

Background: IL-4 signaling through type I IL-4 receptor mediates robust tyrosine phosphorylation of IRS-2.
Results: Replacement of the IL-13Rα1 tail with the γC tail did not augment IRS-2 phosphorylation in response to IL-13.
Conclusion: The extracellular/transmembrane domain, not the cytoplasmic domain, determined outcomes of downstream signaling.
Significance: Altering the cytoplasmic domain of the receptor complex did not change signaling responses.
Keywords: Allergy, Cytokine, Cytokine Action, Interleukin, Signal transduction, IL-13 Receptor, IL-4 Receptor, IRS-2, STAT6, Inflammation
Abstract
Previously, we demonstrated that the γC subunit of type I IL-4 receptor was required for robust tyrosine phosphorylation of the downstream adapter protein, IRS-2, correlating with the expression of genes (ArgI, Retnla, and Chi3l3) characteristic of alternatively activated macrophages. We located an I4R-like motif (IRS-2 docking sequence) in the γC cytoplasmic domain but not in the IL-13Rα1. Thus, we predicted that the γC tail directed enhanced IRS-2 phosphorylation. To test this, IL-4 signaling responses were examined in a mutant of the key I4R motif tyrosine residue (Y325F) and different γC truncation mutants (γ285, γ308, γ318, γ323, and γFULL LENGTH (FL)) co-expressed in L-cells or CHO cells with wild-type (WT) IL-4Rα. Surprisingly, IRS-1 phosphorylation was not diminished in Y325F L-cell mutants suggesting Tyr-325 was not required for the robust insulin receptor substrate response. IRS-2, STAT6, and JAK3 phosphorylation was observed in CHO cells expressing γ323 and γFL but not in γ318 and γ285 mutants. In addition, when CHO cells expressed γ318, γ323, or γFL with IL-2Rβ, IL-2 induced phospho-STAT5 only in the γ323 and γFL clones. Our data suggest that a smaller (5 amino acid) interval than previously determined is necessary for JAK3 activation/γC-mediated signaling in response to IL-4 and IL-2. Chimeric receptor chains of the γC tail fused to the IL-13Rα1 extracellular and transmembrane domain did not elicit robust IRS-2 phosphorylation in response to IL-13 suggesting that the extracellular/transmembrane domains of the IL-4/IL-13 receptor, not the cytoplasmic domains, control signaling efficiency. Understanding this pathway fully will lead to rational drug design for allergic disease.
Introduction
Interleukin (IL)-4 and IL-13 are two cytokines that characterize T-helper (Th)-type 2 inflammatory responses to allergens and to helminth worm infections (1). In the context of an allergic immune response, it is believed that low concentrations of IL-4 secreted from the cells of the innate immune system (mast cells and basophils) initiate differentiation of naive T-cells into Th2 cells and IgE class switching (2). Eventually, a chronic allergic inflammatory state is established, and higher concentrations of the effector cytokine IL-13 are secreted that bring about remodeling, fibrosis, mucus secretion, and airway hyper-responsiveness (3). IL-4 and IL-13 can mediate these responses on a wide array of cell types, both hematopoietic and nonhematopoietic, by binding their cell surface receptors (reviewed in Ref. 4). IL-4 binds to the high affinity IL-4Rα (5) and then heterodimerizes with either the common γ chain (γC) or the IL-13Rα1 chain, forming type I and type II receptor complexes, respectively. Generally, type I complexes are found on hematopoietic cells due to the restricted expression of γC, although there are some exceptions (6–9). IL-13, however, binds with relatively low affinity to the IL-13Rα1 chain and then heterodimerizes with the IL-4Rα chain to form a type II receptor complex. Ligand engagement of receptor complexes activates Janus kinases (JAKs) associated with the cytoplasmic domains of the different chains. JAK3 is constitutively associated with γC through interactions with canonical box 1 and box 2 sequence motifs (JAK-binding motifs (10–13)). The activated kinases initiate downstream signaling cascades by phosphorylating residues in the cytoplasmic tails of receptor subunits that recruit substrate proteins, such as IRS-1 (in nonhematopoietic cells) or IRS-2 (in hematopoietic cells), STAT6, and others, for tyrosine phosphorylation by the JAKs.
Although the two cytokines share a receptor subunit, the IL-4Rα chain, and can bring about some similar effects on cellular responses, it is clear that they can also have different functions and elicit differential signaling. To determine the differences in signaling responses between the two cytokines, we undertook a careful side-by-side comparison of the pathways activated by IL-4 and IL-13 (14). Using cells expressing both type I and II receptors and those only expressing type II receptors on the same cellular background, we compared the kinetics and concentration responses of IL-4 and IL-13 on multiple signal transduction pathways and relative expression of mRNA and protein for a subset of alternatively activated macrophage (AAM)2 genes elicited by IL-4 and IL-13. We found that IL-4 signaling though the type I IL-4 receptor resulted in robust tyrosine phosphorylation of the adapter protein, IRS-2, which led to further recruitment of the p85 regulatory subunit of PI3K and GRB2. Signaling in response to IL-4 or IL-13 through the type II IL-4 receptor induced only weak tyrosine phosphorylation of IRS-2 (typically only 30–50% of that induced by type I). We demonstrated that strong tyrosine phosphorylation of IRS-2 was dependent upon expression of the γC subunit to form functional type I receptor complexes by comparing mouse bone marrow-derived macrophages from wild-type and γC-deficient mice and by restoring expression of the γC subunit in the γC-deficient human monocytic cell line THP-1 (14).
Given our previous observation of strong tyrosine phosphorylation of IRS-2 in response to type I IL-4R signaling and robust induction of a subset of AAM genes, our goal in this study was to determine the mechanism by which γC directs strong tyrosine phosphorylation of IRS-2, compared with type II signaling. We hypothesized that the γC cytoplasmic domain controls this response. Indeed, we located a sequence motif similar to the IRS-docking site, the insulin-IL-4 receptor (I4R) motif, found in the IL-4Rα and insulin receptor. However, mutation of the key tyrosine residue at the heart of this motif did not alter the degree of IRS-1 or STAT6 phosphorylation when mutant chains were overexpressed in L-cells, a mouse fibroblast-like cell line, and IL-4 signaling was examined. Analysis of the IL-4 and IL-13 signaling responses of the γC deletional mutants in Chinese hamster ovary (CHO) cells revealed that the activation of IRS-2, STAT6, JAK3, and STAT5 (for IL-2) phosphorylation depended upon amino acids 319–323. This was a smaller interval than found in previous studies of the γC cytoplasmic domain. We also predicted that fusion of the γC tail to the extracellular and transmembrane domain of IL-13Rα1 would restore robust IRS-2 phosphorylation after IL-13 stimulation, which does not usually occur following engagement of type II receptors. We also hypothesized that stimulation of the reciprocal chimeric receptor chain (γC outside and IL-13Rα1 inside) with IL-4 would result in dampened IRS-2 phosphorylation. However, the results of the chimeric receptor studies revealed that the extracellular/transmembrane domains of the receptor complex dominated the nature of the signaling response inside the cell, i.e. signaling responses resembled the extracellular domains of the receptor, rather than the cytoplasmic domain.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
All cell lines were purchased from the ATCC (Manassas, VA). The mouse L-cell line was cultured in DMEM supplemented with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, 4 mm glutamine as described previously (15); CHO cells were cultured in Ham's F-12 supplemented with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, 2 mm glutamine. RAW 264.7 cells were cultured in DMEM supplemented with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, 4 mm glutamine. M12.4.1 cells were cultured in RPMI 1640 medium supplemented with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, 2 mm glutamine, and 0.05 mm β-mercaptoethanol. G418 was purchased from Invitrogen, and blasticidin was from EMD/Calbiochem. Cytokines were all purchased from R & D Systems (Minneapolis, MN). Specific antibodies to human γC (CD132) conjugated to phycoerythrin (PE) (BD Biosciences), human IL-4Rα (CD124) conjugated to allophycocyanin (R & D Systems), human IL-2Rβ (CD122) conjugated to allophycocyanin (BD Biosciences), mouse CD23-PE (BD Biosciences), and human IL-13-biotin (R & D Systems) were used for analysis of cell surface expression by flow cytometry. For immunoprecipitation, we used commercially available antibodies to IRS-1 (Millipore, Billerica, MA), STAT6 (Santa Cruz Biotechnology, Santa Cruz, CA), JAK3 (Millipore), and STAT5 (Santa Cruz Biotechnology). Rabbit antiserum to human IRS-2 with cross-reactivity to rodent IRS-2 was custom generated by Sigma for immunoprecipitation. Antibodies for Western blot analysis were the same as those used in immunoprecipitation with the exception of anti-IRS-2 (Santa Cruz Biotechnology), and anti-phosphotyrosine antibodies conjugated to horseradish peroxidase were from BD Biosciences.
Plasmids
The wild-type human γC coding region was inserted into the pME18S plasmid using BstXI (gift from Dr. Warren Leonard, NHLBI, National Institutes of Health (13)). A Y325F mutation was introduced in the γC cytoplasmic domain using the QuikChange mutagenesis kit (Stratagene, Santa Clara, CA) as per the manufacturer's instructions, and the mutation was verified by DNA sequencing. The neomycin resistance plasmid was obtained from Dr. Ronald Germain, NIAID, National Institutes of Health (16). The human γC truncation mutants were developed by Dr. Franck Gesbert (described in Ref. 17). The human IL-4Rα cDNA in pDC302 is as described previously (16). The human IL-13Rα1 cDNA was purchased from Open Biosystems/Thermo Fisher (Huntsville, AL) and cloned into pME18S.
