

Background: Large-conductance potassium (BK) channels containing the stress axis regulated insert (STREX) are important regulators of cellular excitability.
Results: Membrane detachment of STREX by protein kinase A (PKA)-mediated phosphorylation or de-palmitoylation established channel inhibition by protein kinase C (PKC).
Conclusion: Membrane association of STREX regulates the access of PKC to its inhibitory phosphorylation site.
Significance: The interplay of phosphorylation by PKA, PKC, and palmitoylation determines BK-STREX channel activity.
Keywords: Membrane Proteins, Potassium Channels, Protein Kinase A (PKA), Protein Kinase C (PKC), Protein Kinase G (PKG), STREX-BK Channels, Palmitoylation
Abstract
Large-conductance, calcium- and voltage-gated potassium (BK) channels play an important role in cellular excitability by controlling membrane potential and calcium influx. The stress axis regulated exon (STREX) at splice site 2 inverts BK channel regulation by protein kinase A (PKA) from stimulatory to inhibitory. Here we show that palmitoylation of STREX controls BK channel regulation also by protein kinase C (PKC). In contrast to the 50% decrease of maximal channel activity by PKC in the insertless (ZERO) splice variant, STREX channels were completely resistant to PKC. STREX channel mutants in which Ser700, located between the two regulatory domains of K+ conductance (RCK) immediately downstream of the STREX insert, was replaced by the phosphomimetic amino acid glutamate (S700E) showed a ∼50% decrease in maximal channel activity, whereas the S700A mutant retained its normal activity. BK channel inhibition by PKC, however, was effectively established when the palmitoylation-mediated membrane-anchor of the STREX insert was removed by either pharmacological inhibition of palmitoyl transferases or site-directed mutagenesis. These findings suggest that STREX confers a conformation on BK channels where PKC fails to phosphorylate and to inhibit channel activity. Importantly, PKA which inhibits channel activity by disassembling the STREX insert from the plasma membrane, allows PKC to further suppress the channel gating independent from voltage and calcium. Our results present an important example for the cross-talk between ion channel palmitoylation and phosphorylation in regulation of cellular excitability.
Introduction
Large-conductance Ca2+- and voltage-activated potassium (BK) channels play an important role in the regulation of membrane excitability in cells of the neuronal, endocrine and vascular system. This is partly due to an unusually high single-channel conductance and the concerted activation by membrane depolarization and elevation of intracellular free calcium which enables BK channels to typically exert a negative feedback influence on cellular excitability by repolarizing cells and turning off voltage-gated Ca2+ channels. In some neuronal cells, however, BK channels are positive feedback regulators which by augmenting the afterhyperpolarization, facilitate recovery from inactivation of sodium channels and promote the discharge frequency of neurons (1). The importance of BK channels in regulation of vascular and non-vascular smooth muscle tone, determination of action potential duration and frequency, tuning of hearing frequencies in cochlear hair cells, the release of hormones and neurotransmitters has been impressively demonstrated in mice with targeted deletion of the pore-forming α-subunit of the BK channel (1–5).
Only one gene (KCNMA1) codes for the pore-forming α-subunits of BK channels in all mammalian tissues (6, 7), but the functional properties of BK channels are exceptionally diverse, not only in different tissues but also in the same tissue or even in the same cell under different hormonal conditions (8–10). This complex phenotypic variation arises from many mechanisms encompassing transcriptional regulation (11), nuclear export of incompletely spliced intron-containing mRNAs (12), extensive alternative exon splicing of the α-subunit (7, 13–15), co-expression with different transmembrane modulatory β-subunits in a tissue-specific manner (16), and reversible protein phosphorylation (17). Several alternative splice sites and alternative exons have been identified in the KCNMA1 gene (18). The stress axis-regulated exon (STREX)4 is by far the most well-characterized alternative exon in the KCNMA1 gene which encodes a 59-residue cysteine-rich insert at the C2 position of alternative splicing in the C terminus of mammalian BK channels (9). Intriguingly, both gonadal (sex) and adrenal (stress) steroids contribute to regulation of the STREX splicing decision as has been demonstrated in uterine smooth muscle (8, 10, 19) and in pituitary as well as in adrenal chromaffin cells (20, 21). Inclusion of the STREX exon has profound effects on BK channel properties as compared with the insertless ZERO splice variant (9). STREX enhances the Ca2+/voltage sensitivity, increases and decreases the rates of channel activation and deactivation, respectively, and by adding an additional phosphorylation site (Ser636), switches the channel from being activated by PKA to a state where BK channels are inhibited by PKA (22).
Recently, we have demonstrated that protein kinase C markedly inhibited the activity of the insertless bovine BK-ZERO channel isoform. This effect was due to phosphorylation of serine 695 (Ser695), which is one of several putative PKC-dependent phosphorylation sites, located between the two regulatory domains of K+ conductance (RCK) (23). We also found that phosphorylation of Ser695 was not only dependent on the preceding phosphorylation of Ser1151, another PKC consensus motif at the C-terminal end, but also prevented the stimulatory effects of PKG and PKA. Here we analyzed the effect of PKC on BK channels expressing the STREX insert which is located immediately upstream of the critical PKC-dependent phosphorylation site (Ser695 in BK-ZERO). Surprisingly, we found that BK channels containing the STREX insert were completely resistant to PKC due to the membrane association of the C terminus via palmitoylation of conserved cysteine residues within STREX. Thus, abolition of palmitoylation or disassembling STREX from the plasma membrane by PKA-mediated phosphorylation resulted in channels which were strongly inhibited by PKC. This so far unrecognized divergent regulation of ZERO- and STREX-BK channels by protein kinase C is likely of physiological importance for cellular excitability as the proportion of STREX-BK channels varies under hormonal influence.
EXPERIMENTAL PROCEDURES
Phorbol-12-myristate-13-acetate (PMA), 4α-phorbol-12-myristate-13-acetate (4α-PMA), protein kinase C catalytic fragment (PKC), protein kinase C 19–31 pseudosubstrate inhibitor (PKC19–31), protein kinase A catalytic subunit (PKA), were obtained from Biomol (Hamburg, Germany). 2-bromopalmitate was purchased from Sigma (Taufkirchen, Germany), iberiotoxin (IbTX) from Alomone Laboratories (Jerusalem, Israel), guanosine-3′,5′-cyclic monophosphate (cGMP) from BioLog (Bremen, Germany). Protein kinase G Iα (PKG) was prepared as described (24) and was consistently used in the presence of 10 μm cGMP. Drugs were either dissolved in physiological saline solution (PSS; see solutions) or in dimethyl sulfoxide (DMSO). The maximum 0.1% final concentration of DMSO in the bath solution did not affect BK currents. Solutions with IbTX contained 0.1% bovine albumin fraction V (Sigma).
Cell Culture and Transfection Procedure
HEK293 cells were cultured in minimum essential medium supplemented with Earle's salts medium (Biochrom, Berlin, Germany) containing 10% fetal calf serum, 2 mm l-glutamine, 100 units ml−1 penicillin, 100 μg ml−1 streptomycin at 37 °C and 6% CO2. For transfection, 105 cells were plated in a 35-mm dish and cultured for another 24 h. Thereafter, the HEK293 cells were transiently co-transfected with EGFP (Clontech, Heidelberg, Germany), cloned into the pcDNA3 vector (Invitrogen, Karlsruhe, Germany), and the pcDNA3 plasmid containing the murine BK channel α subunit either with or without STREX exon (BK-STREX or BK-ZERO channel), or the respective BK mutant. Mouse BK-STREX used in this study is composed of the BK channel with the accession number L16912 (6) starting with MDALII and containing the STREX insert from the BK channel AF156674 (25) instead of IYF from L16912 at STREX splice site C2. The position of amino acids important for phosphorylation and palmitoylation in BK-STREX is as follows: The inhibitory (within the STREX insert) and the stimulatory PKA phosphorylation site is KMS636IY and QPS927IT, respectively, the palmitoylation site is RAC645C646FD (located within STREX), the PKC phosphorylation sites are TLS700PK and PKS1156RE, and the PKG phosphorylation site is KSS1139SV. Mouse BK-ZERO corresponds to the BK channel with the accession number L16912 (6) starting with MDALII but without the IYF insert from L16912 at the STREX splice site C2. Transfection of 1 μg of each cDNA was achieved by calcium phosphate precipitation for 18 h at 35 °C and 3% CO2. After washing, the cells were cultivated for another 24–48 h at 37 °C and 6% CO2. After the medium was exchanged several times with PSS (see below), the cells were transferred in a 35-mm dish to the stage of an inverted microscope (Zeiss Axiovert 200) for electrophysiological measurements. The transfection efficiency varied between 50 and 70% as judged by the expression of EGFP in transfected cells. Cells with similar intensity of EGFP fluorescence were used for the experiments.
