

Background: WBSCR17 is a potential polypeptide N-acetylgalactosaminyltransferase with unknown function.
Results: WBSCR17, induced with N-acetylglucosamine, regulated O-glycosylation, lamellipodium formation, and macropinocytosis.
Conclusion: Mucin-type O-glycosylation may be involved in lamellipodium formation and membrane trafficking through macropinocytosis.
Significance: The data suggest that mucin-type O-glycosylation modulates the dynamic membrane transport of the cell and may be involved in the control of nutrient uptake.
Keywords: Endocytosis, Glycobiology, Glycosyltransferases, Lysosomes, Membrane Trafficking, O-Glycosylation, Lamellipodium, Macropinocytosis
Abstract
We previously identified a novel polypeptide N-acetylgalactosaminyltransferase (GalNAc-T) gene, which is designated Williams-Beuren syndrome chromosome region 17 (WBSCR17) because it is located in the chromosomal flanking region of the Williams-Beuren syndrome deletion. Recent genome-scale analysis of HEK293T cells treated with a high concentration of N-acetylglucosamine (GlcNAc) demonstrated that WBSCR17 was one of the up-regulated genes possibly involved in endocytosis (Lau, K. S., Khan, S., and Dennis, J. W. (2008) Genome-scale identification of UDP-GlcNAc-dependent pathways. Proteomics 8, 3294–3302). To assess its roles, we first expressed recombinant WBSCR17 in COS7 cells and demonstrated that it was N-glycosylated and localized mainly in the Golgi apparatus, as is the case for the other GalNAc-Ts. Assay of recombinant WBSCR17 expressed in insect cells showed very low activity toward typical mucin peptide substrates. We then suppressed the expression of endogenous WBSCR17 in HEK293T cells using siRNAs and observed phenotypic changes of the knockdown cells with reduced lamellipodium formation, altered O-glycan profiles, and unusual accumulation of glycoconjugates in the late endosomes/lysosomes. Analyses of endocytic pathways revealed that macropinocytosis, but neither clathrin- nor caveolin-dependent endocytosis, was elevated in the knockdown cells. This was further supported by the findings that the overexpression of recombinant WBSCR17 stimulated lamellipodium formation, altered O-glycosylation, and inhibited macropinocytosis. WBSCR17 therefore plays important roles in lamellipodium formation and the regulation of macropinocytosis as well as lysosomes. Our study suggests that a subset of O-glycosylation produced by WBSCR17 controls dynamic membrane trafficking, probably between the cell surface and the late endosomes through macropinocytosis, in response to the nutrient concentration as exemplified by environmental GlcNAc.
Introduction
Mucin-type O-glycosylation is a common post-translational modification, which occurs on numerous membrane and secreted proteins. Its functional importance is reported for various physiological activities, such as protection of epithelium, cell adhesion, antigenic properties of some cell surface carbohydrates, and control of the immune system (1–4). The biosynthesis of mucin-type carbohydrates proceeds in a stepwise manner by the ordered successive actions of a number of glycosyltransferases. Among them, a UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase (GalNAc-T)2 is the enzyme responsible for initiating the biosynthetic reaction by catalyzing the transfer of GalNAc from UDP-GalNAc to a hydroxyl group of serine or threonine residues of proteins (5). GalNAc-Ts are important enzymes because they determine the number and the positions of mucin-type sugar chains in a protein. They constitute a large family, with 20 mammalian isozymes (Fig. 1A) (6–8). We previously identified a putative GalNAc-T gene, pt-GalNAc-T (9), which is a member of the “Y subfamily,” a group recently defined by Li et al. (6). The Y isozymes contain several conservative substitutions in the catalytic domain, one of which is represented by the replacement by tyrosine of tryptophan in the Gal/GalNAc-T motif (Fig. 1B) (6). We demonstrated that this substitution in GalNAc-T1 led to a severe decrease in the catalytic activity (10). pt-GalNAc-T is also known as Williams-Beuren syndrome (WBS) chromosome region 17 (WBSCR17) (11), one of the genes identified in the flanking region (7q11.22) of the chromosomal deletion region (7q11.23) in the genome of WBS patients. In addition, different numbers were recently assigned to some of the GalNAc-T family, and pt-GalNAc-T/WBSCR17 was designated GalNAc-T17 and -T19 by Peng et al. (8), and Raman et al. (7), respectively (Fig. 1A).
FIGURE 1.

Molecular analysis of WBSCR17. A, a phylogenetic tree of the human GalNAc-T family. The tree was generated by comparing amino acid sequences of the family with the ClustalW algorithm. Recently, two distinct numbers were assigned to some of the isozymes by Peng et al. (8) and Raman et al. (7), and both numbers separated by a slash are shown in the tree. WBSCR17 is one of the isozymes that belong to the Y subfamily. B, schematic representation of GalNAc-T motifs and comparison of amino acid sequences in the Gal/GalNAc-T motif. The Y subfamily has a tyrosine residue in place of a tryptophan residue in the Gal/GalNAc-T motif of other GalNAc-T family members (indicated by a gray box).
WBS is a neurodevelopmental disorder associated with physical, behavioral, and cognitive abnormalities (11, 12), which is caused by haploinsufficiency of multiple genes at 7q11.23. WBSCR17 mRNA is expressed predominantly in the nervous system (9) and weakly in heart, kidney, liver, lung, and spleen (11). Recently, it was reported to be involved in the uridine 5′-diphospho-N-acetylglucosamine (UDP-GlcNAc)-dependent pathways by genome-scale analysis (13). In this previous study, WBSCR17 was identified as one of the candidate genes that play roles in proliferation, bulk endocytosis, and β1,6GlcNAc-branching of N-glycans under the control of UDP-GlcNAc by microarray analysis and small interfering RNA (siRNA) screening using mouse NMuMG and human HEK293T cells. However, the detailed function of WBSCR17 still remains to be elucidated.
It has been reported that mucin-type sugar chains are involved in endocytosis. For example, inhibition of chain elongation of O-/N-glycosylation with O-benzyl-N-acetyl-α-d-galactosaminide perturbs the endosomal pathway (14). Moreover, the glycosylation state of MUC1, a type I transmembrane protein with a large number of mucin-type glycans, modulates its clathrin-mediated endocytosis (15). MUC1 is also involved in the macropinocytic pathway (16). Macropinocytosis is a form of bulk endocytosis that takes up extracellular solutes and a variety of nutrients or antigens into cytoplasmic vacuoles, called macropinosomes. A macropinosome is a large vesicle, the diameter of which is larger than 0.2 μm, and forms clathrin-independently by actin-driven circular membrane ruffling and fusion of the membrane ruffles at their outermost margins (17). Macropinocytosis significantly contributes to antigen presentation by the immune system and is exploited by a range of pathogens for cellular invasion and avoidance of immune surveillance (18). It was recently reported that newly formed macropinosomes recruit typical early endosome markers, EEA1, mature into vesicles containing Rab7, a late endosome marker, and subsequently fuse with the lysosomes that recruit microtubule-associated protein 1 light chain 3 (LC3), which was previously shown to be an autophagy marker, to hydrolyze the cargo in the vesicles (18–20). Because of the absence of specific markers, the regulatory mechanism of macropinocytosis is not known despite its physiological importance (18).
In this study, we investigated the characteristics and roles of WBSCR17 and found that WBSCR17, which was N-glycosylated, was located predominantly in the Golgi apparatus and had very weak catalytic activity toward typical mucin peptides. The suppression of WBSCR17 in HEK293T cells led to alterations of O-glycan profiles, a decrease in lamellipodium formation, and an unusual accumulation of glycoconjugates in the late endosomes and the lysosomes, which was probably due to enhanced macropinocytosis. These data indicate that O-glycosylation by WBSCR17 is involved in the uptake of extracellular fluid/solutes by a GlcNAc-dependent regulatory mechanism.
EXPERIMENTAL PROCEDURES
Cell Culture
COS7 (African green monkey kidney fibroblast) cells and HEK293T (human embryonic kidney) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum (FCS) at 37 °C under 5% CO2. For HEK293T cells, culture dishes were coated with 20 μg/ml fibronectin (WAKO) for 6 h prior to plating the cells.