The two chimeric receptor chains were generated using a two-step overlapping PCR fusion approach as described previously (18). The first chimera was the human IL-13Rα1 extracellular and transmembrane domain fused to the human γC cytoplasmic domain (referred to as IL-13Rα1/γC, underlining denotes the intracellular domain), and the second was the human γC extracellular and transmembrane domain fused to the human IL-13Rα1 cytoplasmic domain (referred to as γC/IL-13Rα1). Briefly, two primer sets for each chimeric receptor chain were designed. In the case of the IL-13Rα1/γC chimera, the first primer set amplified the extracellular and transmembrane domains of human IL-13Rα1, and the second primer set amplified the cytoplasmic domain γC. One primer in each set incorporated the additional sequence at the end of the reverse primer for the first reaction and the forward primer for the second reaction. This additional sequence was complementary to either the cytoplasmic domain of the other receptor chain for the first reaction or to the extracellular domain of the other receptor chain for the second reaction to allow annealing during the second PCR. Also, a restriction site was incorporated at the end of the 4th primer. PCR was performed, and after PCR cleanup of the amplicons, the two PCR products for each new chimeric receptor were mixed, and PCR was performed again using the outside primers to create a new fusion product for each chimeric receptor. Double digest was performed on each chimeric receptor fusion, as well as on the pME18S γC and IL-13Rα1 plasmids in the presence of calf intestinal alkaline phosphatase to prevent self-ligation. After gel purification of the two digested fusion products and pME18S plasmids, ligation reactions and transformations of competent cells were performed. Correct in-frame chimeric receptor sequences and correct transmembrane-cytoplasmic domain junctions were confirmed by DNA sequencing of mini-prepped DNA from single colonies after selection on ampicillin-containing plates. Specific details are available upon request.
Real Time Quantitative PCR Analysis
Real time PCR methods were as described previously (14). Relative expression of Jak1, Jak2, Jak3, and Tyk2 in CHO cells was calculated relative to that in the control RAW cells, which was equal to 1. The following specific primer pairs were used: Jak1 forward, 5′-CACTGCAGATGCCCACCAT-3′, and Jak1 reverse, 5′-TGGCAGCCGTTCTGTATATTGT-3′; Jak2 forward, 5′-CGGGTAACCAGACTGGACTATATGT-3′, and Jak2 reverse, 5′-CTCAACAGCAAAGGTCAGAAAGTATT-3′; Jak3 forward, 5′-CACACAGTGCATGGCCTATGA-3′, and Jak3 reverse, 5′-TCTGATGTAATGAGGCCGTTGA-3′; and Tyk2 forward, 5′-CCTTCTATGAGACTGCTAGCCTCAT-3′, and Tyk2 reverse, 5′-CACGCACGCAAACACCAT-3′.
Stable Transfection and Selection of Cell Lines
Cell lines were transfected with the γC plasmids in the presence or absence of the huIL-4Rα plasmid by nucleofection using the Amaxa nucleofector II according to manufacturer's instructions. Twenty four hours post-nucleofection, the cells were moved into culture medium containing G418 (for the L-cells) or blasticidin (for the CHO cells). Stable clones were selected after 1–2 weeks in the antibiotic-containing medium, and expression of human γC and human IL-4Rα was determined as appropriate by fluorescence-activated cell sorting (FACS). Briefly, stable clones were incubated on ice with PE-conjugated anti-human γC and allophycocyanin-conjugated anti-human IL-4Rα. After washing, the fluorescence intensity was measured on live cells by back-gating on cells that excluded propidium iodide.
Signaling Analysis
Cells were serum-starved in medium that did not contain FBS for 24 h (L-cells) or 4 h (CHO and M12 cells) prior to addition of the cytokine stimulus. Cells were then stimulated with human IL-4, IL-13, insulin, or IL-2 for 15–30 min at 37 °C. After stimulation, cells were washed in ice-cold PBS containing 1 mm sodium orthovanadate, pelleted, and lysed in lysis buffer (14). Lysis buffer comprised 50 mm HEPES, pH 8.0, 50 mm NaCl, 1% Nonidet P-40, 5 mm EDTA, 10 mm sodium pyrophosphate, 50 mm NaF, 0.25% sodium deoxycholate, 1 mm sodium orthovanadate, 1 mm phenylmethylsulfonyl fluoride (PMSF), pepstatin, leupeptin, and aprotinin. Recombinant protein G-agarose beads (Invitrogen) were used for immunoprecipitation. For phosphotyrosine immunoblots, horseradish peroxidase (HRP)-conjugated anti-phosphotyrosine antibody (PY-20, BD Biosciences) was used, and HyGLOTM (Denville Scientific, Metuchen, NJ) or GE Healthcare was used as a chemiluminescent substrate for visualization of membrane-bound protein on the Western blot membranes. For measuring the amount of total immunoprecipitated protein, blots were stripped and re-probed with specific antibodies to the target protein. Blots with darker protein bands were chosen for the representative figures.
Densitometric Analysis
Lighter exposures of films were chosen for densitometric analysis so that band intensities would be within the linear range of the film. Films were scanned using a flat-bed scanner, and the density of the bands on the captured image was analyzed using the Image software (version 1.63f, National Institutes of Health). The amount of phosphoprotein was calculated as a ratio of the density of the band of the phosphorylated form divided by the density of the band of the unphosphorylated form of the protein for normalization. The amount of phosphoprotein present in the stimulated samples was divided by the amount in the unstimulated samples and plotted as the fold stimulation. Alternatively, the amount of phosphoprotein present was expressed as a percentage of the amount present in the IL-4-stimulated sample (= 100%).
IL-13 Binding Analysis
Cells were washed with cold FACS buffer (Dulbecco's phosphate-buffered saline containing 0.1% bovine serum albumin (BSA) and 0.1% sodium azide) and incubated on ice for 30 min with 100 ng/ml human IL-13. To remove unbound cytokine, the cells were washed three times with FACS buffer and then incubated with anti-human IL-13-biotin for 30 min on ice. After washing three times, cells were incubated with streptavidin-PE on ice in the dark for 30 min, washed again, and then analyzed using the FACSCalibur as described below.
CD23 Induction
M12 cells expressing WT human IL-4Rα and human IL-13Rα1 and the two different clones expressing the IL-13Rα1/γC chimeric receptor chain and WT human IL-4Rα were stimulated with either 5 ng/ml mouse IL-4, human IL-4, and human IL-13 or no cytokine for 24 h. Cell surface expression of CD23 was measured by staining with anti-mouse CD23-PE or isotype IgG-PE control, and fluorescence was analyzed using the FACSCalibur as described below.
Flow Cytometry
Cells were washed with FACS buffer three times, and 3 × 105 cells were stained either with specific antibodies for the surface proteins of interest or isotype-matched immunoglobulin as the negative control. An unstained sample of cells was also used as a negative control; propidium iodide was added to this sample to detect dead cells, so only PI-negative live cells were included in the analysis. The antibodies were allowed to bind for 30 min on ice before an additional three washes to remove unbound antibody. Fluorescently labeled cells were analyzed using the FACSCalibur instrument and CellQuest software to generate FACS histograms showing the fluorescence intensity of the specific surface protein on live cells. The mean fluorescence intensity of these histograms was also measured by the FACSCalibur/CellQuest software.
Statistical Analysis
Averaged data are expressed as the mean ± S.E. from three or more independent experiments. Statistical analysis to evaluate significant differences between samples was performed using the Student's t test or analysis of variance with Tukey's comparison where appropriate. Statistical significance was considered as p < 0.05.
RESULTS
We have shown previously that IL-4 signaling through the type I IL-4 receptor resulted in robust tyrosine phosphorylation of the adapter protein, IRS-2 (14). Signaling in response to IL-4 or IL-13 through the type II IL-4 receptor induced weak tyrosine phosphorylation of IRS-2. We demonstrated that strong tyrosine phosphorylation of IRS-2 was dependent upon expression of the γC subunit. In this study, our goal was to determine the mechanism by which γC directs the enhanced tyrosine phosphorylation of IRS-2. IRS-2 can be tyrosine-phosphorylated in response to cytokines other than IL-4 signaling through γC-containing cytokine receptor complexes such as the IL-2, IL-7, and IL-9 receptors (19). Therefore, we hypothesized that the γC cytoplasmic domain might contain sequence information that allows docking of additional IRS-2 molecules to the activated IL-4 receptor complex, resulting in increased tyrosine phosphorylation of IRS-2. When analyzing the sequences of the cytoplasmic domains, we observed a sequence motif in the γC cytoplasmic domain that resembled the I4R motif in the IL-4Rα and insulin receptor (Fig. 1A, left panel). This sequence is not present in the cytoplasmic tail of the IL-13Rα1. The canonical I4R motif is composed of a phosphotyrosine-binding sequence PLXXXXNPXY and the putative I4R motif in γC incorporates two conservative amino acid changes (boldface) GLXXXXQPXY. We previously showed that a Pro to Gly substitution at the −9 position relative to the critical Tyr residue was able to support the IL-4-induced tyrosine phosphorylation of IRS-2 (20).
FIGURE 1.

Effect of mutation of Tyr-325 in the putative I4R motif in human γC on IL-4 signaling responses. A, left panel, sequences of human and mouse IL-4Rα and insulin receptor surrounding the I4R motif were aligned with human and mouse γC. The amino acid residues that interact with phosphotyrosine-binding domains and Src homology 2 domains are underlined. The amino acid residues known to be important for binding to IRS-2 are boxed in gray. A, right panel, amino acid sequence within the cytoplasmic domain of human γC is shown. The sites for truncation are indicated with a dotted line and gray boxes. Box 1 and box 2 sequences that bind JAK3 (stippled) and the putative I4R motif (underlined) with the I4R central tyrosine mutant (Y325F) are shown. B, left panel, parental L-cells and L-cell clones stably transfected with wild-type γC (L-WT γC) and the mutant Y325F γC construct (L-Y325F γC) were stained with specific antibodies to human γC and mouse IL-4Rα (heavy black lines) as indicated, and isotype-matched controls (dashed lines) conjugated to PE as described under “Experimental Procedures.” Cells were washed to remove unbound antibody and were analyzed by flow cytometry. B, right panel, WT- or Y325F γC-expressing L-cells were serum-starved for 24 h and then cultured in the presence or absence of increasing concentrations of mouse IL-4 for 30 min. Cell lysates were prepared and immunoprecipitated (IP) with antibodies recognizing IRS-1 or STAT6 as indicated followed by WB with anti-phosphotyrosine (PY) or anti-phospho-STAT6 (Y641). WB membranes were then stripped and re-probed for IRS-1 and STAT6 as appropriate. Representative films were from at least three independent experiments with two independent WT and Y325F γC-expressing L-cell clones per experiment. C, densitometric analysis of the WB films was performed as described under “Experimental Procedures.”