GH3/B6 cells, a kind gift from Dr. C. Bauer (University Medical Center Hamburg-Eppendorf, Germany) were cultured in Dulbecco's modified Eagle's medium/nutrient mixture F-12 Ham medium (Sigma) supplemented with 15% horse serum (Invitrogen), 2.5% fetal calf serum (Biochrom), and 2 mm l-glutamine (Biochrom). Cells were maintained at 37 °C in a water-saturated atmosphere of 95% air and 5% CO2. The medium was changed every 2–3 days, and cells were passaged once a week. Patch clamp- or PCR studies were performed 3–5 days after passaging.
Site-directed Mutagenesis
Mutant channels were generated either by extended overlap polymerase chain reaction, or by a one step PCR-based mutation protocol, a modified version of the QuikChange protocol (Invitrogen). PfuUltra DNA Polymerase (Stratagene) was used in the extended overlap polymerase chain reaction with the pcDNA3/BK plasmid as template, and the resulting amplicons were digested with suitable restriction enzymes and cloned back into the pcDNA3/BK background (26). For the one step PCR-based mutation protocol the Phusion High-Fidelity DNA Polymerase (Finnzymes) was used for amplification of the whole pcDNA3/BK plasmid. All mutations were confirmed by sequence analysis over the total length of amplification and the employed restriction sites.
Analysis of BK Isoform Expression in GH3/B6 Cells
mRNA from GH3/B6 cells was isolated using the RNeasy Mini Kit (Qiagen) and transcribed into cDNA with SuperScript Vilo cDNA Synthese Kit (Invitrogen) according to the manufacturers' protocols. The expression of the mRNA encoding rat ZERO and STREX was verified using the primers and a PCR amplification protocol specified before (27).
Electrophysiology
Standard patch-clamp recording techniques were used to measure currents in the whole-cell-, or in the inside-out patch-clamp configuration (28). Patch electrodes were fabricated from borosilicate glass capillaries (MTW 150F; World Precision Instruments, Inc., Sarasota, FL) and filled with pre-filtered solutions of different composition (see below). Currents were recorded at room temperature with an EPC-8 amplifier (HEKA Elektronik, Lambrecht, Germany), connected via a 16 bit A/D interface to a pentium IBM clone computer. The signals were low-pass filtered (1 kHz) before 5 kHz digitization. Data acquisition and analysis were performed with an ISO-3 multitasking patch-clamp program (MFK M. Friedrich, Niedernhausen, Germany). Pipette resistance ranged from 2–3 MΩ in whole-cell-, 2.5–3 MΩ for macroscopic current recording in the excised-patch experiments. A holding potential of −10 mV was used in whole-cell patch-clamp experiments and in inside-out patches when macroscopic currents were recorded. After establishment of the whole cell-, inside-out configurations, the cells or patches were superfused for 5 to 10 min with control solutions until the currents became stable. If the current was not stable after 10 min, the recording was terminated. The membrane conductance (Gm) was calculated as Gm = I/(Em − Erev), where I is macroscopic current, Em is the test membrane potential, and Erev is the reversal potential for potassium. Gm values were plotted against test voltages and fitted to the Boltzmann equation: G = Gmax/(1 + exp((V1/2 − Vm)/k), where Gmax is the maximal conductance, V1/2 is the membrane potential (Vm) required for half-maximal activation of the channels, and k is the logarithmic voltage sensitivity (i.e. ΔV required for an e-fold increase in activity).
Solutions
For whole-cell experiments the cells were superfused with PSS containing (in mm) 127 NaCl, 5.9 KCl, 2.4 CaCl2, 1.2 MgCl2, 11 glucose, and 10 HEPES adjusted to pH 7.4 with NaOH. The pipette solution contained (in mm) 126 KCl, 6 NaCl, 1.2 MgCl2, 5 EGTA, 11 glucose, 1 Mg-ATP, 0.1 Na3GTP, and 10 HEPES adjusted to pH 7.4 with KOH. The free Ca2+ concentration was adjusted to 0.3 μm by adding the appropriate amount of CaCl2 as described earlier (29). For macroscopic current recordings in inside-out patches from HEK293 cells, the extracellular (pipette) solution is PSS. The bath solution (cytosolic surface of the patch) was the same as the pipette solution in whole cell recordings. The free Ca2+ concentration was adjusted to 1 μm. The storage buffers for purified protein kinases were: catalytic fragment of PKC (PKCc): 100 mm NaCl, 1 mm EDTA, 1 mm EGTA, 15 mm DTT, 10% glycerol, 20 mm Tris, pH 7.5; catalytic subunit of PKA (PKAc): 50 mm NaCl, 1 mm EDTA, 10 mm 2-mercaptoethanol, 50% glycerol, 20 mm Tris, pH 7.5; PKG Iα: 10 mm TES, 1.25 mm DTT, 0.25 μg ml−1 leupeptin, 0.05% NaN3, 50% glycerol. When the effect of protein kinases was investigated in inside-out patches, appropriate amounts of storage buffers were diluted into the bath solution. To avoid significant effects of storage buffers on channel activity, patches were first superfused with storage buffer containing solutions alone until channel activity was stable (usually within 5 to 10 min), then superfusion with the same solution containing the respective protein kinase was started.
Statistical Analyses
SigmaPlot for Windows (Jandel Scientific, version 11) was used for statistical analyses. Significance was determined by paired or unpaired t test or by one-way ANOVA. When a significant effect was detected with ANOVA, Student's t test was used for pairwise comparisons. The effects were considered significant if a p < 0.05 was obtained. Results are expressed as means ± S.E. where applicable.
RESULTS
STREX Insert Impedes BK Channel Regulation by PKC
The PKC-dependent regulation of BK channels was investigated with the murine pore-forming BK α-subunit (Kcnma1) containing the 59 amino acid STREX exon (22, 30). When inside-out membrane patches obtained from transfected HEK293 cells were superfused for at least 5 min at the cytosolic side with 20 nm of PKCc, no significant change of macroscopic currents was observed at all voltages tested (−120 to +100 mV; Fig. 1 A). This finding is in contrast to the distinct current decrease obtained with PKCc under otherwise identical experimental conditions from the insertless (ZERO) splice variant of the murine BK α-subunit (Fig. 1B). As reported before the STREX variant exhibited an increased channel activity compared with ZERO (31). The half-maximal activating voltage (V1/2) was 24.2 ± 2.1 mV and 46.2 ± 1.1 mV for STREX and ZERO, respectively. Although the application of 20 nm PKCc in the same patches did not significantly change V1/2 of the ZERO channels 46.7 ± 3.2 mV in the presence of PKCc, the macroscopic currents were decreased at all voltages tested. At +80 mV, PKCc reduced mean membrane conductance (Gm) from 21.9 ± 2.8 to 10.7 ± 1.5 nS, i.e. by 51.5 ± 4.9% (Fig. 1B). Cyclic nucleotide-dependent protein kinases showed the well-known stimulatory effect of PKG and inhibitory effect of PKA on BK-STREX channel currents. PKG (300 nm) enhanced BK currents and shifted the V1/2 to the left from 25.6 ± 1.9 (control) to 0.4 ± 4.1 mV (Fig. 1C). In contrast, PKAc (300 nm) shifted the V1/2 to the right from 24.2 ± 2.1 (control) to 56.5 ± 5.3 mV (Fig. 1D).
