

Background: No known function for the amino acid cystathionine other than as an intermediate in cysteine synthesis.
Results: Cystathionine prevents ER stress induced steatotic liver injury, acute tubular necrosis and apoptosis without changing induction of the unfolded protein response.
Conclusion: Abolition of cystathionine synthesis may contribute to pathogenesis in homocystinuria.
Significance: Cystathionine has therapeutic potential for disease states where ER stress is implicated.
Keywords: Apoptosis, Endoplasmic Reticulum Stress, Homocysteine, Neuroblastoma, Unfolded Protein Response, Cystathionine, Cystathionine β-Synthase, Homocystinuria
Abstract
Cystathionine (R-S-(2-amino-2-carboxyethyl)-l-homocysteine) is a non-proteinogenic thioether containing amino acid. In mammals, cystathionine is formed as an intermediate of the transsulfuration pathway by the condensation of serine and homocysteine (Hcy) in a reaction catalyzed by cystathionine β-synthase (CBS). Cystathionine is subsequently converted to cysteine plus ammonia and α-ketobutyrate by the action of cystathionine γ-lyase (CGL). Pathogenic mutations in CBS result in CBS-deficient homocystinuria (HCU) which, if untreated, results in mental retardation, thromboembolic complications and connective tissue disorders. Currently there is no known function for cystathionine other than serving as an intermediate in transsulfuration and to date, the possible contribution of the abolition of cystathionine synthesis to pathogenesis in HCU has not been investigated. Using both mouse and cell-culture models, we have found that cystathionine is capable of blocking the induction of hepatic steatosis and kidney injury, acute tubular necrosis, and apoptotic cell death by the endoplasmic reticulum stress inducing agent tunicamycin. Northern and Western blotting analysis indicate that the protective effects of cystathionine occur without any obvious alteration of the induction of the unfolded protein response. Our data constitute the first experimental evidence that the abolition of cystathionine synthesis may contribute to the pathology of HCU and that this compound has therapeutic potential for disease states where ER stress is implicated as a primary initiating pathogenic factor.
Introduction
Cystathionine (R-S-(2-amino-2-carboxyethyl)-l-homocysteine) is a non-proteinogenic thioether containing amino acid that is not present in significant amounts in the human diet. Endogenous synthesis of cystathionine is catalyzed by cystathionine β-synthase (CBS)3 via a pyridoxal 5′-phosphate (PLP)-dependent β-replacement reaction condensing serine and homocysteine (Hcy) to form cystathionine, which is subsequently converted to cysteine plus ammonia and α-ketobutyrate by the action of cystathionine γ-lyase (CGL) (1). Fig. 1 illustrates the metabolic relationships between Hcy, cysteine, cystathionine, and related metabolites relevant to this report.
FIGURE 1.

Methionine metabolism in mammals. The transsulfuration pathway and the methionine and folate cycles are shown.
Pathogenic mutations in CBS result in CBS deficient homocystinuria (HCU) which, if untreated, results in mental retardation, thromboembolic complications and a range of connective tissue disorders. The pathogenic mechanisms that underlie HCU are poorly understood but the majority of research to date has focused upon the role of elevated homocysteine (Hcy). In addition to causing elevated tissue and plasma, Hcy, methionine, S-adenosylmethionine (AdoMet), and S-adenosylhomocysteine (AdoHcy), CBS inactivation also completely abolishes synthesis of cystathionine. Currently, there is no known function for cystathionine other than serving as an intermediate in transsulfuration and to date, the possible contribution of the abolition of its synthesis to pathogenesis in HCU has not been investigated.
A previously described Cbs-null mouse model of HCU exhibits a semi-lethal phenotype due to severe steatosis, fibrosis, and neonatal liver failure (2, 3). Recent work in our laboratory has generated a mouse model of HCU designated HO, that lacks any functional copy of the mouse Cbs gene and carries a copy of the human CBS gene under the control of its natural promoter. HO mice only express the human CBS gene at very low levels and exhibit essentially identical levels of Hcy and other methionine cycle metabolites as the previously described Cbs-null mouse model (4). Despite exhibiting identical levels of elevated Hcy, the HO mice exhibit only mild hepatopathy without any detectable steatosis, fibrosis or liver failure. The only metabolite to differ significantly from the Cbs-null model was cystathionine which, due to Hcy mediated inactivation of CGL, accumulated to a level 4-fold higher than that observed in normal control animals (4). Previous work has suggested that hepatopathy in the Cbs-null model is due to the induction of endoplasmic reticulum (ER) stress by Hcy (5). This possibility is consistent with the fact that electron microscopy analysis found markedly distended ER with fine intracisternal proteinaceous precipitates in liver samples of the Cbs-null but not in the HO mice (3, 4). These observations lead to the hypothesis that cystathionine may be able to exert protective effects against hepatic and renal lipid accumulation and tissue injury induced by ER stress.
We report here, that HO HCU mice exhibiting elevated cystathionine show significantly attenuated liver injury and steatosis in the presence of an adipogenic methionine-choline deficient diet. Additionally, we show that elevated cystathionine, independent of its role as a cysteine donor, greatly attenuates hepatic and renal lipid accumulation, tissue injury, and cell death induced by ER stress. Subsequent analysis revealed that cystathionine does not appear to protect against the pathological effects of ER stress by modulating expression levels of the unfolded protein response. Collectively, these findings constitute the first experimental evidence that the abolition of cystathionine synthesis may contribute to the pathology of HCU and that this compound has therapeutic potential for disease states where ER stress is implicated as a primary initiating pathogenic factor.
MATERIALS AND METHODS
Chemicals and Reagents
Unless otherwise stated, all chemicals were obtained from Sigma. All reagents for mammalian tissue culture were obtained from Life Sciences Technologies (Rockville, MD) except for fetal calf serum, which was obtained from HyClone (Logan, UT).
Media and Mammalian Cell Culture
CBS and CGL positive HepG2 hepatocellular carcinoma cells and human embryonic kidney 293AD cells were obtained from the American Type Culture Collection (Manassas, VA) and were cultured as described previously (6). CBS and CGL negative Chinese hamster fibroblast a23 cells were obtained from the University of Colorado Intellectual Developmental Disabilities Research Center central collection and were cultured as described previously (6).
Tunicamycin Treatment of Cultured Cells and Assessment of Cell Death
HepG2, 293AD, and a23 cells were seeded in triplicate at ∼75% confluence and were pre-treated with either propargylglycine (PPG) (5 mm) or cystathionine (1 mm) in complete media (DMEM, 10% FBS). After 24 h, fresh media with 1% FBS was added to each well. Cells were then further treated with tunicamycin (either 5 or 10 μg/ml) for 18 h. Cells were subsequently photographed under phase-contrast with a Zeiss Axiovert 25 microscope for evidence of dead floating cells. To quantify cell death the media supernatant was collected for LDH release assay using a kit from Roche Applied Science (Cat.No.11 644 793 001) according to the manufacturer's standard protocol where cytotoxicity (%) = (experimental value - low control/(high control-low control) × 100.