To test whether this region of the human γC cytoplasmic domain was acting as an I4R motif, we mutated the essential tyrosine (Tyr-325) at the heart of this motif to a phenylalanine (Fig. 1A, right panel). This construct (Y325F) or wild-type human γC was then transfected along with a neomycin resistance plasmid into mouse L-cells. These fibroblast-like cells were chosen for this analysis because they have been utilized in the past to examine the role of γC on signaling transduction (15, 21). They are useful because they express IL-4Rα without either the type I or type II receptor “trigger” chains, γC or IL-13Rα1, respectively. Although interaction of IL-4 with the ligand-binding IL-4Rα is species-specific (22–24), the formation of the ternary complex of IL-4·IL-4Rα with γC is not species-specific, allowing stimulation of the transfected cells with mouse IL-4. Additionally, IRS-1 is the major IRS family member activated downstream of the IL-4 receptor in L-cells transfected with γC (15) via the same I4R-docking motif on IL-4Rα as IRS-2. Stable clones were selected in G418 and surface expression of γC and IL-4Rα was assessed by FACS (Fig. 1B, left panel). To determine the impact of the Y325F mutation on IRS/STAT6 signaling responses, two independent wild-type and mutant γC stable clones and the parental cell line were stimulated with increasing concentrations of mouse IL-4 and IRS-1, and STAT6 tyrosine phosphorylation was evaluated by immunoprecipitation (IP) and Western blot (WB) (Fig. 1B, right panel). Tyrosine phosphorylation of IRS-1 was evaluated in the L-cells because they express and predominantly activate IRS-1 instead of IRS-2 in response to IL-4 stimulation. Tyrosine phosphorylation of IRS-1 and STAT6 increased with increasing concentrations of mouse IL-4, and there were no obvious qualitative differences between the induction of IRS-1 phosphorylation in the clones expressing WT or Y325F γC. There was also no difference in the induction of STAT6 phosphorylation between WT- and Y325F-expressing clones. This was confirmed by densitometric analysis of the bands on the Western blot films. The intensity of the bands was measured, and when the ratio of phosphoprotein/total protein was calculated, mutation of Tyr-325 did not diminish the amount of tyrosine-phosphorylated IRS-1 or phospho-STAT6 induced by IL-4 compared with wild-type γC (Fig. 1C). IRS-2 induction was also unaffected by the Y325F mutation (data not shown). These results suggested that Tyr-325 was not essential for the strong tyrosine phosphorylation of IRS proteins in response to engagement of the type I receptor by IL-4.
Therefore, to determine the region of the γC cytoplasmic domain responsible for the phospho-IRS-2 response, we utilized γC truncation mutants of different lengths of the γC cytoplasmic domain (Fig. 1A, right panel) and transfected the constructs with the human IL-4Rα chain into CHO cells. CHO cells were used to reconstitute a cell-based signaling assay with both human type I receptor chains, as these cells do not express any of the IL-4 receptor chains. CHO cells are of ovarian epithelial origin and are responsive to insulin and IGF-I with strong induction of IRS-2 tyrosine phosphorylation. Stable clones were selected in blasticidin, and cell surface expression of γC and IL-4Rα was measured by FACS (Fig. 2A). After serum-starving for 4 h, the stable clones were then stimulated with 20 ng/ml IL-4 for 15 min, and the amount of tyrosine-phosphorylated IRS-2 and STAT6 was determined by IP and WB as Fig. 1B. A representative Western blot film (Fig. 2B) and densitometric analysis are shown (Fig. 2C). As expected, expression of full-length wild-type γC (γFL) resulted in robust tyrosine phosphorylation of IRS-2. A similar response was observed in the mutant with 40 amino acids remaining in the cytoplasmic domain (γ323). There was no induction of tyrosine phosphorylation of IRS-2 in mutants with 35 amino acids or less of the γC cytoplasmic domain. This was not due to a generalized defect of these clones to phosphorylate tyrosine residues of IRS-2, as stimulation of the γ308 mutant with insulin resulted in strong IRS-2 tyrosine phosphorylation. The induction of STAT6 phosphorylation mirrored the pattern of phospho-IRS-2 induction; only the γC mutants expressing full-length and 40 amino acids (γ323) of the γC cytoplasmic domain mediated induction of phospho-STAT6.
FIGURE 2.

Signaling in response to IL-4 in the γC truncation mutants stably co-expressed with human IL-4Rα in CHO cells. A, FACS analysis for expression of human γC and human IL-4Rα expression in the stable CHO clones of each γC truncation mutant was performed as described (Fig. 1A). B, indicated γC truncation mutant clones were serum-starved for 24 h and then stimulated with human IL-4 (20 ng/ml) or insulin (INS, 20 μg/ml) for 15 min. Tyrosine phosphorylation of IRS-2 and STAT6 was analyzed as described (Fig. 1B). Phosphotyrosine blots were then stripped and re-probed for total IRS-2 and STAT6. Representative films from at least three independent experiments are shown. C, densitometric analysis of the WB films was performed as described under “Experimental Procedures.”
Because the induction of tyrosine phosphorylation of IRS-2 and STAT6 was the same in the truncation mutants and because γC recruits JAK3 to the type I IL-4 receptor complex, we hypothesized that the phosphorylation of IRS-2 and STAT6 observed in the different γC truncation might be due to the ability of the different truncation mutants to activate JAK3. We first assessed expression of JAK3 in the CHO cells, an ovarian epithelial cell line, compared with the mouse macrophage cell line RAW 264.7. Generally, expression of JAK3 is restricted to hematopoietic cells, although it has been reported in nonhematopoietic cells such as type II lung epithelial cells (6), human bronchial epithelial cells (7), and fibroblasts (25). RNA was extracted from CHO and RAW cells, and real time PCR with specific primers to each of the four JAK kinases was performed. The results are graphed as the amount of mRNA expression for each gene, relative to the amount present in the control RAW cell line (equal to 1, Fig. 3A). Expression of mRNA for Jak1 and Jak2 was approximately double that of RAW cells, and CHO cells expressed mRNA for Jak3. Tyk2 mRNA was not detected in either RAW or CHO cells. Induction of tyrosine phosphorylation of JAK3 in the CHO clone expressing full-length γC was examined in response to 10 ng/ml IL-4 over time by IP and WB (Fig. 3B, upper panel). JAK3 phosphorylation was rapidly induced within 1 min after IL-4 stimulation, as expected (26). Next, we compared JAK3 activation in the γ318- and γ323-expressing CHO clones after stimulation for 5 min with 20 ng/ml IL-4 by IP and WB (Fig. 3B, lower left panel). The tyrosine phosphorylation of IRS-2 was also monitored simultaneously (Fig. 3B, lower left panel). There was no induction of tyrosine phosphorylation of JAK3 in response to IL-4 in the γ318 clone, but phosphorylation of JAK3 was strongly induced in the γ323 clone by IL-4. Phospho-JAK3 induced by IL-4 in the γ323 clone was almost 6-fold above that in the IL-4-stimulated γ318 cells by densitometric analysis (Fig. 3B, lower right panel). These data suggest that the critical interval for activation of JAK3 (as well as IRS-2 and STAT6) in response to IL-4 stimulation of type I IL-4 receptor lies between amino acids 318 and 323 of the γC cytoplasmic domain.
FIGURE 3.
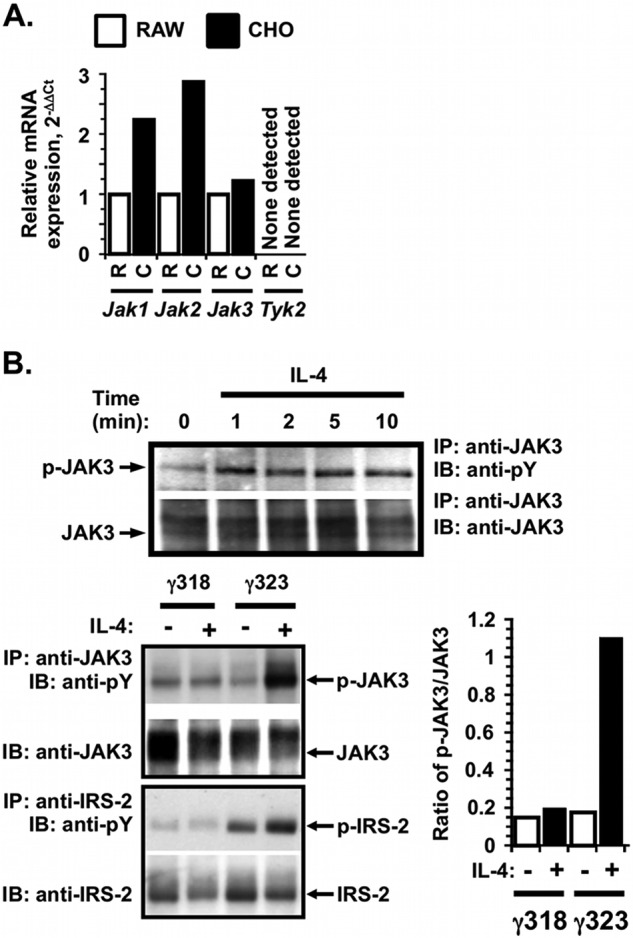
Expression and activation of JAK3 in response to IL-4 in CHO cells expressing γFL, γ323, and γ318. A, total RNA was extracted from CHO (C) cells, and the RAW (R) 264.7 macrophage cell line and cDNA were made. Real time PCR with specific primers for all four JAKs was performed, and the expression of each JAK was calculated using the standard 2−ΔΔCt method, relative to hypoxanthine phosphoribosyltransferase (HPRT). The amount of message found in CHO was expressed as fold change from the amount found in RAW 264.7 ( = 1). B, upper panel, CHO cells expressing human IL-4Rα and γFL were serum-starved prior to stimulation with 10 ng/ml IL-4 for various times. The tyrosine phosphorylation of JAK3 was analyzed by IP and WB as described under “Experimental Procedures.” B, lower left panel, comparison of JAK3 activation in the CHO cells expressing human IL-4Rα and γ318 or γ323. Cells were stimulated as in A for 5 min for JAK3 activation and 15 min for IRS-2 activation. Tyrosine phosphorylation of JAK3 and IRS-2 was analyzed by IP and WB as described in A. Phosphotyrosine blots were then stripped and re-probed for total JAK3 or IRS-2 as appropriate. Representative films from at least three independent experiments are shown. B, lower right panel, densitometric analysis of the JAK3 WB films was performed as described under “Experimental Procedures.”
