

Abstract
Elimination of defective mitochondria is essential for the health of long-lived, postmitotic cells. To gain insight into this process, we examined programmed mitochondrial clearance in reticulocytes. BNIP3L is a mitochondrial outer membrane protein that is required for clearance. It has been suggested that BNIP3L functions by causing mitochondrial depolarization, activating autophagy, or engaging the autophagy machinery. Here we showed in mice that BNIP3L activity localizes to a small region in its cytoplasmic domain, the minimal essential region (MER). The MER is a novel sequence, which comprises three contiguous hydrophobic amino acid residues, and flanking charged residues. Mutation of the central leucine residue causes complete loss of BNIP3L activity, and prevents rescue of mitochondrial clearance. Structural bioinformatics analysis predicts that the BNIP3L cytoplasmic domain lacks stable tertiary structure, but that the MER forms an α-helix upon binding to another protein. These findings support an adaptor model of BNIP3L, centered on the MER.
Keywords: NIX, SLiM, autophagy, mitophagy, reticulocyte
Introduction
Mitochondria are subcellular organelles, and the primary site of oxidative phosphorylation and energy production in animal cells. Energy production is performed by the electron transport chain, located in the inner mitochondrial membrane. Due to intrinsic inefficiencies in electron transport, mitochondria are also a major site of reactive oxygen species generation. Reactive oxygen species mediate cell signaling,1 but in excess can damage mitochondrial proteins and nucleic acids, which impairs electron transport and increases reactive oxygen species production. Unchecked, this process can damage cellular proteins and nucleic acids, and promote cell death, transformation, and disease.2
Mitochondrial health is maintained by mitochondrial biogenesis and elimination, which in turn are coupled to repeated cycles of mitochondrial fusion and fission. Fusion allows mixing of mitochondrial components and functional complementation. Fission produces daughter mitochondria, which can differ in their membrane potential. If a daughter mitochondrion remains polarized, then it will be competent to undergo additional rounds of fusion.3 If it depolarizes, then it will not undergo fusion and is eliminated. Jointly, these processes constitute a mitochondrial quality control mechanism that is required to sustain the vitality of long-lived, postmitotic cells.
Defects in mitochondrial elimination have been implicated in juvenile-onset Parkinson disease. PARK2/PARKIN and PINK1 are frequently mutated in this condition,4,5 and have been placed in the same genetic pathway.6,7 Mitochondrial depolarization causes PARK2 translocation to mitochondria, and PARK2- and PINK1-dependent mitochondrial clearance.8,9 PARK2 is an E3 ligase; the mechanism of clearance involves PINK1-dependent recruitment of PARK2 to depolarized mitochondria, followed by PARK2-mediated ubiquitination of proteins in the mitochondrial outer membrane.10,11
Depolarization is a potent stimulus for mitochondrial clearance, mediated by PARK2 and PINK1; however, mitochondrial clearance also occurs during normal development. Reticulocytes are enucleate cells formed from mature erythroblasts in the bone marrow. As reticulocytes mature into erythrocytes over a period of several days, they eliminate all membrane-bound organelles, including mitochondria, in a coordinated process. Reticulocytes provide one of a few physiological models of mitochondrial clearance. Recently, we showed that an integral mitochondrial outer membrane protein, BNIP3L, is required for mitochondrial clearance in reticulocytes.12,13 Employing an in vivo model, we now show that BNIP3L activity is mediated by a short linear motif in a predicted disordered region in its cytoplasmic domain. Our results are consistent with an adaptor model of BNIP3L, in which its role is the recruitment of an enzymatic or membrane-associated protein to mitochondria.
Results
BNIP3 rescues mitochondrial clearance in Bnip3l−/− reticulocytes
To gain insight into the mechanism of action of BNIP3L, we employed a structure-function approach. The effect of BNIP3L on mitochondrial clearance is not recapitulated in any cell line; therefore, all experiments were performed in vivo, in mice. To accomplish this, we subcloned N-terminal FLAG-tagged BNIP3L (FLAG-BNIP3L), mutants of FLAG-BNIP3L, and BNIP3 into an MSCV-Ires-GFP retroviral vector. We transduced Bnip3l−/− bone marrow, and transplanted the transduced cells into lethally irradiated wild-type recipient mice. We allowed transplanted mice 4–6 weeks for bone marrow reconstitution, and then examined their circulating erythrocytes for evidence of a mitochondrial clearance defect by staining with Mitotracker Red (MTR) and flow cytometry. In these experiments, erythrocytes derived from nontransduced Bnip3l−/− bone marrow cells are GFP negative, whereas erythrocytes from transduced Bnip3l−/− bone marrow cells are GFP positive. We performed experiments in the absence of erythropoietic stress, such as phlebotomy or phenylhydrazine treatment. In this respect, the experiments were designed to reveal major effects of the mutations on BNIP3L activity.