Animal Experiments and Tissue Sample Analysis
All animal experiments were pre-approved by the University of Colorado Health Sciences Center institutional animal care and use committee. Male HO mice (3 to 5 months old) were generated as described previously (4). Male, C57BL/6J mice (3–6 months) were used as wild type (WT) controls and were bred in house. Male mice were used exclusively to conserve females for breeding. To prevent fighting between non-sibling males, mice were kept in individual cages on a 12 h light/-dark cycle at a mean temperature of 22 °C.
Animal Treatments
Propensity for hepatic steatosis was examined in HO and wild type control mice using a methionine-choline deficient amino acid defined and iron supplemented diet (#518810 Dyets inc, Bethlehem, PA) and an isocaloric amino acid defined Lombardi choline sufficient with methionine control diet (#518754, Dyets Inc, Bethlehem, PA) for 19 days as described previously (7). Experimental hypercystathionemia was induced by intra-peritoneal (IP) injection of the CGL inactivator PPG (50 mg·day−1·kg−1) as described previously (8). ER stress was induced in mice by a single IP injection with tunicamycin at 0.5 mg/kg body weight (volume < = 1.0 ml). Control mice were sham treated by injection with an equal volume of phosphate-buffered saline (PBS) (9, 10). After the completion of treatment, all mice were anesthetized (isofluorane/oxygen) and sacrificed by decapitation, and blood samples were collected for plasma analysis. Livers and kidneys were removed, weighed, and then samples were either immersion-fixed overnight in 4% paraformaldehyde in PBS (pH 7.3) for subsequent histological analysis or were snap frozen in liquid nitrogen for subsequent biochemical analyses.
Thiols and Methionine Cycle Metabolites
Determination of plasma levels of methionine cycle metabolites was performed as described previously (11).
Histological Examination of Tissues and Assessment of Hepatopathy
Tissues were immersion-fixed overnight in 4% paraformaldehyde in PBS (pH 7.3). Paraffin embedded sections (5 μm) were stained for examination with hematoxylin and eosin (H and E). Cells undergoing apoptosis were identified by labeling their DNA 3′-OH nick ends using a variant of terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) staining. Staining was performed according to the manufacturer's instruction with ApopTag® Peroxidase in Situ Apoptosis Detection Kit (Millipore, Billerica, MA). Tissue sections were deparaffinized with xylene and washed in succession with different concentrations of ethanol (absolute, 95 and 70%). Tissue sections were then digested with proteinase K (20 μg/ml) for 15 min. Endogenous peroxidase activity was quenched with 3% H2O2 in PBS for 5 min. The slides were immersed in terminal deoxynucleotidyl transferase (TdT) buffer containing digoxigenin-labeled nucleotides at 37 °C for 1 h. After washing, anti-digoxigenin-peroxidase was added to cover the slides and incubated in a humid chamber at room temperature for 30 min. After washing with PBS, the slides were stained with diaminobenzidine and then counterstained with 1% methyl green. Light microscopy was performed to quantify the apoptotic cells, by counting cells in ten randomly selected fields (×100 magnification) of each slide with the following formula: number of apoptotic cells/total number of cells counted × 100. Both histological analysis of stained slides and TUNEL staining were performed by a party unaware of the animal genotype or treatment group. Liver injury was assessed by determining plasma levels of alanine aminotransferase (ALT) activity using an enzyme-coupled assay with lactic dehydrogenase (LDH) as described previously (12) Liver lipid was extracted using the procedure of Bligh and Dyer (13) and after evaporation of the organic solvent, the triacylglycerol content of each sample was measured in duplicate using an enzymatic method (Sigma-Aldrich).
Immmunoblot Analysis
Immunoblot analysis of total cell lysates was performed as described previously (5, 14). Briefly, following incubation with the appropriate primary and horseradish peroxidase-conjugated secondary antibodies (Affinity Biologicals, Ancaster, ON, Canada) membranes were developed using the renaissance Western blot chemiluminescence reagent kit, (Perkin Elmer, Woodbridge, ON, Canada). Proteins were transferred onto a nitrocellulose membrane, and probed with anti-KDEL monoclonal antibody, which recognizes both GRP78 and GRP94 that was purchased from Stressgen Biotechnologies (SPA-827) or anti GADD153 (Santa Cruz Biotech #SC-7351). Anti-β-Actin monoclonal antibody (Sigma) was used as a loading control.
Preparation of Total RNA and Northern Blot Analysis
Total RNA was isolated from 293AD cells using the RNeasy total RNA kit (Qiagen, Santa Clarita, CA) and resuspended in diethylpyrocarbonate (DEPC)-treated water. Quantification and purity of the RNA was assessed by A260/A280 absorption, and RNA samples with ratios greater than 1.6 were stored at −70 °C for further analysis. Total RNA (10 μg/lane) was fractionated on 2.2 mol/liter formaldehyde 1.2% agarose gels and transferred overnight onto Zeta-Probe GT nylon membranes (Bio-Rad) in 10× SSC. The RNA was cross-linked to the membrane using a UV cross-linker (PDI Bioscience, Toronto, Canada) before hybridization. Specific probes were generated by labeling cDNA fragments with [α-32P]dCTP (NEN) using a random primed DNA labeling kit (Roche Applied Science, Laval, Canada). The cDNA probe for GADD153 (no. AA627477) was obtained from Genome Systems (St Louis, MO). The cDNA probe used for GRP78 is a full length cDNA clone that has been described previously (5). After overnight hybridization at 43 °C, the membranes were washed as described by the manufacturer, exposed to x-ray film, and subjected to autoradiography.
Statistical Analysis
All data are presented as means ± S.D. and were compared using the unpaired Student's t test. A p value of less than 0.05 was considered statistically significant. In the graphed data *, **, and *** denote p values of < 0.05, 0.01, and 0.001 respectively.
RESULTS
HO HCU Mice Exhibit Significantly Attenuated Progression of Methionine and Choline-deficient Diet-induced Hepatic Steatosis
To date, all Cbs-null mouse models of HCU have been found to incur severe liver injury and a high degree of neonatal lethality (2, 3, 15). Those Cbs-null mice that survive the neonatal period, invariably incur profound hepatic steatosis (2, 3). Despite having essentially identical levels of plasma and tissue Hcy as the Cbs-null models, the HO HCU mice do not exhibit any discernible hepatic steatosis (4) leading us to speculate that the elevated cystathionine exhibited by HO mice might be exerting protective effects against aberrant hepatic lipid accumulation. To investigate this possibility, we determined the relative degree of hepatic lipid accumulation in HO and WT control mice fed a methionine- and choline-deficient (MCD) diet. This diet rapidly induces hepatic steatosis as methionine and choline are lipotropic precursors of phosphatidylcholine synthesis and their exclusion from the diet acts to inhibit the assembly of very low density lipoprotein (VLDL) causing impairment of the secretion of triglycerides and free fatty acids from hepatocytes (16).