Plasmids and siRNA Transfection
Each of the cDNAs encoding the full-length mouse and human WBSCR17 was inserted into pcDNA6/Myc-His expression vector (Invitrogen). The plasmids were transfected with Effectene transfection reagent (Qiagen). siRNAs against human WBSCR17 (siWBS17 siRNAs 1 and 2) and AllStar negative control siRNA (siNC) were purchased from Qiagen. The target sequences of siWBS17 were as follows: sequence 1, 5′-CTGGTTAGGGTGCACATATTA-3′; sequence 2, 5′-GTGGATGACAACAGCGACGAA-3′. siRNAs were transfected with Lipofectamine RNAiMAX (Invitrogen).
Immunofluorescent Staining and Cell Staining with Lysotracker and Phalloidin
The cells were fixed with 4% paraformaldehyde, washed with phosphate-buffered saline (PBS) three times, and permeabilized with 0.05% saponin in PBS. The cells were then incubated in PBS containing 1% bovine serum albumin (BSA) for 1 h and then with primary antibodies or fluorescein-conjugated lectins in PBS containing 0.05% saponin and 0.1% BSA at 4 °C overnight. After rinsing with PBS three times, the cells were incubated with Alexa Fluor 488- or 594-conjugated secondary antibody (Invitrogen) at room temperature for 1 h. Following staining of the nuclei with Hoechst 33258, the samples were mounted and examined using a fluorescence microscope, Leica DMI 6000B.
Antibodies, Lectins, and Other Probes for Cell Staining
Antibodies and lectins used for Western blot analyses and immunofluorescent staining were as follows: rabbit polyclonal anti-Myc tag and LAMP2 antibodies (Abcam); concanavalin A (ConA) lectin conjugated with Alexa Fluor 488 (Invitrogen); mouse monoclonal anti-Bip/GRP78, GM130, EEA1, paxillin, and caveolin1 antibodies (BD Biosciences); mouse monoclonal anti-Golgi58K and clathrin heavy chain antibodies (Abcam); wheat germ agglutinin (WGA) lectin conjugated with Oregon Green 488 (Invitrogen); Agaricus bisporus agglutinin (ABA) lectin conjugated with biotin (J-Oil Mills); Helix pomatia agglutinin (HPA) lectin conjugated with Alexa Fluor 488 (Invitrogen); Sambucus nigra bark agglutinin (SNA) lectin conjugated with biotin (Vector Laboratories); phytohemagglutinin-L (PHL) lectin conjugated with biotin (J-Oil Mills); Jacalin lectin conjugated with fluorescein (Vector Laboratories); rabbit polyclonal anti-LC3 antibodies (MBL); rabbit polyclonal anti-Rab5, Rab7, Rab4, and Rab11 antibodies (Cell Signaling Technology); and rabbit polyclonal anti-actin antibodies (Sigma). To visualize the lysosomes and the actin filaments, Lysotracker Red DND-99 (Invitrogen) and Alexa Fluor 594 phalloidin (Invitrogen) were used, respectively.
Glycosidase Treatment
The protein lysates (20 μg) were digested with PNGase F (New England BioLabs) or endo-α-N-acetylgalactosaminidase (Seikagaku Corp.). The samples were boiled in 20 μl of denaturing buffer (1% SDS and 1% 2-mercaptoethanol) for 2 min and incubated with 1 unit of enzyme at 37 °C for 12 h.
Assay for Glycosyltransferase Activity
The construction and expression of soluble recombinant human WBSCR17 with His6 and FLAG tags at the N terminus of the truncated WBSCR17 were carried out as described previously (9). The recombinant WBSCR17, which was expressed in High Five cells using a baculovirus expression system and recovered in the conditioned medium, was purified by batch absorption of the tagged recombinant enzyme with anti-FLAG-M2 antibody affinity gel (Sigma) according to the manufacturer's protocol. The assay for glycosyltransferase activity was performed with synthetic peptides as substrate at 37 °C for 40 h as described previously (9). Amino acid sequences of acceptor peptides are as follows: MUC1 (PAPGSTAPPK), MUC5AC-1 (TSAPGTTPSP), MUC5AC-2 (GTTPSPVPTT), MUC5AC-3 (SPVPTTSTTS), and MUC7 (SATTPAPPSS).
RT-PCR and Real-time PCR Analysis
Total RNAs from HEK293T cells were prepared using Sepasol RNA I (Nacalai Tesque) and reverse-transcribed with Superscript III reverse transcriptase (Invitrogen) at 55 °C for 1 h with oligo(dT) as primer. The cDNAs thus obtained were used as template for PCR, which was carried out with PrimeStar GXL DNA polymerase (Takara). Real-time quantitative PCR of WBSCR17 and GAPDH was performed using specific TaqMan probes, which were purchased from Applied Biosystems. Amplifications were run with the StepOnePlus Real-Time PCR system (Applied Biosystems). Primer sequences used were as follows: WBSCR17 (forward, 5′-TCAATCACACGCCCACACAC-3′; reverse, 5′-GGTAGCGTTTGTGGACATAC-3′) and GAPDH (forward, 5′-ATCACTGCCACCCAGAAGAC-3′; reverse, 5′-TCGCTGTTGAAGTCAGAGGAG-3′).
Quantification of Cell Surface Area
The cells were observed by phase-contrast microscopy, and their digital images were captured. A hundred cells in an arbitrarily chosen area of the images were analyzed with ImageJ software to quantify cell surface area. The average area was obtained from three independent data sets.
Cell Surface Biotinylation
The cells were washed with ice-cold PBS and incubated with 1 mg/ml sulfosuccinimidyl biotin reagent (Thermo Scientific) in PBS at 4 °C for 30 min. To remove excess biotin reagent, the cells were incubated with DMEM at 4 °C for 15 min. The cells were then incubated in DMEM containing 10% FCS at 37 °C for 4 h. After the incubation, the cells were placed on ice, and biotin that had non-specifically bound to the plasma membrane was removed by adding 60 mm glutathione in PBS. After fixation of the cells with 4% PFA, biotin-conjugated cell surface proteins were detected with FITC-streptavidin.
Dextran and Transferrin Incorporation Assays
For dextran incorporation, the cells were cultured in 24-well plates for 72 h. Then the growth medium was removed, and the cells were cultured in medium containing 0.2 mg/ml FITC-dextran with Mr 3,000 or 10,000 (Invitrogen) at 4 or 37 °C for 30 min. For transferrin incorporation, the cells were preincubated in serum-free medium at 37 °C for 30 min and then cultured in medium containing 50 μg/ml FITC-transferrin (Invitrogen) at 4 or 37 °C for 15 min. Immediately after the incubation, the cells were washed with ice-cold PBS and then incubated with acid wash buffer (50 mm glycine/HCl, pH 3, containing 100 mm NaCl) at 4 °C to release cell surface-bound transferrin. For observation with a fluorescein microscope, the cells were fixed with 4% PFA, washed with PBS, and then stained with Hoechst 33258. The samples were mounted and examined using a fluorescence microscope, Leica DMI 6000B. To measure the dextran and transferrin incorporation, the cells were dissociated with enzyme-free cell dissociation buffer (Invitrogen), pelleted, and washed with PBS. Fluorescein intensity was determined with a FACSCalibur flow cytometer (BD Biosciences). The median fluorescence value was determined for 10,000 cells.
Metabolic Labeling of Glycoproteins with N-Azidoacetylgalactosamine (GalNAz)
GalNAz (Invitrogen) labeling of glycoproteins was performed as reported previously (21). Briefly, 10 μl of 100 mm GalNAz (Invitrogen) in ethanol was added to a 10-cm culture dish, and the ethanol was evaporated at room temperature. GalNAz was dissolved in 10 ml of DMEM containing 10% FCS and incubated for 2 h. The growth medium containing GalNAz was used for metabolic labeling of HEK293T cells cultured in the fibronectin-coated dish. The cells were collected, lysed, and visualized with the Click-IT tetramethylrhodamine protein analysis detection kit (Invitrogen) following the manufacturer's instructions.