BNIP3 is closely related to BNIP3L (56% identical overall), but not to any other gene. BNIP3 is implicated in mitochondrial clearance caused by hypoxia,14 which suggested it may be able to mediate mitochondrial clearance during reticulocyte maturation. Indeed, we found that BNIP3 is effective in promoting mitochondrial clearance in Bnip3l−/− reticulocytes (Fig. 1). Thus, BNIP3L and BNIP3 exhibit functionally redundancy. BNIP3 is not normally expressed in the erythroid lineage, explaining its failure to complement BNIP3L in this tissue. The ability of BNIP3 to compensate for the absence of BNIP3L is useful, since it means the active sequences in BNIP3L are likely to be conserved in BNIP3.
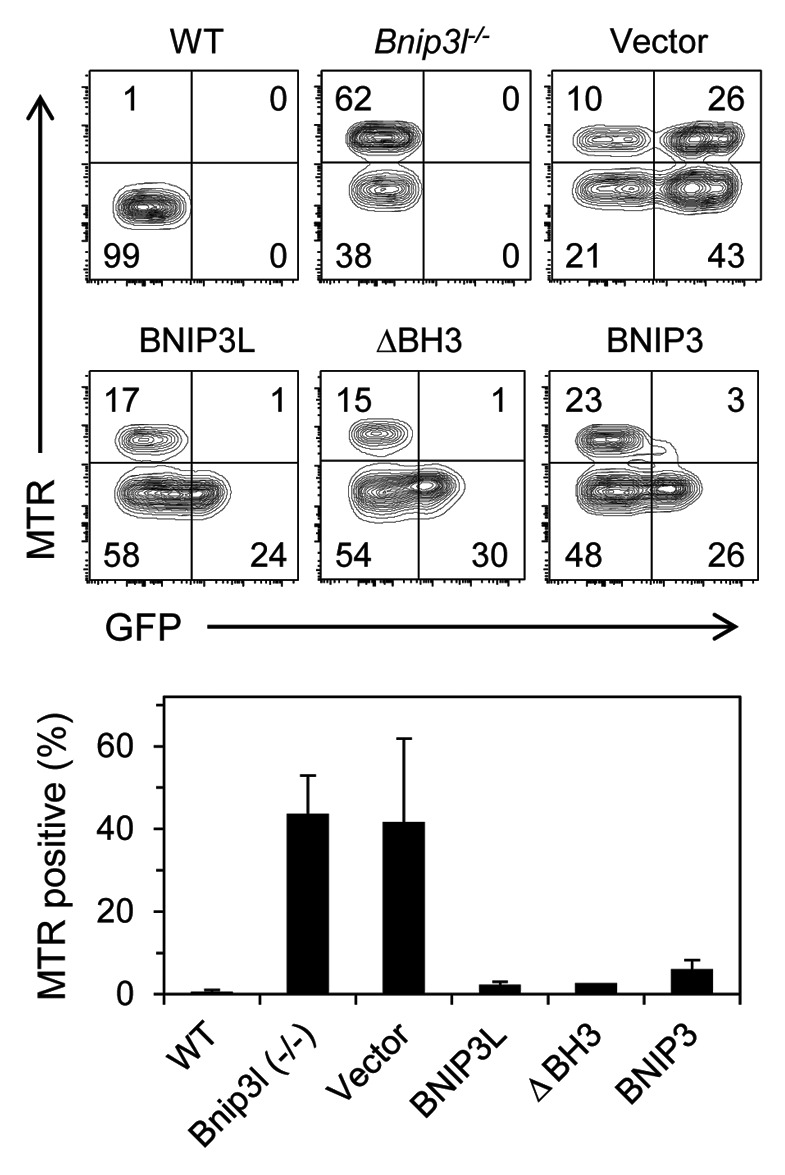
Figure 1. BNIP3 rescues mitochondrial clearance in Bnip3l−/− reticulocytes. Bnip3l−/− bone marrow cells were transduced with viral vector, which expressed GFP (Vector), or virus containing N-terminal FLAG-tagged BNIP3L (BNIP3L), FLAG-tagged BNIP3L lacking its BH3 domain (ΔBH3), or BNIP3. Mitochondrial clearance is assessed by the percentage of Mitotracker Red positive erythrocytes in the GFP positive fraction, except in wild type and non-transduced Bnip3l−/− mice, where it is assessed in the GFP negative fraction.