One group of HO mice and one group of WT control mice (n = 5 for each) were pair-fed an MCD diet as described in “Materials and Methods.” Control groups of both genotypes (n = 5) were pair-fed a chow diet that matched the composition of the MCD diet except that it contained normal levels of choline and methionine. All groups were kept on their respective diets for a total of 19 days. At the end of the trial, the mice were sacrificed and the livers of these animals were examined. Histological analysis was performed by a party unaware of the animal genotype or treatment group. No hepatic steatosis was observed in either of the control groups (data not shown). Histological examination of the livers after H+E staining found that the MCD diet induced severe macrovesicular hepatic steatosis, ballooning degeneration of hepatocytes and multiple inflammatory foci in the WT mice. These sequelae were greatly attenuated in the HO HCU mice (Fig. 2A). To quantitate the development of hepatic steatosis induced by the MCD diet, we measured the triglyceride content in the livers of the MCD fed HO and WT mice and in the animals fed the control diet (Fig. 2B). The triglyceride levels of both the HO and WT animals on the control chow diet did not differ significantly from each other (p = 0.707). The MCD diet induced an approximate 6-fold increase in the hepatic triglyceride levels of WT animals but only a 2-fold increase in HO mice. This observation is consistent with our histological analysis and collectively, these data support the possibility that elevated cystathionine can exert hepatoprotective effects.
FIGURE 2.

Hepatic steatosis induced by a methionine-choline-deficient diet is significantly attenuated in HO HCU mice. A, H and E staining from representative liver sections of C57BL/6 WT control (upper panel) and HO (lower panel) mice fed an MCD diet for 17 days. Data are representative of 5 animals per group. Scale bar denotes 200 micrometers. B, hepatic triglyceride levels from HO and C57BL/6 WT mice fed an MCD or control diet (n = 5). In this and all subsequent figures *, **, and *** denote p values of < 0.05., 0.01, and 0.001 respectively.
HO Mice Exhibit Ablated Hepatic and Renal Injury after Tunicamycin Treatment
The ER is the primary site for the synthesis, folding and modification of proteins in the eukaryotic cell and exposure of cells to conditions such as inhibition of protein glycosylation, nutrient/oxygen deprivation or perturbation of Ca2+ homeostasis, results in the accumulation of unfolded proteins causing ER stress (17). Previous work has indicated that the MCD diet induces hepatic steatosis at least in part by inducing ER stress (7). Similarly, previous work has suggested that the steatosis observed in Cbs null mice is directly related to the action of elevated Hcy inducing ER stress causing an alteration of serum response element-binding protein 1 (SREBP1) function that subsequently results in dysfunctional lipid metabolism (5). The ability of the HO mice to resist hepatic steatosis in the presence of an MCD diet and our previous observation of distended ER in the steatotic Cbs-null mice but not the HO mice raises the possibility that elevated cystathionine in HO mice is exerting its hepatoprotective effects by blocking the induction of ER stress.
To examine this possibility, we used a tunicamycin mediated model of ER stress induced tissue injury on groups of HO and WT mice as described in “Materials and Methods.” The nucleoside antibiotic, tunicamycin, produced by the actinomycete, Streptomyces lysosuperifcus, causes ER stress by inhibiting UDP-N-acetylglucosamine:dolichol phosphate N-acetylglucosamine-1-P transferase and thus blocking protein N-glycosylation. Previous work has shown that a single sub-lethal IP injection with 0.5 mg/kg body weight of tunicamycin results in a reproducible pattern of lassitude, lack of grooming, weight loss, renal injury, and hepatic steatosis that peaks between day 4 and 5 post-injection (9, 10). Groups of WT and HO mice (n = 5 for all groups) were given either a single intraperitoneal injection of tunicamycin or sham injected with an identical volume of the vehicle solution PBS. All mice treated with tunicamycin became markedly inappetant shortly after injection and marked weight loss was clearly noticeable and progressive during the course of the experiment. Three days after the administration of treatment, mice were sacrificed and tissues were extracted and processed for histological analysis. In the sham-injected control animals, no evidence of tissue injury or steatosis was observed (data not shown). In the WT control mice we observed, as expected, significant weight loss, lassitude, and altered fur. Additionally, we observed that the livers of the tunicamycin treated wild type animals were severely enlarged and yellowish in color. Histologically, H and E staining revealed a clearly discernible pattern of periportal hepatocellular damage with severe vacuolation of hepatocytes, much of which appeared to result from lipid accumulation. Single, condensed, rounded, or ovoid cytoplasmic bodies, apparently derived from degenerating hepatocytes, were scattered throughout the liver, but especially prevalent in the periportal region (Fig. 3). The most severe injury was in the kidney, where we observed significant enlargement and accumulation of fat around the organ. Histological analysis revealed severe vacuolation, steatosis, and acute tubular necrosis with swelling of proximal tubular epithelial cells with pyknotic nuclei and tubules with focal areas of denuded basal lamina. The histological alterations were primarily located in the proximal tubular epithelium. (Fig. 3). These morphological changes are very similar to those described in mice and other species following tunicamycin injection (9, 18, 19).
FIGURE 3.

Tunicamycin induced hepatic steatosis and renal tubule damage is greatly attenuated in HO HCU mice. Representative H and E staining of liver and kidney sections from HO and C57BL/6 WT mice harvested 3 days after treatment with Tuc as described in “Materials and Methods.” Data shown are representative of n = 5 per group. Scale bar denotes 200 micrometers.
HO mice treated with tunicamycin incurred essentially identical weight loss, lassitude and altered grooming as the WT mice but strikingly, did not exhibit any significant liver enlargement with only minimal hepatic steatosis and no discernible alteration of renal morphology (Fig. 3). These data are consistent with the possibility that elevated cystathionine acts to protect against ER stress-mediated tissue injury in both the liver and kidney.
Daily Injection with the CGL Inactivating Compound PPG Causes Temporary Hypercystathionemia in Normal Mice
Although the data described above indirectly supports the possibility that cystathionine exerts tissue protective effects induced by prolonged ER stress, there are multiple biochemical differences between the HO mice and WT mice (3, 4, 20, 21). To provide more direct evidence of a protective role for cystathionine, we proposed to induce elevated cystathionine in normal mice to examine its protective properties in isolation from the severely elevated Hcy, methionine, AdoMet, AdoHcy, and decreased cysteine that is observed in the HO mice.
Inducing elevated cystathionine in normal mice is complicated by the fact that this compound has relatively low solubility in water and is rapidly excreted in the urine of mice. Previous work by Abeles and Walsh synthesized and tested a range of acetylenic substrates capable of irreversible inactivation of CGL (22).
I.P. injection of one of these inactivators, d,l-2-amino-4-pentynoic acid also known as PPG has been shown to rapidly inactivate hepatic CGL activity and results in a concomitant accumulation of cystathionine within 1 h that lasts for approximately 1 day (23). For our purposes, this method has the additional benefit of allowing us to investigate the possible protective effects of elevated cystathionine in isolation from its role as a cysteine donor. To our knowledge, the longevity of this cystathionine accumulation with repeated injections of PPG over time in mice has not been examined. We gave daily injections of PPG to WT mice (n = 7) each morning over a period of 4 days. Plasma samples were taken from these mice in the evening of each day by non-lethal tail bleed and the levels of cystathionine, Hcy, methionine, and cysteine were determined as described in “Materials and Methods” (Fig. 4, A and B). We observed that inactivation of CGL was accompanied by a 56-fold increase in the plasma level of cystathionine on day 1. This maximal accumulation persisted for the second day and was reduced to 30-fold by day 3. By day 4 of the experiment, the level of cystathionine accumulation was reduced to 8-fold compared with untreated animals indicating that despite daily re-administration of this compound, the effect of PPG diminishes over time. Blocking transsulfuration also induced a relatively mild elevation in total Hcy (tHcy) and methionine that declined back to normal as the efficacy of the PPG treatment diminished. As expected, blocking transsulfuration via PPG treatment resulted in a concomitant decrease in plasma cysteine levels during the first and second days of PPG treatment. Cysteine levels subsequently returned to near normal as the effects of the PPG treatment diminished. These experiments allowed us to define an optimal time frame for examining the possible protective effects of PPG mediated cystathionine accumulation in WT mice. The effect of PPG on liver health was assessed by determining ALT values before PPG treatment and at each subsequent time point studied. PPG treatment did not induce any significant increase in plasma ALT levels at any of the time points analyzed indicating that this treatment is essentially benign over the time period studied (data not shown).