RESULTS
Biological Characterization of WBSCR17
To analyze the biological roles of WBSCR17, we expressed recombinant WBSCR17 in COS7 cells and characterized its properties. For this purpose, mouse WBSCR17 cDNA was inserted into a pcDNA6/Myc-His expression vector and transfected into COS7 cells. To detect the expression of the recombinant molecule, both the culture medium and the cell lysate were examined by reducing SDS-PAGE followed by Western blotting with anti-Myc tag antibodies, identifying two bands of ∼71 and 80 kDa only in the lysate of the transfected cells (Fig. 2A). With the calculated molecular mass of recombinant WBSCR17 being 71 kDa, the lower and the upper bands are most likely molecular forms without and with post-translational modifications, respectively. Because in silico analyses predicted N- and O-glycosylation sites in WBSCR17 (NetNGlyc 1.0 Server (22)3 and NetOGlyc 3.1 Server (23)), we examined whether it was glycosylated by lectin blotting analyses. ConA, which recognizes high mannose N-glycans, bound to the upper band (supplemental Fig. 1) of the transfected cells. When the cell lysate was digested with O-glycosidase or N-glycosidase PNGase F and detected with anti-Myc antibodies, only PNGase F affected the pattern, with the upper band decreased and the lower band concomitantly increased (Fig. 2B). In addition, PNGase treatment resulted in loss of the ConA-positive upper band. Thus, the upper and lower bands were those for N-glycosylated and unglycosylated forms of WBSCR17, respectively. We then examined its intracellular localization by immunostaining (Fig. 2C). The majority of recombinant WBSCR17 detected by anti-Myc antibody was co-localized with the Golgi markers, Golgi58K and GM130. In addition, GalNAc-T1, a cis-Golgi-localized isozyme (24), gave an almost superimposable staining pattern with WBSCR17. There was a small amount of WBSCR17 in the perinuclear region, where it partially co-localized with an endoplasmic reticulum (ER) marker, GRP78. No signal for WBSCR17 was found in the trans-Golgi network, which was positive for WGA. The immunostaining clearly demonstrated that WBSCR17 was predominantly localized in the Golgi apparatus, as is the case for the other GalNAc-Ts.
FIGURE 2.

Characterization of recombinant WBSCR17 protein in COS7 cells. A, the culture medium and cell lysates were collected and analyzed by Western blotting with an anti-Myc antibody. Only in cell lysate of the WBSCR17 transfectant, two bands of about 80 and 71 kDa were observed. Black and white arrowheads indicate the upper and the lower bands, respectively. B, treatment of N-glycosidase PNGase F, but not O-glycosidase, led to a decrease in the 80-kDa form and increase in the 71-kDa form (top). The upper band positive for ConA staining was lost upon digestion with PNGase F (bottom). Black and white arrowheads indicate the upper and lower bands, respectively. C, subcellular localization of recombinant WBSCR17 in COS7 cells. The recombinant WBSCR17 was detected with anti-Myc antibody, and its subcellular localization was compared with that of several organelle markers: Golgi58K, Golgi; GM130, cis-Golgi; GalNAc-T1, cis-Golgi and medial Golgi; WGA, trans-Golgi; and Grp78, ER. The rightmost panels are magnified images of the merged panels. WBSCR17 was predominantly localized in the Golgi apparatus. Scale bars, 10 μm. IB, immunoblot.
Assay of Recombinant WBSCR17 for Its Catalytic Activity
To characterize human WBSCR17 biochemically, we expressed it as a soluble recombinant form in insect cells using a baculovirus expression system. High Five cells were transfected with baculoviruses containing cDNA for the soluble WBSCR17 with His6 and FLAG tags at its N terminus, and, 3 days later, the conditioned medium was recovered. Recombinant WBSCR17 in the medium was purified with anti-FLAG antibody affinity gel to homogeneity, as determined by silver staining of the SDS-polyacrylamide gel (Fig. 3A) and also by detection of the FLAG tag (Fig. 3B) on a PVDF membrane after Western blotting. We then assayed for enzymatic activity of the purified recombinant WBSCR17 using several mucin peptides as acceptor substrates. WBSCR17 exhibited very low but detectable activity toward some of the substrates. Among them, MUC7 peptide was the most efficient substrate, and MUC5AC-1 and -2 peptides were glycosylated as well but with less efficiency (Fig. 3C).
FIGURE 3.

Purification and enzyme assay of WBSCR17. Recombinant soluble WBSCR17 was purified with anti-FLAG-M2 agarose. A, purified WBSCR17 was subjected to SDS-PAGE and silver-stained. An arrowhead indicates purified recombinant WBSCR17. B, Western blotting of purified recombinant WBSCR17 with an anti-FLAG antibody. C, assay for the enzymatic activity of purified WBSCR17 using several mucin peptides as acceptor substrates. WBSCR17 exhibited very low but detectable activity toward MUC5AC-1, MUC5AC-2, and MUC7 peptides. Data shown represent means ± S.D. (error bars) of three independent experiments.
Roles of WBSCR17 in HEK293T Cells
To elucidate the roles of WBSCR17, we suppressed endogenous WBSCR17 in HEK293T cells with two siRNAs targeting at the distinct sequences. RT-PCR revealed that both siRNAs, but not control siRNA, suppressed the WBSCR17 mRNA expression (Fig. 4A). When HEK293T cells were cultured in the fibronectin (FN)-coated dish, they showed an epithelial-like phenotype and formed lamellipodium-like plasma membrane structures more efficiently than those in the absence of FN (Fig. 4B and supplemental Fig. 2). By contrast, the cells with WBSCR17 suppressed (WBS17KD cells) showed an altered morphology with rounder and smaller appearances and almost completely lost the lamellipodium-like structures in the FN-coated dish (Fig. 4B and supplemental Fig. 2). Cells expressing protrusive structures, such as lamellipodia and filopodia, are generally more adhesive to a solid substratum and have a larger cell surface under microscopic observation than those without them (25). We therefore carried out morphometric analysis of cell surface area to measure cell spreading (Fig. 4C). The analysis showed that the average cell surface area of the control cells (siNC) cultured in the presence of FN was larger than that of the cells without FN. Knocking down WBSCR17 in the cells with or without FN resulted in a decreased area, suggesting that WBSCR17 positively modulates the lamellipodium-like structure and increases the adhesive nature of the cells. These findings were further corroborated by studies of WBSCR17 overexpression. Overexpressed WBSCR17, in turn, increased the cell surface area, indicating the enhanced formation of protrusive structures.
FIGURE 4.
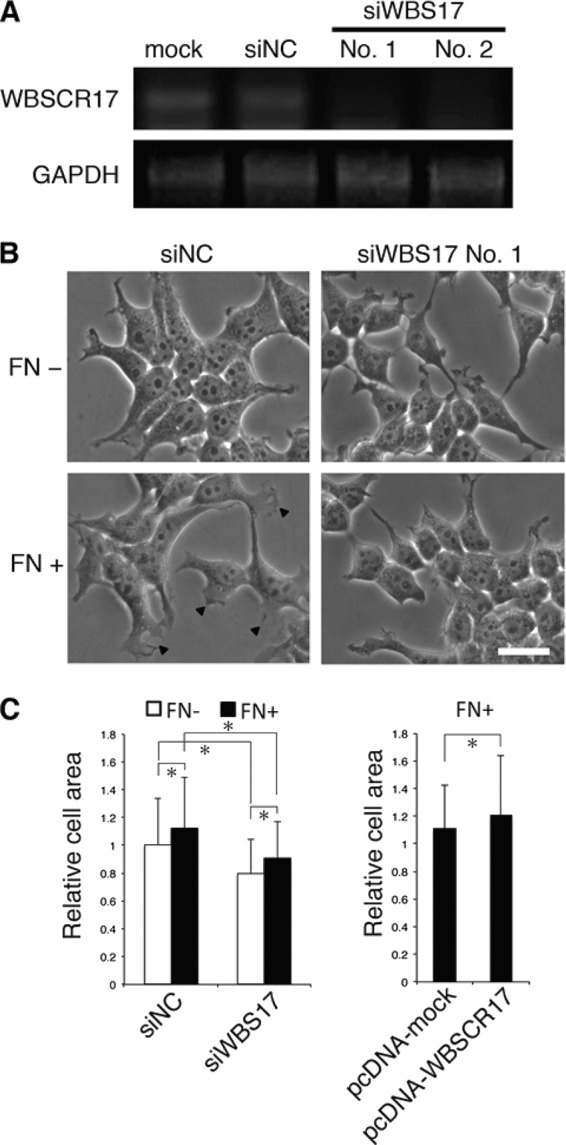
Suppression of WBSCR17 in HEK293T cells inhibits lamellipodium formation. A, validation of siRNA suppression by RT-PCR analysis. WBSCR17 mRNA levels were evaluated by RT-PCR using primers for WBSCR17. GAPDH mRNA was used as a control for equal gel loading. Two siRNAs against WBSCR17 (No. 1 and No. 2), but not control siRNA (siNC), suppressed the WBSCR17 mRNA expression. B, change of cell morphology by knockdown of WBSCR17. A dish coated with FN induced cell spreading and lamellipodium formation (bottom left, lamellipodia are indicated by arrowheads). HEK293T cells transfected with WBSCR17 siRNA remained round-shaped and did not form lamellipodia on the FN-coated dish. Scale bar, 20 μm. C, measurement of cell surface area revealed the involvement of WBSCR17 in cell spreading. Cell surface area was quantified using ImageJ software. Suppression of WBSCR17 led to decreased cell area, whereas WBSCR17 overexpression gave rise to an increased area. Data shown represent means ± S.D. (error bars) of three independent experiments. *, p < 0.05.