Previous studies in the mid-1990s of the γC cytoplasmic domain necessary for mediating IL-2 signaling responses suggested that a larger interval, amino acids 322–335, was required for JAK3 activation (27). However, these studies were conducted using artificial chimeric receptor constructs of other cytokine receptor extracellular domains (erythropoietin (Epo) and granulocyte-macrophage colony-stimulating factor (GM-CSF)) coupled to the γC cytoplasmic domain in overexpression systems (27, 28). Therefore, we tested whether the γC sequence requirements for transmission of IL-2 signaling responses in our CHO model were the same or different from those of IL-4 signaling. In these experiments, the truncation mutants γ323 and γ318 were transfected with a construct encoding the human IL-2Rβ chain, and stable clones were identified by FACS (Fig. 4A). STAT5 signaling responses to IL-2 stimulation were determined in the stable clones following stimulation of the cells for 30 min with IL-2 by IP and Western blot (Fig. 4B, left panel). Densitometric analysis of the films was performed, and the ratio of phospho-STAT5/total STAT5 was calculated and is shown (Fig. 4B, right panel). Tyrosine phosphorylation of STAT5 was induced in the clones expressing γ323 and full-length γC but not in the clone expressing only 35 amino acids of the γC cytoplasmic domain. These data indicate that the critical interval in the γC tail for activation of JAK3 and other signaling pathways downstream of both IL-2 and type I IL-4 receptor complexes lies between amino acids 318 and 323 in the γC cytoplasmic domain.
FIGURE 4.

Signaling in response to IL-2 in the γ318, γ323, truncation mutants and WT γC stably co-expressed with human IL-2Rβ in CHO cells. A, CHO cells were transfected with cDNA encoding human IL-2Rβ and various constructs of human γC (γC FL, γ318, or γ323); stable clones were selected. These cells were evaluated for expression of human γC and IL-2Rβ by FACS analysis as described (Fig. 1A). B, CHO cells expressing human IL-2Rβ and γC truncations were serum-starved for 24 h and then stimulated with human IL-2 (28 ng/ml) for 30 min. Cell lysates were prepared and immunoprecipitated with anti-STAT5 followed by WB with anti-phosphotyrosine (PY). WB membranes were then stripped and re-probed with anti-STAT5. Densitometric analysis of the WB films was performed as described under “Experimental Procedures.” These results are representative of two independent experiments.
Finally, to demonstrate the requirement of the γC cytoplasmic domain for activation of strong tyrosine phosphorylation of IRS-2, we undertook a cytoplasmic domain swap experiment. We hypothesized that replacement of the IL-13Rα1 cytoplasmic domain with the cytoplasmic domain of γC would facilitate robust IRS-2 phosphorylation upon engagement of the chimeric receptor complex. Using a PCR fusion approach, two chimeric receptor chains were generated (Fig. 5). The IL-13Rα1 extracellular and transmembrane (TM) domains were fused to the γC cytoplasmic domain (IL-13Rα1/γC, underlining denotes the intracellular domain), and the γC extracellular and TM domains were fused to the IL-13Rα1 cytoplasmic domain (γC/IL-13Rα1). We compared the effect of the cytoplasmic domain swaps in two cell models. Based on the hypothesis that the cytoplasmic tail of γC controlled the robust tyrosine phosphorylation of IRS-2, we predicted a reduced tyrosine phosphorylation of IRS-2 as a result of signaling through the type I chimeric complex (Fig. 5, upper box) and increased tyrosine phosphorylation of IRS-2 from the type II chimeric complex (Fig. 5, lower box).
FIGURE 5.

Chimeric receptor chains γC/IL-13Rα1 and IL-13Rα1/γC and expected signaling responses in cells co-expressing human IL-4Rα and either of chimeric receptor chains. The cytoplasmic domains of the γC chain (black) or the IL-13Rα1 chain (white) were swapped (arrow) by a two-step overlapping PCR fusion approach as described under “Experimental Procedures.” The cytokine-binding homology regions of the extracellular domains are indicated by the ovals and the transmembrane domain is shown by a zig-zag line. Two chimeric receptor chains were created, γC/IL-13Rα1 and IL-13Rα1/γC (the underline denotes the cytoplasmic domain). The signaling responses of wild-type receptors are shown below the receptor diagram, as well as the predicted signaling responses of receptor complexes containing the respective chimeric receptor chains (boxed).
First, we compared the type I signaling responses in the CHO cells expressing WT human type I receptors (IL-4Rα + WT γC) to CHO expressing the chimeric type I complex (IL-4Rα + γC/IL-13Rα1). Expression of the receptor chains on WT type I-expressing CHO and two independent chimeric receptor-expressing clones was measured by FACS (Fig. 6A), and then signaling responses to IL-4 and IL-13 were analyzed (Fig. 6B).
FIGURE 6.
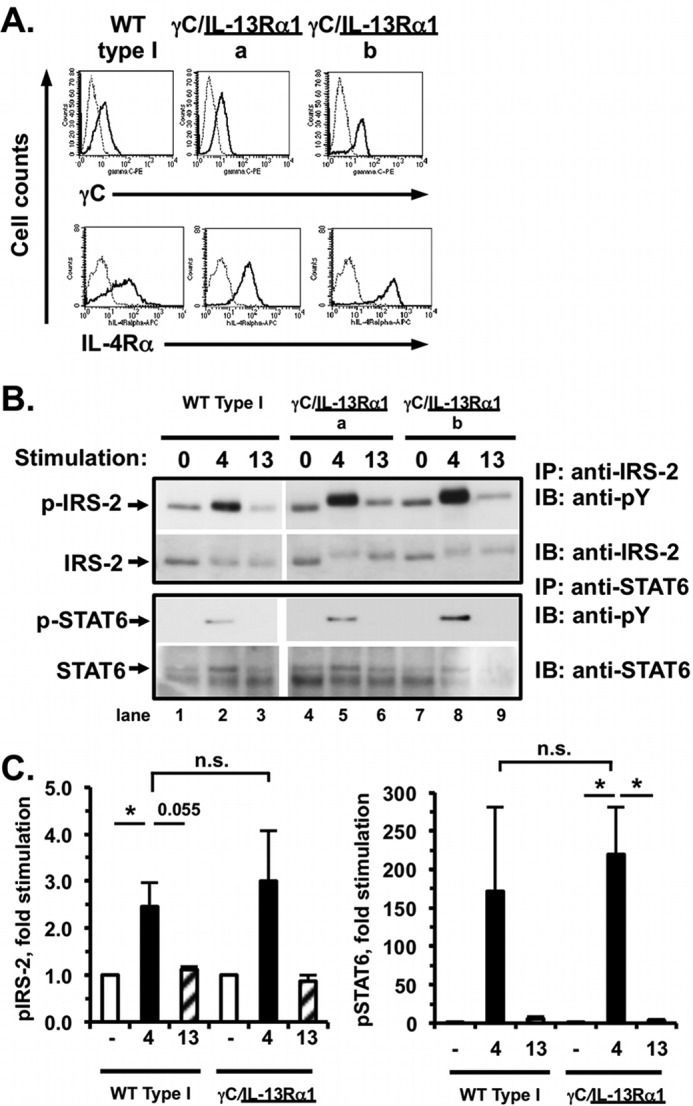
Signaling responses of CHO cells expressing human IL-4Rα and either γC (WT type I CHO) or chimeric receptor γC/IL-13Rα1 to IL-4 and IL-13. A, CHO cells were transfected with human IL-4Rα and either WT γC or γC/IL-13Rα1, and stable clones were selected. Expression of human γC and IL-4Rα on the surface of the indicated clones was evaluated by FACS as described (Fig. 1A). B, stable CHO cell clones expressing human IL-4Rα and either WT γC (WT Type I) or γC/IL-13Rα1 were serum-starved for 4 h and then either stimulated with human IL-4 (4) or human IL-13 (13, 5 ng/ml) for 15 min or were not stimulated with cytokine (0). Tyrosine phosphorylation of IRS-2 and STAT6 was analyzed as described (Fig. 1B). Phosphotyrosine blots were then stripped and re-probed for total IRS-2 and STAT6. Representative films from four independent experiments with one wild-type clone and two different chimeric clones are shown. C, densitometric analysis of the WB films was performed as described under “Experimental Procedures.” The amount of phosphoprotein is graphed as the fold induced above unstimulated cells. n = 5 (WT type I) and n = 7 (chimeric receptor), *, p < 0.05; n.s., not significant.