FIGURE 4.
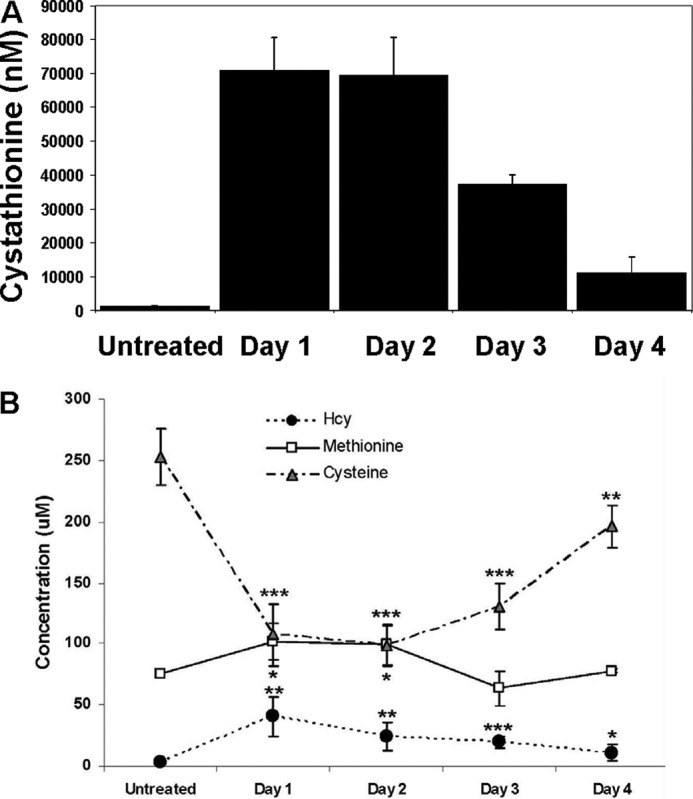
Daily PPG injection induces elevated plasma cystathionine in WT mice. Plasma levels in C57BL/6 WT mice of (A) cystathionine and (B) Hcy, methionine, and cysteine before and 1, 2, 3, and 4 days after treatment with the CGL inactivating compound PPG. Values are means ± S.D. at each time point (n = 7). Amino acid levels were determined as described in “Materials and Methods.”
Elevated Cystathionine Protects against Hepatic and Renal Injury Induced by ER Stress in WT Mice
To investigate the effects of elevated cystathionine upon the response to ER stress, we exposed wild type mice (n = 6) to a single sub-lethal IP injection of tunicamycin in the presence and absence of PPG. WT mice were randomly assigned to one of four treatment groups (n = 6 for each group): 1) control, IP injected with PBS; 2) IP injection with PPG; 3) single IP injection with tunicamycin and 4) tunicamycin + PPG. In groups 2 and 4, PPG treatment was maintained by daily injection until sacrifice at 3 days postvehicle/tunicamycin injection. After completion of this treatment regime, the livers and kidneys of all of the sham injected and PPG alone control animals were found to be normal in size and appearance. The livers from the animals treated solely with tunicamycin were enlarged, yellowish, and fatty in appearance (Fig. 5A). Similarly, the kidneys of these animals were also swollen and covered in fat (Fig. 6A). This enlargement was reflected in increased liver and kidney weights compared with the controls (Figs. 5B and 6C). Determination of triglyceride content in the liver and kidneys found a highly significant increase in the liver (p < 0.001) and kidneys (p < 0.001) of the animals treated solely with tunicamycin compared with the sham and PPG control groups confirming the induction of hepatic and renal steatosis by this toxin (Figs. 5C and 6B). The induction of significant liver injury by tunicamycin treatment was also confirmed by determination of plasma ALT levels. (Fig. 5D). Strikingly, livers and kidneys from the hypercystathionemic mice that were treated with tunicamycin appeared ostensibly normal and did not differ significantly from the control groups in weight, triglycerides, or plasma ALT levels.
FIGURE 5.

PPG induced elevated cystathionine significantly protects WT mice against tunicamycin induced steatosis and liver injury. A, representative examples of whole livers dissected out of C57BL/6 WT mice from the four experimental groups. Livers from the Tuc group were all severely enlarged, yellowish, and fatty in appearance. Liver morphology appeared ostensibly normal in mice pretreated with PPG prior to tunicamycin administration. B, liver weight as an index of hepatic enlargement. C, hepatic trigycerides as an index of steatosis and D, plasma ALT as an index of liver injury were determined as described in “Materials and Methods.” Values are means ± S.D. (n = 6).
FIGURE 6.

PPG induced elevated cystathionine significantly attenuates steatotic renal enlargement in WT mice treated with tunicamycin. A, representative examples of whole kidneys dissected out of C57BL/6 WT mice from the four experimental groups. Kidneys from the Tuc group were all severely enlarged and were encased in a relatively large amount of fat. In mice treated with PPG prior to tunicamycin administration, this effect was not apparent. B, kidney trigycerides as an index of steatosis were determined as described in “Materials and Methods.” C, kidney weight as an index of enlargement. Values shown represent means ± S.D. n = 6.
Histological analysis revealed that the sham injection and PPG control groups showed no obvious morphological alteration while the tunicamycin alone group incurred severe morphological changes in the liver and kidney that were essentially identical to those described above. These morphological changes were effectively ablated in the mice that were induced into hypercystathionemia by PPG treatment during the tunicamycin treatment (Fig. 7, A and B).
FIGURE 7.

PPG induced elevated cystathionine significantly attenuates steatotic hepatopathy, renal tubular injury and apoptotic cell death in C57BL/6 WT mice treated with tunicamycin. H and E staining of representative sections from (A) liver and (B) kidneys from WT mice treated with Tuc in the presence and absence of elevated cystathionine induced by PPG. Tunicamycin induced hepatic steatosis and acute tubular necrosis with swelling of proximal tubular epithelial cells. These tunicamycin induced morphological alterations were virtually ablated in mice treated with PPG. Sections shown are representative of each experimental group (n = 6). C, assessment of apoptosis by TUNEL staining of representative kidney sections from WT mice treated with Tuc in the presence and absence of elevated cystathionine induced by PPG. Apoptotic cells appear dark brown. Scale bars denote 200 micrometers.