We then investigated profiles in mucin-type O-glycosylation with lectin blotting, using total cell lysates prepared from the control and the WBS17KD cells. The lectins used were Jacalin, ABA, HPA, and SNA lectins, which predominantly recognize (Sia)Galβ1–3GalNAcα1-Ser/Thr ((sialyl) T-antigen), (sialyl) T-antigen, non-reducing terminal αGalNAc, and Neu5Acα2–6Gal(NAc)-R, respectively (Fig. 5A). Analyses with Jacalin and ABA lectins revealed enhanced expression of ∼100-kDa glycoproteins in the WBS17KD cells (Fig. 5, A and A′). HPA also detected the elevated expression of a band with similar but, to some extent, higher molecular weight (Fig. 5, A and A′). Decreased expressions of ∼150- and ∼170-kDa glycoproteins were recognized by ABA but not by the other lectins (Fig. 5A). By contrast, the control and the WBS17KD cells had essentially the same glycoprotein profiles as examined with SNA (Fig. 5A). Metabolic labeling of glycoconjugates containing GalNAc with GalNAz, an azide-modified GalNAc, demonstrated alterations in glycoprotein profiles of the WBS17KD cells (Fig. 5B). There were high molecular mass (150–180 kDa) bands with decreased expression, together with the changes in the expression of 70–90-kDa glycoproteins. The increase in ∼100-kDa glycoproteins recognized by Jacalin and ABA lectins was not observed by GalNAz labeling (Fig. 5B). Next, we overexpressed WBSCR17 and carried out lectin blot analysis with an ABA lectin (Fig. 5, C and C′). The overexpression of WBSCR17 revealed a weak but significant increase in the expression of ∼150-kDa glycoproteins, which may correspond to some of the decreased bands in the WBS17KD cells. No other significant changes were observed (Fig. 5C). Some of the changes observed for the carbohydrate profiles were subtle, but these changes were highly reproducible. We repeated the same experiments more than three times, and the representative data are presented.
FIGURE 5.

Alterations of O-glycan profiles and intracellular accumulation of glycoconjugates in the WBS17KD cells. A and A′, profiles of glycoproteins in the cell lysates from control and WBS17KD cells were examined with Jacalin, ABA, HPA, and SNA lectins. Black and white arrowheads indicate down-regulated and up-regulated glycoproteins, respectively. The magnified images of bands of ∼100 kDa that were up-regulated in the knockdown cells (A) are shown in A′. B, metabolic labeling of glycoproteins with GalNAz, an azide-modified GalNAc. Cell lysates were prepared from the cells that were labeled with GalNAz. GalNAz-labeled glycoproteins in cell lysate were chemically conjugated with tetramethylrhodamine, subjected to SDS-PAGE, and detected by fluorescence observation. Total proteins were stained by Coomassie Brilliant Blue (CBB). There were changes in O-glycan profiles in the WBS17KD cells. Black and white arrowheads indicate down-regulated and up-regulated O-glycans, respectively. C and C′, lectin blot analysis with ABA detected enhanced expressions of ∼150-kDa O-glycoproteins (indicated by an asterisk) in the cells with WBSCR17 overexpressed. C′, a magnified image of C. D and E, lectin staining of HEK293T cells with a PHL lectin (D) and Jacalin, ConA, and WGA lectins (E). Intracellular accumulations of glycoconjugates that were positive for all of the lectins used were detected in WBSCR17 siRNA transfectants. The bottom panels in both D and E are magnified images of the boxed areas in the top panels.
These studies demonstrated that the modulation of WBSCR17 expression in the cells alters their glycoprotein expression profiles. It should be noted that minor but significant changes in O-glycosylation profiles were observed in the WBS17KD cells, under the conditions where only WBSCR17, whose catalytic activity is very low, was suppressed, with the other isozymes remaining unchanged. This indicates that WBSCR17 is involved in O-glycosylation in the cells.
To investigate the localization of glycoproteins in the cells, lectin staining was performed using fluorescence-labeled lectins. We first used a PHL lectin, which binds to complex N-glycans (Fig. 5D and supplemental Fig. 3). This lectin stained the trans-Golgi network most strongly in the control cells, but, interestingly, unusually large vesicles with a diameter of ∼2 μm, which were distributed widely in the cytoplasm, were stained in the WBS17KD cells. Fig. 5E and supplemental Fig. 3 show the staining with Jacalin, ConA, and WGA lectins. In the control cells, Jacalin and WBA predominantly stained the Golgi apparatus, whereas ER was strongly stained with ConA. We found that all of these lectins similarly stained the large vesicles in the cytoplasm in the WBS17KD cells. Taken together, the data so far suggest that WBSCR17 is involved in lamellipodium formation, O-glycosylation, and the intracellular transport of glycoconjugates.
Cytoplasmic Vesicles Are Positive for a Lysosome Marker
To characterize the cytoplasmic vesicles, double labeling with fluorescein-labeled lectins (PHL, Jacalin, and WGA) and antibodies toward organelle markers was performed. The large vesicles positive for the PHL lectin staining in the WBS17KD cells did not overlap with the Golgi or ER markers, Golgi58K and GRP78, the cellular localization of which was not affected (supplemental Fig. 4). They also did not co-localize with an early endosome marker, EEA1 (Fig. 6A), but a lysosome marker, LAMP2, was found in the large vesicles that were positive for PHL in the knockdown cells (Fig. 6B). Interestingly, in the WBS17KD cells, the phenotypes of the vesicles positive for both EEA1 and LAMP2 were affected, with a larger structure than those in the control (Fig. 6, A and B). These data suggest that WBSCR17 is involved in regulation of the endosome/lysosome pathway and that its suppression disturbs the normal membrane trafficking, resulting in the accumulation of glycoconjugates incorporated from the plasma membrane in the transport vesicles and the enlargement of the vesicles. To examine this possibility, we analyzed the transport of membrane proteins by the cell surface biotinylation assay. In the assay, cell surface proteins of HEK293T cells were biotinylated, the cells were cultured for 4 h, and then the biotinylated proteins were reacted with FITC-streptavidin and analyzed. In the WBS17KD cells, most of the biotinylated membrane proteins were internalized into the cells and accumulated in the large vesicles (Fig. 6C), whereas those of the control cells were mainly associated on the cell surface with a small portion incorporated into small vesicles. These data clearly demonstrate that, upon the suppression of WBSCR17, the cell surface glycoconjugates were incorporated into the cells and accumulated in the large vesicles.
FIGURE 6.

Accumulation of glycoconjugates in the lysosomes in the WBS17KD cells. Two days after the transfection of siRNAs, HEK293T cells were stained with fluorescence-labeled PHL lectin and antibodies against organelle markers: EEA1 for early endosomes (A) and LAMP2 for lysosomes (B). The glycoconjugates positive for PHL accumulated in the vesicles that are labeled with LAMP2 but not with EEA1. The vesicles that were positive for either EEA1 or LAMP2 were enlarged in the WBS17KD cells. The rightmost panels are magnified images of the merged panels. C, cell surface proteins of HEK293T cells were biotinylated, the cells were cultured for 4 h, and then the biotinylated proteins were reacted with FITC-streptavidin and analyzed. The analysis revealed increased internalization of plasma membrane proteins to large vesicles (indicated by an arrowhead).
