

Abstract
Objective
Apoptosis of smooth muscle cells (SMCs) is a prominent pathological characteristic of Abdominal Aortic Aneurysm (AAA). We have previously shown that SMC apoptosis stimulates proinflammatory signaling in a mouse model of AAA. Here, we test whether Protein Kinase C-delta (PKCδ), an apoptotic mediator, participates in the pathogenesis of AAA by regulating apoptosis and proinflammatory signals.
Methods and Results
Mouse experimental AAA is induced by perivascular administration of CaCl2. Mice deficient in PKCδ exhibit a profound reduction in aneurysmal expansion, SMC apoptosis, and transmural inflammation as compared to wildtype littermates. Delivery of PKCδ to the aortic wall of PKCδ−/− mice restores aneurysm, while overexpression of a dominant negative PKCδ mutant in the aorta of wildtype mice attenuates aneurysm. In vitro, PKCδ−/− aortic SMCs exhibit significantly impaired monocyte chemoattractant protein-1 (MCP-1) production. Ectopic administration of recombinant MCP-1 to the arterial wall of PKCδ−/− mice restores inflammatory response and aneurysm development.
Conclusions
PKCδ is an important signaling mediator for SMC apoptosis and inflammation in a mouse model of AAA. By stimulating MCP-1 expression in aortic SMCs, upregulated PKCδ exacerbates the inflammatory process, in turn perpetuating elastin degradation and aneurysmal dilatation. Inhibition of PKCδ may serve as a potential therapeutic strategy for AAA.
Abdominal aortic aneurysm (AAA), a progressive aortic dilation, is a common vascular disease associated with high mortality. Aneurysm results from the culmination of a series of events that lead to disruption of structural integrity and segmental weakening of the abdominal aortic wall. An incomplete understanding of the biological mechanisms underlying the disease has limited the development of therapeutic treatment and diagnostic strategies, thus leaving surgical and endovascular procedures as the only treatment options for patients with abdominal aortic aneurysm.
Histologically, aneurysmal tissues are characterized by disruption of the elastic fibers in the aortic wall and extensive transmural infiltration of macrophages and lymphocytes1–3. These features have been consistently duplicated in animal models of AAA4. The prevailing view is that inflammatory cells, mainly macrophages, are the major source of matrix-degrading enzymes such as matrix metalloproteinases5–9 and proinflammatory cytokines10–12. Anti-inflammatory strategies such as those that deplete neutrophils, lymphocytes, mast cells, or proinflammatory cytokines have been shown to prevent the upregulation of matrix metalloproteinases and attenuate aneurysm formation in mouse models of AAA13–16.
Although the depletion of vascular smooth muscle cells (SMCs) is well documented in human aneurysmal tissues17, potential interactions between SMCs and infiltrating inflammatory cells remain unclear. We have recently demonstrated that blocking apoptosis with a pan caspase inhibitor protected mice from angiotensin II-induced aneurysm expansion18. The caspase inhibitor not only prevented SMC depletion but also diminished infiltration of macrophages and lymphocytes, suggesting a potential link between the apoptotic process and inflammatory signaling in the pathogenesis of aneurysm.
Protein kinase C-delta (PKCδ), a member of the PKC family of serine and threonine kinases, is a crucial mediator of SMC apoptosis19–21. Studies of PKCδ knockout (KO) mice reveal that mice lacking PKCδ develop normally but exhibit an apoptosis-resistant phenotype when subjected to models of vascular injury such as vein graft or carotid artery ligation22, 23. Conversely, gene transfer of PKCδ via an adenoviral vector led to excessive apoptosis of vascular SMCs in a rat carotid balloon injury model23. More recently, we showed that PKCδ may also be involved in the regulation of chemokine expression. Inhibition of PKCδ with rottlerin profoundly decreases the production of monocyte chemoattractant protein-1 (MCP-1) by aortic vascular SMCs and subsequently inhibits chemotaxis of inflammatory cells toward SMC-conditioned media21.
We have previously shown that the expression of PKCδ is significantly higher in human aneurysmal aortic tissues as compared to normal arteries21. The collection of these tissues at the time of surgical repair precluded analysis of a potential causal relationship between PKCδ and aneurysm; specifically, whether PKCδ upregulation contributes to the pathophysiology of aneurysm or is merely a resultant phenomenon. To determine whether PKCδ is an integral mediator of SMC apoptosis and vascular inflammation during aneurysm pathogenesis, the current study tests the effects of PKCδ gene deficiency on aneurysm formation using the calcium chloride (CaCl2) mouse model. In addition, we explored the potential molecular mechanisms by which PKCδ regulates the pro-inflammatory signals produced by apoptotic SMCs.
METHODS
The detailed methods are shown in online supplements.
Mouse Models of AAA
The generation of PKCδ target deletion in mice was described elsewhere24. PKCδ knockout mice and their wildtype littermates were generated by mating heterozygous pairs. C57BL/6 mice and ApoE−/− mice were purchased from Harlan Laboratories (Madison, WI) and Jackson Laboratory (Bar Harbor, ME), respectively. GFP transgenic mice were gifted from Dr. William Burlingham at the University of Wisconsin-Madison.
Male mice, 12 weeks of age, underwent a CaCl2 –induced abdominal aortic aneurysm model as described previously25–28. Briefly, the infrarenal region of the aorta was isolated and treated with 0.5M CaCl2 perivascularly via gauze for 20 minutes. Control mice were similarly treated with 0.5M of sodium chloride (NaCl). Tissues were fixed in 4% formaldehyde in phospho-buffered saline (PBS), embedded and cut to 6 or 8μm sections for OCT and paraffin-imbedded arteries, respectively.
Immunohistochemistry
Antibodies were purchased from Abcam (Cambridge, MA; IFN-γ, IL-6, MOMA2, MHC, and CD45), Santa Cruz (Santa Cruz, CA; CD3, MCP-1, Mac3, PKCδ, Ly6G, and CD68), Sigma-Aldrich (SMA), and Cell Signaling (Danvers, MA; Cleaved caspase-3). TUNEL staining kit was purchased from Roche (Madison, WI). Van Geison stains were carried out using Chromaview Van Gieson kit (Richard Allan Scientific, Kalamazoo, MI).
Cell Culture
The murine macrophage cell line RAW264.7 cells were obtained from American Type Culture Collection (ATCC, Manassas, VA). Primary mouse aortic SMCs from the aorta of both PKCδ KO and WT mice were isolated based on a protocol described by Clowes et al.29.
Migration Assay
In vitro migration assay was carried out as previously described30. Briefly, RAW264.7 macrophages, or CD11b+ cells isolated from bone marrow, were placed in a 5μm pore transwell insert. Conditioned and/or treated media were used as chemoattractants. Following 6 hours incubation, inserts were removed and stained with hematolxylin to facilitate nuclei visualization. The mean value of migrated cells was counted in eight high-power fields per membrane.