BNIP3L acts independently of its BH3 domain and BCL-XL
BNIP3L and BNIP3 possess a BH3-like domain, and their expression causes mitochondrial dysfunction and cell death in specific settings.15 In this regard, BH3-only proteins can also activate autophagy by competing with the multidomain antiapoptotic proteins BCL2 and BCL-XL for binding to the autophagy regulator BECN1.16,17 Specifically, BNIP3L and BNIP3 activate autophagy by this mechanism.18 Given the established role of autophagy in mitochondrial clearance in reticulocytes,19-21 we sought to determine the contribution of BNIP3L BH3-like domain. We generated a mutant of BNIP3L in which the BH3-like domain was deleted; our results indicate that the BH3-like domain of BNIP3L is not required for mitochondrial clearance (Fig. 1).
BNIP3L and BCL-XL are coordinately upregulated during terminal erythroid maturation;22 however, they are not co-required for mitochondrial clearance.12 Given BCL-XL can inhibit autophagy, we considered the opposite notion, namely that BNIP3L mediates mitochondrial clearance by antagonizing BCL-XL. Although the BH3-like domain of BNIP3L is dispensable for mitochondrial clearance, BNIP3L could inhibit BCL-XL through a different domain or protein. To address this possibility, we employed a genetic approach. The development of erythroid cells triply deficient for BCL-XL, BAX and BAK is essentially normal;12 therefore, we generated erythroid cells quadruply deficient for BNIP3L, BCL-XL, BAX and BAK. If the model is correct, then BCL-XL deficiency should correct the mitochondrial clearance defect caused by BNIP3L deficiency. We found that BAK deficiency partially corrects the mitochondrial clearance defect; however, BCL-XL deficiency had almost no independent effect (Fig. 2). We conclude that BCL-XL does not have a significant role, positive or negative, in mitochondrial clearance in reticulocytes.
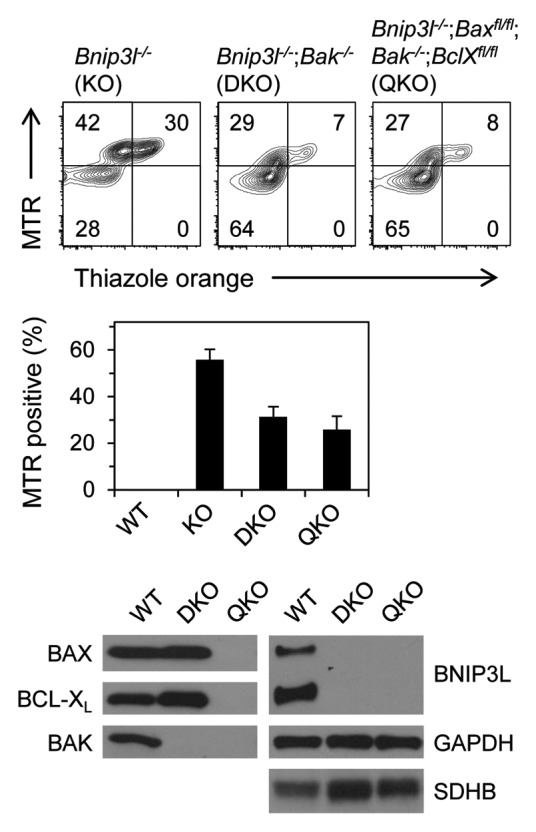
Figure 2. BNIP3L functions independently of BCL-XL. Mitochondrial clearance is assessed in mice that are deficient for BNIP3L (Bnip3l−/− KO); BNIP3L and BAK (Bnip3l−/−;Bak−/− DKO); and BNIP3L, BAX, BAK and BCL-XL (Bnip3l−/−;Baxfl/fl;Bak−/−;BclXfl/fl QKO). Deletion of floxed genes is driven by an MMTV-Cre transgene, and is highly efficient in the erythroid lineage (see western blot in this figure). Erythrocytes were stained with MTR and thiazole orange, which stains nucleic acids in nascent reticulocytes. Mitochondrial clearance is assessed in the thiazole orange negative fraction. Western blots are of WT, DKO and QKO reticulocytes probed for BNIP3L, BAX, BAK, BCL-XL. GAPDH and SDHB are loading controls.
