 dynes/cm2) for 6 h in the presence or absence of 15d-PGJ2 (2 μm), the EP2 receptor antagonist AH6809 (3 μm), the EP3 receptor agonist sulprostone (1 μm), the PI3K/Akt inhibitor LY294002 (10 μm), the ERK1/2 inhibitor U0126 (10 μm), or the JNK inhibitor SP600125 (20 μm) for 6 h (D). In separate experiments, T/C-28a2 chondrocytes were treated with 15d-PGJ2 (2 μm), the PI3K/Akt inhibitor LY294002 (10 μm), the ERK1/2 inhibitor U0126 (10 μm), or the JNK inhibitor SP600125 (20 μm) in the absence or presence of IL-1β (100 ng/ml) for 48 h (E). Cross-linked chromatin was immunoprecipitated using an anti-p65 antibody. In ChIP assays, the anti-RNA polymerase II antibody was used as a positive control. DNA purified from both immunoprecipitated (IP) and preimmune (Input) specimens were subjected to qPCR amplification using primers for MMP-9 promoter genes. All experiments are representative of three independent experiments, all revealing similar results. Data represent the mean ± S.E. of three independent experiments. *, p < 0.05 as compared with static or vehicle treatment control; ▴, p < 0.05 with respect to shear stress or IL-1β treatment specimens.
ChIP assays were performed to demonstrate the binding of phosphorylated NF-κB to its putative site on the mmp-9 promoter. As shown in Fig. 9C, the binding of the NF-κB p65 subunit to the mmp-9 promoter markedly increased following exposure of T/C-28a2 chondrocytes to high fluid shear for 6 h. Interestingly, this binding was reduced after prolonged (48 h) shear exposure times. It is noteworthy that T/C-28a2 chondrocytes treated with exogenous 15d-PGJ2 (2 μm) displayed reduced shear-induced binding of NF-κB p65 subunit to mmp-9 promoter at 6 h (Fig. 9D). Similar observations were made upon treatment of T/C-28a2 cells with the EP2 receptor antagonist AH6809 (3 μm), the EP3 receptor agonist sulprostone (1 μm), the PI3K/Akt inhibitor LY294002 (10 μm), the ERK1/2 inhibitor U0126 (10 μm), and the JNK inhibitor SP600125 (20 μm) (Fig. 9D). Similar data were also obtained using IL-1β-stimulated chondrocytes (Fig. 9E). Collectively, these data support the notion that NF-κB p65 subunit functions as a critical transcriptional factor to mediate shear-regulated MMP-9 expression.
DISCUSSION
Excessive mechanical loading can directly damage the articular cartilage, adversely affect chondrocyte function, and precipitate OA (2). MMP-9 up-regulation has been detected in the early stage of OA-affected cartilage (11). In view of the involvement of MMP-9 in cartilage erosion (7, 8) and the role of mechanical forces in the pathogenesis and progression of OA (2), here we have delineated the signaling pathway of MMP-9 regulation in mechanically stimulated human T/C-28a2 chondrocytes. We demonstrate that high fluid shear induces IL-1β and 15d-PGJ2 synthesis, which antagonistically regulate MMP-9 expression via PI3K-, ERK1/2-, ERK1/2-PPARγ-, and JNK-dependent NF-κB-activating pathways in human T/C-28a2 chondrocytes (Fig. 10).
FIGURE 10.