Bone Marrow Isolation and Sorting
Bone marrow was isolated from long bones, washed with PBS, and counted. Monocytes were collected from bone marrow by magnetic sorting using CD11b microbeads (Miltenyi Biotec, Boston, MA). Purity of the resulting CD11b+ cells was assessed by flow cytometry using antibodies to CD3, CD11b, and CD45/B220 (Miltenyi Biotec).
Statistical Analysis
Values were expressed as mean ± standard error. Experiments were repeated at least three times unless stated otherwise. Differences between 2 groups were analyzed by Student's t test. For time course comparison, one-way ANOVA analysis was followed by Bonferroni correction to adjust for multiple comparisons. Values of p<0.05 were considered significant.
More detailed methods are provided in Supplemental Methods
RESULTS
PKCδ expression in experimental aneurysms
We subjected C57BL/6 male mice to perivascular treatment of 0.5M CaCl2 (or equal concentration of NaCl) to the infrarenal region of the aorta and sacrificed the animals at selected time points. Administration of CaCl2 led to gradual aortic dilatation associated with elastin fragmentation (Supplemental Figure 1). Immunohistochemical analysis showed a profound upregulation of PKCδ protein in the aortic media 3 and 7 days after CaCl2 treatment (Figure 1A), a time frame at which aortic expansion was barely visible. The temporal and spatial pattern of PKCδ expression mirrored that of TUNEL positivity (Figure 1B). Confocal images confirmed the colocalization between PKCδ upregulation and apoptosis (TUNEL). Furthermore, PKCδ positive cells were primarily SMCs, as identified by Myosin Heavy Chain (MHC) (Figure 1C). A similar expression pattern of PKCδ and its association with apoptosis was also observed in angiotensin II-induced aneurysm in apoE−/∓ (Supplemental Figure 2). Western blot analysis confirmed the elevated level of PKCδ protein in CaCl2-treated aortas as compared to the NaCl-treated controls (Figure 1D, E). Additionally, levels of the apoptosis-associated catalytic fragment of PKCδ became readily detectable in CaCl2-treated group (Figure 1D).
Figure 1. PKCδ expression correlates with apoptosis in an experimental aneurysm model.
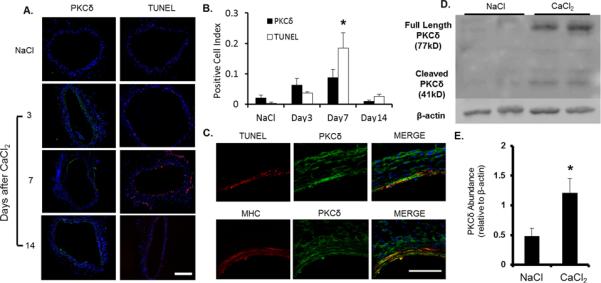
Aortas of C57B/6 mice were treated with CaCl2 or NaCl and harvested 3, 7 and 14 days (CaCl2 group) or 7 days (NaCl group) after surgery. (A) Cross sections stained for PKCδ (green) or apoptosis (TUNEL, red), and nuclei (DAPI, blue). Scale bar=200μm. (B) Positive cell ratio calculated as number of apoptotic (TUNEL+) or PKCδ positive cells divided by respective number of DAPI positive cells. *p<0.05 compared to NaCl control, n=6. (C) Representative confocal images for co-localization analysis in cross sections harvested 7 days after CaCl2 treatment. Top panel: Co-stain TUNEL (red) and PKCδ (green). Bottom panel: Co-stain for SMCs (MHC, red) and PKCδ (green). Scale bar=50 μm, Overlay with DAPI (blue). (D) Representative Western blot analysis of PKCδ expression in tissues harvested from two different aortas of C57B/6 mice 7 days after NaCl or CaCl2 treatment. (E) Quantification of PKCδ expression from Western blot, normalized to β-actin. PKCδ expression shown as a total of both cleaved and full-length portions. *p<0.05, n=4.
Mice lacking PKCδ are resistant to AAA induction
To prove a potential role of PKCδ in AAA formation, we subjected PKCδ knockout (KO) mice and their wildtype (WT) littermates to aneurysm induction by CaCl2. Forty-two days after the CaCl2 treatment, abdominal aorta of WT mice were visibly inflamed and dilated while the arteries of KO mice appeared minimally affected (Figure 2A). The maximal external diameter of the abdominal aorta was measured immediately prior to the CaCl2 application and at the time of tissue harvest. As seen in Figure 2B, the baseline aortic diameters are comparable in PKCδ WT and KO mice. Six weeks after the CaCl2 treatment, arteries of WT mice expanded to 1.04±0.08mm (96.6±31%), while arteries of KO mice expanded only to 0.74±0.06mm (39.7±9%) (Figure 2B). Similarly, PKCδ was shown to play a role in the elastase perfusion model of murine AAA. Inactive elastase produced minimal dilation of the artery in both WT and KO animals (0.77±0.06mm and 0.76±0.02mm, respectively; n=2), while active elastase treatment produced a more severe dilation in WT animals (1.47±0.16mm) compared to KO (0.97±0.29mm) (Supplemental Figure 3).
Figure 2. PKCδ knockout mice are resistant to aneurysm induction.
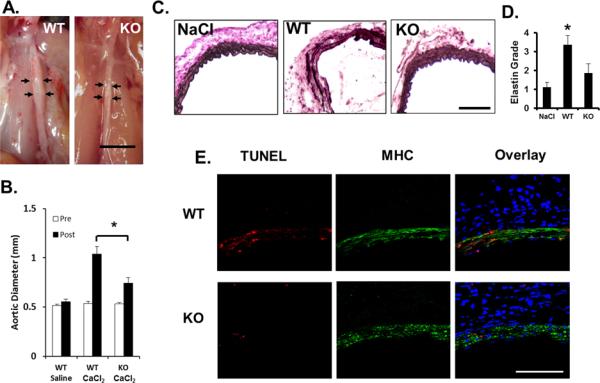
(A) Representative photos of abdominal aortas of PKCδ wildtype (WT) and knockout (KO) mice, taken 42 days after the CaCl2 treatment. Scale bar=5 mm. (B) Aortic diameter measured prior to (Pre, white bars), and 42 days after (Post, black bars), CaCl2 treatment. *p<0.05 compared to the CaCl2 treated KO arteries, n=6. (C) Representative photos of 42 day aortic sections stained for elastin (Van-Gieson), scale bar=100 μm. (D) Grading of elastin degradation in Van Gieson stained arteries harvested 42 days after surgery. *p<0.05 WT compared to KO, n=4. (E) Representative confocal images of arterial sections co-immunostained for TUNEL (red) and Myosin Heavy Chain (MHC, green) overlay with DAPI (blue); arteries harvested 7 days after surgery, scale bar = 50μm.