FIGURE 1.
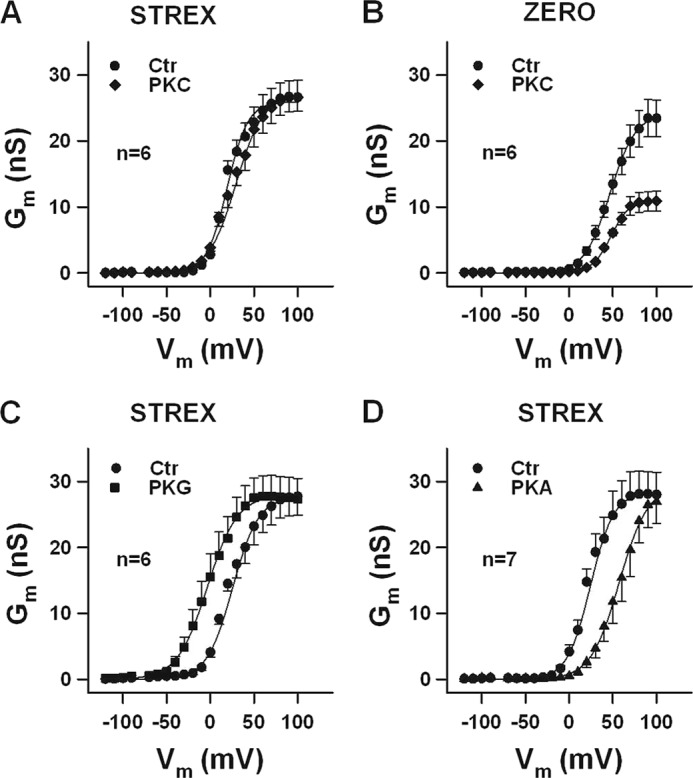
The STREX insert prevents the inhibitory effect of PKC on BK channel activity. A–D, conductance-voltage relationships obtained from inside-out membrane patches of HEK293 cells expressing STREX or ZERO BK channels. A and B, curves before (Ctr) and in the presence of 20 nm PKCc are shown. Note that PKC reduced ZERO BK channel activity at all voltages tested. C and D, 300 nm PKG produces a leftward- and 300 nm PKA a rightward shift of the conductance-voltage relationships (Ctr, curves before application of either PKG or PKA). Means ± S.E. when exceeding the size of the symbol are shown. The intracellular (bath) Ca2+ concentration was 1 μm.
We have shown earlier that phosphorylation of Ser695 in the ZERO splice variant of the bovine BK α-subunit is responsible for the PKC-dependent channel inhibition (23). To investigate whether the STREX insert prevents PKC to phosphorylate Ser700, which corresponds to Ser695 in the bovine ZERO variant and is located immediately downstream of the STREX exon, we constructed mutants in which Ser700 was replaced by either the phosphomimetic amino acid glutamate (S700E) or by alanine (S700A). Introducing a negative charge at position 700 (S700E), resulted in channels with a strongly reduced membrane conductance compared with wild-type BK-STREX channels. At 20 mV, the mean membrane conductance was 6.5 ± 0.6 nS (Ctr in Fig. 2A), which is 43% of the conductance measured in the wild-type BK channel at the same potential (Wt: 15.0 ± 1.5 nS in Fig. 2A). No significant change in membrane conductance was observed by superfusion with 20 nm PKCc for 5 min (6.1 ± 0.7 nS). Interestingly, exposure of the intracellular face of the patch to 300 nm PKG, failed to influence Gm in patches carrying the S700E mutant channel, whereas PKAc decreased Gm from 6.1 ± 0.9 to 1.9 ± 0.5 nS (Fig. 2A). The basal Gm of STREX channels with the mutation of Ser700 to alanine (S700A), was similar to the wild type and not altered by PKCc (Fig. 2B). PKG enhanced the mean Gm at 20 mV from 18.6 ± 2.6 to 29.6 ± 2.7 nS, whereas PKAc decreased the mean membrane conductance from 16.7 ± 1.2 to 7.1 ± 1.4 nS. Altogether, these findings suggest that the STREX insert prevents BK channel phosphorylation at position Ser700 by PKC, and thus, in contrast to the insertless ZERO splice variant, no decrease in membrane conductance can occur. A phosphomimetic negative charge in Ser700, however, reduces the channel conductance to a similar extent as PKC phosphorylation does in the ZERO variant and abolishes the enhancing effect of PKG on channel activity. The inhibitory effect of PKA, mediated via phosphorylation of Ser636 within the STREX insert, is apparently independent from the charge at the Ser700 position.
FIGURE 2.
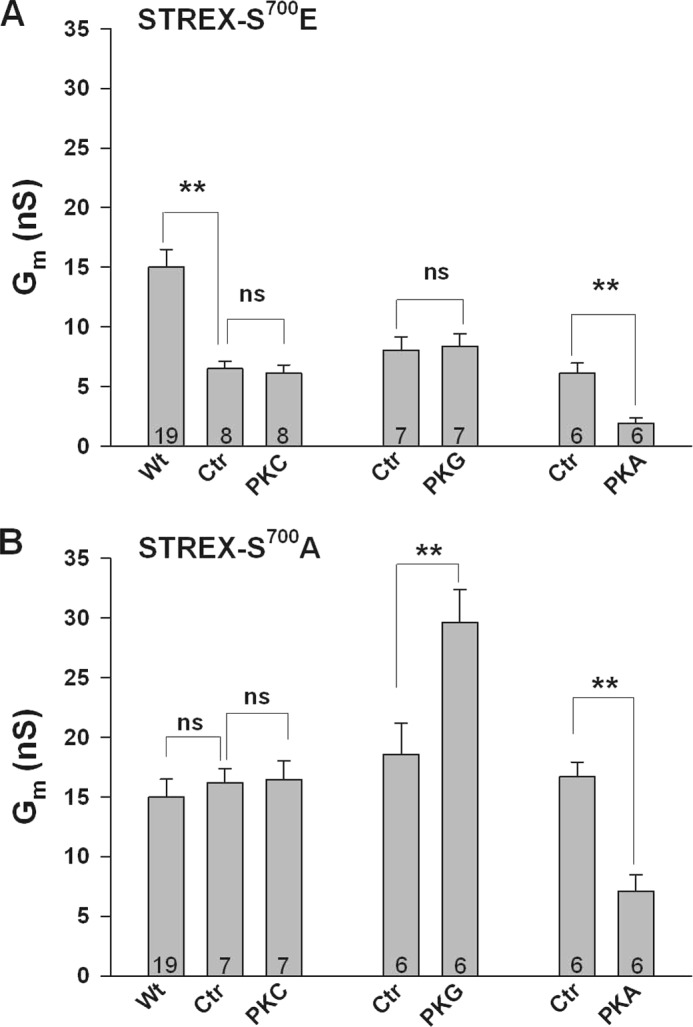
The phosphomimetic amino acid glutamate in position 700 simulates PKC-dependent inhibition of STREX and prevents PKG-dependent activation. Bars represent conductances from inside-out membrane patches expressing either the S700E (A) or the S700A (B) mutant STREX channel at +20 mV. (A) Note that control Gm of the mutant channel (Ctr) is ∼50% smaller than Gm of the wild type channel (Wt). 300 nm PKG failed to enhance Gm whereas the inhibitory effect of 300 nm PKA is preserved in this mutant. B, S700A mutant shows the same characteristics as the wild type STREX channel (compare with Fig. 1). Means ± S.E. are shown. Numbers within bars represent the number of membrane patches. The intracellular (bath) Ca2+ concentration was 1 μm. **, p < 0.01; ns, not significant.