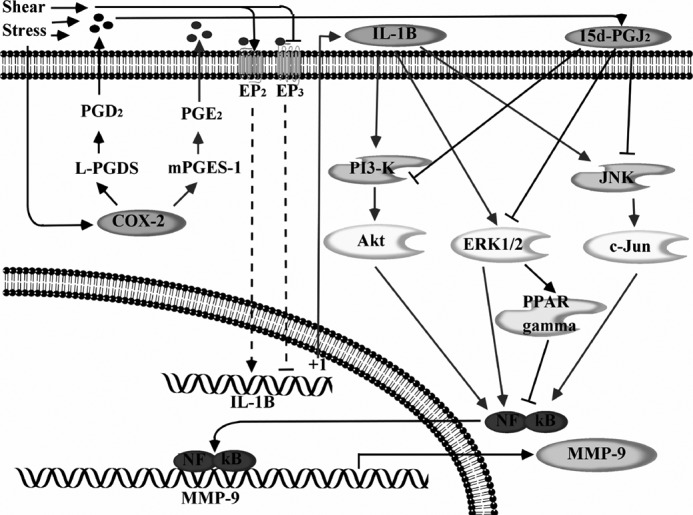
Proposed cascade of signaling events regulating the temporal synthesis of MMP-9 in human chondrocytes stimulated with high shear stress. High fluid shear stress induces the rapid and sustained synthesis of IL-1β in human chondrocytes. 15d-PGJ2 synthesis is detected only after prolonged exposure of human chondrocytes to high fluid shear. IL-1β and 15d-PGJ2 exert antagonistic effects on PI3K-, ERK1/2-, ERK1/2-PPARγ-, and JNK-dependent NF-κB p65 subunit activation, thereby leading to the temporal regulation of MMP-9 expression in chondrocytes.
MMP-9 is tightly regulated and kept at basal levels under physiological conditions. Conflicting reports reveal that MMP-9 is either markedly (7, 8) or marginally (9) up-regulated or even significantly suppressed (10) in osteoarthritic relative to healthy articular cartilage. These disparate results may be reconciled by findings showing that MMP-9 expression varies not only with the zone of the chondrocyte in the cartilage but also with the stage of OA (11). It is reported that MMP-9 is markedly elevated at early but not late stages of OA relative to normal cartilage (11). In accord with these in vivo observations, our in vitro data reveal that high fluid shear induces a pronounced up-regulation of MMP-9 expression in human chondrocytes at short shear exposure times, but this expression falls below basal (control) levels after prolonged shear exposure.
Prior work has shown that proinflammatory cytokines such as tumor necrosis factor-α, IL-6, and IL-1β direct MMP-mediated digestion of the cartilage matrix in OA (29). In agreement with these observations, exogenous IL-1β is capable of up-regulating MMP-9 expression in human OA chondrocytes and human chondrosarcoma cells (6, 30). Of note, Söder et al. (7) report that low concentrations of exogenous IL-1β (0.01–10 ng/ml) are not sufficient to stimulate MMP-9 expression or activity in human OA chondrocytes. These observations (7) are in accord with our data showing that a threshold concentration of 50 ng/ml exogenous IL-1β is needed to induce MMP-9 production in human T/C-28a2 chondrocytes. Furthermore, our study has revealed the key role of endogenous IL-1β induced by high fluid shear stress (20 dynes/cm2) in mediating MMP-9 expression at short shear exposure times (3–6 h) in human chondrocytes.