Histological analysis performed at 7 days after CaCl2-treatment revealed similar elastin degradation in KO and WT arteries (Supplemental Figure 4). However, the same histological analysis 42 days after CaCl2 treatment showed elastin fibers in arteries harvested from KO mice appeared continuous and organized, similar to NaCl-treated controls, whereas elastin fibers in CaCl2-treated WT arteries appeared fragmented and disoriented (Figure 2C, D). Furthermore, PKCδ KO tissue harvested at 7 days displayed significantly reduced SMC apoptosis, as evidenced by confocal staining showing colocalization of MHC and apoptosis (TUNEL), as compared to WT samples (Figure 2E). Accordingly, cleaved Caspase-3 was nearly undetectable in PKCδ KO arteries, while it was abundant in the WT arteries (Supplemental Figure 5A, B). Furthermore, the percentage of nuclei staining positive for TUNEL was decreased from 24.1±3.4 in WT arteries to 12.5±2.9 in KO arteries (Data not shown). In vitro analysis of cultured SMCs confirmed the apoptosis-resistant phenotype. The lack of PKCδ diminished the ability of SMCs to undergo apoptosis in response to TNFα, which was rescued by restoration of PKCδ expression with an adenoviral vector (Supplemental Figure 5C, D).
PKCδ is critical for the inflammatory response
Next, we analyzed macrophage infiltration, another important characteristic of aneurysm, in the aortas of both WT and KO animals. Immunohistochemical analysis revealed a profound reduction in the number of macrophages (Mac-3+, CD68+) detected in the aorta of PKCδ KO mice as compared to their WT littermates (Figure 3A, B). Additionally, neutrophils (Ly6G+), leukocytes (CD45+), and T cells (CD3+) were shown to be present in the aortic samples of the PKCδ WT mice, mostly prevalent in the adventitia, and almost entirely absent in KO aortas (Supplemental Figure 6). Similarly, levels of AAA-associated inflammatory cytokines IL-6 and monocyte chemoattractant protein-1 (MCP-1) were markedly decreased by PKCδ gene deficiency (Figure 3C). To better quantify the altered cytokine expression, we analyzed aortic tissues using real-time (RT)-PCR analysis. As shown in Figure 3D, PKCδ gene deficiency caused a 50.7% and 48.1% reduction in mRNA levels of IL-6 and MCP-1 in TNFα-treated SMCs, respectively. Additionally, aneurysm-associated induction of IL-1β and INFγ was also significantly blunted in PKCδ KO mice (Supplemental Figure 7). There was also a small but statistically insignificant trend of reduction in TNF-α mRNA abundance.
Figure 3. PKCδ gene deficiency attenuates the inflammatory response in experimental aneurysm.
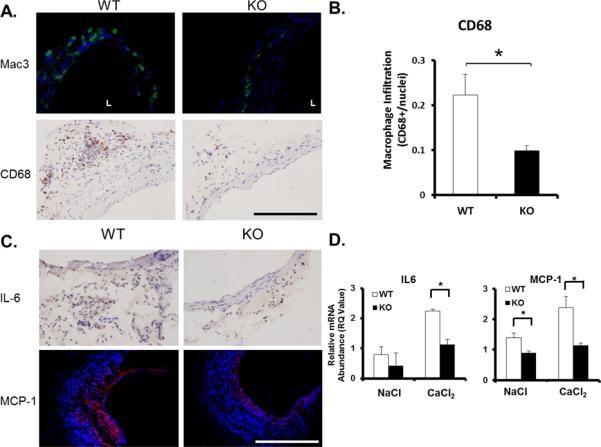
(A) Macrophage infiltration as measured by immunofluorescent stain (IFC) for Mac3 (green), overlay with DAPI (blue) or by immunohistochemical stain (IHC) for CD68; scale bar = 200μm. (B) Quantification of macrophage infiltration in aneurysm tissue as identified by CD68 stain, expressed as CD68 positive cells/nuclei. *p<0.05 compared to CaCl2 treated KO arteries, n=4. (C) IHC for inflammatory cytokine interleukin 6 (IL-6) and IFC for monocyte chemoattractant protein-1 (MCP-1). Scale bar=200 μm. (D) RT-PCR analysis of IL-6 and MCP-1 mRNA abundance in aorta of WT and KO animals 7 days after surgery. *p<0.05 compared to KO arteries, n=4.
PKCδ-deficient aortic SMCs are impaired in MCP-1 expression
The diminished inflammatory infiltrate in PKCδ KO mice could be caused by a lack of PKCδ-mediated chemokine production in the aortic wall or diminished migratory property of monocytes. A complete blood count (CBC) performed on WT and KO animals showed no significant difference in white blood cell or red blood cell populations between the two genotypes (Supplemental Table 1). Furthermore, the percentage of monocytes (CD11b+) in the bone marrow was comparable between the genotypes (Supplemental Figure 8A, B). In a chemotaxis assay, CD11b+ monocytes isolated from KO and WT animals migrated with equal efficiency toward recombinant MCP-1 protein (Supplemental Figure 8C). Together, these data suggest that neither number nor migratory capability of bone marrow monocytes are affected by PKCδ gene deficiency.
RT-PCR analysis of aortic SMCs showed KO cells to have a nearly absolute impairment of MCP-1 production. Expression of IFNγ and IL-6 also appeared to be modulated by PKCδ, albeit to a lesser degree (Figure 4A). The dependence of MCP-1 expression on PKCδ was further demonstrated by ELISA measurement of MCP-1 production by cultured SMCs. Following treatment with TNFα, WT SMCs are shown to produce significantly more MCP-1 as compared to KO SMCs (Figure 4B). Furthermore, overexpression of PKCδ using adenoviral-mediated gene delivery (AdPKCδ) further enhanced the production of MCP-1 in WT SMCs (Figure 4C).
Figure 4. PKCδ mediates production of MCP-1 by vascular SMCs.
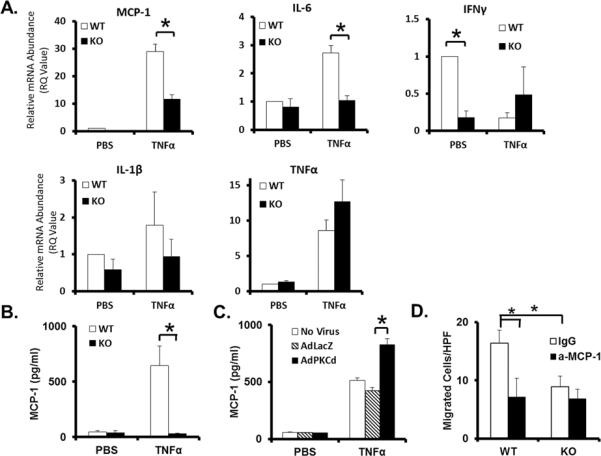
Cultured aortic SMCs isolated from PKCδ KO or WT mice were treated with TNFα (50ng/mL, for 6h) (A–C). (A) RTPCR analysis of selected cytokines. (B) Levels of secreted MCP-1 protein, measured by ELISA. (C) Levels of MCP-1 protein secreted by WT cells infected with AdLacZ or AdPKC, followed by treatment with TNFα. (D) Migration of RAW264.7 macrophages in response to conditioned media harvested from TNFα treated WT or KO SMCs in the presence of an anti-MCP-1 antibody (a-MCP-1) or hamster IgG (IgG) as control. *p<0.05, n=3.