WBSCR17 Regulates Macropinocytosis
The observations that the knockdown of WBSCR17 led to the accumulation of the glycoconjugates in the vesicles that were positive for LAMP2 staining (Fig. 6B), together with a previous report showing the involvement of WBSCR17 in bulk endocytosis (13), prompted us to investigate whether WBSCR17 regulates fluid phase endocytosis. For this purpose, a dextran incorporation assay was carried out, in which fluorescein-conjugated dextran (Mr 3,000 and 10,000) was added to the culture medium, and endocytosed dextran was observed by fluorescence microscopy (Fig. 7A) and quantified by flow cytometry (Fig. 7, B–E). Dextran Mr 3,000 (small) is a marker for all types of pinocytosis, such as receptor-mediated endocytosis and macropinocytosis, and Mr 10,000 dextran (large) is endocytosed primarily through macropinocytosis (26, 27). Fluorescent microscopic observations of the incorporated small Mr 3,000 dextran in the control cells revealed that the fluorescent signals were associated with small vesicles (Fig. 7A). However, in the WBS17KD cells, the incorporated small dextran was found in large vesicles as well as in small ones (Fig. 7A and supplemental Fig. 5). Flow cytometry showed that the amount of incorporated small dextran was essentially the same between the control and the WBS17KD cells (Fig. 7, B and D). Contrary to this, the incorporation of the large dextran into the WBS17KD cells was ∼1.4 times higher than that in the control cells (Fig. 7D), and there was a population of cells with stronger fluorescent signals that appeared as a small shoulder in flow cytometry analysis (Fig. 7C). We then overexpressed WBSCR17 to confirm that the elevated macropinocytosis is due to its suppression. Fig. 7E shows that WBSCR17 expressed in the cells suppressed the macropinocytosis (i.e. the incorporation of large dextran) to a level as low as about half that of the control. The macropinocytosis increased by the siRNA fell to ∼70% of that of the control after the overexpression of WBSCR17. Thus, WBSCR17 negatively regulates macropinocytosis. We then labeled the cells with large dextran and Lysotracker, a lysosome marker. In the control cells, the incorporated dextran generally did not overlap with Lysotracker (Fig. 8A). The WBS17KD cells, on the other hand, contained large vesicles that were positive for both dextran and Lysotracker (Fig. 8A), which clearly demonstrates the fusion of macropinosomes with lysosomes after their entry into and migration in the cells. We also found large vesicles that were positive for dextran and negative for Lysotracker, which indicates that the vesicles on the way to the lysosomes were already enlarged. Furthermore, we investigated whether the macropinocytosis-related events were influenced by the WBSCR17 knockdown. Because macropinosomes are formed from actin-rich ruffles of the plasma membrane, we examined the localization of actin filaments in the WBS17KD cells (Fig. 8B). Compared with the control cells, where actin filaments were distributed throughout the cytoplasm, the WBS17KD cells showed an overall decrease in F-actin, which corresponds to decreased lamellipodia, and had characteristic actin-rich rufflings (see Fig. 8B, inset), the structures typical of macropinocytosis. Paxillin is a scaffold protein localized to the intracellular surface of cell adhesion sites to the extracellular matrix. The decrease in paxillin in the WBS17KD cells suggests the less adhesive nature of the knockdown cells (Fig. 8B and supplemental Fig. 6). No overlapping expression of paxillin and actin filaments indicates that the actin-rich extensions were of dynamic organization. We also investigated the distribution of LC3, a major constituent of autophagosomes, which had been recently reported to be recruited to macropinosomes and facilitate their fusion with lysosomes (19). Although LC3 was uniformly distributed in the control cells, the WBS17KD cells had LC3 accumulated in the large vesicles positive for PHL staining (Fig. 8C). This indicates the fusion of dextran-positive vesicles incorporated by macropinocytosis with the lysosomes.
FIGURE 7.

WBSCR17 regulates fluid phase endocytosis. A, labeling with fluorescein-conjugated dextran Mr 3,000. The cells were labeled with Mr 3,000 dextran to visualize the fluid phase endocytosis. Fluorescence microscopic observation revealed large vesicles containing dextran in the WBS17KD cells. The bottom panels are magnified images of the boxed areas in the top panels. B and C, flow cytometric analysis of cells labeled with Mr 3,000 (B) and Mr 10,000 (C) dextran. Black lines, incorporation at 37 °C; gray areas, incorporation at 4 °C; black areas, without dextran. Histograms are representative of three independent experiments. D and E, fluorescence intensity of the cells as determined by flow cytometry. Data shown represent means ± S.D. (error bars) of three independent experiments. *, p < 0.05. D, knocking down WBSCR17 enhanced uptake of Mr 10,000 dextran but not that of Mr 3,000 dextran. E, overexpression of WBSCR17 inhibited incorporation of Mr 10,000 dextran and rescued the effect of WBSCR17 siRNA.
FIGURE 8.

WBSCR17 regulates macropinosome formation. A, co-labeling of the cells with Mr 10,000 dextran and Lysotracker, a lysosome marker. In the control cells, most of the vesicles positive for dextran were small and not stained with Lysotracker. The WBS17KD cells, on the other hand, had large vesicles co-labeled with dextran and Lysotracker. The rightmost panels are magnified images of the boxed areas in the merged panels. B, staining of HEK293T cells with phalloidin, which binds to F-actin, and with anti-paxillin antibody. The arrowheads indicate actin-rich round-shaped rufflings in the WBS17KD cells. The top left corner of the merged image of the knockdown cells is a magnified image of the boxed area. C, staining of HEK293T cells with antibody to LC3 and a PHL lectin. In the WBS17KD cells, LC3 was associated with the large vesicles positive for PHL staining.
Suppression of WBSCR17 Does Not Affect Clathrin- and Caveolin-dependent Endocytosis
To investigate how WBSCR17 is involved in other membrane transport and trafficking, we examined the expression of endosomal and lysosomal markers. The fluorescein-conjugated transferrin (Tf) incorporation assay was employed to investigate the clathrin-dependent endocytic pathways. After incubating the cells with fluorescent Tf, the incorporated Tf was analyzed by microscopy (Fig. 9A) and by flow cytometry (Fig. 9B). There were no significant differences in the Tf localization or in the amount of Tf incorporation between the control and the knockdown cells. We then investigated the expression of several markers and molecules for membrane trafficking. To examine the receptor-dependent endosome formation, the expression of clathrin heavy chain (Fig. 9, C and D) and caveolin 1 (Fig. 9D) was investigated by Western blotting and/or immunostaining. They had similar expression patterns in the control and WBS17KD cells, demonstrating that the macropinocytosis elevated by WBSCR17 suppression was independent of the clathrin- and caveolin-dependent endosome formation. We also investigated expression of marker molecules involved in intracellular membrane trafficking by Western blotting. Concerning EEA1 and Rab5, early endosome markers that are required for endosome fusion, we observed that EEA1 was decreased in the knockdown cells (Fig. 9C), but Rab5 exhibited unchanged expression (Fig. 9C). Immunofluorescent staining of Rab5, however, demonstrated that it was associated with the large vesicles in the knockdown cells (Fig. 9D). Despite the EEA1 decrease, the fusion required to form endosomes seemed unaffected in the knockdown cells because they had large EEA1-positive vesicles (Fig. 6A). Rab4 is an early recycling endosome marker, which is involved in membrane retrieval from the early endosome to the plasma membrane, and its expression was decreased in the WBS17KD cells (Fig. 9, C and D). By contrast, Rab7 is a late endosomal marker that mediates the fusion of the vesicles from early endosomes with late endosomes, and its expression was markedly enhanced (Fig. 9C) and was associated with large PHL-positive vesicles (Fig. 9D) in the knockdown cells. The imbalance of Rab4/Rab7 expression may direct the vesicle transport more to late endosomes/lysosomes than to the plasma membrane in the WBS17KD cells, generating the large endosomal/lysosomal vesicles. There were no significant changes in the amount of LAMP2 (a lysosome marker) (Fig. 9C) and Rab11 (a late recycling endosome marker) (Fig. 9, C and D). These findings indicate that WBSCR17 is not involved in the clathrin- and caveolin-dependent endosome formation but in the receptor-independent macropinocytosis and the following intracellular membrane trafficking above all between the early endosomes and the late endosomes/lysosomes.