The intracellular signaling pathways that transduce mechanical stimuli in human chondrocytes may involve the activation of L-PGDS and release of endogenous PGD2 and its metabolite, 15d-PGJ2 (15). Interestingly, 15d-PGJ2 has been reported to counteract the induction of MMP1 and MMP13 in cytokine-activated chondrocytes (31, 32). In view of our recent observations showing that high fluid shear suppresses 15d-PGJ2 production by human chondrocytes at short exposure times (2–6 h), whereas it stimulates 15d-PGJ2 release at long (48 h) times (14, 15), we examined the potential interplay between 15d-PGJ2 and IL-1β on MMP-9 synthesis. Our work reveals that both exogenous and endogenous 15d-PGJ2 suppresses MMP-9 expression. In concert with our data, Hashimoto et al. (25) and Marx et al. (26) found that 15d-PGJ2 represses MMP-9 activity in pancreatic cancer cells and human smooth muscle cells. It has also been reported that 15d-PGJ2 is capable of mitigating MMP-9 activity in diabetic placental tissues (33). Along with these observations, 15d-PGJ2 inhibits the IL-1β-induced inflammatory responses in rat chondrocytes via a PPARγ-independent pathway (34). Similar to the aforementioned results, we report herein the antagonistic actions of 15d-PGJ2 and IL-1β on MMP-9 synthesis; 15d-PGJ2 appears to exert its effects on MMP-9 inhibition partially via ERK1/2-regulated PPARγ. In contrast, 15d-PGJ2 has been reported to inhibit IL-1β-induced inducible nitric-oxide synthase (iNOS) and MMP-13 expression in human chondrocytes in a PPARγ-dependent manner (31). These studies suggest that PPARγ ligands exert two opposing effects on gene expression: 1) their direct binding transactivates PPARγ, which results in gene expression; and 2) they simultaneously regulate the ERK1/2-mediated PPARγ phosphorylation, which suppresses PPARγ-DNA binding and facilitates the interactions of PPARγ with p65, thereby leading to the inhibition of NF-κB and gene expression (35). In our case, 15d-PGJ2 suppresses MMP-9 mRNA expression and activity levels in sheared chondrocytes not as a natural PPARγ ligand but through an ERK1/2-regulated PPARγ pathway.
Prior studies have identified the three major MAPKs (ERK1/2, JNK, and p38) (36, 37) and PI3K (37) in transmitting IL-1β-dependent signaling. In light of these findings (36, 37), we evaluated the potential contributions of these signaling pathways to shear-induced MMP-9 synthesis. Shear stress rapidly up-regulates the activities of PI3K/Akt, ERK1/2, and JNK/c-Jun (but not p38) pathways in human T/C-28a2 chondrocytes via an IL-1β-dependent mechanism. This activation pattern is in line with previously published data showing that IL-1β induces MMP-9 expression via a PI3K (38)-, ERK1/2 (39)-, and JNK (40)-dependent pathway in rat brain astrocytes and rat cardiac fibroblasts. Interestingly, IL-1β has been reported to up-regulate MMP-9 expression via a p38-/ERK1/2-/JNK-dependent pathway in human tracheal smooth muscle cells (41). The lack of involvement of p38 in shear-induced MMP-9 expression in human chondrocytes as opposed to IL-1β-treated smooth muscle cells may be due to cell type differences. Both exogenous and endogenous 15d-PGJ2 suppress the activation of PI3K, ERK1/2, and JNK pathways in human chondrocytes. Consistent with our observations, previous studies have shown that 15d-PGJ2 inhibits JNK and c-Jun activation in human colon carcinoma and human umbilical vein endothelial cells (42, 43). Interestingly, 15d-PGJ2 interferes with the DNA binding activity of AP-1 by forming a covalent adduct with c-Jun (44).
In light of our prior work showing that exposure of human chondrocytes to high fluid shear transactivates the NF-κB p65 subunit via phosphorylation at Ser-276 and Ser-536 (13, 14), and given that the expression of MMP-9 is dependent on NF-κB, we have established that the canonical NF-κB pathway plays an essential role in shear-induced MMP-9 expression. This finding is in concert with an earlier report that suggests a key role for NF-κB in the regulation of MMP-9 expression induced by different inflammatory mediators in chondrocytes (45, 46). Through the use of pharmacological inhibitors, promoter constructs, and ChIP assays, we demonstrated here the functional role of the NF-κB p65 subunit in shear-induced MMP-9 expression in human chondrocytes.
In summary, we have elucidated the signaling pathway by which high fluid shear mediates the temporal regulation of MMP-9 expression in human chondrocytes. Specifically, high shear stress induces the rapid and sustained synthesis of IL-1β; 15d-PGJ2 synthesis is detected only after prolonged exposure of human chondrocytes to high fluid shear. IL-1β and 15d-PGJ2 exert antagonistic effects on PI3K-, ERK1/2-PPARγ-, and JNK-dependent NF-κB activation, thereby leading to the temporal regulation of MMP-9 expression in chondrocytes. These findings provide new insights into the mechanism of MMP-9 regulation in shear-activated human chondrocytes.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 AR053358 from NIAMS.