Identification of a discrete region in the cytoplasmic domain of BNIP3L required for activity
BNIP3L has an LC3-interaction region (LIR) near its N-terminus.23,24 Consistent with its low affinity for mammalian Atg8 homologs, mutation of this motif has a measurable but modest effect on the rescue of mitochondrial clearance in Bnip3l−/− reticulocytes.23 Apart from the LIR and the BH3-like domain there are no other known functional motifs in the cytoplasmic domain of BNIP3L; therefore, we employed an unbiased approach. We generated amino- and C-terminal truncation mutants of FLAG-BNIP3L (N1-N5 and ΔC) (Fig. S1). Murine BNIP3L has 218 amino acids; truncation of its N-terminal 69 amino acids including the LIR (mutant N1), caused a slight decrease in activity, whereas removal of 95 amino acids or more (mutants N2-N5) caused complete loss of activity (Fig. 3A). Truncation of its C-terminal 43 amino acids including the transmembrane domain (mutant ΔC), also caused complete loss of activity. BNIP3L mutants were expressed at similar levels to the endogenous protein in erythroid cells (Fig. S2). N-terminal truncation mutants localized to mitochondria, like wild-type BNIP3L, whereas the ΔC BNIP3L mutant exhibited diffuse cytoplasmic staining (Fig. S3). Thus, to define the C-terminal boundary of the active cytoplasmic region, and preserve mitochondrial localization, we made internal deletion (ID) mutants. Deletion of amino acids from 87–185 had no effect on activity (ID1-ID4). By contrast, deletion of amino acids 58–86 caused a complete loss of BNIP3L activity (ID5) (Fig. 3B). These studies define the boundaries of a minimal essential region (MER) of BNIP3L as amino acids 70–86. The MER does not interact with LC3 (data not shown).
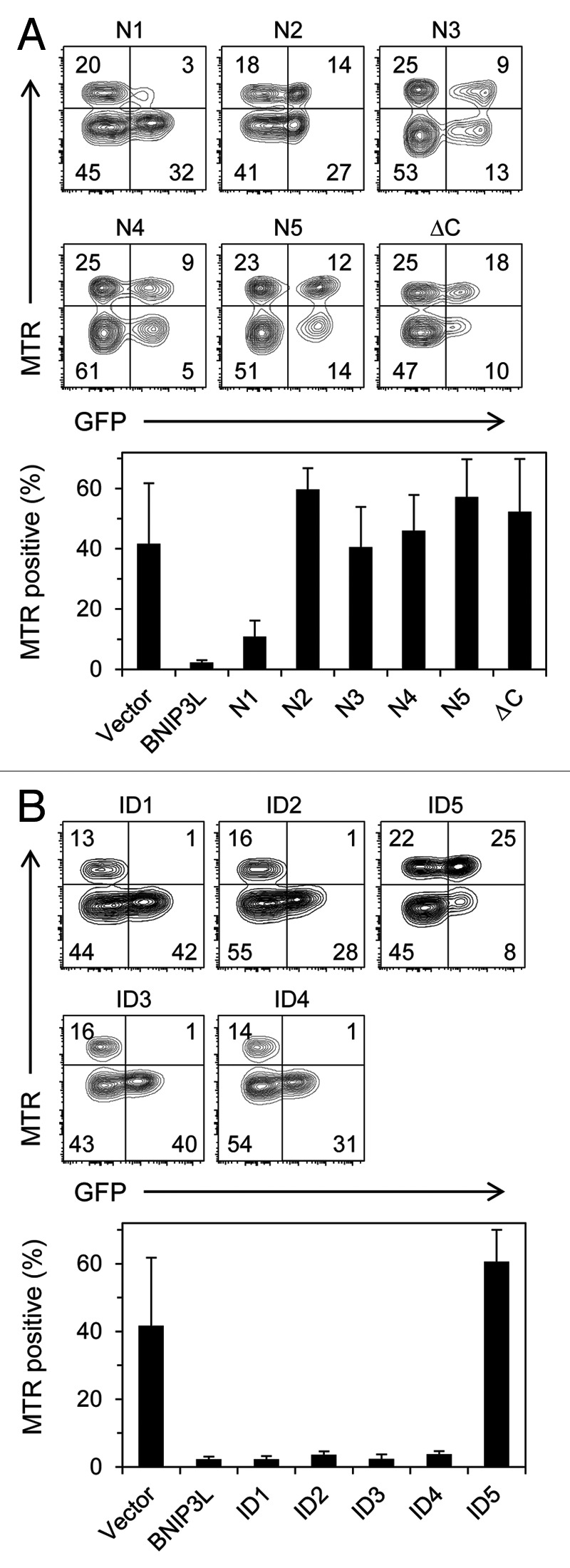
Figure 3. A discrete region in the cytoplasmic domain of BNIP3L is required for its activity. (A) FLAG-tagged amino- and C-terminal truncation mutants of BNIP3L. (B) FLAG-tagged internal deletion (ID) mutants of BNIP3L.