Prevention of BK Channel Inhibition by PKC Depends on Palmitoylation of STREX
To examine whether palmitoylation, and thus association of the STREX domain with the plasma membrane (31), prevents PKC from inhibiting BK channel gating, we abolished STREX palmitoylation and membrane localization by 1) pharmacological inhibition of palmitoyl transferases, 2) site-directed mutagenesis of palmitoylation sites, and 3) detachment of STREX from the cell membrane by PKA phosphorylation. In accordance with published data on the role of STREX palmitoylation (31) pre-treatment of transfected HEK293 cells with the palmitoyl transferase inhibitor 2-bromopalmitate (100 μm 2-BP) for 24 h, resulted in the expression of BK-STREX channels with reduced activity (supplemental Fig. S1). Most important, these channels were sensitive to PKC. At 40 mV, 20 nm PKCc decreased Gm from 14.8 ± 1.1 to 4.7 ± 0.6 nS, i.e. by 58.6 ± 3.6% (Fig. 3A). Additional application of 300 nm PKG produced no further effect (Gm = 4.8 ± 0.8 nS). In the absence of PKCc, PKG enhanced Gm to 28.9 ± 2.3 nS, whereas PKAc was no longer able to decrease BK channel activity (Gm = 15.2 ± 1.3 nS; Fig. 3A). Representative macroscopic current recordings from 2-BP pretreated inside-out patches exposed to PKCc, PKCc plus PKG, PKG alone and PKAc are also shown in Fig. 3A. No inhibitory PKCc effect was obtained in patches from cells pretreated with 2-BP and expressing the S700A mutant instead of the wild type channel (Fig. 3B). Pretreatment of cells expressing the phosphomimetic S700E mutant with 2-BP revealed that the inhibitory effect of this mutation is not influenced by palmitoylation. Compared with STREX-Wt the S700E mutant exhibited a similar reduced membrane conductance (about 50% of the Wt conductivity) in 2-BP treated as well as untreated cells (see supplemental Fig. S1). These data indicate that membrane association of the STREX insert prevents the inhibitory phosphorylation by PKC, which however still exerts a strong inhibitory effect when Ser700 is accessible after membrane detachment.
FIGURE 3.

Prevention of STREX palmitoylation established PKC-dependent channel inhibition. Bars represent conductances at +40 mV from inside-out membrane patches obtained from HEK293 cells expressing STREX (A), the S700A- (B) or the C645A/C646A (C) mutant channel. A and B, HEK293 cells were treated for 24 h with 100 μm 2-BP before patches were obtained. A, inhibition of palmitoylation by 2-BP established PKC-dependent inhibition of STREX and abolished PKA-dependent inhibition. The enhancing effect of PKG was inhibited in the presence of PKC. B, in the S700A STREX mutant treated with 2-BP, PKC failed to inhibit Gm. C, in the palmitoylation-deficient double mutant C645A/C646A PKC inhibited Gm, and the inhibitory effect of PKA was abolished. As in A, the enhancing effect of PKG was blocked in the presence of PKC. The upper parts of A–C show representative macroscopic current recordings with the interventions as indicated. Bars in the lower parts represent means ± S.E. The numbers within bars represent the number of membrane patches. The intracellular (bath) Ca2+ concentration was 1 μm. **, p < 0.01; ns, not significant.
It has been demonstrated that the palmitoylation of cysteine Cys645 and Cys646 within the STREX insert is required for membrane attachment of the STREX C terminus at the plasma membrane (31). We therefore studied the STREX palmitoylation-deficient double mutant C645A/C646A in inside-out membrane patches from transfected HEK293 cells (Fig. 3C). This mutant showed similar characteristics as the wild type STREX channel treated with 2-BP. At 40 mV, 20 nm PKCc decreased Gm by 57.0 ± 6.9% and additional application of PKG had no further effect. In the absence of PKC, PKG enhanced Gm from 15.8 ± 3.0 (control) to 25.7 ± 4.4 nS, whereas PKA had lost its inhibitory action (15.5 ± 1.8 nS). Original current records from the C645A/C646A double mutant are also shown in Fig. 3C. To investigate whether BK channels become sensitive toward PKC when the STREX domain is detached from the plasma membrane by PKA-mediated phosphorylation, we superfused inside-out patches with 300 nm PKA first. When the decrease of Gm was stable for at least 1 min, the additional application of 20 nm PKCc induced a further decline of current amplitude (Fig. 4A). At 60 mV, PKCc reduced Gm from 16.9 ± 1.4 (PKAc) to 9.2 ± 1.0 nS (PKAc plus PKCc). Additional application of 5 μm of the PKC pseudosubstrate inhibitor peptide PKC19–31 reversed the PKC effect completely. When we applied PKG in the presence of PKAc plus PKCc (Fig. 4B), no change in channel activity was observed. In conclusion, the inhibitory effect of PKC via phosphorylation of Ser700 requires that the STREX insert is not membrane associated via palmitoylation.
FIGURE 4.
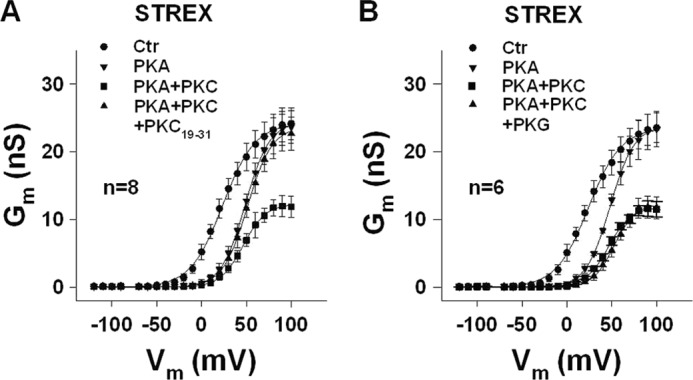
Removal of STREX membrane localization by PKA re-established PKC-dependent channel inhibition. A and B, conductance-voltage relationships obtained from inside-out membrane patches of HEK293 cells expressing STREX BK channels. Control patches (Ctr) were first superfused with 300 nm PKA, and when the decrease of Gm was stable for at least 1 min, 20 nm PKCc (PKC) was additionally applied. The further decline of Gm induced by PKC was completely reversed by the PKC pseudosubstrate inhibitor peptide PKC19–31 (5 μm; A), or remained unchanged when the patch was additionally superfused with 300 nm PKG (B). Means ± S.E. when exceeding the size of the symbol are shown. n, number of patches. The intracellular (bath) Ca2+ concentration was 1 μm.