To further assess the protective effects of cystathionine, we analyzed the tissues of all of these mice for evidence of apoptosis using TUNEL staining. No apoptosis was observed in either of the control groups. In the tunicamycin-treated animals, the most affected organ was the kidney. In this tissue, the proximal tubule epithelium in particular showed clear evidence of apoptotic cell death (Fig. 7C). No apoptotic cells were observed in the kidneys of tunicamycin treated mice that had been pretreated with PPG. Collectively, these results indicate that cystathionine can exert significant protective effects against ER stress-mediated lipid accumulation, tissue injury, and apoptotic cell death.
Cells Treated with Cystathionine Have Increased Resistance to the Death-promoting Effects of ER Stress
The results from the in vivo model presented above indicate that in addition to protecting against ER stress induced lipid accumulation and tissue injury, cystathionine may also play a role in promoting cell survival by attenuating the induction of apoptosis. Examination of a direct effect of exogenously added cystathionine is not currently possible in a whole animal model because of complications with the solubility of this compound and the rapid kinetics of its excretion. To investigate the direct effects of cystathionine against ER stress-induced cell death, we examined the cytotoxicity of tunicamycin in the human embryonic kidney cell line 293AD and the human hepatoma cell line HepG2 in the presence and absence of both PPG and exogenously added cystathionine. Preliminary experiments confirmed that both of these cell lines express both CBS and CGL (data not shown). To investigate the possible protective effects of cystathionine in the absence of transsulfuration, we repeated these experiments with the CBS and CGL negative Chinese hamster fibroblast line a23 (6).
Cells were seeded in triplicate at ∼75% confluence and pre-treated with either PPG) (5 mm), cystathionine (1 mm), or with media alone (DMEM, 10% FBS) as a control. After 24 h pre-treatment, cells appeared normal, with minimal floating cells. Fresh media with 1% FBS was then added to each well, the cells were further treated with tunicamycin (either 5 or 10 μg)/ml. After 18 h treatment with tunicamycin, cells were photographed under phase-contrast for evidence of dead floating cells. Cells from the group treated solely with tunicamycin became strikingly hyper-refringent, detached from the substratum and floated in the media. Conversely, No floating dead cells were observed in either the PPG or cystathionine pre-treated cells (Fig. 8A).
FIGURE 8.

Cystathionine protects against ER stress induced cell death in 293AD and a23 cells. A, photomicrograph of 293AD cells treated with tunicamycin in the presence and absence of PPG. Detached floating cells were clearly visible in the tunicamycin group and were not evident in the PPG plus tunicamycin group indicating that this compound protects cells against the cytotoxic effects of tunicamycin treatment. B, relative survival of transsulfuration-positive 293AD (upper graph) and transsulfuration-deficient a23 cells (lower graph) treated with tunicamycin (5 or 10 μg/ml) in the presence or absence of either PPG (5 mm) or cystathionine (1 mm). Cell viability was assessed by LDH release assay as described in “Materials and Methods.” Shown are means ± S.D. of a typical experiment performed in triplicate that is representative of three independent experiments. The ability of cystathionine, but not PPG, to protect against ER stress-induced cell death in transsulfuration negative a23 cells indicates that cystathionine itself is directly cytoprotective and is not simply serving as a cysteine prodrug.
To further evaluate a role for cystathionine in resisting the induction of cell death by tunicamycin, cell lysis was quantified by assaying lactate dehydrogenase (LDH) release. LDH enzyme activity was determined spectrophotometrically and the amount of enzyme leak was expressed as a percentage of the LDH activity observed in untreated control cells. Treatment of 293AD cells with 5 μg/ml of tunicamycin resulted in significant cell death that was largely prevented by pre-treatment with PPG (Fig. 8A). Treatment of the cells with a higher dose of tunicamycin (10 μg/ml) resulted in an even more pronounced protective effect with PPG and cystathionine inducing an 11- and 27-fold reduction in cell death respectively (p < 0.0001 for both). We observed essentially identical results when this experiment was repeated using HepG2 cells (data not shown). When this experiment was repeated with the transsulfuration negative a23 cells, cystathionine was still clearly protective with a 16-fold reduction in tunicamycin induced cell death (p < 0.0001) but PPG conferred no significant cytoprotection (p = 0.7301). This latter observation allows us to discount the possibility that PPG itself or a metabolite thereof is responsible for the protection independent of its role in inducing elevated cystathionine. From these results, we conclude that cystathionine promotes increased survival of cells exposed to toxic levels of ER stress.
Cystathionine Exerts Its Protective Effects Against ER Stress Induced Cell Death without Modulating Induction of the Unfolded Protein Response
Eukaryotic cells respond to ER stress by activating a set of pathways known collectively as the unfolded protein response (UPR). The UPR is transmitted through the activation of ER resident proteins, such as protein kinase-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-required enzyme 1 (Ire1α/β). These signaling pathways induce the expression of a number of molecular chaperones such as the KDEL amino acid signature containing proteins glucose-regulated protein (GRP) 78 (also Known as Bip) and 70 that play a role in assisting the correct folding of proteins in an effort to try and ameliorate the accumulation of misfolded proteins and thus alleviate ER stress (24). If ER stress is prolonged and the burden of misfolded proteins in the ER exceeds its folding capacity, cells are typically induced into apoptosis. Prolonged perturbation of the ER is a powerful inducer of the C/EBP family member GADD153 (also known as CHOP) which plays a key role in the induction of programmed cell death by ER stress (10). To assess the effect of cystathionine upon induction of the UPR and GADD153 by ER stress we performed Northern and Western blotting experiments as described in “Materials and Methods.” Neither PPG nor exogenously added cystathionine appeared to markedly induce expression of the KDEL containing proteins Grp78 or Grp94 or lower expression of the cell death promoting gene GADD15 in the control cells (Fig. 9, A and B). Nor did either of these treatments appear to drastically alter the scale of induction of these proteins in the presence of tunicamycin treatment.
FIGURE 9.

Cystathionine does not exert its cytoprotective effects by modulating the induction levels of either Grp78 or GADD153. A, Northern blot determination of Grp78 and GADD153 mRNA levels induced by 5 and 10 μg/ml tunicamycin in the presence and absence of 1 mm PPG in 293AD cells. Cells treated with 5 mm Hcy were included as a positive control for Grp78 induction. Cell treatment conditions, transfer, probe generation, and detection were performed as described in “Materials and Methods.” B, Western blotting analysis of the tunicamycin mediated induction of the UPR molecular chaperones GRP78 and GRP94 in the presence and absence of PPG (5 mm) and cystathionine (1 mm) in 293AD cells. Cell treatment conditions, transfer, antibody, and detection were performed as described in “Materials and Methods.”
DISCUSSION
To our knowledge, the studies described here, constitute the first direct experimental evidence of a possible function for cystathionine independent of its role as an intermediate in transsulfuration. Previous work has shown that cystathionine can exert protective effects in a mouse model of acetaminophen-induced liver injury by serving as a cysteine prodrug (25). The use of PPG in our experiments, which induces hypercystathionemia by blocking the conversion of cystathionine to cysteine (26) coupled with the observation that cystathionine exerts significant protective effects in a23 cells that are incapable of converting this compound to cysteine, indicate that the protective effects observed in our experiments are not a result of cystathionine acting as a precursor for cysteine synthesis.