FIGURE 9.

Suppression of WBSCR17 does not affect clathrin- and caveolin-dependent endocytic pathways. A, clathrin-dependent endocytic pathways. A fluorescein-conjugated Tf incorporation assay was performed to detect clathrin-dependent endocytosis. B, determination of Tf incorporation by flow cytometry. The suppression of WBSCR17 did not affect the Tf incorporation significantly. Data shown represent means ± S.D. (error bars) of three independent experiments. C, Western blot analysis of endosomal and lysosomal markers. clathrin, clathrin heavy chain; Rab5, an early endosomal marker; EEA1, an early endosomal marker 1; Rab4, an early recycling endosome marker; Rab11, a late recycling endosome marker; Rab7, a late endosomal marker; LAMP2, a lysosomal marker; actin, a loading control. The expressions of clathrin heavy chain, Rab5, and Rab11 were not affected by the knockdown of WBSCR17. By contrast, in the WBS17KD cells, there were an increase in Rab7 and a decrease in Rab4. D, labeling of the cells with a lectin and antibodies to the endosome markers. No changes were observed for clathrin and caveolin. Rab5 was associated with the large vesicles in WBS17KD cells (indicated by arrowheads). The top left corner of the Rab5 image of the knockdown cells is a magnified image of the boxed area. PHL-positive staining in the WBS17KD cells was positive for Rab7.
A High Concentration of GlcNAc Induces WBSCR17 Expression in HEK293T Cells
A previous report showed that, using microarray analysis, HEK293T cells cultured in 60 mm GlcNAc have enhanced expression of several genes, including WBSCR17 (13). We carried out RT-PCR and real-time PCR analyses to make sure of the enhanced WBSCR17 expression. HEK293T cells were cultured in medium including 0, 50, and 100 mm GlcNAc for 2 days, and the expression of WBSCR17 mRNA was measured, showing its ∼10- and ∼5-fold increases for 50 and 100 mm GlcNAc, respectively (Fig. 10, A and B). Furthermore, we examined the influences of the high concentration GlcNAc treatment on mucin-type O-glycosylation. We prepared total cell lysates from the cells with or without the GlcNAc treatment and carried out lectin blot analyses with an ABA lectin. The analysis revealed enhanced expression of several glycoproteins with relatively high molecular weights (Fig. 10C). This may indicate the up-regulation of several glycosyltransferases involved in mucin-type carbohydrates, including WBSCR17 (13). An elevated concentration of UDP-GalNAc may account for the increased O-glycosylation as well because GlcNAc incorporated into the cells is metabolized into UDP-GlcNAc, which is convertible to UDP-GalNAc (13). Decreases in cell proliferation and dextran incorporation by the GlcNAc treatment were also observed as reported previously (Fig. 10D) (13). These data suggest that WBSCR17 regulates macropinocytosis through O-glycosylation under the control of GlcNAc concentration.
FIGURE 10.

Up-regulation of WBSCR17 mRNA and O-glycosylation in response to concentration of GlcNAc. The expression of WBSCR17 was examined by RT-PCR (A) and real-time PCR (B) in the HEK 293T cells that were treated with or without a high concentration of GlcNAc. The treatment with GlcNAc up-regulated WBSCR17 mRNA. Data shown represent means ± S.D. (error bars) of three independent experiments. C, ABA lectin blot analysis of glycoproteins in the cell lysates. The GlcNAc treatment enhanced the expression of ABA-positive proteins with relatively high molecular weight as indicated by arrowheads (left). Total proteins were stained with Coomassie Brilliant Blue (CBB) (right). D, a high concentration of GlcNAc inhibited proliferation and fluid phase endocytosis. The bottom panels show incorporated high molecular weight dextran through macropinocytosis. E, a hypothetical scheme of the role of WBSCR17. The expression of WBSCR17 corresponds to the nutrient concentration as exemplified by environmental GlcNAc and may work as a modulator of nutrient uptake through controlling macropinocytosis in the cells.
DISCUSSION
It has been widely believed that mucin-type O-glycosylation is initiated in the Golgi apparatus (24). This idea was supported by reports showing the Golgi localization of GalNAc-T1, -T2, -T3, and -T6 (24, 28). A recent report, however, demonstrated that GalNAc-T2 is redistributed from the Golgi to the ER after the activation of a Src kinase by EGF and that the relocalization may be related to the increased O-glycosylation in cancer cells (29, 30). More recently, GalNAc-T18, a member of the Y subfamily, was reported to be localized in the ER in lung carcinoma cells (6). Our present study on WBSCR17 demonstrated its predominant localization in the Golgi (Fig. 2C). Our preliminary experiments showed that the EGF treatment of the cells did not change its localization (data not shown). This may reflect the isoform-specific localization and redistribution of GalNAc-Ts.
Our previous study on the structure-function relationship of GalNAc-T1 indicated that the substitutions of several amino acid residues resulted in loss or decrease of the activity (10). The Trp residue in the Gal/GalNAc-T motif (Fig. 1) is one of the most important elements in the motif, the replacement of which by Tyr led to reduced activity by ∼70% for GalNAc-T1 (10). Li et al. (6) recently designated a GalNAc-T subfamily consisting of GalNAc-T8, -T9, -T18, and WBSCR17 as the Y subfamily because they all have the Trp (W)-to-Tyr (Y) replacement. They also reported that the Y isozymes are inactive under the classical assay using peptide substrates and raised the possibility that GalNAc-T18, which is co-localized with GalNAc-T2 in the ER, functions as a chaperone that modulates the activity of the other W subfamily GalNAc-Ts. In agreement with their hypothesis, the catalytic activity of WBSCR17 detected was less than 1% of that of GalNAc-T1 when assayed with typical mucin peptides as substrates (Fig. 3C). This suggests that WBSCR17 might be a chaperone or otherwise function as an enzyme glycosylating defined substrates with sequences distinct from mucins. It would therefore be reasonable to speculate that WBSCR17 is involved in the glycosylation of a small subset of glycoproteins rather than overall glycosylation in the cell. In fact, the impact of suppression/overexpression of WBSCR17 was restricted to a few proteins (Fig. 5, A–C). Taken together, altered O-glycosylation in association with the WBSCR17 suppression/overexpression indicated that it is involved in regulation or synthesis of O-glycans.
In the lectin blot analyses of O-glycosylation in the WBS17KD cells, we observed elevated expression of glycoproteins in addition to the decreased bands. The cause of this increase is not clear, but it may be ascribed to enhanced expression of O-glycosylated proteins that are responsible for the novel properties acquired in the knockdown cells, such as decreased lamellipodia and modulated membrane trafficking. To clarify the roles of WBSCR17 in detail, we are identifying the glycoproteins, the expressions of which were affected by the suppression/overexpression of WBSCR17. In addition, an extensive analysis of the WBSCR17 activity toward random peptides including glycopeptides is under way.
Mucin-type glycans modulate the adhesion between the cells and between cells and extracellular matrix. There are some reports describing the involvement of O-glycosylation in integrin-mediated cell adhesion. For example, sialyl-Tn epitopes on β1 integrin impair the mammalian carcinoma cell migration on fibronectin (31). In addition, in Drosophila, O-glycosylation of tiggrin, an integrin ligand, is essential for its proper secretion and localization to interact with integrin (32). The treatment of HT29 cells with O-benzyl-N-acetyl-α-d-galactosaminide, a competitive substrate for the elongation of O-glycans, induces the predominant accumulation of β1 integrin in the late endosomes as a result of perturbing the endocytic pathway (14). It is also possible that WBSCR17 regulates the cell adhesion through the formation of focal adhesion between integrins and fibronectin because it was necessary for the lamellipodium formation induced by fibronectin (Fig. 4B) and that its suppression led to the decrease in the actin filaments and paxillin (Fig. 8B and supplemental Fig. 6). We therefore examined the subcellular distribution of β1 integrin, a subunit of fibronectin receptors, which is expressed in HEK293T cells, and found that its expression in both the control and the WBS17KD cells was essentially the same, located on the plasma membrane without accumulation in the intracellular vesicles (data not shown). The suppression of WBSCR17 did not affect the expression of Rab11 (Fig. 9, C and D), which recycles β1 integrin from the late endosomes to the plasma membrane (33). Judging from the subcellular localization of integrin, the impact of the WBSCR17 suppression on the membrane trafficking seems different from the influences by the O-benzyl-N-acetyl-α-d-galactosaminide treatment.