To further test the notion that PKCδ gene deficiency reduces the presence of proinflammatory aneurysm signals produced by the aortic wall, we examined the ability of aortic SMCs to attract RAW264.7 monocyte/macrophages. As shown in Figure 4D, the number of RAW264.7 cells that migrated toward media conditioned by KO SMCs was ~50% less than that toward media conditioned by WT SMCs. Furthermore, administration of an MCP-1 neutralizing antibody completely eliminated the ability of WT SMCs to stimulate migration, suggesting MCP-1 to be a critical proinflammatory signal released by aortic SMCs (Figure 4D).
Exogenous PKCδ reverses the aneurysm-resistant phenotype of KO mice
Data derived from the above in vitro analyses suggest that PKCδ gene deletion attenuates aneurysm development primarily through preventing aortic SMCs from undergoing apoptosis and/or producing proinflammatory chemokines, specifically MCP-1. To test this hypothesis, we developed an aortic tissue-specific gene transfer method to restore PKCδ expression in the arterial wall of KO mice. As described in Methods, adenovirus was administered to the aortic wall immediately following the removal of CaCl2. This gene transfer method produced a localized transgene expression as illustrated by using an adenovirus encoding EGFP (AdGFP). While producing abundant GFP expression in the infrarenal region of the aortic wall, aortic administration of AdGFP did not produce transgene expression in circulating white blood cells (Supplemental Figure 9A, B).
To restore PKCδ expression in aortas of PKCδ KO mice, adenovirus expressing either PKCδ or β-galactosidase (AdPKCδ or AdLacZ) was administered to the infrarenal aorta of PKCδ KO mice. Mice were sacrificed after 7 or 42 days for histological and morphological analyses, respectively. Delivery of AdPKCδ successfully induced localized aortic expression of PKCδ in KO mice, mostly in the perivascular region and to a lesser degree in the SMA+ media (Supplemental Figure 9C, D). Forty-two days after the CaCl2 treatment, AdLacZ-treated PKCδ KO mice displayed minimum aortic expansion, with a final diameter measurement of 0.67±0.07mm (29.2±15.9%), indicating that viral infection alone did not alter the aneurysm-resistant phenotype of KO mice. In comparison, delivery of AdPKCδ produced significant aortic expansion in KO mice (final diameter 1.11±0.21mm; 114.8±28.3%), an induction comparable to that seen in wildtype mice (Figure 5A, B). The apparent restoration of aneurysm formation shown to accompany aortic gene transfer of PKCδ is further evidenced by fragmented elastin fibers as well as TUNEL positive apoptotic cells and infiltrating monocytes/macrophages at 7 days after surgery (Figure 5C).
Figure 5. Acute manipulation of PKCδ by adenovirus alters aneurysm phenotype in mice.

Adenoviruses encoding PKCδ (AdPKCδ) (A–C) or a dominant negative PKCδ mutant (AdPKCδ-DN) (D–F) were delivered to aortas of PKCδKO mice or C57B/6 mice, respectively, immediately following CaCl2 treatment. AdLacZ was used as a control. (A&D) Representative photos of aortas taken 42 days after surgery. Scale bar=5mm. (B&E) Aortic diameter measured prior to (Pre, white bars), and 42 days after (Post, black bars), CaCl2 treatment. *p<0.05, n=4 and n=5 in B and E, respectively. (C&F) Aortic sections stained for Van Gieson (elastin) at 42 days, TUNEL (red, 7 days), or monocytes and macrophages (MOMA, green, 7 days). Nuclei were stained by DAPI (blue). `L' delineates arterial lumen. Scale bar=200μm.
Aortic inhibition of PKCδ attenuates aneurysm formation in C57B/6 mice
To further demonstrate the importance of aortic PKCδ expression in aneurysm development, we tested the effect of aortic inhibition of PKCδ. Following the routine CaCl2 treatment, C57BL/6 mice were subjected to local infection with either a dominant negative PKCδ mutant adenovirus (AdPKCδDN) or AdLacZ as control. Mice were sacrificed after 7 or 42 days for histological and morphological analyses, respectively. As shown in Figure 5D and E, treatment with AdPKCδDN produced a moderate but significant attenuation in aneurysm formation in C57B/6 mice as reflected by a reduction in aortic expansion as compared to the AdLacZ-treated mice (final aortic diameter measurement 0.74±0.11mm or 54.7±28.3%, and 1.05±0.06mm or 126.5±15.7%, respectively). Accordingly, local inhibition of PKCδ activity diminished elastin degradation, apoptotic activity, and infiltration of monocytes/macrophages in the arterial wall (Figure 5F).
Exogenous MCP-1 protein restores aneurysm to PKCδ KO animals
Various studies have implicated an important role for both MCP-1/CCR2 signaling in vascular diseases including atherosclerosis and AAA31–34. Both in vivo and in vitro analyses within the current study indicate a reduction of MCP-1 expression caused by PKCδ gene deficiency; this prompted us to test whether delivery of exogenous MCP-1 to the arterial wall of PKCδ KO mice could restore aneurysm formation. Immediately following CaCl2 treatment, recombinant MCP-1 protein suspended in pluronic gel was delivered to the infrarenal aortic region of KO mice. As vehicle controls, parallel groups of KO and WT mice were treated with pluronic gel + solvent. As shown in Figure 6A and B, solvent treated WT aortas developed aneurysmal expansion comparable to those previously observed in these mice at 42 days after CaCl2 treatment (0.84±0.04mm, 73.7±8.2%). At this same time point, pluronic gel + solvent treated PKCδ KO aortas maintained their aneurysm-resistant phenotype despite the administration of pluronic gel, a stark contrast to the KO aortas treated with recombinant MCP-1 (Aortic diameter 0.62±0.06mm or 27.1±12.5%, and 0.91±0.04mm or 89.6±9.4%, respectively). Administration of recombinant MCP-1 in KO aorta created elastin degradation similar to that seen in solvent treated WT aorta, while solvent treated KO aortas remained largely unaffected. Further histological analysis of these samples revealed a marked increase of macrophage infiltration in the MCP-1 treated mice as compared to solvent-treated PKCδ KO mice. Of note, the level of aortic SMC apoptosis in PKCδ KO mice was not significantly altered by the MCP-1 administration (Figure 6C).