PKC-dependent Inhibition in Cells Co-expressing STREX and ZERO BK Channels
Many native cells express both, the ZERO and the STREX splice variant of the BK channel. Moreover, as the BK channel tetramerization domain is conserved in splice-variant α-subunits (32), heterotetramerization of ZERO and STREX α-subunits may occur in these cells with an unpredictable response to PKC. To investigate the PKC-dependent channel gating in cells co-expressing ZERO and STREX α-subunits, we used a clonal rat somatomammotrope pituitary cell line (GH3/B6) endogenously expressing both variants and HEK293 cells co-transfected with both isoforms. In GH3/B6 cells, PCR products without (ZERO) and with the 174 bp STREX exon could be detected (Fig. 5A). When we elicited whole-cell outward currents (Iout) in GH3/B6 cells, the PKC activator phorbol 12-myristate 13 acetate (PMA, 100 nm) decreased current densities at a potential of +80 mV from 227.6 ± 18.1 (control) to 160.8 ± 17.5 pA pF−1, i.e. by 35.3 ± 5.8%. The inactive phorbol ester analog 4α-PMA (100 nm) had no significant effect on Iout (203.0 ± 16.3 pA pF−1; Fig. 5B). Addition of the specific BK channel-blocking peptide iberiotoxin (IbTX, 300 nm) revealed that under our experimental conditions more than 90% of Iout was conducted by BK channels. To inhibit plasma membrane attachment of the STREX domain via palmitoylation, we treated GH3/B6 cells for 24 h with 100 μm 2-BP. In these cells, 100 nm PMA reduced Iout from 171.4 ± 22.3 (control) to 82.7 ± 13.6 pA pF−1 which is a significantly larger decrease (by 48.0 ± 2.8%; p < 0.05) than that in cells not treated with 2-BP (compare Fig. 5, B and C). Again, 4α-PMA was ineffective, and 300 nm IbTX reduced Iout to less than 10% of the control current. In a second series of experiments, we co-transfected HEK293 cells with the same amount of plasmid DNA encoding for the ZERO and the STREX variant of the BK channel, respectively. To examine the PKC effect in these cells, inside-out patches were obtained and the cytosolic side was superfused with 20 nm PKCc. The inhibitory effect of PKCc could be detected at potentials from −120 to +100 mV. At 80 mV, PKCc reduced Gm from 35.1 ± 2.6 to 25.4 ± 2.1 nS, i.e. by 25.3 ± 4.8% (Fig. 5D). As observed in the GH3/B6 cells, the prevention of the membrane-anchoring of the STREX domain by 2-BP treatment, resulted in an enhanced sensitivity of the BK channels toward PKC. At +80 mV, 20 nm PKCc decreased Gm from 38.3 ± 3.5 to 19.1 ± 1.9 nS, i.e. by 49.3 ± 2.2% (Fig. 5E). This decrease is significantly (p < 0.01) larger than the reduction induced by PKCc in the absence of 2-BP (compare Fig. 5, D and E). As the dependence of the observed inhibitory effect of PKCc on 2-BP treatment in HEK293 cells largely resembles the situation in GH3/B6 cells, the data demonstrate that both BK channel isoforms co-expressed in GH3/B6 cells are incorporated into the cell membrane and are functionally active.
FIGURE 5.

Abolition of STREX palmitoylation by 2-BP augments PKC-dependent inhibition in cells co-expressing STREX and ZERO BK channels. A, PCR products without (ZERO) and with the 174 bp STREX exon are detected in GH3/B6 cells. B and C, whole-cell outward currents (Iout) were elicited from a holding potential of −10 mV by depolarizing GH3/B6 cells every 5 s for 300 ms from −60 to +80 mV. Bars represent means ± S.E. of current densities at +80 mV. It is shown that the inhibitory effect of the PKC activator PMA (100 nm) is markedly enhanced in 2-BP-treated cells, whereas the inactive phorbol ester analog 4α-PMA is ineffective. Note that more than 90% of Iout is blocked by the specific BK channel blocking peptide iberiotoxin (IbTX, 300 nm). Numbers within bars indicate number of cells. The original current recordings are at +80 mV with interventions as indicated. The pipette solution contained 0.3 μm Ca2+. D and E, conductance-voltage relationships obtained from inside-out membrane patches of HEK293 cells co-transfected with equal amounts of plasmid DNA encoding for the ZERO and the STREX variant of the BK channel, respectively. The PKC-dependent inhibition (20 nm PKCc, D) of Gm was almost doubled in cells treated with 2-BP (E). Means ± S.E. when exceeding the size of the symbol are shown. Insets, representative macroscopic currents at +80 mV reflecting interventions as indicated by the symbols in the corresponding figure. Numbers within bars represent the number of membrane patches. The intracellular (bath) Ca2+ concentration was 1 μm. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus control (Ctr) in (B–E).
Ser1156 as the Master Switch for STREX BK Channel Regulation by Protein Kinases
Phosphorylation and dephosphorylation of Ser1151, located close to the C-terminal end of the bovine ZERO channel, plays a key role in the complex regulation of BK channels by PKC, PKG, and PKA (23). To examine the role of Ser1156 in STREX channels which corresponds to Ser1151 in the bovine ZERO isoform, we constructed mutants in which Ser1156 was replaced either by the phosphomimetic amino acid aspartate (S1156D) or by alanine (S1156A). In inside-out patches from HEK293 cells expressing the S1156D mutant, we found a channel-behavior identical to that of the non-mutated STREX channel shown in Fig. 1. Application of 20 nm PKCc to the intracellular face of the patch was ineffective with respect to Gm (Fig. 6A), whereas PKG enhanced (by 117.0 ± 35% at 20 mV), and PKA inhibited Gm (by 68.8 ± 11.5%). In the S1156A mutant, neither PKC nor PKG was effective, whereas PKA retained its inhibitory action on Gm (decrease by 61.6 ± 8.1%; Fig. 6B). 2-BP treatment (100 μm, 24 h), resulted in STREX S1156D mutant channels in which superfusion of inside-out patches with PKCc, reduced Gm by 52.1 ± 2.5%. PKG enhanced Gm by 73.7 ± 9.6% and PKAc was ineffective (Fig. 6C). In the STREX S1156A mutant treated with 2-BP, BK channel conductance remained unchanged in the presence of PKCc and PKG, but PKAc, surprisingly, enhanced Gm by 101.0 ± 37%; Fig. 6D). Taken together, the data indicate that phosphorylation of Ser1156 of STREX BK channels is the precondition for PKG to enhance and for PKC to inhibit the activity of the membrane-detached channel. The inhibitory effect of PKA is independent from Ser1156, but depends on the attachment of the STREX domain to the cell membrane. When the STREX insert is detached from the plasma membrane, PKA enhances channel activity if Ser1156 is dephosphorylated, a regulation which has been described before for the bovine ZERO variant not phosphorylated at Ser1151 (23).
FIGURE 6.
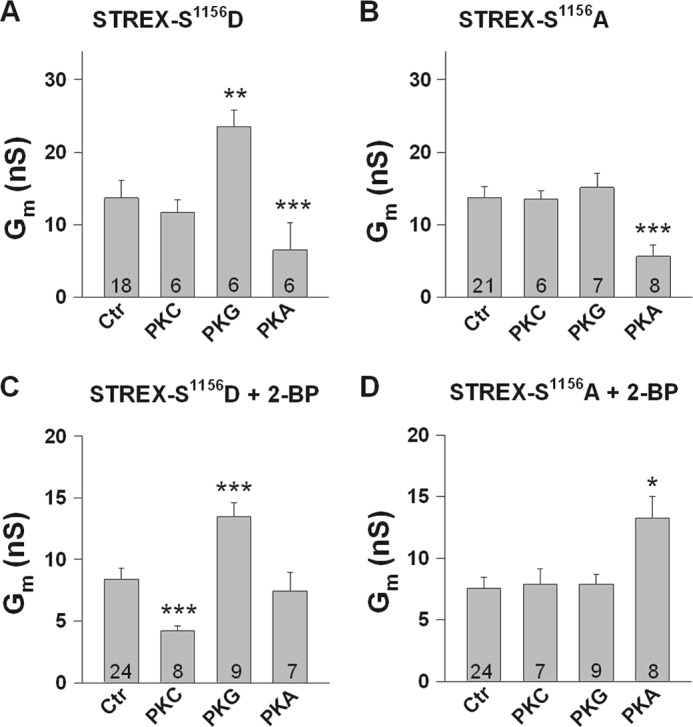
Phosphorylation of serine 1156 determines the sensitivity of STREX channels to protein kinases. A–D, bars represent conductances at +20 mV from inside-out membrane patches obtained from HEK293 cells expressing either the phosphomimetic S1156D (A and C) or the phosphoresistant S1156A STREX mutant channels (B and D). Cells in C and D were treated with the palmitoyl transferase inhibitor 2-BP (100 μm). Phosphorylation of Ser1156 is a precondition for PKG-dependent STREX activation (A and B) and for PKCc-dependent STREX inhibition which, however, occurs only when STREX is detached from the plasma membrane (+2-BP in C and D). The PKA-induced inhibition of STREX is independent of Ser1156 (A and B) but relies on the membrane-attached STREX insert (C). In the S1156A mutant that mimics the dephosphorylated channel at this position, PKAc enhances Gm when STREX is detached from the membrane (+2-BP in D). PKCc was applied at 20 nm and PKG and PKAc at 300 nm, respectively. Means ± S.E. are shown. Numbers within bars represent the number of membrane patches. The intracellular (bath) Ca2+ concentration was 1 μm. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
DISCUSSION
In the present study we examined the PKC-dependent regulation of BK channels from mouse brain expressing the 59-amino acid insert at splice site 2 (STREX) of the C-terminal domain of the channel pore-forming α subunit (22, 30). The STREX exon is widely expressed in excitable cells with a predominant distribution in neuroendocrine cells (9, 25), including pancreatic islets and adrenal chromaffin cells (15, 33) and it confers a higher Ca2+/voltage sensitivity on the channel compared with the ZERO splice variant which contains no inserts at the alternative splice sites (9, 13, 25). This well-known functional difference between the two channel isoforms is illustrated by the respective control current-voltage-relationship shown in Fig. 1, A and B. In addition, STREX speeds and slows rates of channel activation and deactivation, respectively, effects which may have profound effects on the intrinsic firing properties of excitable cells (9, 15, 34). Phorbol esters as activators of PKC have been shown to inhibit BK channel activity in arterial- (35, 36) and tracheal smooth muscle cells (23) that mainly express the BK channel ZERO isoform. In inside-out patches from HEK293 cells transiently transfected with the pore-forming BK channel α-subunit cloned from bovine trachea, it was shown that PKC decreased BK channel open probability, NPo, by shortening channel open time and prolonging the closed state of the channel (23). PKC neither affected single-channel conductance, nor the voltage dependence or the calcium sensitivity of the channel. Surprisingly, we found that the catalytically active fragment of PKC, when applied directly to the intracellular side of inside-out patches expressing the STREX channel, was completely ineffective (Fig. 7, A and B).