This article contains supplemental Fig. S1.
- OA
- osteoarthritis
- MMP
- matrix metalloproteinase
- PG
- prostaglandin
- 15d-PGJ2
- 15-deoxy-Δ12,14-prostaglandin J2
- L-PGDS
- lipocalin-type prostaglandin D synthase
- qRT-PCR
- quantitative real-time PCR
- PPARγ
- peroxisome proliferator-activated receptor γ.
REFERENCES
- 1. Carter D. R., Beaupré G. S., Wong M., Smith R. L., Andriacchi T. P., Schurman D. J. (2004) The mechanobiology of articular cartilage development and degeneration. Clin. Orthop. Relat. Res. 427, (suppl.) S69–S77 [DOI] [PubMed] [Google Scholar]
- 2. Buckwalter J. A., Martin J. A., Brown T. D. (2006) Perspectives on chondrocyte mechanobiology and osteoarthritis. Biorheology 43, 603–609 [PubMed] [Google Scholar]
- 3. Zhu F., Wang P., Lee N. H., Goldring M. B., Konstantopoulos K. (2010) Prolonged application of high fluid shear to chondrocytes recapitulates gene expression profiles associated with osteoarthritis. PLoS ONE 5, e15174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goldring M. B. (2000) Osteoarthritis and cartilage: the role of cytokines. Curr. Rheumatol. Rep. 2, 459–465 [DOI] [PubMed] [Google Scholar]
- 5. Daheshia M., Yao J. Q. (2008) The interleukin 1β pathway in the pathogenesis of osteoarthritis. J. Rheumatol. 35, 2306–2312 [DOI] [PubMed] [Google Scholar]
- 6. Akhtar N., Miller M. J., Haqqi T. M. (2011) Effect of a herbal-leucine mix on the IL-1β-induced cartilage degradation and inflammatory gene expression in human chondrocytes. BMC Complement. Altern. Med. 11, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Söder S., Roach H. I., Oehler S., Bau B., Haag J., Aigner T. (2006) MMP-9/gelatinase B is a gene product of human adult articular chondrocytes and increased in osteoarthritic cartilage. Clin. Exp. Rheumatol. 24, 302–304 [PubMed] [Google Scholar]
- 8. Tsuchiya K., Maloney W. J., Vu T., Hoffman A. R., Huie P., Sibley R., Schurman D. J., Smith R. L. (1997) Osteoarthritis: differential expression of matrix metalloproteinase-9 mRNA in nonfibrillated and fibrillated cartilage. J. Orthop. Res. 15, 94–100 [DOI] [PubMed] [Google Scholar]
- 9. Tetlow L. C., Adlam D. J., Woolley D. E. (2001) Matrix metalloproteinase and proinflammatory cytokine production by chondrocytes of human osteoarthritic cartilage: associations with degenerative changes. Arthritis Rheum. 44, 585–594 [DOI] [PubMed] [Google Scholar]
- 10. Naito K., Takahashi M., Kushida K., Suzuki M., Ohishi T., Miura M., Inoue T., Nagano A. (1999) Measurement of matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinases-1 (TIMP-1) in patients with knee osteoarthritis: comparison with generalized osteoarthritis. Rheumatology (Oxford) 38, 510–515 [DOI] [PubMed] [Google Scholar]
- 11. Freemont A. J., Hampson V., Tilman R., Goupille P., Taiwo Y., Hoyland J. A. (1997) Gene expression of matrix metalloproteinases 1, 3, and 9 by chondrocytes in osteoarthritic human knee articular cartilage is zone- and grade-specific. Ann. Rheum. Dis. 56, 542–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pelletier J. P., Martel-Pelletier J., Abramson S. B. (2001) Osteoarthritis, an inflammatory disease: potential implication for the selection of new therapeutic targets. Arthritis Rheum. 44, 1237–1247 [DOI] [PubMed] [Google Scholar]