Figure 6. Delivery of exogenous MCP-1 to PKCδ KO mice restores aneurysm formation.
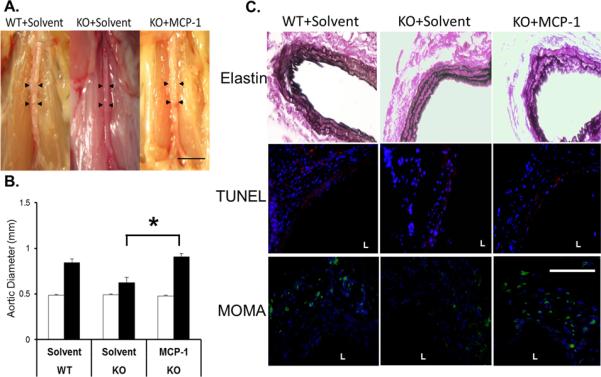
(A) Representative photos of abdominal aortas of WT or KO mice that received solvent or MCP-1 (80μM), taken 42 days after the CaCl2 procedure; scale bar=5mm. (B) Aortic diameter measured prior to (Pre, white bars), and 42 days after (Post, black bars), CaCl2 treatment. *p<0.05, n=4. (C) Aortic sections stained for Van Gieson (42 days), TUNEL (red, 7 days), or monocytes and macrophages (MOMA, green, 7 days). Nuclei were stained by DAPI (blue). `L' delineates arterial lumen. Scale bar=200 μm.
DISCUSSION
Our data for the first time provide direct evidence that PKCδ is an integral signaling molecule in the pathogenesis of abdominal aortic aneurysm. We showed that inhibition of PKCδ, either through targeted gene deletion or overexpression of a dominant negative mutant, protected mice from developing characteristic features of aneurysm including inflammation, disruption of elastin fibers, and loss of vascular SMCs. Additionally, the aneurysm-resistant phenotype was accompanied by diminished inflammatory infiltration, cytokine production, and medial apoptosis. These results not only confirm the importance of PKCδ in regulation of SMC apoptosis but also indicate a novel role for this kinase in the proinflammatory signaling cascade, at least in the aneurysm setting.
Although PKCδ is ubiquitously expressed in many tissues and cell types, results reported here suggest the role of this signaling protein in aneurysm pathophysiology may be primarily localized in vascular SMCs. Furthermore, our evidence suggests PKCδ may act largely through regulating expression of proinflammatory chemokines and cytokines, notably MCP-1. This notion is supported by several in vivo and in vitro findings: (1) PKCδ gene deficiency reduced the production of MCP-1 and other cytokines by aortic SMCs, but did not significantly alter the ability of monocytes to migrate; (2) an adenovirus-mediated delivery of PKCδ locally to the arterial wall was sufficient to rescue aneurysm development in PKCδ KO mice; (3) aorta-specific inhibition of PKCδ delivered a moderate but significant level of protection in C57B/6 mice; and (4) ectopic administration of MCP-1 to the aortic wall of PKCδ KO mice sufficiently rescued aneurysm development.
It has been postulated that vascular SMCs are the “soil” of AAA development35. Being a major source of ECM proteins, SMCs would be critical in counter balancing the upregulated proteolytic activities present in aneurysmal tissue. As such, the depletion of medial SMCs eliminates a cell population capable of directing connective tissue repair and may thus potentiate the degradation of the arterial wall and facilitate eventual rupture36. This study contains data supporting the idea that the dearth of connective tissue in AAA can be reversed in the presence of healthy SMCs, thus possibly either preventing or even reversing aneurysm growth. Specifically, we showed that arteries of CaCl2-treated KO mice sustained a similar degree of initial damage to aortic elastin fibers as WT aorta, but by 42 days elastin integrity is restored in KO arteries.
Results from the current study further illustrate another important function of SMCs in vascular disease, i.e. as providing proinflammatory signals. The potential link between SMC apoptosis and the production of pro-inflammatory chemokines has been previously indicated in atherosclerosis. Using a mouse atherosclerosis model, Clarke and colleagues convincingly demonstrated that SMC apoptosis induces MCP-1 expression, inflammatory infiltrate, and other features of plaque rupture37. Recently, our own group demonstrated that blocking apoptosis with a pan-caspase inhibitor protected mice from angiotensin II-induced vascular inflammation and aneurysm expansion18. These data suggest that, while apoptosis and inflammation are most commonly considered unrelated events, apoptosis in an aneurysm setting may promote the inflammatory response. Such interaction between apoptosis and inflammation has been explored in atherosclerosis. Clarke and colleagues suggest that the pro-inflammatory property of apoptotic SMCs may be attributed to inhibited phagocytosis generated in the hyperlipidemic environment in atherosclerotic arteries (31). Although abdominal aortic aneurysm is commonly associated with atherosclerosis, these two diseases are believe to be caused by distinct pathological processes. However, deficient phagocytosis is also being investigated as an underlying pathophysiological event in other types of inflammatory disorders, including autoimmue diseases (32), thus warranting the exploration of this process in the pathogenesis of AAA.
Another important finding described in this work is the apparent critical role of PKCδ in MCP-1 function during formation and progression of aneurysm. In vitro and in vivo evidence suggests that impaired production of MCP-1 expression by aortic SMCs was the primary mechanism underlying the aneurysm-resistant phenotype of PKCδ KO mice. Importantly, this notion is further supported by the evidence that localized aortic administration of recombinant MCP-1 to the aorta of PKCδ KO mice restored vascular inflammation, elastin degradation, and aneurysmal expansion. While several groups have explored the role of the CCR2/MCP-1 signaling axis in the aneurysm progression31, 38–40, this work provides what we believe to be the first evidence suggesting MCP-1 to be a critical downstream effector of PKCδ signaling in the pathogenesis of aneurysm.
Although our study has implicated a critical role for MCP-1 in AAA, it is important to consider the large number of cytokines that likely play a role in AAA development and progression. Our RT-PCR analysis identified additional cytokines that may be regulated by PKCδ and require further investigation.