Endogenous BNIP3L is present in a full-length and short form in erythroid tissues.12 A methionine residue at position 40 in BNIP3L, may serve as alternative translation initiation site. To determine its functional significance, we generated two point mutants of BNIP3L: an M1A mutant that only initiates at M40, and an M40A mutant that only initiates at M1. Consistent with the preceding analysis, both forms of BNIP3L were fully active (Fig. S4).
Structural bioinformatics analysis of BNIP3L
BLAST analysis of BNIP3L sequence against the Protein Databank of solved protein structures reveals that the C-terminal region of BNIP3L is 77% identical to a region of BNIP3 that has been characterized in lipid bilayers by nuclear magnetic resonance spectroscopy.25 This region of BNIP3 forms a kinked α-helical structure 32 residues in length. Analysis done on the Phyre structure prediction server indicates that this structure is a good model for the C-terminal domain of BNIP3L.26 Structural models of other domains of BNIP3L were not identified. Secondary structure and disorder analysis using the PredictProtein server indicates that except for its C-terminal transmembrane helix (residues 186–203), BNIP3L lacks consolidated secondary structure that would be associated with a stably folded, globular protein.27 Notably, the LIR and amino acids 68–79, overlapping the MER, are predicted to have secondary structure (Fig. 4). Specifically, the MER is predicted to form an α-helix with a high degree of reliability. Analysis using the PONDR server predicts that amino acids 1–125 of BNIP3L are disordered, amino acids 126–172 are partially structured, and amino acids 173–208, corresponding to the transmembrane helix, are structured.28 These secondary structure and disorder analyses suggest that the N-terminal cytoplasmic domain of BNIP3L lacks secondary and tertiary structure in isolation, and that the LIR and MER, which are predicted to have secondary structure, may fold upon binding to a biological target.

Figure 4. Amino acid sequence and structural bioinformatics analysis of the LIR-MER region. The MER (underlined, starting at 70), as defined by N-terminal truncation and internal deletion mutants overlaps a region of predicted α-helical (H) structure. The algorithm produces a reliability index (REL IN), on a scale of 0–9. Nine is the highest degree of reliability. The BNIP3L LC3-interaction region (LIR)(underlined, starting at 35), overlaps a predicted region of β-strand (E). PredictProtein secondary structure, PP SEC.
BNIP3L activity localizes to a single leucine residue in a hydrophobic triplet flanked by charged amino acid residues
The MER comprises a stretch of hydrophobic amino acid residues (M70, I73, L74 and L75), flanked by charged residues (K72, D76, H79 and E80) and serines (S81, S84, S85 and S86). Analysis employing alanine substitution mutations shows that the three contiguous hydrophobic residues are essential, and that the charged residues also contribute significantly to activity. By contrast, mutation of amino acid K72 or the serines had no effect. These results define the boundaries of the MER as amino acids 70–80, which correspond closely to the region of predicted α-helical structure. Scanning alanine mutations further showed that hydrophobic residue leucine 74 makes a critical contribution to BNIP3L activity (Fig. 5).
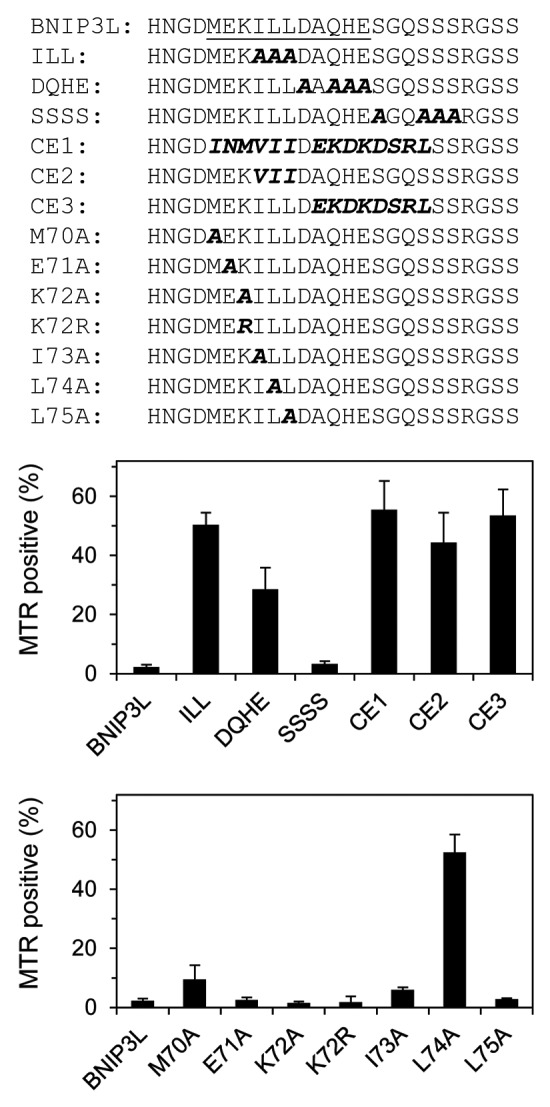
Figure 5. Leucine 74 is essential for BNIP3L activity. Details of the MER mutants are shown. Mutations include the hydrophobic core (ILL), flanking charged residues (DQHE), flanking serine residues (SSSS), C. elegans substitutions (CE1-3) and scanning alanine mutations.