- 13. Wang P., Zhu F., Lee N. H., Konstantopoulos K. (2010) Shear-induced interleukin-6 synthesis in chondrocytes: roles of E prostanoid (EP) 2 and EP3 in cAMP/protein kinase A- and PI3K/Akt-dependent NF-κB activation. J. Biol. Chem. 285, 24793–24804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang P., Zhu F., Tong Z., Konstantopoulos K. (2011) Response of chondrocytes to shear stress: antagonistic effects of the binding partners Toll-like receptor 4 and caveolin-1. FASEB J. 25, 3401–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu F., Wang P., Kontrogianni-Konstantopoulos A., Konstantopoulos K. (2010) Prostaglandin (PG)D(2) and 15-deoxy-Δ(12,14)-PGJ(2), but not PGE(2), mediate shear-induced chondrocyte apoptosis via protein kinase A-dependent regulation of Polo-like kinases. Cell Death Differ. 17, 1325–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tacon C. E., Wiehler S., Holden N. S., Newton R., Proud D., Leigh R. (2010) Human rhinovirus infection up-regulates MMP-9 production in airway epithelial cells via NF-κB. Am. J. Respir. Cell Mol. Biol. 43, 201–209 [DOI] [PubMed] [Google Scholar]
- 17. Abulencia J. P., Gaspard R., Healy Z. R., Gaarde W. A., Quackenbush J., Konstantopoulos K. (2003) Shear-induced cyclooxygenase-2 via a JNK2/c-Jun-dependent pathway regulates prostaglandin receptor expression in chondrocytic cells. J. Biol. Chem. 278, 28388–28394 [DOI] [PubMed] [Google Scholar]
- 18. Healy Z. R., Lee N. H., Gao X., Goldring M. B., Talalay P., Kensler T. W., Konstantopoulos K. (2005) Divergent responses of chondrocytes and endothelial cells to shear stress: cross-talk among COX-2, the phase 2 response, and apoptosis. Proc. Natl. Acad. Sci. U.S.A. 102, 14010–14015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Healy Z. R., Zhu F., Stull J. D., Konstantopoulos K. (2008) Elucidation of the signaling network of COX-2 induction in sheared chondrocytes: COX-2 is induced via a Rac/MEKK1/MKK7/JNK2/c-Jun-C/EBPβ-dependent pathway. Am. J. Physiol. Cell Physiol. 294, C1146–C1157 [DOI] [PubMed] [Google Scholar]
- 20. Wang P., Zhu F., Konstantopoulos K. (2010) Prostaglandin E2 induces interleukin-6 expression in human chondrocytes via cAMP/protein kinase A- and phosphatidylinositol 3-kinase-dependent NF-κB activation. Am. J. Physiol. Cell Physiol. 298, C1445–C1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang P., Zhu F., Konstantopoulos K. (2011) Interleukin-6 synthesis in human chondrocytes is regulated via the antagonistic actions of prostaglandin (PG)E2 and 15-deoxy-Δ(12,14)-PGJ2. PLoS ONE 6, e27630. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22. Hu D., Man Z., Wang P., Tan X., Wang X., Takaku S., Hyuga S., Sato T., Yao X., Yamagata S., Yamagata T. (2007) Ganglioside GD1a negatively regulates matrix metalloproteinase-9 expression in mouse FBJ cell lines at the transcriptional level. Connect. Tissue Res. 48, 198–205 [DOI] [PubMed] [Google Scholar]