Interestingly, localized aortic delivery of exogenous MCP-1 failed to reverse the apoptosis-resistant phenotype of PKCδ KO mice. A similar number of TUNEL+ cells were found in MCP-1 and solvent-treated PKCδ KO mice. In contrast, a similar rescue experiment delivering exogenous PKCδ to the arterial wall restored all aneurysm-related cellular events, i.e. inflammation, apoptosis, and elastin degradation, in CaCl2 PKCδ KO arteries. These results not only underscore the critical role of PKCδ in the apoptotic response of SMCs, but also provide support for a novel relationship between PKCδ, MCP-1, and AAA. In the absence of this “master” mediator, apoptosis would be inhibited even when aortic SMCs are surrounded by infiltrating inflammatory cells, their inflammatory byproducts, and degraded elastin fibers. Based on rescue experiments presented here, as well as other data from the current and prior reports19, 20, we propose a model in which PKCδ-mediated MCP-1 functions as a molecular link through which apoptotic SMCs stimulate the inflammatory process. Importantly, our model suggests that SMC apoptosis may contribute to aneurysm development primarily through the induction of inflammatory cytokines. That is, in the presence of abundant pro-inflammatory cytokines, such as the environment created by delivery of exogenous MCP-1 protein, the inflammatory and proteolytic events can proceed in full force without the participation of apoptosis.
Using a rat carotid balloon-injury model of intimal hyperplasia, our group recently showed that PKCδ mediated the expression of MCP-1, which was critical for the migration of adventitial fibroblasts to the media and neo-intima41. In the CaCl2-treated aorta, we noted a marked expansion of the adventitia associated with abundant infiltration of macrophages and other inflammatory cells. While the presence of macrophages in the adventitia is a prominent feature of AAA31, 32, 35 and the role of adventitial fibroblasts in aneurysm has been explored to some extent by several groups42–45, the precise relationship between adventitial fibroblasts, SMCs, and inflammatory cells in the context of AAA remains a highly interesting subject for future study. Evidence presented here shows the localization of IL-6 and macrophages predominantly in the adventitia, whereas MCP-1 production and apoptosis appears to occur primarily, though not exclusively, in the medial layer. It is also important to note that PKCδ, being a ubiquitously expressed protein, is also found in the adventitia. Whether PKCδ also contributes to aneurysm pathogenesis through adventitia cells should be explored in future studies given the prominent inflammatory response in the adventitia. However, several key questions remain to be addressed in models of AAA, for example, how adventitial fibroblasts may respond to medial SMC depletion, matrix degradation, and inflammatory cell infiltration.
Being a major signaling molecule, PKCδ can be activated by multiple extracellular and intracellular signals including growth factors, inflammatory cytokines, mechanical stimuli, and oxidative stress. Not all of these signals are able to induce apoptosis or the production of MCP-1. It remains to be determined whether the pro-apoptotic and proinflammatory functions of PKCδ are exerted through the same or partially overlapping pathways. We have previously shown that MAP kinase pathways are affected by PKCδ gene deficiency20, 46. Although the involvement of MAP kinases in the regulation of MCP-1 expression has been demonstrated47 the precise molecular interaction between PKCδ and MAP kinases and how this interaction may influence MCP-1 expression remains to be determined. Additionally, Liu and colleagues recently demonstrated that PKCδ mediates the stability of MCP-1 mRNA in vascular SMCs using a chemical inhibitor of PKCδ48. Our group previously described the role of MAP kinases in regulation of mRNA stability in vascular SMCs, leading us to speculate that PKCδ may control MCP-1 mRNA turnover through a MAP kinase-mediated mechanism49.
Taken together, our data show that the stress response regulating apoptosis and inflammatory signaling in the arterial wall may be largely dependent upon PKCδ upregulation. Accordingly, inhibition of PKCδ attenuated vascular inflammation and preserved tissue integrity, resulting in the prevention of aneurysm development in a CaCl2-induced model of AAA. Further, PKCδ gene deficiency appears to protect mice from developing aneurysm in the elastse model of AAA, as shown in Supplemental Figure 3. Unfortunately, the potential role of PKCδ in the Angiotensin II model is yet to be explored, as our attempts to breed PKCδ−/−ApoE−/− double knockout mice were unsuccessful. However, we did show that levels of PKCδ were significantly elevated in aortas of ApoE−/− mice treated with AngII. Taken together, we believe the elevated expression of this stress gene in human aneurysmal tissues, as well as the role we have shown it to play in mouse models, suggest it to be an attractive candidate for therapeutic target(s).
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Drs. K. Craig Kent and Jon Mutsumura of the University of Wisconsin, Madison for intellectual inputs, Dr. Keiichi I Nakayama of Kyushu University in Japan for the generous gift of PKCδ knockout mice, Dr. T. Biden at the Garvan Institute of Australia for AdPKCδDN, and Drew Roenneburg of the University of Wisconsin, Madison for technical assistance in histology.
This work was supported by the National Institute of Health R01HL088447 (BL), American Heart Association 10GRNT3020052 (BL), Howard Hughes Medical Institute MSN135276 (SS), and the Ruth L. Kirschstein National Research Service Award T32 HL 07936 from the National Heart Lung and Blood Institute to the University of Wisconsin-Madison Cardiovascular Research Center (SM).
Footnotes
No disclosures to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Ailawadi G, Eliason JL, Upchurch GR., Jr Current concepts in the pathogenesis of abdominal aortic aneurysm. J Vasc Surg. 2003;38:584–588. doi: 10.1016/s0741-5214(03)00324-0. [DOI] [PubMed] [Google Scholar]
- 2.Nordon I, Hinchliffe R, Holt P, Loftus I, Thompson M. Review of current theories for abdominal aortic aneurysm pathogenesis. Vascular. 2009;17:253–263. doi: 10.2310/6670.2009.00046. [DOI] [PubMed] [Google Scholar]
- 3.Wills A, Crowther MTM, Sayers R, Bell P. Pathogenesis of abdominal aortic aneurysms - cellular and biochemical mechanisms. Eur J Vasc Endovasc Surg. 1996;12:391–400. doi: 10.1016/s1078-5884(96)80002-5. [DOI] [PubMed] [Google Scholar]