The amino acids in BNIP3L and BNIP3 are identical in the region around L74, and similar but not identical in the C. elegans ortholog of BNIP3.29,30 To see if the differences would affect BNIP3L function, we replaced 15 amino acids in the MER with the corresponding amino acids in C. elegans BNIP3. This substitution resulted in complete loss of BNIP3L activity (Fig. 5, mutant CE1). Compared with BNIP3L, the changes in the hydrophobic residues in C. elegans BNIP3 are relatively conservative (ILL vs. VII). The presence of flanking charged amino acid residues is also conserved; however, the specific charged residues are not conserved, and in some cases result in a substitution with an amino acid of the opposite charge. When we separated the effect of the conservative hydrophobic substitutions from the charged substitutions, we found that both still caused loss of BNIP3L activity (CE2 and CE3). This shows that BNIP3L is sensitive to substitutions within the hydrophobic amino acid triplet. Further, it reinforces our results obtained with the alanine substitution mutants, which showed that the charged residues contribute to BNIP3L activity.
Discussion
Remarkably little is known about the processes by which cells control the quality and abundance of their subcellular organelles. Postmitotic cells, such as neurons and cardiomyocytes, maintain a cohort of healthy mitochondria for decades in humans, but the basis of this stability is poorly understood. At least in part, it is thought to require the removal of damaged or defective mitochondria. In this regard, impaired mitochondrial clearance has been implicated in aging, degenerative disease and cancer. Mitochondria are also eliminated in response to metabolic change and during differentiation. For example, hypoxia induces mitochondrial clearance in mammalian cells, which is dependent on BNIP3 and FUNDC1.31 To gain insight into the mechanism of mitochondrial clearance, we examined the BNIP3L-dependent elimination of mitochondria that occurs during reticulocyte maturation.
Mitochondrial depolarization is a potent stimulus for mitochondrial clearance, and both BNIP3L and BNIP3 can cause mitochondrial depolarization. Given chemical mediators of mitochondrial outer membrane permeabilization and depolarization can rescue mitochondrial clearance in Bnip3l−/− reticulocytes, it has been suggested that BNIP3L promotes clearance by causing depolarization.13 Against this model, mitochondrial clearance is normal in the absence of BAX and BAK, and in the presence of blockers of the mitochondrial permeability transition pore.12 Also, in contrast to BNIP3, the evidence is weak that BNIP3L can directly cause mitochondrial depolarization.32 Further, experiments in autophagy-defective reticulocytes suggest that mitochondrial depolarization follows rather than precedes degradative vacuole formation.19 Finally, our identification of a short linear motif in the cytoplasmic domain of BNIP3L, which mediates mitochondrial clearance, points to an adaptor mechanism.
Mitochondrial clearance in mature erythroblasts and reticulocytes was first linked to autophagy in ultrastructural studies, which showed mitochondria inside double-membraned structures, undergoing degradation and exocytosis.33-35 Autophagy increases during terminal erythroid differentiation, but is unaffected by the loss of BNIP3L. Thus, BNIP3L is not a global regulator of autophagy in erythroid cells. Therefore, we considered the possibility that BNIP3L has a local effect on autophagy, mediated by BCL-XL. BECN1 is an upstream regulator of autophagy, which itself is regulated by BCL2 family members. BCL-XL represses BECN1 through a direct interaction; when this interaction is disrupted by the BH3-like domain in BNIP3L or BNIP3, autophagy is activated.18 Ambra1 is another autophagy protein that is repressed by BCL2.36 Our studies indicate that BNIP3L functions independent of its BH3-like domain and BCL-XL. Thus, BNIP3L does not mediate mitochondrial clearance in reticulocytes through a direct or indirect effect on BCL-XL.