The WBS17KD cells exhibited the accumulation of glycoconjugates in the lysosomes (Fig. 5, D and E), which were enlarged and positive for LAMP2 (Fig. 6B). Glycoconjugate accumulation is a characteristic phenotype of lysosomal storage diseases that are caused by defects of lysosomal enzymes (34). It is not clear why the glycoconjugates remain in the WBS17KD lysosomes, escaping from degradation by lysosomal enzymes, but WBSCR17 may be involved in regulation and/or transport of the enzymes, and its suppression may cause dysfunction of the lysosomes. We also observed that the EEA1-positive vesicles were enlarged in the WBS17KD cells (Fig. 6A). WBSCR17 may be involved in regulation of the endosome/lysosome pathway. In accordance with this idea, its suppression enhanced macropinocytosis, which may produce enlarged EEA1-positive vesicles that take up the cell surface glycoconjugates. The amount of glycoconjugates in the EEA1-positive vesicles seems insufficient to be detected by lectin staining (Fig. 6A). The EEA1-positive vesicles are usually transported and fused with the lysosomes. The lysosomes after the fusion, if their function were disabled, would accumulate a large amount of glycoconjugates and be positive for the lectin staining (Fig. 6B). The formation of an excess amount of macropinosomes and their fusion with the lysosomes may have deleterious effects on lysosome function, resulting in the accumulation of glycoconjugates within them.
Macropinocytosis is clathrin-independent actin-driven large size endocytosis (18). It is sometimes hard to analyze macropinocytosis due to the absence of useful markers or cargo. To confirm that macropinocytosis is elevated in the WBS17KD cells, we obtained four lines of evidence: 1) the occurrence of large EEA1-positive vesicles (Fig. 6A); 2) the increase in fluid phase endocytosis, as demonstrated by the elevated uptake of large dextran, a macropinocytosis marker (Fig. 7, D and E); 3) the formation of membrane ruffling positive for actin filaments (Fig. 8B), which is a hallmark of macropinocytosis; and 4) the accumulation of LC3 in the large vesicles (Fig. 8C). LC3, which has been known as an autophagy marker, was recently reported to be recruited to macropinosomes (19). We also examined if autophagy was elevated in the knockdown cells and found that, in both the control and the WBS17KD cells, no recruitment of Atg12, which is involved in autophagosome formation (35), to the vesicles was seen; no double-membrane structures of autophagosome were found by electron microscopic observation (data not shown). WBSCR17 thus controls the membrane trafficking by negatively regulating macropinocytosis.
We confirmed that the treatment of HEK293T cells with a high concentration of GlcNAc induces WBSCR17 mRNA expression, as reported previously (13), and demonstrated that overexpression of WBSCR17 negatively regulates macropinocytosis. Extracellular GlcNAc is incorporated into the cytosol by bulk phase endocytosis and salvaged into UDP-GlcNAc, which leads to the increase in branched N-glycans on cell surface chemokine receptors. This enhances affinities between the glycoproteins and galectins, thereby forming the galectin lattice and protecting the glycoproteins from endocytosis (13, 36). Our study suggests that the mucin-type O-glycans generated by WBSCR17 also regulate the glycoprotein incorporation through macropinocytosis in response to the GlcNAc concentration in the cell. Because UDP-GlcNAc, which is convertible to UDP-GalNAc by an epimerase, can be metabolically produced from nutrients other than GlcNAc, such as glucose, amino acids, fatty acids, and nucleic acids (37, 38), WBSCR17 may work as a modulator of macropinocytosis in the cells in a nutrition concentration-dependent manner; excess nutrition enhances the expression of WBSCR17, giving rise to the reduced macropinocytosis, whereas nutrition shortage, in turn, reduces WBSCR17 and promotes macropinocytosis to take up the nutrients from the environmental solutes (Fig. 10E).
In this study, we found that a vertebrate-specific WBSCR17 regulates cell adhesion and macropinocytosis in response to the nutrient concentration. This finding provides a novel hypothesis that mucin-type O-glycosylation is involved in the regulation of dynamic membrane trafficking. Because WBSCR17 is mainly expressed in the nervous system (6, 9, 11) and is related to WBS (11, 12), it should be functionally important in the brain. It was recently reported that the endocytic pathways, including macropinocytosis as well as cell adhesion, are involved in neuronal development (27, 39, 40). Moreover, we found that WBSCR17 in zebrafish is involved in the regulation of axonal projections in the hindbrain.4 Thus, it would be tempting to speculate that WBSCR17 regulates neuronal events, such as axonal guidance and filopodium/lamellipodium formation, through the control of membrane trafficking. A study to examine the roles of mucin-type O-glycosylation produced by WBSCR17 in cell adhesion or macropinocytosis is under way.
Acknowledgment
We thank Professor Nobuhiro Nakamura (Kyoto Sangyo University) for helpful advice and discussions.
This work was in part supported by Grants-in-Aid for Scientific Research on Innovative Areas 24110516 (to Y. N.), and for Young Scientists (B) 23770159 (to Y. N.), the Protein 3000 Project from the Japan Society for the Promotion of Science (to A. K.), and the Private University Strategic Research Foundation Support Program (to A. K.) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), Japan.

This article contains supplemental Figs. 1–6.
R. Gupta, E. Jung, and S. Brunak, manuscript in preparation.
N. Nakamura, Y. Nakayama, M. Tawara, K. Nishimura, A. Miyake, N. Itoh, and A. Kurosaka, manuscript in preparation.
- GalNAc-T
- UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase
- WBS
- Williams-Beuren syndrome
- WBSCR17
- Williams-Beuren syndrome chromosome region 17
- GalNAz
- N-azidoacetylgalactosamine
- FN
- fibronectin
- ER
- endoplasmic reticulum
- Tf
- transferrin
- siNC
- negative control siRNA
- ConA
- concanavalin A
- WGA
- wheat germ agglutinin
- ABA
- A. bisporus agglutinin
- HPA
- H. pomatia agglutinin
- SNA
- S. nigra bark agglutinin
- PHL
- phytohemagglutinin-L
- PNGase F
- peptide:N-glycosidase F.