Tyrosine phosphorylation of IRS-2 was strongly induced in the WT type I-expressing CHO cells by IL-4 but not by IL-13 as expected (Fig. 6, B, lanes 1–3 and quantitated densitometrically in C, left panel). Surprisingly, the two clones expressing the chimeric γC/IL-13Rα1 did not exhibit diminished IRS-2 phosphorylation in response to stimulation with IL-4 (Fig. 6B, lanes 5 and 8). There was no significant difference between the amount of IL-4-stimulated phospho-IRS-2 elicited by the WT and chimeric type I receptor complexes (Fig. 6C, left panel, n = 5 (WT) and 7 (chimeric)). The presence of the IL-13Rα1 tail in the activated chimeric receptor complexes did not diminish tyrosine phosphorylation of IRS-2 as we had hypothesized. Thus, the relative robustness of IRS-2 phosphorylation correlated with the nature of the extracellular and transmembrane domain (γC) of the chimeric receptor, rather than the cytoplasmic tail (IL-13Rα1). Phosphorylation of STAT6 was induced equally by IL-4 stimulation of the WT and chimeric type I receptor complexes (Fig. 6, B, lower blots and quantitated in C, n = 5).
Second, to examine the effect of the reciprocal domain swap on type II signaling responses, we performed complementary experiments in the mouse B-cell line, M12.4.1. We chose this cell line because it lacks endogenous IL-13Rα1, allowing us to examine the impact of the chimeric IL-13Rα1/γC on type II signaling responses. Because of the species specificity of IL-4 binding to IL-4Rα and IL-13Rα1 association with IL-4Rα (29), we could generate stable M12 clones expressing WT human type II receptors and chimeric receptors (IL-4Rα + IL-13Rα1/γC) and compare their signaling responses.
We verified that M12 cells responded predictably to cytokine stimulation when transfected with each individual human chain (huIL-4Rα and huIL-13Rα1) separately and both human chains together, prior to carrying out the chimeric receptor experiment (Fig. 7). Parental M12 cells and all the transfectants responded to mouse IL-4 with phosphorylation of STAT6 because they express endogenous mouse IL-4Rα and mouse γC. However, parental M12 cells did not respond to IL-13, as they do not express mIL-13Rα1 (Fig. 7A, lanes 1–3). Transfection with human IL-13Rα1 (designated “M12-huIL-13Rα1”) also did not support STAT6 activation after stimulation with either mouse or human IL-13, as huIL-13Rα1 cannot pair with mouse IL-4Rα (Fig. 7A, lanes 4–8) (29). Transfection of M12 cells with human IL-4Rα (creating “M12-huIL-4Rα”) resulted in the tyrosine phosphorylation of STAT6 in response to human IL-4 due to formation of functional interspecies type I receptors (huIL-4/huIL-4Rα with mouse γC, Fig. 7A, lanes 9–13). Transfection of both human IL-4Rα and IL-13Rα1 (“M12-type II”) resulted in activation of STAT6 phosphorylation in response to mouse IL-13, human IL-4, and human IL-13 (Fig. 7A, lanes 14–18).
FIGURE 7.

Signaling responses of M12 cells expressing either human IL-4Rα, human IL-13Rα1, or both human IL-4Rα and IL-13Rα1 (type II M12) to IL-4 and IL-13. The murine B-cell lymphoma cell line M12.4.1 was transfected with human (hu) IL-13Rα1, huIL-4Rα, or both as indicated, and stable clones were selected. A, activated receptor complexes present in these transfectants following stimulation with mouse or human IL-4 and IL-13 are shown. These cells were deprived of serum for 2 h before treating with murine IL-4 (m4), murine IL-13 (m13), human IL-4 (h4), or human IL-13 (h13) (20 ng/ml) for 30 min. Cell lysates were prepared and precipitated with anti-STAT6 (A) or anti-IRS-2 (B) followed by Western blotting with anti-phosphotyrosine (pY). The blots were stripped and reprobed with anti-STAT6 or anti-IRS-2 as appropriate. C, parental M12 cells and M12 cells stably transfected with both human IL-13Rα1 and human IL-4Rα (M12-type II) were cultured in the presence (thick black line) or absence (thin line) of the indicated cytokine (20 ng/ml) for 24 h, and CD23 expression was evaluated by FACS.
Phosphorylation of IRS-2 was also examined in the parental M12 and M12 cells expressing human type II receptor chains (Fig. 7B,). Parental M12 responded predictably with robust tyrosine phosphorylation of IRS-2 after engagement of mouse type I receptor by mouse IL-4 (Fig. 7B, lane 2). There was no phosphorylation of IRS-2 in response to human IL-13 (Fig. 7B, lane 3). Expression of both human IL-4Rα and IL-13Rα1 (M12-type II) leads to robust phosphorylation of IRS-2 in response to human IL-4 (Fig. 7B, right panel, lane 6), due to formation of interspecies type I receptor complexes between huIL-4/huIL-4Rα and mouse γC. There was reduced tyrosine phosphorylation of IRS-2 induced by IL-13 in these cells (Fig. 7B, lane 9) via the human type II receptor complexes consistent with our studies in macrophages demonstrating that the type II receptor is less efficient at inducing the tyrosine phosphorylation of IRS-2 than the type I receptor (14).
The expression of CD23, the low affinity IgE receptor, on the surface of M12 cells is a sensitive measure of STAT6 transcriptional function. Therefore, we examined CD23 expression on the parental M12 cells and the M12-type II cells following stimulation with mouse IL-4 and human IL-13 for 24 h (Fig. 7C). Stimulation with mouse IL-4 induced CD23 expression on both parental and human type II receptor M12 cells (Fig. 7C, left panels). Stimulation of the parental M12 cells with human IL-13 did not induce cell-surface CD23 expression as expected, but CD23 was induced on the M12-expressing human type II receptor (Fig. 7C, right panels), correlating with the induction of STAT6 phosphorylation.
After fully characterizing the signaling responses of M12 cells transfected with each WT human IL-4Rα and IL-13Rα1 chain individually, as well as both human chains together, we then compared signaling responses in M12 cells expressing WT human IL-4Rα and WT human IL-13Rα1 or the chimeric IL-13Rα1/γC receptor chain (Fig. 8A). The effect of replacement of the γC tail with the IL-13Rα1 cytoplasmic domain on IL-4-induced type I responses could not be evaluated in the M12 cells, because these cells express endogenous WT mouse γC. Mouse γC could pair with either endogenous mouse IL-4Rα or the transfected human IL-4Rα, and this would make the signaling results uninterpretable. Stable clones were selected in G418, and surface expression of human IL-13Rα1 and human IL-4Rα was measured by FACS (Fig. 8B). Two independent WT and three different chimeric clones were selected for subsequent signaling analyses.
FIGURE 8.

Signaling responses of M12 cells expressing human IL-4Rα and either IL-13Rα1 (type II M12) or chimeric receptor IL-13Rα1/γC to IL-4 and IL-13. A, M12.4.1 cells were transfected with human (hu) IL-4Rα and either huIL-13Rα1 or the huIL-13Rα1/γC chimera. Activated receptor complexes formed following stimulation with human IL-4 and IL-13 are shown. B, stable clones were evaluated for expression of huIL-13Rα1 and huIL-4Rα by FACS. C, indicated cells were serum-starved for 2 h and then stimulated with human IL-4 (h4) or IL-13 (h13) (5 ng/ml) for 15 min. Tyrosine phosphorylation of IRS-2 and STAT6 was analyzed as described above. Phosphotyrosine blots were then stripped and re-probed for total IRS-2 and STAT6. Representative films from two independent experiments using two independent WT and three chimeric type II clones. D, densitometric analysis of the WB films was performed as described under “Experimental Procedures.” The amount of phosphoprotein is expressed as a percentage of the IL-4-stimulated amount. n = 5 (WT type II) and n = 7 (chimeric receptor); **, p < 0.0001; *, p < 0.05. E, human IL-13 binding by the parental M12, M12-type II, and IL-13Rα1/γC-expressing chimeric receptor-expressing clones and cells. Cells were incubated in the presence or absence of human IL-13 on ice for 30 min in buffer containing 0.1% azide. After washing away unbound cytokine, bound IL-13 was measured by incubating the cells with anti-human IL-13-biotin, followed by streptavidin-PE and performing FACS analysis as described under “Experimental Procedures.” A representative set of FACS histograms is shown from two independent experiments performed with two IL-13Rα1/γC-expressing chimeric receptor-expressing clones. F, CD23 induction on the type II M12 cells and chimeric receptor IL-13Rα1/γC-expressing clones. Cells were cultured in the presence or absence (dotted line) of murine IL-4 (thick black line), human IL-4 (thin line), or human IL-13 (dashed line) (5 ng/ml) for 24 h. CD23 expression was determined by FACS analysis as described under “Experimental Procedures.”
Stimulation of the M12 cells expressing WT human IL-4Rα and IL-13Rα1 with human IL-4 resulted in robust induction of IRS-2 phosphorylation due to interspecies type I receptor complex formation with endogenous mouse γC (Fig. 8, C, upper panel, lane 2, and quantitated in D, n = 5, p < 0.0001). The intensity of the phospho-IRS-2 band was substantially weaker upon stimulation with human IL-13 (Fig. 8, C, lane 3, and quantitated in D, striped bar, n = 5, p < 0.05). This is consistent with our previous observations (Fig. 7B) (30). Stimulation of M12 cells expressing the chimeric IL-13Rα1/γC receptor with human IL-4 also induced robust tyrosine phosphorylation of IRS-2 (Fig. 8, C, lane 5, and quantitated in D, n = 7, p < 0.0001). Surprisingly, IL-13 stimulation of these cells also induced weaker tyrosine phosphorylation of IRS-2 (Fig. 8C, lane 6) when compared with the IL-4-induced signaling via the endogenous type I receptor (lane 5 and quantitated in Fig. 8D, n = 7; p < 0.01). There was no enhancement of IRS-2 phosphorylation in response to IL-13 in the chimeric clones despite the presence of the γC cytoplasmic tail in the chimeric receptor chain/complex (Fig. 8D, compare striped bars, p = no significant difference). There was also no increase in IRS-2 phosphorylation at higher concentrations of IL-13 (20 and 50 ng/ml, data not shown). Thus, similar to our observations in the CHO cells, the relative robustness of IRS-2 phosphorylation correlated with the extracellular and transmembrane domains (IL-13Rα1) of the chimeric receptor, rather than the cytoplasmic tail (γC).