Autophagy in yeast depends on ubiquitin-like conjugation pathways, which contribute to the growth of the phagophore membrane.37 These pathways, which are conserved in mammals, conjugate Atg12 to Atg5 and Atg8 to phosphatidylethanolamine. Atg7 is an E1-like ligase, which catalyzes a critical intermediate step in both conjugation pathways. Surprisingly, ATG7-deficient reticulocytes clear their mitochondria, albeit at a reduced rate, despite inactivation of both pathways.19 Further, autophagy is induced in Atg5−/− murine embryonic fibroblasts subjected to starvation or stress.38 Thus, alternative autophagy pathways exist in mammalian cells, which operate independent of the ubiquitin-like conjugation pathways, specifically ATG5 and ATG7. Given the MER does not interact with LC3, perhaps the function of BNIP3L is to recruit a protein from one of these alternative pathways.
No other sequence in BNIP3L can substitute for the MER; consequently, it is central to our understanding of BNIP3L mechanism of action. Consistent with the ability of BNIP3 to rescue mitochondrial clearance, the MER is conserved in this protein. Structural bioinformatic algorithms predict that the cytoplasmic domain of BNIP3L is disordered, and lacks extensive secondary and tertiary structure. However, two short sequences, corresponding to the MER and the LIR, are predicted to exhibit α-helical and β-strand secondary structure, respectively, and may assume these conformations through functional interactions with one or more binding partners. Within the MER, in the center of a predicted α-helix, a single hydrophobic amino acid is critical for BNIP3L activity; adjacent charged amino acids also contribute. These residues appear to constitute a conserved linear motif, which often mediate the function of disordered regions of proteins.39
Short linear motifs mediate cellular signaling, proteolytic cleavage, post-translational modification, and trafficking. Due to their compact linear structure, these motifs are copied by viruses and intracellular pathogens.40 The nonredundant role of the MER in erythroid development suggests that the MER-target protein interaction is a candidate for the regulation of mitochondrial clearance. At present, the biologically relevant protein that interacts with the MER is not known. It does not appear to be LC3, but could be another autophagy-related protein. Alternatively, vesicle trafficking proteins, which participate in autophagy,41,42 could be recruited by BNIP3L to mitochondria. Considering the effect of mitochondrial dynamics on clearance,43 another possibility is that BNIP3L interacts with a protein that regulates mitochondrial fission or one that enzymatically modifies mitochondrial outer membrane proteins. Identification of the MER-interacting protein will likely lead to insights into mitochondrial clearance and cellular quality control.
Materials and Methods
Generation of BNIP3L mutants
Mouse BNIP3L cDNA was obtained from ATCC IMAGE collection in Bluescript vector. For all mutants with an N-terminal FLAG tag, a single FLAG (DYKDDDDK) epitope was inserted between the initial methionine and the second serine residue by PCR. All deletion mutants and long region swap mutants were generated by recombinant PCR with two exceptions: (1) the ΔC deletion was generated by introducing premature stop codon ahead of the transmembrane domain; and (2) the M1A mutant was generated by mutating the initial methionine. All single amino acid and triple amino acids mutants were made with the QuikChange Site-directed mutagenesis kit (Agilent Technologies).
Virus producer cells
Wild-type and BNIP3L mutant cDNAs were subcloned into MSCV-Ires-GFP. Viral and helper plasmids were cotransfected into 293T cells, as previously described.23,44 Transient viral supernatants were applied to GPE86 (+) ecotropic packaging cells six times, at 12 h intervals. Infected, GFP high-expressing, GPE86 (+) cells were obtained by sorting and plated for use in bone marrow transduction experiments.
Animal studies
Donor Bnip3l−/− mice12 were injected with a single dose of 5-fluorouracil (5-FU)(150 mg/kg), intraperitoneally. Four days later, viral producer cells were irradiated (3,000 rad) and plated in 10 cm tissue culture dishes coated with 1% gelatin. The next day, bone marrow was harvested from the 5-FU-treated Bnip3l−/− mice. Bone marrow cells were treated with erythrocyte lysis buffer (BD Biosciences), washed once with phosphate-buffered saline, resuspended in DMEM with 15% fetal bovine serum, and plated at a density of 1 × 106 cells per ml in 10 cm suspension dishes. Bone marrow cells were prestimulated with cytokines [murine stem cell factor, 50 ng/ml (Peprotech Inc.); murine interleukin-3, 20 ng/ml (Peprotech, Inc.); interleukin-6 (BioVision Research Products)] for 36 h. After prestimulation, 5 × 105 bone marrow cells were resuspended in 10 ml fresh medium, cytokines and polybrene (6 μg/ml), and added to dishes of irradiated producer cells. The bone marrow and viral producer cells were co-cultured for 36 h. Transduced bone marrow cells were harvested and 1–2 × 106 cells injected, intravenously, into lethally irradiated (1,000 rad) wild-type recipients.