- 4.Daugherty A, Cassis LA. Mouse models of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2004;24:429–434. doi: 10.1161/01.ATV.0000118013.72016.ea. [DOI] [PubMed] [Google Scholar]
- 5.Aziz F, Kuivaniemi H. Role of matrix metalloproteinase inhibitors in preventing abdominal aortic aneurysm. Ann Vasc Surg. 2007;21:392–401. doi: 10.1016/j.avsg.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. The Journal of Clinical Investigation. 2002;110:625–632. doi: 10.1172/JCI15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petrinec D, Liao S, Holmes DR, Reilly JM, Parks WC, Thompson RW. Doxycycline inhibition of aneurysmal degeneration in an elastase-induced rat model of abdominal aortic aneurysm: Preservation of aortic elastin associated with suppressed production of 92 kd gelatinase. Journal of Vascular Surgery. 1996;23:336–346. doi: 10.1016/s0741-5214(96)70279-3. [DOI] [PubMed] [Google Scholar]
- 8.Pyo R, Lee JK, Shipley JM, Curci JA, Mao D, Ziporin SJ, Ennis TL, Shapiro SD, Senior RM, Thompson RW. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase b) suppresses development of experimental abdominal aortic aneurysms. The Journal of Clinical Investigation. 2000;105:1641–1649. doi: 10.1172/JCI8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thompson RW, Baxter BT. Mmp inhibition in abdominal aortic aneurysms. Rationale for a prospective randomized clinical trial. Ann N Y Acad Sci. 1999;878:159–178. doi: 10.1111/j.1749-6632.1999.tb07682.x. [DOI] [PubMed] [Google Scholar]
- 10.Golledge ALV, Walker P, Norman PE, Golledge J. A systematic review of studies examining inflammation associated cytokines in human abdominal aortic aneurysm samples. Disease Markers. 2009;26:181–188. doi: 10.3233/DMA-2009-0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Golledge J, Tsao PS, Dalman RL, Norman PE. Circulating markers of abdominal aortic aneurysm presence and progression. Circulation. 2008;118:2382–2392. doi: 10.1161/CIRCULATIONAHA.108.802074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shimizu K, Mitchell RN, Libby P. Inflammation and cellular immune responses in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2006;26:987–994. doi: 10.1161/01.ATV.0000214999.12921.4f. [DOI] [PubMed] [Google Scholar]
- 13.Eliason JL, Hannawa KK, Ailawadi G, Sinha I, Ford JW, Deogracias MP, Roelofs KJ, Woodrum DT, Ennis TL, Henke PK, Stanley JC, Thompson RW, Upchurch GR., Jr Neutrophil depletion inhibits experimental abdominal aortic aneurysm formation. Circulation. 2005;112:232–240. doi: 10.1161/CIRCULATIONAHA.104.517391. [DOI] [PubMed] [Google Scholar]
- 14.Xiong W, Zhao Y, Prall A, Greiner TC, Baxter BT. Key roles of cd4+ t cells and ifn-gamma in the development of abdominal aortic aneurysms in a murine model. J Immunol. 2004;172:2607–2612. doi: 10.4049/jimmunol.172.4.2607. [DOI] [PubMed] [Google Scholar]
- 15.Sun J, Sukhova GK, Yang M, Wolters PJ, MacFarlane LA, Libby P, Sun C, Zhang Y, Liu J, Ennis TL, Knispel R, Xiong W, Thompson RW, Baxter BT, Shi GP. Mast cells modulate the pathogenesis of elastase-induced abdominal aortic aneurysms in mice. J Clin Invest. 2007;117:3359–3368. doi: 10.1172/JCI31311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimizu K, Shichiri M, Libby P, Lee RT, Mitchell RN. Th2-predominant inflammation and blockade of ifn-gamma signaling induce aneurysms in allografted aortas. J Clin Invest. 2004;114:300–308. doi: 10.1172/JCI19855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez-Candales A, Holmes D, Liao S, Scott M, Wickline S, Trompson R. Decreased vascular smooth muscle cell density in medial degeneration of human abdominal aortic aneurysms. Am J Pathol. 1997;150:993–1007. [PMC free article] [PubMed] [Google Scholar]
- 18.Yamanouchi D, Morgan S, Kato K, Lengfeld J, Zhang F, Liu B. Effects of caspase inhibitor on angiotensin ii-induced abdominal aortic aneurysm in apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol. 2010;30:702–707. doi: 10.1161/ATVBAHA.109.200527. [DOI] [PubMed] [Google Scholar]
- 19.Kato K, Yamanouchi D, Esbona K, Kamiya K, Zhang F, Kent KC, Liu B. Caspase-mediated protein kinase c-{delta} cleavage is necessary for apoptosis of vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2009;297:H2253–2261. doi: 10.1152/ajpheart.00274.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ryer EJ, Sakakibara K, Wang C, Sarkar D, Fisher PB, Faries PL, Kent KC, Liu B. Protein kinase c delta induces apoptosis of vascular smooth muscle cells through induction of the tumor suppressor p53 by both p38-dependent and p38-independent mechanisms. Journal of Biological Chemistry. 2005;280:35310–35317. doi: 10.1074/jbc.M507187200. [DOI] [PubMed] [Google Scholar]
- 21.Schubl S, Tsai S, Ryer EJ, Wang C, Hu J, Kent KC, Liu B. Upregulation of protein kinase c[delta] in vascular smooth muscle cells promotes inflammation in abdominal aortic aneurysm. Journal of Surgical Research. 2009;153:181–187. doi: 10.1016/j.jss.2008.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leitges M, Mayr M, Braun U, Mayr U, Li C, Pfister G, Ghaffari-Tabrizi N, Baier G, Hu Y, Xu Q. Exacerbated vein graft arteriosclerosis in protein kinase cδ null mice. The Journal of Clinical Investigation. 2001;108:1505–1512. doi: 10.1172/JCI12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamanouchi D, Kato K, Ryer EJ, Zhang F, Liu B. Protein kinase c delta mediates arterial injury responses through regulation of vascular smooth muscle cell apoptosis. Cardiovasc Res. 2010;85:434–443. doi: 10.1093/cvr/cvp328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miyamoto A, Nakayama K, Imaki H, Hirose S, Jiang Y, Abe M, Tsukiyama T, Nagahama H, Ohno S, Hatakeyama S, Nakayama KI. Increased proliferation of b cells and auto-immunity in mice lacking protein kinase c[delta] Nature. 2002;416:865–869. doi: 10.1038/416865a. [DOI] [PubMed] [Google Scholar]
- 25.Gertz SD, Kurgan A, Eisenberg D. Aneurysm of the rabbit common carotid artery induced by periarterial application of calcium chloride in vivo. The Journal of Clinical Investigation. 1988;81:649–656. doi: 10.1172/JCI113368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ikonomidis JS, Gibson WC, Butler JE, McClister DM, Sweterlitsch SE, Thompson RP, Mukherjee R, Spinale FG. Effects of deletion of the tissue inhibitor of matrix metalloproteinases-1 gene on the progression of murine thoracic aortic aneurysms. Circulation. 2004;110:II-268–II-273. doi: 10.1161/01.CIR.0000138384.68947.20. [DOI] [PubMed] [Google Scholar]