STAT6 phosphorylation was equally induced by human IL-4 and IL-13 in the M12 cells expressing WT human type II receptors (Fig. 8, C, lower panels, lanes 2 and 3, and quantitated in D, n = 5, p = no significant difference between IL-4 and IL-13). Human IL-4 and IL-13 induced equal amounts of phospho-STAT6 in the chimeric IL-13Rα1/γC cells (Fig. 8, C, lower panels, lanes 5 and 6, and quantitated in D, n = 5, p = no significant difference between IL-4 and IL-13). There was no difference in the amount of phospho-STAT6 induced by WT type II compared with chimeric type II receptor complexes.
We verified that the lack of robust tyrosine phosphorylation of IRS-2 was not due to a defect in the ability of the chimeric receptor to bind IL-13. Using a FACS-based IL-13 binding assay, human IL-13 was bound by cells expressing the chimeric receptor complex and cells expressing the WT type II receptors (positive control, Fig. 8E, 2nd panel) but not by the parental M12 cells (negative control, Fig. 8E, 1st panel). Confirmation of activation of signaling downstream of the cytokine receptor complex containing the chimeric IL-13Rα1/γC chain was assessed by induction of CD23 expression by human and mouse IL-4/IL-13 on the stable IL-13Rα1/γC clones compared with WT IL-13Rα1 (Fig. 8F). All three cytokines (5 ng/ml) induced CD23 surface expression after 24 h on the type II M12 cells (positive control cells) and on the IL-13Rα1/γC chimeric cells (Fig. 8E).
Taken together, the results from the analysis of the chimeric type I and type II receptor complexes suggest that the extracellular and TM domains of γC and IL-13Rα1, and not their cytoplasmic tails, dictate the magnitude of IL-4/IL-13-induced IRS-2 signaling responses inside the cell.
DISCUSSION
The goal of this study was to understand the mechanism by which IL-4 engagement of the type I IL-4 receptor was able to induce strong tyrosine phosphorylation of the downstream adapter protein IRS-2, whereas engagement of type II receptor by either IL-4 or IL-13 resulted in weak tyrosine phosphorylation of IRS-2. We had previously demonstrated that activation of IRS-2 by type I receptor resulted in robust activation of expression of a subset of genes characteristic of alternative activation of macrophages, which are thought to play a role in the remodeling and fibrotic changes that occur in asthmatic lungs. Therefore, understanding the molecular mechanism by which type I receptor triggered by IL-4 initiates downstream IRS-2 signaling and subsequent AAM gene expression will be critical in designing therapies to block this pathway.
The relative role of type I and II IL-4 receptors in the development of Th2 inflammation in vivo has also been examined. Studies of IL-4- and IL-13-deficient mice have suggested that eosinophilic infiltration of the lung is heavily dependent on IL-4 (31, 32). Studies of characteristic parameters of allergic lung inflammation in mice deficient in the IL-13Rα1 chain have revealed that type I and II IL-4 receptors have both distinct and overlapping roles in the setting of allergen challenge (ovalbumin, Aspergillus, or house dust mite), worm infection, or intratracheal instillation of each cytokine (33–35). For example, type II receptor was dispensable for AAM development in the context of worm infection (33), yet both receptors play a role in AAM development in ovalbumin or IL-4-induced allergic lung inflammation (34). The exact cellular and molecular mechanism(s) by which AAM/AAM genes might contribute to or suppress the development of Th2 allergic lung inflammation has not been fully defined.
Our data have shown that activation of IRS-2, STAT6, and JAK3 by IL-4 required a five-amino acid interval (amino acids 319–323) of the cytoplasmic domain of the γC subunit. This interval did not include a tyrosine (Tyr-325) that we initially hypothesized was part of a putative I4R motif.
The data presented here contrast with earlier studies of chimeric IL-2Rγ that showed that truncation mutants of 48 amino acids (γ323) or more could not bind JAK3 in response to activating ligand (11, 13, 28, 36) suggesting JAK3 binding was after Gln-322. Subsequent analysis using a receptor chimera of the GM-CSF extracellular domain coupled to the γC chain suggested that, in addition to the membrane-proximal box 1 motif, a distal interval between Pro-324 and Ile-345 was critical for JAK3 and JAK1 phosphorylation and activation (27). These earlier studies examining the signaling responses to IL-2 or IL-7 were conducted using chimeric receptors of the GM-CSF, Epo, or CD4 extracellular domains linked to the γC cytoplasmic domain transfected with the respective ligand binding subunit, and the chimeric complex was activated by GM-CSF, Epo, or even antibody cross-linking in the case of the CD4 chimeras. We chose to use the physiological binding partner of γC, IL-4Rα (or IL-2Rβ), and the natural ligand, IL-4 (or IL-2), in our study to recapitulate any essential intersubunit or ternary complex interactions necessary for proper activation of the receptor complex when the chains heterodimerize around the ligand. The difference that we observed in the size of the interval necessary for activation of downstream signaling may be due to these differences in utilizing “natural” receptor chains, as opposed to chimeric receptors, as well as engagement by their physiological ligands to initiate activation.
Although we were surprised by the lack of effect of the Y325F mutation on IL-4-induced signal transduction, Goldsmith et al. (37) had previously noted that the growth response of an Epo extracellular domain-γC cytoplasmic domain chimera lacking all four γC cytoplasmic tyrosines was no different from that of chimera with the WT γC domain. This was also recapitulated using the same Epo-γC chimeric receptor chain in the IL-7 receptor system (28). However, Lindemann et al. (38) were able to uncover an anti-apoptotic signaling role for γC tyrosines that was only revealed in the absence of the partner IL-2Rβ chain tyrosines. The γC tyrosines were required for activation of Akt signaling, possibly by PI3K, and Bcl-2 induction to mediate the anti-apoptotic effect. It is possible that the effect of the Y325F mutation on signaling responses to IL-4 would also only be exposed in the absence of the IL-4Rα tyrosines.
From the earlier studies indicating lack of JAK3 binding in the Gln-322 mutants (27) and our data here that the Pro-323 mutant can activate JAK3 phosphorylation but the shorter Ala-318 mutant does not, it seems likely that Pro-323 may be an essential amino acid involved in membrane-distal JAK3 binding/activation. Although we have not formally tested this hypothesis, future experiments using a truncation mutant that removes this proline and a Pro to Ala substitution mutant could test this hypothesis in our CHO model system. Proline residues introduce kinks into helical secondary structure, and such a “helix distorter” may be involved in aligning JAK3 into the correct orientation within the cytoplasmic domains of the IL-4 receptor complex for transphosphorylation of JAK1 (39), IL-4Rα, or γC cytoplasmic domains and downstream signaling intermediates (IRS-2, STAT6, etc.). The extracellular WSXWS domain is hypothesized to play a role in rotating the kinase domains of the JAKs into alignment (40), but the structure of the cytoplasmic domain may also be important as well. There are no structural studies to date that have determined conformation of the cytoplasmic domains of the IL-4 receptor with the associated JAKs. However, a recent paper has given some insight into the structure of JAK1 associated with the cytoplasmic domains of gp130 full-length receptor (41).
Understanding the requirements for JAK3 activation is critical for designing inhibitors to prevent activation of this kinase. Clinical trials using pharmacological inhibitors of JAK3 kinase activity are currently underway for many immune-based diseases, such as arthritis (42), cancer (43), transplantation (44), and psoriasis (45). Targeting this kinase in allergic disease may provide another potential therapeutic target to explore in asthma, which is often refractory in many patients to standard therapies, such as inhaled steroids. Furthermore, the JAK3 inhibitor CP-690,550 demonstrated potent anti-inflammatory activity in the ovalbumin mouse model of allergic lung inflammation (46). In particular, a striking reduction (90%) in pulmonary eosinophilia was noted in the mice treated with the JAK3 inhibitor. This finding may be a result of inhibiting the type I receptor (JAK3-dependent) signaling in mouse eosinophils. Indeed, data from our laboratory demonstrated that IL-4 priming of mouse eosinophil chemotaxis was dependent on the type I receptor, as γC-deficient eosinophils did not show enhanced migration following IL-4 pretreatment (47). Type II IL-4 receptor signaling had little impact on eosinophil chemotaxis consistent with studies on IL-13Rα1-deficient mice (33–35). However, JAK3 inhibitors that we have tested in our laboratory lack specificity, as they also suppress IL-13 signaling in vitro.3 Novel peptide-based inhibitors that mimic the JAK3 interaction sites on γC could pull the kinase away from receptor complexes that utilize γC and thus prevent activation when ligand is present.