FIGURE 7.

Schematic overview of the different conformations of the STREX variant of the BK channel and their susceptibility to regulation by phosphorylation and palmitoylation. A, palmitoylated but unphosphorylated STREX channel, B, palmitoylated STREX channel conditionally phosphorylated by PKC on Ser1156, C, STREX channel at which the membrane attachment of the STREX insert has been removed by depalmitoylation and/or phosphorylation of Ser636 by PKA, D, STREX channel fully phosphorylated by PKC at Ser1156 and Ser700; E, depalmitoylated and dephosphorylated STREX channel. The consequences of treatment of the individual conformations with PKA, PKC, and PKG are indicated. The relative conductivity at intermediate potentials (+40 to +50 mV) are shown. +, low, ++, intermediate, +++, high. The conditional phosphorylation at Ser1156 is depicted in green, whereas the inhibitory phosphorylation sites Ser700 and Ser636 are shown in red.
The STREX insert contains an additional PKA consensus motif located at Ser636 in the mouse channel (Fig. 7C). Phosphorylation of this serine has been shown to cause inhibition of channel activity by producing a ∼20 mV rightward shift of the voltage-dependent activation curve (Fig. 1D; Ref. 22). In contrast, PKC reduced the maximal BK channel activity in the mouse ZERO mutant without affecting the apparent voltage sensitivity (Fig. 1B). These findings indicate that both PKA and PKC are principally able to inhibit BK channels but by different mechanisms. It has been shown before that inhibition of the bovine ZERO-BK channel by PKC depends on phosphorylation of Ser695 located within the linker between the two regulators of K conductance RCK1 and RCK2 (23). Ser695 corresponds to Ser700 in the STREX isoform and when we replaced Ser700 by glutamate (S700E) to mimic the negative charge induced by PKC-dependent phosphorylation, Gm was markedly reduced, resembling the effect of PKC in the ZERO channel. Obviously, the inhibitory effect of the negative charge at the Glu700 position is not dependent on membrane attachment of the STREX insert as it similarly occurred in cells treated with and without 2-BP. Furthermore the S700E mutant did not respond to either PKC or PKG (Figs. 2A and 7D). PKG usually increases BK channel activity not only in the ZERO (26) but also in the STREX isoform (Fig. 1C) by phosphorylation of a serine residue within an optimal consensus sequence close to the C-terminal end of the BK channel (Ser1139 in STREX; (26, 37)). This stimulatory effect was abolished when Ser695 in bovine ZERO was either phosphorylated by PKC or replaced by a phosphomimetic amino acid (23). Since PKC treatment did not inhibit the STREX channel (Figs. 1A and 7B), our findings in the S700E mutant strongly indicate that the STREX insert induces a conformation of the C terminus of the BK channel in which the access of PKC to Ser700 is hindered. This interpretation is further supported by the data in which we interfered with the membrane attachment of the STREX insert. A palmitoylated cysteine-rich domain (CRD) (15) targets the large intracellular C terminus of the STREX channel to the plasma membrane (31). After treatment with the largely irreversible palmitoyl transferase inhibitor 2-BP (38) or mutation of the two critical cysteines for palmitoylation to alanine (C645A/C646A; Ref. 31), the STREX channels became sensitive to PKC, i.e. PKC markedly decreased Gm at all potentials. In addition, PKA lost its inhibitory action and PKG failed to enhance channel activity in the presence of PKC (Figs. 3 and 7D). As PKC did not inhibit Gm in the S700A mutant treated with 2-BP (Fig. 3B), Ser700 would appear to be accessible to phosphorylation by PKC only when the anchoring of the C terminus in the cell membrane was prevented. Interestingly, palmitoylation impairs PKC-dependent phosphorylation not only in STREX channels but also in some ligand-gated ion channels. A palmitoylation resistant mutant of the GluR6 subunit of the kainate receptor was a better substrate for PKC-dependent phosphorylation than the wild-type GluR6 receptor (39). Depalmitoylation of a specific cysteine residue of the GluR1subunit of the AMPA receptor facilitated phosphorylation by PKC, which in turn enhanced the interaction with binding partners promoting the GluR1 insertion in the plasma membrane (40). All these C-terminal PKC consensus sites are near the palmitoylation sites and therefore it is attractive to speculate that palmitoylation, determining the accessibility of PKC to its target sites, is a mechanism to regulate these ion channels. Protein palmitoylation is a reversible process depending on a variety of protein palmitoyl transferases and thioesterases (41, 42). Thus it is likely that both palmitoylated and depalmitoylated BK channels may exist in the same cell. With regard to the STREX channel the complete resistance to PKC in our electrophysiological assay, however, indicates that the membrane-attached C terminus is the predominant form. Therefore, the question arises, whether a physiological mechanism exists that disassembles the C terminus of STREX channels from the membrane to allow PKC-induced inhibition. PKA-dependent phosphorylation of Ser636 immediately upstream of the conserved palmitoylated cysteine residues within the STREX insert dissociates the C terminus from the plasma membrane and thereby inhibits the channel activity (31). When we used PKAc to remove the C terminus from the membrane, we found that the additional application of PKCc further reduced the channel activity, which contrary to PKA resulted in a decrease of maximal channel gating that was insurmountable by voltage or [Ca2+]I (Fig. 4 and Fig. 7D). Such an effect could be of physiological relevance in particular in endocrine cells of the anterior pituitary gland that express the STREX variant such as somatomamotropes and corticotrophs. In both these cell types cellular excitability and hormone secretion is regulated by neuropeptides that activate both the PKA- and PKC-dependent signaling cascades resulting in inhibition of endogenous BK channels (43–45). When we investigated cells which co-express STREX and ZERO BK channels such as GH3/B6 cells, a clonal rat somatomamotrope pituitary cell line, we found that the maximal PKC-dependent channel inhibition was significantly amplified in cells treated with 2-BP. In HEK293 cells co-transfected with equal amounts of ZERO- and STREX cDNA, the maximal PKC-dependent channel inhibition was only half compared with cells exclusively expressing the ZERO variant. These results indicate that co-expressed ZERO and STREX channels are incorporated in the plasma membrane and are functionally active. The sensitivity to PKC can be regulated in these cells by mechanisms that increase or decrease the expression of either channel isoform or lead to the detachment of the STREX C terminus from the plasma membrane such as PKA-mediated phosphorylation or depalmitoylation.