REFERENCES
- 1. Fukuda M. (2002) Roles of mucin-type O-glycans in cell adhesion. Biochim. Biophys. Acta 1573, 394–405 [DOI] [PubMed] [Google Scholar]
- 2. Gerloni M., Castiglioni P., Zanetti M. (2005) The cooperation between two CD4 T cells induces tumor protective immunity in MUC.1 transgenic mice. J. Immunol. 175, 6551–6559 [DOI] [PubMed] [Google Scholar]
- 3. Ogata S., Uehara H., Chen A., Itzkowitz S. H. (1992) Mucin gene expression in colonic tissues and cell lines. Cancer Res. 52, 5971–5978 [PubMed] [Google Scholar]
- 4. Varki A., Angata T. (2006) Siglecs. The major subfamily of I-type lectins. Glycobiology 16, 1R–27R [DOI] [PubMed] [Google Scholar]
- 5. Elhammer A. P., Kézdy F. J., Kurosaka A. (1999) The acceptor specificity of UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferases. Glycoconj. J. 16, 171–180 [DOI] [PubMed] [Google Scholar]
- 6. Li X., Wang J., Li W., Xu Y., Shao D., Xie Y., Xie W., Kubota T., Narimatsu H., Zhang Y. (2012) Characterization of ppGalNAc-T18, a member of the vertebrate-specific Y subfamily of UDP-N-acetyl-α-d-galactosamine. Polypeptide N-acetylgalactosaminyltransferases. Glycobiology 22, 602–615 [DOI] [PubMed] [Google Scholar]
- 7. Raman J., Guan Y., Perrine C. L., Gerken T. A., Tabak L. A. (2012) UDP-N-Acetyl-α-d-galactosamine:polypeptide N-acetylgalactosaminyltransferases. Completion of the family tree. Glycobiology 22, 768–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Peng C., Togayachi A., Kwon Y. D., Xie C., Wu G., Zou X., Sato T., Ito H., Tachibana K., Kubota T., Noce T., Narimatsu H., Zhang Y. (2010) Identification of a novel human UDP-GalNAc transferase with unique catalytic activity and expression profile. Biochem. Biophys. Res. Commun. 402, 680–686 [DOI] [PubMed] [Google Scholar]
- 9. Nakamura N., Toba S., Hirai M., Morishita S., Mikami T., Konishi M., Itoh N., Kurosaka A. (2005) Cloning and expression of a brain-specific putative UDP-GalNAc:Polypeptide N-acetylgalactosaminyltransferase gene. Biol. Pharm. Bull. 28, 429–433 [DOI] [PubMed] [Google Scholar]
- 10. Tenno M., Saeki A., Elhammer A. P., Kurosaka A. (2007) Function of conserved aromatic residues in the Gal/GalNAc-glycosyltransferase motif of UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase 1. FEBS J. 274, 6037–6045 [DOI] [PubMed] [Google Scholar]
- 11. Merla G., Ucla C., Guipponi M., Reymond A. (2002) Identification of additional transcripts in the Williams-Beuren syndrome critical region. Hum. Genet. 110, 429–438 [DOI] [PubMed] [Google Scholar]
- 12. Hinsley T. A., Cunliffe P., Tipney H. J., Brass A., Tassabehji M. (2004) Comparison of TFII-I gene family members deleted in Williams-Beuren syndrome. Protein Sci. 13, 2588–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lau K. S., Khan S., Dennis J. W. (2008) Genome-scale identification of UDP-GlcNAc-dependent pathways. Proteomics 8, 3294–3302 [DOI] [PubMed] [Google Scholar]
- 14. Ulloa F., Real F. X. (2003) Benzyl-N-acetyl-α-d-galactosaminide induces a storage disease-like phenotype by perturbing the endocytic pathway. J. Biol. Chem. 278, 12374–12383 [DOI] [PubMed] [Google Scholar]
- 15. Altschuler Y., Kinlough C. L., Poland P. A., Bruns J. B., Apodaca G., Weisz O. A., Hughey R. P. (2000) Clathrin-mediated endocytosis of MUC1 is modulated by its glycosylation state. Mol. Biol. Cell 11, 819–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hisatsune A., Kawasaki M., Nakayama H., Mikami Y., Miyata T., Isohama Y., Katsuki H., Kim K. C. (2009) Internalization of MUC1 by anti-MUC1 antibody from cell membrane through the macropinocytotic pathway. Biochem. Biophys. Res. Commun. 388, 677–682 [DOI] [PubMed] [Google Scholar]
- 17. Swanson J. A., Watts C. (1995) Macropinocytosis. Trends Cell Biol. 5, 424–428 [DOI] [PubMed] [Google Scholar]
- 18. Kerr M. C., Teasdale R. D. (2009) Defining macropinocytosis. Traffic 10, 364–371 [DOI] [PubMed] [Google Scholar]
- 19. Florey O., Kim S. E., Sandoval C. P., Haynes C. M., Overholtzer M. (2011) Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat. Cell Biol. 13, 1335–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kerr M. C., Lindsay M. R., Luetterforst R., Hamilton N., Simpson F., Parton R. G., Gleeson P. A., Teasdale R. D. (2006) Visualisation of macropinosome maturation by the recruitment of sorting nexins. J. Cell Sci. 119, 3967–3980 [DOI] [PubMed] [Google Scholar]
- 21. Laughlin S. T., Bertozzi C. R. (2007) Metabolic labeling of glycans with azido sugars and subsequent glycan profiling and visualization via Staudinger ligation. Nat. Protoc. 2, 2930–2944 [DOI] [PubMed] [Google Scholar]
- 22.Deleted in proof [Google Scholar]
- 23. Julenius K., Mølgaard A., Gupta R., Brunak S. (2005) Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology 15, 153–164 [DOI] [PubMed] [Google Scholar]
- 24. Röttger S., White J., Wandall H. H., Olivo J. C., Stark A., Bennett E. P., Whitehouse C., Berger E. G., Clausen H., Nilsson T. (1998) Localization of three human polypeptide GalNAc-transferases in HeLa cells suggests initiation of O-linked glycosylation throughout the Golgi apparatus. J. Cell Sci. 111, 45–60 [DOI] [PubMed] [Google Scholar]
- 25. Suraneni P., Rubinstein B., Unruh J. R., Durnin M., Hanein D., Li R. (2012) The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J. Cell Biol. 197, 239–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Araki N., Johnson M. T., Swanson J. A. (1996) A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J. Cell Biol. 135, 1249–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kabayama H., Takeuchi M., Taniguchi M., Tokushige N., Kozaki S., Mizutani A., Nakamura T., Mikoshiba K. (2011) Syntaxin 1B suppresses macropinocytosis and semaphorin 3A-induced growth cone collapse. J. Neurosci. 31, 7357–7364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Park J. H., Nishidate T., Kijima K., Ohashi T., Takegawa K., Fujikane T., Hirata K., Nakamura Y., Katagiri T. (2010) Critical roles of mucin 1 glycosylation by transactivated polypeptide N-acetylgalactosaminyltransferase 6 in mammary carcinogenesis. Cancer Res. 70, 2759–2769 [DOI] [PubMed] [Google Scholar]
- 29. Gill D. J., Clausen H., Bard F. (2011) Location, location, location. New insights into O-GalNAc protein glycosylation. Trends Cell Biol. 21, 149–158 [DOI] [PubMed] [Google Scholar]
- 30. Gill D. J., Chia J., Senewiratne J., Bard F. (2010) Regulation of O-glycosylation through Golgi-to-ER relocation of initiation enzymes. J. Cell Biol. 189, 843–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Clément M., Rocher J., Loirand G., Le Pendu J. (2004) Expression of sialyl-Tn epitopes on β1 integrin alters epithelial cell phenotype, proliferation and haptotaxis. J. Cell Sci. 117, 5059–5069 [DOI] [PubMed] [Google Scholar]
- 32. Zhang L., Tran D. T., Ten Hagen K. G. (2010) An O-glycosyltransferase promotes cell adhesion during development by influencing secretion of an extracellular matrix integrin ligand. J. Biol. Chem. 285, 19491–19501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Powelka A. M., Sun J., Li J., Gao M., Shaw L. M., Sonnenberg A., Hsu V. W. (2004) Stimulation-dependent recycling of integrin β1 regulated by ARF6 and Rab11. Traffic 5, 20–36 [DOI] [PubMed] [Google Scholar]
- 34. Cantz M., Ulrich-Bott B. (1990) Disorders of glycoprotein degradation. J. Inherit. Metab. Dis. 13, 523–537 [DOI] [PubMed] [Google Scholar]
- 35. Mizushima N., Sugita H., Yoshimori T., Ohsumi Y. (1998) A new protein conjugation system in human. J. Biol. Chem. 273, 33889–33892 [DOI] [PubMed] [Google Scholar]
- 36. Lau K. S., Partridge E. A., Grigorian A., Silvescu C. I., Reinhold V. N., Demetriou M., Dennis J. W. (2007) Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 129, 123–134 [DOI] [PubMed] [Google Scholar]
- 37. Marshall S., Nadeau O., Yamasaki K. (2004) Dynamic actions of glucose and glucosamine on hexosamine biosynthesis in isolated adipocytes. Differential effects on glucosamine 6-phosphate, UDP-N-acetylglucosamine, and ATP levels. J. Biol. Chem. 279, 35313–35319 [DOI] [PubMed] [Google Scholar]
- 38. Slawson C., Copeland R. J., Hart G. W. (2010) O-GlcNAc signaling. A metabolic link between diabetes and cancer? Trends Biochem. Sci. 35, 547–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kawauchi T., Sekine K., Shikanai M., Chihama K., Tomita K., Kubo K., Nakajima K., Nabeshima Y., Hoshino M. (2010) Rab GTPases-dependent endocytic pathways regulate neuronal migration and maturation through N-cadherin trafficking. Neuron 67, 588–602 [DOI] [PubMed] [Google Scholar]
- 40. Shirane M., Nakayama K. I. (2006) Protrudin induces neurite formation by directional membrane trafficking. Science 314, 818–821 [DOI] [PubMed] [Google Scholar]