- 23. Goldring M. B., Birkhead J. R., Suen L. F., Yamin R., Mizuno S., Glowacki J., Arbiser J. L., Apperley J. F. (1994) Interleukin-1β-modulated gene expression in immortalized human chondrocytes. J. Clin. Invest. 94, 2307–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martel-Pelletier J. (2004) Pathophysiology of osteoarthritis. Osteoarthr. Cartil. 12, Suppl. A, S31–S33 [DOI] [PubMed] [Google Scholar]
- 25. Hashimoto K., Ethridge R. T., Evers B. M. (2002) Peroxisome proliferator-activated receptor γ ligand inhibits cell growth and invasion of human pancreatic cancer cells. Int. J. Gastrointest. Cancer 32, 7–22 [DOI] [PubMed] [Google Scholar]
- 26. Marx N., Schönbeck U., Lazar M. A., Libby P., Plutzky J. (1998) Peroxisome proliferator-activated receptor γ activators inhibit gene expression and migration in human vascular smooth muscle cells. Circ. Res. 83, 1097–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wada T., Penninger J. M. (2004) Mitogen-activated protein kinases in apoptosis regulation. Oncogene 23, 2838–2849 [DOI] [PubMed] [Google Scholar]
- 28. Forman B. M., Tontonoz P., Chen J., Brun R. P., Spiegelman B. M., Evans R. M. (1995) 15-Deoxy-Δ12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ. Cell 83, 803–812 [DOI] [PubMed] [Google Scholar]
- 29. Pearle A. D., Warren R. F., Rodeo S. A. (2005) Basic science of articular cartilage and osteoarthritis. Clin. Sports Med. 24, 1–12 [DOI] [PubMed] [Google Scholar]
- 30. Lu Y. C., Jayakumar T., Duann Y. F., Chou Y. C., Hsieh C. Y., Yu S. Y., Sheu J. R., Hsiao G. (2011) Chondroprotective role of sesamol by inhibiting MMPs expression via retaining NF-κB signaling in activated SW1353 cells. J. Agric. Food Chem. 59, 4969–4978 [DOI] [PubMed] [Google Scholar]
- 31. Fahmi H., Di Battista J. A., Pelletier J. P., Mineau F., Ranger P., Martel-Pelletier J. (2001) Peroxisome proliferator-activated receptor γ activators inhibit interleukin-1β-induced nitric oxide and matrix metalloproteinase-13 production in human chondrocytes. Arthritis Rheum. 44, 595–607 [DOI] [PubMed] [Google Scholar]
- 32. Zayed N., Afif H., Chabane N., Mfuna-Endam L., Benderdour M., Martel-Pelletier J., Pelletier J. P., Motiani R. K., Trebak M., Duval N., Fahmi H. (2008) Inhibition of interleukin-1β-induced matrix metalloproteinases 1 and 13 production in human osteoarthritic chondrocytes by prostaglandin D2. Arthritis Rheum. 58, 3530–3540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pustovrh M. C., Capobianco E., Martínez N., Higa R., White V., Jawerbaum A. (2009) MMP/TIMP balance is modulated in vitro by 15dPGJ(2) in fetuses and placentas from diabetic rats. Eur. J. Clin. Investig. 39, 1082–1090 [DOI] [PubMed] [Google Scholar]
- 34. Boyault S., Bianchi A., Moulin D., Morin S., Francois M., Netter P., Terlain B., Bordji K. (2004) 15-Deoxy-Δ(12,14)-prostaglandin J(2) inhibits IL-1β-induced IKK enzymatic activity and IκBα degradation in rat chondrocytes through a PPARγ-independent pathway. FEBS Lett. 572, 33–40 [DOI] [PubMed] [Google Scholar]
- 35. Papageorgiou E., Pitulis N., Msaouel P., Lembessis P., Koutsilieris M. (2007) The non-genomic cross-talk between PPAR-γ ligands and ERK1/2 in cancer cell lines. Expert Opin. Ther. Targets 11, 1071–1085 [DOI] [PubMed] [Google Scholar]