- 27.Kimura T, Yoshimura K, Aoki H, Imanaka-Yoshida K, Yoshida T, Ikeda Y, Morikage N, Endo H, Hamano K, Imaizumi T, Hiroe M, Aonuma K, Matsuzaki M. Tenascin-c is expressed in abdominal aortic aneurysm tissue with an active degradation process. Pathology International. 2011;61:559–564. doi: 10.1111/j.1440-1827.2011.02699.x. [DOI] [PubMed] [Google Scholar]
- 28.Yoshimura K, Aoki H, Ikeda Y, Fujii K, Akiyama N, Furutani A, Hoshii Y, Tanaka N, Ricci R, Ishihara T, Esato K, Hamano K, Matsuzaki M. Regression of abdominal aortic aneurysm by inhibition of cjun n-terminal kinase. Nat Med. 2005;11:1330–1338. doi: 10.1038/nm1335. [DOI] [PubMed] [Google Scholar]
- 29.Clowes A, Clowes M, Fringerle J, Reidy M. Kinetics of cellular poliferation after arterial injury: Role of acute distension in the induciton of smooth muscle proliferation. Lab Invest. 1989;60:360–364. [PubMed] [Google Scholar]
- 30.Shimizu K, Shichiri M, Libby P, Lee RT, Mitchell RN. Th2-predominant inflammation and blockade of ifn-γ signaling induce aneurysms in allografted aortas. The Journal of Clinical Investigation. 2004;114:300–308. doi: 10.1172/JCI19855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daugherty A, Rateri DL, Charo IF, Owens AP, Howatt DA, Cassis LA. Angiotensin ii infusion promotes ascending aortic aneurysms: Attenuation by ccr2 deficiency in apoe-/- mice. Clinical Science. 2010;118:681–689. doi: 10.1042/CS20090372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Waard V, Bot I, de Jager SCA, Talib S, Egashira K, de Vries MR, Quax PHA, Biessen EAL, van Berkel TJC. Systemic mcp1/ccr2 blockade and leukocyte specific mcp1/ccr2 inhibition affect aortic aneurysm formation differently. Atherosclerosis. 2010;211:84–89. doi: 10.1016/j.atherosclerosis.2010.01.042. [DOI] [PubMed] [Google Scholar]
- 33.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 34.Zhang L, Li HY, Li H, Zhao J, Su L, Zhang Y, Zhang SL, Miao JY. Lipopolysaccharide activated phosphatidylcholine-specific phospholipase c and induced il-8 and mcp-1 production in vascular endothelial cells. Journal of Cellular Physiology. 2011;226:1694–1701. doi: 10.1002/jcp.22500. [DOI] [PubMed] [Google Scholar]
- 35.Curci JA. Digging in the "soil" of the aorta to understand the growth of abdominal aortic aneurysms. Vascular. 2009;17:S21–29. doi: 10.2310/6670.2008.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thompson RW, Liao S, Curci JA. Vascular smooth muscle cell apoptosis in abdominal aortic aneurysms. Coron Artery Dis. 1997;8:623–631. doi: 10.1097/00019501-199710000-00005. [DOI] [PubMed] [Google Scholar]
- 37.Clarke MC, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD, Bennett MR. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006;12:1075–1080. doi: 10.1038/nm1459. [DOI] [PubMed] [Google Scholar]
- 38.Ishibashi M, Egashira K, Zhao Q, Hiasa K-i, Ohtani K, Ihara Y, Charo IF, Kura S, Tsuzuki T, Takeshita A, Sunagawa K. Bone marrow-derived monocyte chemoattractant protein-1 receptor ccr2 is critical in angiotensin ii-induced acceleration of atherosclerosis and aneurysm formation in hypercholesterolemic mice. Arterioscler Thromb Vasc Biol. 2004;24:e174–178. doi: 10.1161/01.ATV.0000143384.69170.2d. [DOI] [PubMed] [Google Scholar]
- 39.MacTaggart JN, Xiong W, Knispel R, Baxter BT. Deletion of ccr2 but not ccr5 or cxcr3 inhibits aortic aneurysm formation. Surgery. 2007;142:284–288. doi: 10.1016/j.surg.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 40.Moehle CW, Bhamidipati CM, Alexander MR, Mehta GS, Irvine JN, Salmon M, Upchurch GR, Jr, Kron IL, Owens GK, Ailawadi G. Bone marrow-derived mcp1 required for experimental aortic aneurysm formation and smooth muscle phenotypic modulation. The Journal of Thoracic and Cardiovascular Surgery. 2011;142:1567–1574. doi: 10.1016/j.jtcvs.2011.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Si Y, Ren J, Wang P, Rateri DL, Daugherty A, Shi X-D, Kent KC, Liu B. Protein kinase c-delta mediates adventitial cell migration through regulation of monocyte chemoattractant protein-1 expression in a rat angioplasty model. Arteriosclerosis, Thrombosis, and Vascular Biology. 2012;32:943–954. doi: 10.1161/ATVBAHA.111.244921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maiellaro K, Taylor WR. The role of the adventitia in vascular inflammation. Cardiovascular Research. 2007;75:640–648. doi: 10.1016/j.cardiores.2007.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sakata N, Nabeshima K, Iwasaki H, Tashiro T, Uesugi N, Nakashima O, Ito H, Kawanami T, Furuya K, Kojima M. Possible involvement of myofibroblast in the development of inflammatory aortic aneurysm. Pathology - Research and Practice. 2007;203:21–29. doi: 10.1016/j.prp.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 44.Tedesco MM, Terashima M, Blankenberg FG, Levashova Z, Spin JM, Backer MV, Backer JM, Sho M, Sho E, McConnell MV, Dalman RL. Analysis of in situ and ex vivo vascular endothelial growth factor receptor expression during experimental aortic aneurysm progression. Arteriosclerosis, Thrombosis, and Vascular Biology. 2009;29:1452–1457. doi: 10.1161/ATVBAHA.109.187757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tieu BC, Lee C, Sun H, LeJeune W, Recinos A, Ju X, Spratt H, Guo D-C, Milewicz D, Tilton RG, Brasier AR. An adventitial il-6/mcp1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. The Journal of Clinical Investigation. 2009;119:3637–3651. doi: 10.1172/JCI38308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu B, Ryer EJ, Kundi R, Kamiya K, Itoh H, Faries PL, Sakakibara K, Kent KC. Protein kinase c-[delta] regulates migration and proliferation of vascular smooth muscle cells through the extracellular signal-regulated kinase 1/2. Journal of Vascular Surgery. 2007;45:160–168. doi: 10.1016/j.jvs.2006.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fischer S, Weishaupt A, Troppmair J, Martini R. Increase of mcp-1 (ccl2) in myelin mutant schwann cells is mediated by mek-erk signaling pathway. Glia. 2008;56:836–843. doi: 10.1002/glia.20657. [DOI] [PubMed] [Google Scholar]
- 48.Liu B, Dhawan L, Blaxall B, Taubman M. Protein kinase cδ mediates mcp-1 mrna stabilization in vascular smooth muscle cells. Molecular and Cellular Biochemistry. 2010;344:73–79. doi: 10.1007/s11010-010-0530-6. [DOI] [PubMed] [Google Scholar]
- 49.Sakakibara K, Kubota K, Worku B, Ryer EJ, Miller JP, Koff A, Kent KC, Liu B. Pdgf-bb regulates p27 expression through erk-dependent rna turn-over in vascular smooth muscle cells. Journal of Biological Chemistry. 2005;280:25470–25477. doi: 10.1074/jbc.M502320200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.