A number of indirect lines of evidence support possible physiological roles for either cystathionine or a derivative thereof. In primate brain and CNS, there appears to be an imbalance between the relative activity levels of CBS and CGL leading to an accumulation of cystathionine (27). Similarly, during embryonic development, CBS is expressed in multiple tissues such as the heart and lungs that do not express any detectable CBS in adult tissues (28). A range of data indicates that, CGL, is not expressed during early mammalian development. In human liver samples for instance, CGL activity is only detected in post-natal tissue whereas activity in fetal, premature, and full-term neonatal liver tissue is essentially undetectable (29–31). Collectively, these results indicate that at certain stages of development and in adult neural tissues, CBS is expressed specifically for the production of cystathionine distinct from its role as an intermediate in cysteine synthesis.
The studies described here, using both tissue culture and whole animal models strongly point to a role for cystathionine in protecting against ER stress induced tissue damage and cell death. However, this interpretation must be qualified somewhat as previous research has shown that in addition to the transsulfuration pathway, there are other metabolic fates for cystathionine, the biochemistry of which is only partially understood. Previous investigations have observed that inducing elevated cystathionine with PPG in rats also results in a detectable increase in the cystathionine metabolites perhydro-1,4-thiazepine-3,5-dicarboxylic acid, cystathionine mono-oxo acids [S-(3-oxo-3-carboxy-n-propyl)cysteine, and S-(2-oxo-2-carboxyethyl)homocysteine], cystathionine ketimines, cystathionine sulfoxide and N-acetylcystathionine sulfoxide. The level of these compounds increase in direct proportion to the scale of the accumulation of cystathionine (32). The increase in the levels of these cystathionine metabolites is not an artifact of the PPG compound itself as all of these metabolites been identified previously at elevated levels in the urine of patients with cystathioninuria due to mutational inactivation of the human CGL gene (33). This is a generic concern in interpreting protective effects of a compound where the biochemistry, regulation, and tissue distribution of its metabolism are incompletely understood. Consequently, at this stage, we cannot unequivocally rule out the possibility that one or more of the metabolites of cystathionine are responsible for the protective effects observed in this study. Further work is required to see if any of these cystathionine metabolites are capable of exerting cytoprotective effects.
HCU is unique among the genetic homocystinurias in that in addition to elevated Hcy, methionine, AdoMet, and AdoHcy, it also incurs a concomitant abolition of cystathionine and cysteine synthesis. While cysteine can be obtained from the diet, HCU acts to effectively remove cystathionine. The finding that cystathionine exerts cytoprotective effects raises the possibility that the abolition of its synthesis might contribute to pathogenesis in HCU. The observation that the HO mouse model of HCU exhibits a hypercoagulative phenotype in the presence of 4-fold higher than normal levels of cystathionine (4, 20, 21) and the fact that increased risk of thromboembolic complications is a common feature of all of the genetic homocystinurias indicates that the abolition of cystathionine synthesis is unlikely to be contributing to this aspect of pathogenesis.
The connective tissue defects of HCU are unique among the genetic homocystinurias. Because of the strong similarity between these sequelae and those observed in the fibrillinopathy Marfan syndrome, the leading candidate mechanism for connective tissue disturbances in HCU is impairment of the folding/function of fibrillin-1. This protein is highly cysteine rich, much of which appears in the free reactive sulfhydryl form. The functional significance of these vulnerable cysteines is underscored by the fact that many of the fibrillin-1 mutations found in Marfan syndrome delete or alter the spacing of these residues (34). Because of this high cysteine content, it is likely that the folding pathway and assembly of this protein is relatively complicated to prevent the inappropriate juxtaposition of cysteine residues and subsequent formation of erroneous disulfide bonds. Consequently, fibrillin-1 may be particularly vulnerable to the protein misfolding that is typically associated with ER stress. Recent work has suggested homocysteinylation of cysteine residues in fibrillin-1 as a mechanism for impairing folding and inducing fibrillinopathy in HCU (35, 36). One possibility is that cystathionine protects against ER stress by serving as a chemical chaperone and could thus conceivably assist with the correct folding of fibrillin-1 in the presence of elevated Hcy.
The mechanisms that underlie mental retardation in HCU are unknown but as described above, CBS and CGL expression in the mammalian brain appeared to be specifically tailored toward the accumulation of cystathionine. Enhanced levels of ER stress are implicated in various neuropathological conditions. Examples include brain ischemia and excitotoxicity in neurons (37) and neurodegenerative conditions such as Parkinson disease (38). It is therefore possible that cystathionine specifically accumulates in the normal mammalian brain to serve as a cytoprotectant and that the abolition of its synthesis could contribute to mental retardation in HCU by increasing sensitivity of neural tissues to the toxic insult of elevated Hcy and/or derivatives thereof.
The process of apoptosis can be functionally divided into two distinct phases, induction and execution. A key a point of convergence of the many different apoptogenic stimuli in the induction phase has been shown to be the specific extrusion of cellular thiols including the antioxidant glutathione (GSH) in the reduced form prior to any plasma membrane leakage (39–41). Cystathionine has previously been shown to be able to inhibit apoptosis, independent of its role as a cysteine donor compound, by inhibiting the specific sinusoidal type carrier mediated efflux of GSH in two different cell lines. The ability of cystathionine to effect a rescue to perfect viability upon the removal of the apoptogenic stimuli, indicated that this compound exerts its protective role at an early step of the induction phase, before any irreversible involution of cellular structure occurs. Given the almost normal appearance of the liver and kidney in both HO and PPG treated hypercystathionemic mice treated with tunicamycin and the almost complete lack of cell death in the tissue culture studies, it appears likely that cystathionine is exerting its protective effects in the induction phase of apoptosis in these models. If thiol extrusion is a point of convergence for multiple apoptogenic stimuli it is quite possible that cystathionine could exert protective effects against multiple intracellular disturbances in addition to ER stress. This possibility is currently the subject of investigation in our laboratory.
In addition to HCU, the observation that elevated cystathionine promotes cell survival in the face of cytotoxic challenge has wider clinical implications. Neuroblastoma is the commonest and most deadly solid tumor in children under the age of 5 years (50% of cases before 2 years, 90% before 5) (42, 43). Most children older than 1 year have extensive or metastatic disease at diagnosis and their prognosis is generally poor. Massive accumulation of cystathionine and subsequent cystathionuria has been observed as a frequent and highly specific marker for neuroblastoma by multiple independent groups (44–47). The data presented here indicates that the cystathionine accumulated in neuroblastoma has the potential to allow the cells to resist cytotoxic chemotherapy. The use of a CBS inactivating drug might therefore serve to increase chemosensitization and improve clinical outcome in neuroblastoma. This possibility is the subject of current research in our laboratory.
This work was supported in part by research grants (to R. C. A.) from the Heart and Stroke Foundation of Ontario (T-6146), the Canadian Institutes of Health Research (MOP-74477), and the Ontario Research and Development Challenge Fund. This work was also supported by the William R. Hummel Homocystinuria Research Fund, the Jerome LeJeune Foundation, Special Products U.K., and research award 6703 from the Denver Children's Hospital (to K. N. M.). Financial support from St. Joseph's Healthcare Hamilton is acknowledged.