Our cytoplasmic domain swap experiments were designed to test whether the γC tail could be placed on the extracellular + TM domain of a receptor chain that does not ordinarily induce strong tyrosine phosphorylation of IRS-2 (i.e. IL-13Rα1) and render it capable of accomplishing such a response. We hypothesized that the reverse switch of placing the IL-13Rα1 cytoplasmic domain on the γC extracellular + TM domain would result in reduced phosphorylation of IRS-2. However, our data in CHO and M12 using the two reciprocal domain swap chimeras suggests that the extracellular + TM portion of the receptor, rather than the cytoplasmic tail, dictated the magnitude of the IRS-2 response. The γC/IL-13Rα1 chimera signaled like the full-length γC, and the IL-13Rα1/γC signaled like the full-length IL-13Rα1. It is possible that contacts between the transmembrane region and cytoplasmic domain influence the activation of signaling inside the cell. If the cytoplasmic domain is chimeric, then perhaps signaling responses are lost if required intra- or interchain TM-cytoplasmic domain-JAK interactions are missing or conformational changes cannot be conveyed appropriately because the cytoplasmic domain is different. Indeed, the structure of the gp130·IL-6·IL-6Rα·JAK1 holocomplex suggests that the extracellular and TM domains of the receptor complex lock around the ligand and relay signal to JAK1 bound to the extreme juxtamembrane portion of the intracellular domain (41). Earlier studies with erythropoietin (48) and insulin receptor (49, 50) also concluded that the orientation of the extracellular domains in a heterodimeric receptor have a direct influence on the organization of the receptor cytoplasmic domains and the outcome of signaling responses (51). Subtle differences in the ternary complexes of IL-4 and IL-13 bound to the extracellular receptor domains have been noted and may help to explain differences in signaling intensity (52).
We constructed chimeras exactly at the TM-cytoplasmic domain junction to test the contribution of the cytoplasmic domain to signaling responses. The earlier studies characterizing the function of the γC cytoplasmic domain utilized both chimeric receptors, which included the γC TM domain or had TM domains that matched the extracellular domain (11, 27). However, it is possible that altering the precise location of the chimeric switch may alter the outcome of signaling responses. The outcome of signaling may be different if the TM domain matched the intracellular domain or extended into the membrane-proximal region of the extracellular domain. In fact, Zhong et al. (53) found that the TM and juxtamembrane domains of a chimeric receptor/thymic stromal lymphopoietin receptor exert a critical influence on the outcome of downstream signaling pathways. In the recent gp130/IL-6/JAK1 paper (41), the authors speculated that JAK1 interacts with the inner leaflet of the plasma membrane; it is also possible that the JAKs could make important interactions with amino acids of the TM domain by nuzzling into the lipid bilayer. Future studies will address whether altering the transmembrane domain to match the cytoplasmic domain or extending the chimera up into the membrane-proximal region of the extracellular domain of the receptor can alter the outcome of signaling in this IL-4/IL-13 receptor system.
In conclusion, our truncation analyses defined a small five-amino acid interval of the γC chain (amino acids 319–323) necessary for type I receptor-mediated tyrosine phosphorylation of IRS-2, STAT6, and JAK3 in response to IL-4. Our mutational and chimeric studies demonstrated that the cytoplasmic domain of the γC likely did not dictate the nature of the signaling pathways activated. Rather, we found that the extracellular and transmembrane domains of γC and IL-13Rα1 controlled the magnitude of the intracellular signaling response to IL-4 and IL-13.
N. M. Heller, unpublished observations.
- AAM
- alternatively activated macrophage
- PE
- phycoerythrin
- IP
- immunoprecipitation
- WB
- Western blot
- Epo
- erythropoietin
- TM
- transmembrane
- hu
- human
- h
- human.
REFERENCES
- 1. Barnes P. J. (2011) Pathophysiology of allergic inflammation. Immunol. Rev. 242, 31–50 [DOI] [PubMed] [Google Scholar]
- 2. Paul W. E., Zhu J. (2010) How are T(H)2-type immune responses initiated and amplified? Nat. Rev. Immunol. 10, 225–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Al-Muhsen S., Johnson J. R., Hamid Q. (2011) Remodeling in asthma. J. Allergy Clin. Immunol. 128, 451–462 [DOI] [PubMed] [Google Scholar]
- 4. Chatila T. A. (2004) Interleukin-4 receptor signaling pathways in asthma pathogenesis. Trends Mol. Med. 10, 493–499 [DOI] [PubMed] [Google Scholar]
- 5. Lowenthal J. W., Castle B. E., Christiansen J., Schreurs J., Rennick D., Arai N., Hoy P., Takebe Y., Howard M. (1988) Expression of high affinity receptors for murine interleukin 4 (BSF-1) on hemopoietic and nonhemopoietic cells. J. Immunol. 140, 456–464 [PubMed] [Google Scholar]
- 6. Lesur O., Arsalane K., Bérard J., Mukuna J. P., de Brum-Fernandes A. J., Lane D., Rola-Pleszczynski M. (1997) Functional IL-2 receptors are expressed by rat lung type II epithelial cells. Am. J. Physiol. 273, L495–L503 [DOI] [PubMed] [Google Scholar]
- 7. van der Velden V. H., Naber B. A., Wierenga-Wolf A. F., Debets R., Savelkoul H. F., Overbeek S. E., Hoogsteden H. C., Versnel M. A. (1998) Interleukin 4 receptors on human bronchial epithelial cells. An in vivo and in vitro analysis of expression and function. Cytokine 10, 803–813 [DOI] [PubMed] [Google Scholar]
- 8. Corrigall V. M., Arastu M., Khan S., Shah C., Fife M., Smeets T., Tak P. P., Panayi G. S. (2001) Functional IL-2 receptor β (CD122) and γ (CD132) chains are expressed by fibroblast-like synoviocytes. Activation by IL-2 stimulates monocyte chemoattractant protein-1 production. J. Immunol. 166, 4141–4147 [DOI] [PubMed] [Google Scholar]
- 9. Reinecker H. C., Podolsky D. K. (1995) Human intestinal epithelial cells express functional cytokine receptors sharing the common γc chain of the interleukin 2 receptor. Proc. Natl. Acad. Sci. U.S.A. 92, 8353–8357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Murakami M., Narazaki M., Hibi M., Yawata H., Yasukawa K., Hamaguchi M., Taga T., Kishimoto T. (1991) Critical cytoplasmic region of the interleukin 6 signal transducer gp130 is conserved in the cytokine receptor family. Proc. Natl. Acad. Sci. U.S.A. 88, 11349–11353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miyazaki T., Kawahara A., Fujii H., Nakagawa Y., Minami Y., Liu Z. J., Oishi I., Silvennoinen O., Witthuhn B. A., Ihle J. N., et al. (1994) Functional activation of Jak1 and Jak3 by selective association with IL-2 receptor subunits. Science 266, 1045–1047 [DOI] [PubMed] [Google Scholar]
- 12. Boussiotis V. A., Barber D. L., Nakarai T., Freeman G. J., Gribben J. G., Bernstein G. M., D'Andrea A. D., Ritz J., Nadler L. M. (1994) Prevention of T cell anergy by signaling through the γ c chain of the IL-2 receptor. Science 266, 1039–1042 [DOI] [PubMed] [Google Scholar]
- 13. Russell S. M., Johnston J. A., Noguchi M., Kawamura M., Bacon C. M., Friedmann M., Berg M., McVicar D. W., Witthuhn B. A., Silvennoinen O., et al. (1994) Interaction of IL-2R β and γ c chains with Jak1 and Jak3. Implications for XSCID and XCID. Science 266, 1042–1045 [DOI] [PubMed] [Google Scholar]
- 14. Heller N. M., Qi X., Junttila I. S., Shirey K. A., Vogel S. N., Paul W. E., Keegan A. D. (2008) Type I IL-4Rs selectively activate IRS-2 to induce target gene expression in macrophages. Sci. Signal. 1, ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang L. M., Michieli P., Lie W. R., Liu F., Lee C. C., Minty A., Sun X. J., Levine A., White M. F., Pierce J. H. (1995) The insulin receptor substrate-1-related 4PS substrate but not the interleukin-2R γ chain is involved in interleukin-13-mediated signal transduction. Blood 86, 4218–4227 [PubMed] [Google Scholar]
- 16. Keegan A. D., Nelms K., White M., Wang L. M., Pierce J. H., Paul W. E. (1994) An IL-4 receptor region containing an insulin receptor motif is important for IL-4-mediated IRS-1 phosphorylation and cell growth. Cell 76, 811–820 [DOI] [PubMed] [Google Scholar]
- 17. Malardé V., Proust R., Dautry-Varsat A., Gesbert F. (2009) NEDD4-2 associates with γ (c) and regulates its degradation rate. Biochem. Biophys. Res. Commun. 387, 409–413 [DOI] [PubMed] [Google Scholar]
- 18. Wang H. Y., Paul W. E., Keegan A. D. (1996) IL-4 function can be transferred to the IL-2 receptor by tyrosine containing sequences found in the IL-4 receptor α chain. Immunity 4, 113–121 [DOI] [PubMed] [Google Scholar]
- 19. Johnston J. A., Wang L. M., Hanson E. P., Sun X. J., White M. F., Oakes S. A., Pierce J. H., O'Shea J. J. (1995) Interleukins 2, 4, 7, and 15 stimulate tyrosine phosphorylation of insulin receptor substrates 1 and 2 in T cells. Potential role of JAK kinases. J. Biol. Chem. 270, 28527–28530 [DOI] [PubMed] [Google Scholar]