In analogy to ZERO BK channels (23), we found that besides Ser700 a second PKC-dependent phosphorylation site, Ser1156 at the C-terminal end, was critical for STREX channel regulation by PKC, PKG, and PKA (see Fig. 7). When we introduced a negative charge by replacing Ser1156 by aspartate (S1156D) to mimic PKC-dependent phosphorylation at this site, PKCc was ineffective as in the wild type channel, whereas after detachment of the C terminus from the membrane by prior inhibition of palmitoyl transferases, STREX was effectively inhibited by PKCc. This inhibitory effect was lost in the S1156A mutant, indicating that phosphorylation of Ser700 is conditional, depending on the preceding phosphorylation of Ser1156 which is the default setting in our electrophysiological assay. Activation of STREX channels by PKG was also dependent on phosphorylation of Ser1156 and was not dependent on the membrane association of the STREX domain. As outlined above, the inhibitory effect of PKA was dependent of the association of the STREX domain with the plasma membrane but independent of Ser1156. Interestingly, when Ser1156 was dephosphorylated as in the mutant S1156A and the STREX C terminus dissociated from the cell membrane by 2-BP, PKAc enhanced BK channel activity (Fig. 7E). This activating PKA effect is dependent on phosphorylation of Ser927 which is a second PKA-dependent phosphorylation site located in the C terminus downstream of the STREX insert (22, 26).
In summary, we show in this study that STREX BK channels are insensitive to PKC due to the membrane attachment of its C terminus to the plasma membrane which in turn most likely poses a steric hindrance for PKC to phosphorylate the critical Ser700. Abolition of STREX palmitoylation or membrane localization resulted in STREX channels that were inhibited by PKC similar to the insertless ZERO channel. Disassembling the C terminus from the cell membrane via PKA-mediated phosphorylation enables PKC to further inhibit BK channel activity suspending the negative feedback regulation on voltage-dependent Ca2+ influx. Such an effect may be important in neuroendocrine cells where PKA- and PKC-dependent regulations of BK channel activity synergize in hormone secretion. As highlighted in Fig. 7, the activity of BK channels can thus obviously be modulated by a concert of regulatory mechanisms including palmitoylation and phosphorylation at various sites. In individual cells and specialized tissues this complex regulation offers adaptive mechanisms to achieve fine tuning of cellular excitability.
This work was supported the German Center for Cardiovascular Research (grants to D. D. and T. W.) and the Deutsche Forschungsgemeinschaft (SFB 773, Project A2 to P. R.)

This article contains supplemental Fig. S1.
- STREX
- stress-axis-regulated insert
- CRD
- cysteine-rich domain
- IbTX
- iberiotoxin
- PKG
- protein kinase G
- PMA
- phorbol-12-myristate-13-acetate
- PKCc
- catalytic subunit of protein kinase C.
REFERENCES
- 1. Sausbier M., Hu H., Arntz C., Feil S., Kamm S., Adelsberger H., Sausbier U., Sailer C. A., Feil R., Hofmann F., Korth M., Shipston M. J., Knaus H. G., Wolfer D. P., Pedroarena C. M., Storm J. F., Ruth P. (2004) Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc. Natl. Acad. Sci. U.S.A. 101, 9474–9478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Meredith A. L., Thorneloe K. S., Werner M. E., Nelson M. T., Aldrich R. W. (2004) Overactive bladder and incontinence in the absence of the BK large conductance Ca2+-activated K+ channel. J. Biol. Chem. 279, 36746–36752 [DOI] [PubMed] [Google Scholar]
- 3. Rüttiger L., Sausbier M., Zimmermann U., Winter H., Braig C., Engel J., Knirsch M., Arntz C., Langer P., Hirt B., Müller M., Köpschall I., Pfister M., Münkner S., Rohbock K., Pfaff I., Rüsch A., Ruth P., Knipper M. (2004) Deletion of the Ca2+-activated potassium (BK) α-subunit but not the BKβ1-subunit leads to progressive hearing loss. Proc. Natl. Acad. Sci. U.S.A. 101, 12922–12927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sausbier M., Arntz C., Bucurenciu I., Zhao H., Zhou X. B., Sausbier U., Feil S., Kamm S., Essin K., Sailer C. A., Abdullah U., Krippeit-Drews P., Feil R., Hofmann F., Knaus H. G., Kenyon C., Shipston M. J., Storm J. F., Neuhuber W., Korth M., Schubert R., Gollasch M., Ruth P. (2005) Elevated blood pressure linked to primary hyperaldosteronism and impaired vasodilation in BK channel-deficient mice. Circulation 112, 60–68 [DOI] [PubMed] [Google Scholar]
- 5. Werner M. E., Zvara P., Meredith A. L., Aldrich R. W., Nelson M. T. (2005) Erectile dysfunction in mice lacking the large-conductance calcium-activated potassium (BK) channel. J. Physiol. 567, 545–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Butler A., Tsunoda S., McCobb D. P., Wei A., Salkoff L. (1993) mSlo, a complex mouse gene encoding “maxi” calcium-activated potassium channels. Science 261, 221–224 [DOI] [PubMed] [Google Scholar]
- 7. Tseng-Crank J., Foster C. D., Krause J. D., Mertz R., Godinot N., DiChiara T. J., Reinhart P. H. (1994) Cloning, expression, and distribution of functionally distinct Ca2+-activated K+ channel isoforms from human brain. Neuron 13, 1315–1330 [DOI] [PubMed] [Google Scholar]
- 8. Benkusky N. A., Fergus D. J., Zucchero T. M., England S. K. (2000) Regulation of the Ca2+-sensitive domains of the maxi-K channel in the mouse myometrium during gestation. J. Biol. Chem. 275, 27712–27719 [DOI] [PubMed] [Google Scholar]
- 9. Xie J., McCobb D. P. (1998) Control of alternative splicing of potassium channels by stress hormones. Science 280, 443–446 [DOI] [PubMed] [Google Scholar]
- 10. Zhu N., Eghbali M., Helguera G., Song M., Stefani E., Toro L. (2005) Alternative splicing of Slo channel gene programmed by estrogen, progesterone and pregnancy. FEBS Lett. 579, 4856–4860 [DOI] [PubMed] [Google Scholar]
- 11. Kundu P., Alioua A., Stefani E., Toro L. (2007) Regulation of mouse Slo gene expression: multiple promoters, transcription start sites, and genomic action of estrogen. J. Biol. Chem. 282, 27478–27492 [DOI] [PubMed] [Google Scholar]
- 12. Bell T. J., Miyashiro K. Y., Sul J. Y., McCullough R., Buckley P. T., Jochems J., Meaney D. F., Haydon P., Cantor C., Parsons T. D., Eberwine J. (2008) Cytoplasmic BKCa channel intron-containing mRNAs contribute to the intrinsic excitability of hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 1901–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen L., Tian L., MacDonald S. H., McClafferty H., Hammond M. S., Huibant J. M., Ruth P., Knaus H. G., Shipston M. J. (2005) Functionally diverse complement of large conductance calcium- and voltage-activated potassium channel (BK) α-subunits generated from a single site of splicing. J. Biol. Chem. 280, 33599–33609 [DOI] [PubMed] [Google Scholar]
- 14. Lagrutta A., Shen K. Z., North R. A., Adelman J. P. (1994) Functional differences among alternatively spliced variants of Slowpoke, a Drosophila calcium-activated potassium channel. J. Biol. Chem. 269, 20347–20351 [PubMed] [Google Scholar]