- CBS
- cystathionine β-synthase
- ALT
- alanine aminotransferase
- HCU
- CBS-deficient homocystinuria
- CGL
- cystathionine γ-lyase
- ER
- endoplasmic reticulum
- H and E
- hematoxylin and eosin
- Hcy
- homocysteine
- LDH
- lactate dehydrogenase
- MCD
- methionine choline-deficient
- PPG
- propargylglycine
- tHcy
- total homocysteine
- Tuc
- tunicamycin.
REFERENCES
- 1. Mudd S. H., Levy H. L., Kraus J. P. (2001) in The Metabolic and Molecular Bases of Inherited Disease (Scriver C. R., Beaudet A. L., Sly W. S., Valle D., Childs B., Kinzler K., Vogelstein B., eds), pp. 2007–2056, 8 Ed., McGraw-Hill, New York [Google Scholar]
- 2. Watanabe M., Osada J., Aratani Y., Kluckman K., Reddick R., Malinow M. R., Maeda N. (1995) Mice deficient in cystathionine β-synthase: animal models for mild and severe homocyst(e)inemia. Proc. Natl. Acad. Sci. U.S.A. 92, 1585–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maclean K. N., Sikora J., Kožich V., Jiang H., Greiner L. S., Kraus E., Krijt J., Crnic L. S., Allen R. H., Stabler S. P., Elleder M., Kraus J. P. (2010) Cystathionine beta-synthase null homocystinuric mice fail to exhibit altered hemostasis or lowering of plasma homocysteine in response to betaine treatment. Mol. Genet. Metab. 101, 163–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maclean K. N., Sikora J., Kožich V., Jiang H., Greiner L. S., Kraus E., Krijt J., Overdier K. H., Collard R., Brodsky G. L., Meltesen L., Crnic L. S., Allen R. H., Stabler S. P., Elleder M., Rozen R., Patterson D., Kraus J. P. (2010) A novel transgenic mouse model of CBS-deficient homocystinuria does not incur hepatic steatosis or fibrosis and exhibits a hypercoagulative phenotype that is ameliorated by betaine treatment. Mol. Genet. Metab. 101, 153–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Werstuck G. H., Lentz S. R., Dayal S., Hossain G. S., Sood S. K., Shi Y. Y., Zhou J., Maeda N., Krisans S. K., Malinow M. R., Austin R. C. (2001) Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J. Clin. Invest. 107, 1263–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maclean K. N., Kraus E., Kraus J. P. (2004) The dominant role of Sp1 in regulating the cystathionine β-synthase-1a and -1b promoters facilitates potential tissue-specific regulation by Kruppel-like factors. J. Biol. Chem. 279, 8558–8566 [DOI] [PubMed] [Google Scholar]
- 7. Rahman S. M., Schroeder-Gloeckler J. M., Janssen R. C., Jiang H., Qadri I., Maclean K. N., Friedman J. E. (2007) CCAAT/enhancing binding protein beta deletion in mice attenuates inflammation, endoplasmic reticulum stress, and lipid accumulation in diet-induced nonalcoholic steatohepatitis. Hepatology 45, 1108–1117 [DOI] [PubMed] [Google Scholar]
- 8. Barber T., Triguero A., Martínez-López I., Torres L., García C., Miralles V. J., Viña J. R. (1999) Elevated expression of liver γ-cystathionase is required for the maintenance of lactation in rats. J. Nutr. 129, 928–933 [DOI] [PubMed] [Google Scholar]
- 9. Finnie J. W., O'Shea J. D. (1989) Acute hepatotoxicity with resultant pulmonary and cerebral embolism in guinea pigs given tunicamycin. Pathology 21, 194–199 [DOI] [PubMed] [Google Scholar]
- 10. Zinszner H., Kuroda M., Wang X., Batchvarova N., Lightfoot R. T., Remotti H., Stevens J. L., Ron D. (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 12, 982–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Allen R. H., Stabler S. P., Lindenbaum J. (1993) Serum betaine, N,N-dimethylglycine and N-methylglycine levels in patients with cobalamin and folate deficiency and related inborn errors of metabolism. Metabolism 42, 1448–1460 [DOI] [PubMed] [Google Scholar]
- 12. Bergmeyer H. U., Hørder M. (1980) International federation of clinical chemistry. Scientific committee. Expert panel on enzymes. IFCC document stage 2, draft 1; 1979–11-19 with a view to an IFCC recommendation. IFCC methods for the measurement of catalytic concentration of enzymes. Part 3. IFCC method for alanine aminotransferase. J. Clin. Chem. Clin. Biochem. 18, 521–534 [PubMed] [Google Scholar]
- 13. Bligh E. G., Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 14. Dickhout J. G., Hossain G. S., Pozza L. M., Zhou J., Lhoták S., Austin R. C. (2005) Peroxynitrite causes endoplasmic reticulum stress and apoptosis in human vascular endothelium: implications in atherogenesis. Arterioscler. Thromb. Vasc. Biol. 25, 2623–2629 [DOI] [PubMed] [Google Scholar]
- 15. Wang L., Chen X., Tang B., Hua X., Klein-Szanto A., Kruger W. D. (2005) Expression of mutant human cystathionine beta-synthase rescues neonatal lethality but not homocystinuria in a mouse model. Hum. Mol. Genet. 14, 2201–2208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vance J. E., Vance D. E. (1985) The role of phosphatidylcholine biosynthesis in the secretion of lipoproteins from hepatocytes. Can J. Biochem. Cell Biol. 63, 870–881 [DOI] [PubMed] [Google Scholar]
- 17. Schröder M., Kaufman R. J. (2005) The mammalian unfolded protein response. Annu. Rev. Biochem. 74, 739–789 [DOI] [PubMed] [Google Scholar]
- 18. Koj A., Bereta J., Dubin A., Kurdowska A., Chindemi P., Regoeczi E. (1986) Alpha-1-acute phase globulin in the blood of tunicamycin-injected rats. Isolation of the non-glycosylated form, its inhibitory properties and synthesis in liver slices. Folia Histochem. Cytobiol. 24, 7–14 [PubMed] [Google Scholar]
- 19. Finnie J. W., O'Shea J. D. (1988) Pathological and pathogenetic changes in the central nervous system of guinea pigs given tunicamycin. Acta Neuropathol. 75, 411–421 [DOI] [PubMed] [Google Scholar]
- 20. Maclean K. N., Jiang H., Greiner L. S., Allen R. H., Stabler S. P. (2012) Long-term betaine therapy in a murine model of cystathionine β-synthase deficient homocystinuria: decreased efficacy over time reveals a significant threshold effect between elevated homocysteine and thrombotic risk. Mol. Genet. Metab. 105, 395–403 [DOI] [PubMed] [Google Scholar]