- 20. Wang H. Y., Zamorano J., Keegan A. D. (1998) A role for the insulin-interleukin (IL)-4 receptor motif of the IL-4 receptor α-chain in regulating activation of the insulin receptor substrate 2 and signal transducer and activator of transcription 6 pathways. Analysis by mutagenesis. J. Biol. Chem. 273, 9898–9905 [DOI] [PubMed] [Google Scholar]
- 21. Russell S. M., Keegan A. D., Harada N., Nakamura Y., Noguchi M., Leland P., Friedmann M. C., Miyajima A., Puri R. K., Paul W. E., et al. (1993) Interleukin-2 receptor γ chain. A functional component of the interleukin-4 receptor. Science 262, 1880–1883 [DOI] [PubMed] [Google Scholar]
- 22. Mosmann T. R., Yokota T., Kastelein R., Zurawski S. M., Arai N., Takebe Y. (1987) Species specificity of T cell stimulating activities of IL 2 and BSF-1 (IL 4). Comparison of normal and recombinant mouse and human IL 2 and BSF-1 (IL 4). J. Immunol. 138, 1813–1816 [PubMed] [Google Scholar]
- 23. Park L. S., Friend D., Sassenfeld H. M., Urdal D. L. (1987) Characterization of the human B cell stimulatory factor 1 receptor. J. Exp. Med. 166, 476–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Park L. S., Friend D., Grabstein K., Urdal D. L. (1987) Characterization of the high affinity cell-surface receptor for murine B-cell-stimulating factor 1. Proc. Natl. Acad. Sci. U.S.A. 84, 1669–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ozawa A., Tada H., Tamai R., Uehara A., Watanabe K., Yamaguchi T., Shimauchi H., Takada H., Sugawara S. (2003) Expression of IL-2 receptor β and γ chains by human gingival fibroblasts and up-regulation of adhesion to neutrophils in response to IL-2. J. Leuk. Biol. 74, 352–359 [DOI] [PubMed] [Google Scholar]
- 26. Witthuhn B. A., Silvennoinen O., Miura O., Lai K. S., Cwik C., Liu E. T., Ihle J. N. (1994) Involvement of the Jak-3 Janus kinase in signaling by interleukins 2 and 4 in lymphoid and myeloid cells. Nature 370, 153–157 [DOI] [PubMed] [Google Scholar]
- 27. Nelson B. H., Lord J. D., Greenberg P. D. (1996) A membrane-proximal region of the interleukin-2 receptor γ c chain sufficient for Jak kinase activation and induction of proliferation in T cells. Mol. Cell. Biol. 16, 309–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lai S. Y., Molden J., Goldsmith M. A. (1997) Shared γ (c) subunit within the human interleukin-7 receptor complex. A molecular basis for the pathogenesis of X-linked severe combined immunodeficiency. J. Clin. Invest. 99, 169–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Andrews R., Rosa L., Daines M., Khurana Hershey G. (2001) Reconstitution of a functional human type II IL-4/IL-13 receptor in mouse B cells. Demonstration of species specificity. J. Immunol. 166, 1716–1722 [DOI] [PubMed] [Google Scholar]
- 30. Heller N., Dasgupta P., Dorsey N., Chapoval S., Keegan A. D. (2012) in Allergic Diseases. Highlights in the Clinic, Mechanisms, and Treatments (Pereira C., ed) pp. 43–82, InTech, Rijeka, Croatia [Google Scholar]
- 31. Walter D. M., McIntire J. J., Berry G., McKenzie A. N., Donaldson D. D., DeKruyff R. H., Umetsu D. T. (2001) Critical role for IL-13 in the development of allergen-induced airway hyper-reactivity. J. Immunol. 167, 4668–4675 [DOI] [PubMed] [Google Scholar]
- 32. Wills-Karp M., Luyimbazi J., Xu X., Schofield B., Neben T. Y., Karp C. L., Donaldson D. D. (1998) Interleukin-13. Central mediator of allergic asthma. Science 282, 2258–2261 [DOI] [PubMed] [Google Scholar]
- 33. Ramalingam T. R., Pesce J. T., Sheikh F., Cheever A. W., Mentink-Kane M. M., Wilson M. S., Stevens S., Valenzuela D. M., Murphy A. J., Yancopoulos G. D., Urban J. F., Jr., Donnelly R. P., Wynn T. A. (2008) Unique functions of the type II interleukin 4 receptor identified in mice lacking the interleukin 13 receptor α1 chain. Nat. Immunol. 9, 25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Munitz A., Brandt E. B., Mingler M., Finkelman F. D., Rothenberg M. E. (2008) Distinct roles for IL-13 and IL-4 via IL-13 receptor α1 and the type II IL-4 receptor in asthma pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 105, 7240–7245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rothenberg M. E., Wen T., Shik D., Cole E. T., Mingler M. M., Munitz A. (2011) IL-13 receptor α1 differentially regulates aeroallergen-induced lung responses. J. Immunol. 187, 4873–4880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Asao H., Okuyama C., Kumaki S., Ishii N., Tsuchiya S., Foster D., Sugamura K. (2001) Cutting edge. The common γ-chain is an indispensable subunit of the IL-21 receptor complex. J. Immunol. 167, 1–5 [DOI] [PubMed] [Google Scholar]
- 37. Goldsmith M. A., Lai S. Y., Xu W., Amaral M. C., Kuczek E. S., Parent L. J., Mills G. B., Tarr K. L., Longmore G. D., Greene W. C. (1995) Growth signal transduction by the human interleukin-2 receptor requires cytoplasmic tyrosines of the β chain and non-tyrosine residues of the γc chain. J. Biol. Chem. 270, 21729–21737 [DOI] [PubMed] [Google Scholar]
- 38. Lindemann M. J., Benczik M., Gaffen S. L. (2003) Anti-apoptotic signaling by the interleukin-2 receptor reveals a function for cytoplasmic tyrosine residues within the common γ (γC) receptor subunit. J. Biol. Chem. 278, 10239–10249 [DOI] [PubMed] [Google Scholar]
- 39. Witthuhn B. A., Williams M. D., Kerawalla H., Uckun F. M. (1999) Differential substrate recognition capabilities of Janus family protein tyrosine kinases within the interleukin 2 receptor (IL2R) system. Jak3 as a potential molecular target for treatment of leukemias with a hyperactive Jak-Stat signaling machinery. Leuk. Lymphoma 32, 289–297 [DOI] [PubMed] [Google Scholar]
- 40. Weidemann T., Höfinger S., Müller K., Auer M. (2007) Beyond dimerization. A membrane-dependent activation model for interleukin-4 receptor-mediated signaling. J. Mol. Biol. 366, 1365–1373 [DOI] [PubMed] [Google Scholar]
- 41. Lupardus P. J., Skiniotis G., Rice A. J., Thomas C., Fischer S., Walz T., Garcia K. C. (2011) Structural snapshots of full-length Jak1, a transmembrane gp130/IL-6/IL-6Rα cytokine receptor complex, and the receptor-Jak1 holocomplex. Structure 19, 45–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Coombs J. H., Bloom B. J., Breedveld F. C., Fletcher M. P., Gruben D., Kremer J. M., Burgos-Vargas R., Wilkinson B., Zerbini C. A., Zwillich S. H. (2010) Improved pain, physical functioning, and health status in patients with rheumatoid arthritis treated with CP-690,550, an orally active Janus kinase (JAK) inhibitor. Results from a randomized, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 69, 413–416 [DOI] [PubMed] [Google Scholar]
- 43. Kimura S. (2010) AT-9283, a small molecule multitargeted kinase inhibitor for the potential treatment of cancer. Curr. Opin. Investig. Drugs 11, 1442–1449 [PubMed] [Google Scholar]
- 44. Quaedackers M. E., Mol W., Korevaar S. S., van Gurp E. A., van Ijcken W. F., Chan G., Weimar W., Baan C. C. (2009) Monitoring of the immunomodulatory effect of CP-690,550 by analysis of the JAK/STAT pathway in kidney transplant patients. Transplantation 88, 1002–1009 [DOI] [PubMed] [Google Scholar]
- 45. West K. (2009) CP-690550, a JAK3 inhibitor as an immunosuppressant for the treatment of rheumatoid arthritis, transplant rejection, psoriasis and other immune-mediated disorders. Curr. Opin. Investig. Drugs 10, 491–504 [PubMed] [Google Scholar]
- 46. Kudlacz E., Conklyn M., Andresen C., Whitney-Pickett C., Changelian P. (2008) The JAK-3 inhibitor CP-690550 is a potent anti-inflammatory agent in a murine model of pulmonary eosinophilia. Eur. J. Pharmacol. 582, 154–161 [DOI] [PubMed] [Google Scholar]
- 47. Heller N. M., Gwinn W. M., Donnelly R. P., Constant S. L., Keegan A. D. (2012) IL-4 Engagement of the type I IL-4 receptor complex enhances mouse eosinophil migration to eotaxin-1 in vitro. PloS One 7, e39673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Muthukumaran G., Kotenko S., Donnelly R., Ihle J. N., Pestka S. (1997) Chimeric erythropoietin-interferon γ receptors reveal differences in functional architecture of intracellular domains for signal transduction. J. Biol. Chem. 272, 4993–4999 [DOI] [PubMed] [Google Scholar]
- 49. Böni-Schnetzler M., Scott W., Waugh S. M., DiBella E., Pilch P. F. (1987) The insulin receptor. Structural basis for high affinity ligand binding. J. Biol. Chem. 262, 8395–8401 [PubMed] [Google Scholar]
- 50. Lee J., O'Hare T., Pilch P. F., Shoelson S. E. (1993) Insulin receptor autophosphorylation occurs asymmetrically. J. Biol. Chem. 268, 4092–4098 [PubMed] [Google Scholar]
- 51. Livnah O., Stura E. A., Johnson D. L., Middleton S. A., Mulcahy L. S., Wrighton N. C., Dower W. J., Jolliffe L. K., Wilson I. A. (1996) Functional mimicry of a protein hormone by a peptide agonist. The EPO receptor complex at 2.8 Å. Science 273, 464–471 [DOI] [PubMed] [Google Scholar]
- 52. LaPorte S. L., Juo Z. S., Vaclavikova J., Colf L. A., Qi X., Heller N. M., Keegan A. D., Garcia K. C. (2008) Molecular and structural basis of cytokine receptor pleiotropy in the interleukin-4/13 system. Cell 132, 259–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhong J., Liu X., Pandey A. (2010) Effects of transmembrane and juxtamembrane domains on proliferative ability of TSLP receptor. Mol. Immunol. 47, 1207–1215 [DOI] [PubMed] [Google Scholar]