- 15. Saito M., Nelson C., Salkoff L., Lingle C. J. (1997) A cysteine-rich domain defined by a novel exon in a slo variant in rat adrenal chromaffin cells and PC12 cells. J. Biol. Chem. 272, 11710–11717 [DOI] [PubMed] [Google Scholar]
- 16. Orio P., Rojas P., Ferreira G., Latorre R. (2002) New disguises for an old channel: MaxiK channel β-subunits. News Physiol. Sci. 17, 156–161 [DOI] [PubMed] [Google Scholar]
- 17. Schubert R., Nelson M. T. (2001) Protein kinases: tuners of the BKCa channel in smooth muscle. Trends Pharmacol. Sci. 22, 505–512 [DOI] [PubMed] [Google Scholar]
- 18. Shipston M. J. (2001) Alternative splicing of potassium channels: a dynamic switch of cellular excitability. Trends Cell Biol. 11, 353–358 [DOI] [PubMed] [Google Scholar]
- 19. Holdiman A. J., Fergus D. J., England S. K. (2002) 17β-Estradiol upregulates distinct maxi-K channel transcripts in mouse uterus. Mol. Cell Endocrinol. 192, 1–6 [DOI] [PubMed] [Google Scholar]
- 20. Lai G. J., McCobb D. P. (2006) Regulation of alternative splicing of Slo K+ channels in adrenal and pituitary during the stress-hyporesponsive period of rat development. Endocrinology 147, 3961–3967 [DOI] [PubMed] [Google Scholar]
- 21. Mahmoud S. F., McCobb D. P. (2004) Regulation of Slo potassium channel alternative splicing in the pituitary by gonadal testosterone. J. Neuroendocrinol. 16, 237–243 [DOI] [PubMed] [Google Scholar]
- 22. Tian L., Duncan R. R., Hammond M. S., Coghill L. S., Wen H., Rusinova R., Clark A. G., Levitan I. B., Shipston M. J. (2001) Alternative splicing switches potassium channel sensitivity to protein phosphorylation. J. Biol. Chem. 276, 7717–7720 [DOI] [PubMed] [Google Scholar]
- 23. Zhou X. B., Wulfsen I., Utku E., Sausbier U., Sausbier M., Wieland T., Ruth P., Korth M. (2010) Dual role of protein kinase C on BK channel regulation. Proc. Natl. Acad. Sci. U.S.A. 107, 8005–8010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ruth P., Pfeifer A., Kamm S., Klatt P., Dostmann W. R., Hofmann F. (1997) Identification of the amino acid sequences responsible for high affinity activation of cGMP kinase Iα. J. Biol. Chem. 272, 10522–10528 [DOI] [PubMed] [Google Scholar]
- 25. Shipston M. J., Duncan R. R., Clark A. G., Antoni F. A., Tian L. (1999) Molecular components of large conductance calcium-activated potassium (BK) channels in mouse pituitary corticotropes. Mol. Endocrinol. 13, 1728–1737 [DOI] [PubMed] [Google Scholar]
- 26. Zhou X. B., Arntz C., Kamm S., Motejlek K., Sausbier U., Wang G. X., Ruth P., Korth M. (2001) A molecular switch for specific stimulation of the BKCa channel by cGMP and cAMP kinase. J. Biol. Chem. 276, 43239–43245 [DOI] [PubMed] [Google Scholar]
- 27. Mahmoud S. F., Bezzerides A. L., Riba R., Lai G. J., Lovell P. V., Hara Y., McCobb D. P. (2002) Accurate quantitative RT-PCR for relative expression of Slo splice variants. J. Neurosci. Methods 115, 189–198 [DOI] [PubMed] [Google Scholar]
- 28. Hamill O. P., Marty A., Neher E., Sakmann B., Sigworth F. J. (1981) Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391, 85–100 [DOI] [PubMed] [Google Scholar]
- 29. Zhou X. B., Ruth P., Schlossmann J., Hofmann F., Korth M. (1996) Protein phosphatase 2A is essential for the activation of Ca2+-activated K+ currents by cGMP-dependent protein kinase in tracheal smooth muscle and Chinese hamster ovary cells. J. Biol. Chem. 271, 19760–19767 [DOI] [PubMed] [Google Scholar]
- 30. Tian L., Coghill L. S., MacDonald S. H., Armstrong D. L., Shipston M. J. (2003) Leucine zipper domain targets cAMP-dependent protein kinase to mammalian BK channels. J. Biol. Chem. 278, 8669–8677 [DOI] [PubMed] [Google Scholar]
- 31. Tian L., Jeffries O., McClafferty H., Molyvdas A., Rowe I. C., Saleem F., Chen L., Greaves J., Chamberlain L. H., Knaus H. G., Ruth P., Shipston M. J. (2008) Palmitoylation gates phosphorylation-dependent regulation of BK potassium channels. Proc. Natl. Acad. Sci. U.S.A. 105, 21006–21011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Quirk J. C., Reinhart P. H. (2001) Identification of a novel tetramerization domain in large conductance KCa channels. Neuron 32, 13–23 [DOI] [PubMed] [Google Scholar]
- 33. Ferrer J., Wasson J., Salkoff L., Permutt M. A. (1996) Cloning of human pancreatic islet large conductance Ca(2+)-activated K+ channel (hSlo) cDNAs: evidence for high levels of expression in pancreatic islets and identification of a flanking genetic marker. Diabetologia 39, 891–898 [DOI] [PubMed] [Google Scholar]
- 34. Lovell P. V., McCobb D. P. (2001) Pituitary control of BK potassium channel function and intrinsic firing properties of adrenal chromaffin cells. J. Neurosci. 21, 3429–3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Barman S. A., Zhu S., White R. E. (2004) PKC activates BKCa channels in rat pulmonary arterial smooth muscle via cGMP-dependent protein kinase. Am. J. Physiol. Lung. Cell Mol. Physiol. 286, L1275–L1281 [DOI] [PubMed] [Google Scholar]
- 36. Schubert R., Noack T., Serebryakov V. N. (1999) Protein kinase C reduces the KCa current of rat tail artery smooth muscle cells. Am. J. Physiol. 276, C648–658 [DOI] [PubMed] [Google Scholar]
- 37. Fukao M., Mason H. S., Britton F. C., Kenyon J. L., Horowitz B., Keef K. D. (1999) Cyclic GMP-dependent protein kinase activates cloned BKCa channels expressed in mammalian cells by direct phosphorylation at serine 1072. J. Biol. Chem. 274, 10927–10935 [DOI] [PubMed] [Google Scholar]
- 38. Jennings B. C., Nadolski M. J., Ling Y., Baker M. B., Harrison M. L., Deschenes R. J., Linder M. E. (2009) 2-Bromopalmitate and 2-(2-hydroxy-5-nitro-benzylidene)-benzothiophen-3-one inhibit DHHC-mediated palmitoylation in vitro. J. Lipid Res. 50, 233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pickering D. S., Taverna F. A., Salter M. W., Hampson D. R. (1995) Palmitoylation of the GluR6 kainate receptor. Proc. Natl. Acad. Sci. U.S.A. 92, 12090–12094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lin D. T., Makino Y., Sharma K., Hayashi T., Neve R., Takamiya K., Huganir R. L. (2009) Regulation of AMPA receptor extrasynaptic insertion by 4.1N, phosphorylation and palmitoylation. Nat. Neurosci. 12, 879–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. El Husseini A.-E. D., Bredt D. S. (2002) Protein palmitoylation: a regulator of neuronal development and function. Nat. Rev. Neurosci. 3, 791–802 [DOI] [PubMed] [Google Scholar]
- 42. Fukata Y., Fukata M. (2010) Protein palmitoylation in neuronal development and synaptic plasticity. Nat. Rev. Neurosci. 11, 161–175 [DOI] [PubMed] [Google Scholar]
- 43. Hall S. K., Armstrong D. L. (2000) Conditional and unconditional inhibition of calcium-activated potassium channels by reversible protein phosphorylation. J. Biol. Chem. 275, 3749–3754 [DOI] [PubMed] [Google Scholar]
- 44. Shipston M. J., Armstrong D. L. (1996) Activation of protein kinase C inhibits calcium-activated potassium channels in rat pituitary tumour cells. J. Physiol. 493, 665–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. White R. E., Schonbrunn A., Armstrong D. L. (1991) Somatostatin stimulates Ca2+ activated K+ channels through protein dephosphorylation. Nature 351, 570–573 [DOI] [PubMed] [Google Scholar]