- 21. Keating A. K., Freehauf C., Jiang H., Brodsky G. L., Stabler S. P., Allen R. H., Graham D. K., Thomas J. A., Van Hove J. L., Maclean K. N. (2011) Constitutive induction of pro-inflammatory and chemotactic cytokines in cystathionine β-synthase deficient homocystinuria. Mol. Genet. Metab. 103, 330–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Abeles R. H., Walsh C. T. (1973) Acetylenic enzyme inactivators. Inactivation of gamma-cystathionase, in vitro and in vivo, by propargylglycine. J. Am. Chem. Soc. 95, 6124–6125 [DOI] [PubMed] [Google Scholar]
- 23. Shinozuka S., Tanase S., Morino Y. (1982) Metabolic consequences of affinity labeling of cystathionase and alanine aminotransferase by l-propargylglycine in vivo. Eur. J. Biochem. 124, 377–382 [DOI] [PubMed] [Google Scholar]
- 24. Malhotra J. D., Kaufman R. J. (2007) The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 18, 716–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hazelton G. A., Hjelle J. J., Klaassen C. D. (1986) Effects of cysteine pro-drugs on acetaminophen-induced hepatotoxicity. J. Pharmacol. Exp. Ther. 237, 341–349 [PubMed] [Google Scholar]
- 26. Houghton C. B., Cherian M. G. (1991) Effects of inhibition of cystathionase activity on glutathione and metallothionein levels in the adult rat. J. Biochem. Toxicol. 6, 221–228 [DOI] [PubMed] [Google Scholar]
- 27. Volpe J. J., Laster L. (1972) Transsulfuration in fetal and postnatal mammalian liver and brain. Cystathionine synthase, its relation to hormonal influences, and cystathionine. Biol. Neonate 20, 385–403 [DOI] [PubMed] [Google Scholar]
- 28. Quéré I., Paul V., Rouillac C., Janbon C., London J., Demaille J., Kamoun P., Dufier J. L., Abitbol M., Chassé J. F. (1999) Spatial and temporal expression of the cystathionine β-synthase gene during early human development. Biochem. Biophys. Res. Commun. 254, 127–137 [DOI] [PubMed] [Google Scholar]
- 29. Sturman J. A., Gaull G., Raiha N. C. (1970) Absence of cystathionase in human fetal liver: is cystine essential? Science 169, 74–76 [DOI] [PubMed] [Google Scholar]
- 30. Gaull G., Sturman J. A., Räihä N. C. (1972) Development of mammalian sulfur metabolism: absence of cystathionase in human fetal tissues. Pediatric Research 6, 538–547 [DOI] [PubMed] [Google Scholar]
- 31. Levonen A. L., Lapatto R., Saksela M., Raivio K. O. (2000) Human cystathionine γ-lyase: developmental and in vitro expression of two isoforms. Biochem. J. 347, 291–295 [PMC free article] [PubMed] [Google Scholar]
- 32. Machida Y., Zhang J., Hashimoto K., Wakiguchi H., Kurashige T., Masuoka N., Ubuka T., Kodama H. (1995) Identification of perhydro-1,4-thiazepine-3,5-dicarboxylic acid, cystathionine mono-oxo acids, cystathionine ketimines, cystathionine sulfoxide, and N-acetylcystathionine sulfoxide in the urine sample of d,l-propargylglycine-treated rats. Physiol. Chem. Phys. Med. NMR 27, 203–216 [PubMed] [Google Scholar]
- 33. Zhang J., Masuoka N., Ubuka T., Sugahara K., Kodama H. (1995) Identification of N-acetyl-S-(3-oxo-3-carboxy-n-propyl)cysteine in the urine of a patient with cystathioninuria using LC/APCI-MS. J. Inherit. Metab. Dis. 18, 675–681 [DOI] [PubMed] [Google Scholar]
- 34. Pyeritz R. E. (2000) The Marfan syndrome. Annu. Rev. Med. 51, 481–510 [DOI] [PubMed] [Google Scholar]
- 35. Hutchinson S., Aplin R. T., Webb H., Kettle S., Timmermans J., Boers G. H., Handford P. A. (2005) Molecular effects of homocysteine on cbEGF domain structure: insights into the pathogenesis of homocystinuria. J. Mol. Biol. 346, 833–844 [DOI] [PubMed] [Google Scholar]
- 36. Hubmacher D., Tiedemann K., Bartels R., Brinckmann J., Vollbrandt T., Bätge B., Notbohm H., Reinhardt D. P. (2005) Modification of the structure and function of fibrillin-1 by homocysteine suggests a potential pathogenetic mechanism in homocystinuria. J. Biol. Chem. 280, 34946-34955 [DOI] [PubMed] [Google Scholar]
- 37. Kuznetsov G., Bush K. T., Zhang P. L., Nigam S. K. (1996) Perturbations in maturation of secretory proteins and their association with endoplasmic reticulum chaperones in a cell culture model for epithelial ischemia. Proc. Natl. Acad. Sci. U.S.A. 93, 8584–8589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ryu E. J., Harding H. P., Angelastro J. M., Vitolo O. V., Ron D., Greene L. A. (2002) Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J. Neurosci. 22, 10690–10698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Aw T. Y., Ookhtens M., Kaplowitz N. (1984) Inhibition of glutathione efflux from isolated rat hepatocytes by methionine. J. Biol. Chem. 259, 9355–9358 [PubMed] [Google Scholar]
- 40. Ghibelli L., Fanelli C., Rotilio G., Lafavia E., Coppola S., Colussi C., Civitareale P., Ciriolo M. R. (1998) Rescue of cells from apoptosis by inhibition of active GSH extrusion. Faseb J. 12, 479–486 [DOI] [PubMed] [Google Scholar]
- 41. Ghibelli L., Coppola S., Rotilio G., Lafavia E., Maresca V., Ciriolo M. R. (1995) Non-oxidative loss of glutathione in apoptosis via GSH extrusion. Biochem. Biophys. Res. Commun. 216, 313–320 [DOI] [PubMed] [Google Scholar]
- 42. Gurney J. G., Davis S., Severson R. K., Fang J. Y., Ross J. A., Robison L. L. (1996) Trends in cancer incidence among children in the U.S. Cancer 78, 532–541 [DOI] [PubMed] [Google Scholar]
- 43. Gurney J. G., Ross J. A., Wall D. A., Bleyer W. A., Severson R. K., Robison L. L. (1997) Infant cancer in the U.S.: histology-specific incidence and trends, 1973 to 1992. J. Pediatr Hematol. Oncol. 19, 428–432 [DOI] [PubMed] [Google Scholar]
- 44. Gjessing L. R. (1963) Studies of Functional Neural Tumors Isolation and Identification of Urinary Cystathionine. Scand J. Clin. Lab. Invest. 15, 601–602 [DOI] [PubMed] [Google Scholar]
- 45. Geiser C. F., Efron M. L. (1968) Cystathioninuria in patients with neuroblastoma or ganglioneuroblastoma. Its correlation to vanilmandelic acid excretion and its value in diagnosis and therapy. Cancer 22, 856–860 [DOI] [PubMed] [Google Scholar]
- 46. Helson L., Fleisher M., Bethune V., Murphy M. L., Schwartz M. K. (1972) Urinary cystathionine, catecholamine, and metabolites in patients with neuroblastoma. Clin. Chem. 18, 613–615 [PubMed] [Google Scholar]
- 47. Klein C. E., Roberts B., Holcenberg J., Glode L. M. (1988) Cystathionine metabolism in neuroblastoma. Cancer 62, 291–298 [DOI] [PubMed] [Google Scholar]