- 36. Ahmed S., Wang N., Hafeez B. B., Cheruvu V. K., Haqqi T. M. (2005) Punica granatum L. extract inhibits IL-1β-induced expression of matrix metalloproteinases by inhibiting the activation of MAP kinases and NF-κB in human chondrocytes in vitro. J. Nutr. 135, 2096–2102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lin Y. C., Liang Y. C., Sheu M. T., Lin Y. C., Hsieh M. S., Chen T. F., Chen C. H. (2008) Chondroprotective effects of glucosamine involving the p38 MAPK and Akt signaling pathways. Rheumatol. Int. 28, 1009–1016 [DOI] [PubMed] [Google Scholar]
- 38. Wu C. Y., Hsieh H. L., Sun C. C., Tseng C. P., Yang C. M. (2008) IL-1β induces proMMP-9 expression via c-Src-dependent PDGFR/PI3K/Akt/p300 cascade in rat brain astrocytes. J. Neurochem. 105, 1499–1512 [DOI] [PubMed] [Google Scholar]
- 39. Xie Z., Singh M., Singh K. (2004) Differential regulation of matrix metalloproteinase-2 and -9 expression and activity in adult rat cardiac fibroblasts in response to interleukin-1β. J. Biol. Chem. 279, 39513–39519 [DOI] [PubMed] [Google Scholar]
- 40. Wu C. Y., Hsieh H. L., Sun C. C., Yang C. M. (2009) IL-1β induces MMP-9 expression via a Ca2+-dependent CaMKII/JNK/c-JUN cascade in rat brain astrocytes. Glia 57, 1775–1789 [DOI] [PubMed] [Google Scholar]
- 41. Liang K. C., Lee C. W., Lin W. N., Lin C. C., Wu C. B., Luo S. F., Yang C. M. (2007) Interleukin-1β induces MMP-9 expression via p42/p44 MAPK, p38 MAPK, JNK, and nuclear factor κB signaling pathways in human tracheal smooth muscle cells. J. Cell. Physiol. 211, 759–770 [DOI] [PubMed] [Google Scholar]
- 42. Funovics P., Brostjan C., Nigisch A., Fila A., Grochot A., Mleczko K., Was H., Weigel G., Dulak J., Jozkowicz A. (2006) Effects of 15d-PGJ(2) on VEGF-induced angiogenic activities and expression of VEGF receptors in endothelial cells. Prostaglandins Other Lipid Mediat. 79, 230–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grau R., Iñiguez M. A., Fresno M. (2004) Inhibition of activator protein 1 activation, vascular endothelial growth factor, and cyclooxygenase-2 expression by 15-deoxy-Δ12,14-prostaglandin J2 in colon carcinoma cells: evidence for a redox-sensitive peroxisome proliferator-activated receptor γ-independent mechanism. Cancer Res. 64, 5162–5171 [DOI] [PubMed] [Google Scholar]
- 44. Pérez-Sala D., Cernuda-Morollón E., Cañada F. J. (2003) Molecular basis for the direct inhibition of AP-1 DNA binding by 15-deoxy-Δ12,14-prostaglandin J2. J. Biol. Chem. 278, 51251–51260 [DOI] [PubMed] [Google Scholar]
- 45. Lianxu C., Hongti J., Changlong Y. (2006) Lianxu, C., Hongti, J., and Changlong, Y. (2006) NF-κBp65-specific siRNA inhibits expression of genes of COX-2, NOS-2, and MMP-9 in rat IL-1β-induced and TNF-α-induced chondrocytes. Osteoarthr. Cartil. 14, 367–376 [DOI] [PubMed] [Google Scholar]
- 46. Shakibaei M., John T., Schulze-Tanzil G., Lehmann I., Mobasheri A. (2007) Suppression of NF-κB activation by curcumin leads to inhibition of expression of cyclo-oxygenase-2 and matrix metalloproteinase-9 in human articular chondrocytes: implications for the treatment of osteoarthritis. Biochem. Pharmacol. 73, 1434–1445 [DOI] [PubMed] [Google Scholar]