Transplant recipients were allowed 4–6 weeks for bone marrow recovery. Mice were not subjected to any additional erythropoietic stress. Approximately 20 μl of blood was obtained by retroorbital puncture for analysis. Mitochondrial clearance was determined by Mitotracker Red staining and flow cytometry, as previously described.45 Mitochondrial clearance was assessed in the GFP-positive cells (corresponding to virally-transduced cells). At least two transplanted mice were studied per BNIP3L mutant. All animal studies were conducted in accordance with the guidelines set by the St. Jude Children’s Research Hospital Institutional Animal Care and Use Committee.
Expression of BNIP3L mutants in fetal liver cells
Bnip3l−/− fetal liver cells were harvested at embryonic day 14.5 and cultured in erythroid expansion medium (EEM) [friend virus anemia (FVA) medium,46 plus erythropoietin, 2 U/ml; murine stem cell factor, 100 ng/ml; human insulin-like growth factor-1, 40 ng/ml; and dexamethasone, 1 μM] for 4–6 d. Fresh medium was added every 2 d to maintain the cell density less than 2 × 106 cells per ml. Expanded erythroblast cells were co-cultured with irradiated GPE86 (+) producer cells in EEM at a density of 2 × 105 cells per ml for 48 h, in the presence of 6 μg/ml polybrene. Erythroblasts were collected, washed twice in FVA medium, resuspended in FVA medium with erythropoietin 4 U/ml, and cultured an additional 48 h. Protein was extracted with RIPA buffer in the presence of protease inhibitors. For fetal liver cells, 30 μg of protein was resolved by electrophoresis [12% Tris-Bis NuPAGE gel (Invitrogen)], and transferred to a 0.45 micron PVDF membrane, except for BNIP3L, which was transferred to nitrocellulose. For reticulocytes, we loaded 100 μg of protein.
Subcellular localization of BNIP3L mutants in fibroblasts
Primary MEFs were cultured in a slide chamber (BD Falcon, 354104) for 24 h prior to transfection. FLAG-tagged BNIP3L mutants were subcloned into pcDNA3.1 and transfected into primary MEFs through the use of FuGENE 6 (Roche), in accordance with the manufacturer’s protocol; 48 h post-transfection, fresh medium with MTR (Invitrogen) at a concentration of 100 nM was applied. The cells were incubated for 45 min at 37°C, washed with cold PBS, fixed at room temperature (RT) with freshly made 4% paraformaldehyde in PBS (Electron Microscopy Sciences, 15710) for 10 min, and then quenched with 0.1 M glycine for 5 min. Next, the cells were incubated with 3% bovine serum albumin in PBS (PBS-BSA) with 0.25% Triton X-100 for 1 h at RT to permeabilize and block nonspecific binding. The cells were incubated overnight at 4°C with rabbit anti-FLAG antibody, 1:500 (Sigma, F7425) in PBS-BSA with 0.25% Triton X-100. They were washed once with PBS-BSA with 0.5% Triton X-100 for 5 min at RT, and twice with PBS-BSA with 0.1% Triton X-100 for 5 min at RT. Next, the cells were incubated with 300 μl Alexa Fluor-488 anti-rabbit IgG, 1:1,000 (Invitrogen, A21206), in PBS-BSA with 0.25% Triton X-100 for 45 min at RT. They were washed twice with PBS with 0.1% Triton X-100 for 5 min at RT, and incubated DAPI (Invitrogen, D21490) in PBS for 5 min at RT. After two more washes with PBS, the slides were mounted with Fluoromount-G (Southern Biotech) and a coverslip and sealed with nail polish.
Cells were imaged with a Zeiss 510 Meta NLO laser scanning microscope with dedicated Zen 2009 software. We used a 100× Plan Apochromat oil objective (N.A. 1.4). The cells were excited with 488 nm (Alexa Fluor-488), 543 nm (MTR) and 740 nm (DAPI) lasers; the latter was provided by a Coherent Chameleon Ti:Sapphire laser tuned to 740 nm. The emitted signal was directed to a Meta Spectral detector.
Primary antibodies
FLAG M2, 1:1,000 (Sigma, F1804), BNIP3L, 0.5 μg/ml (Exalpha Biologicals, X1120P), GFP 1:1,000 (Clontech); ACTB/β-actin, 1:10,000 (Sigma, A5441); BAX, 1:3,000 (gift of Joseph Opferman and the Korsmeyer laboratory); BAK, 0.5 μg/ml (Upstate Cell Signaling Solutions, 06-536); BCL-XL, 0.5 μg/ml (BD Biosciences, 610209); GAPDH, 1:5,000 (Santa Cruz Biotechnology, sc-32233); SDHB, 0.1 μg/ml (Santa Cruz Biotechnology, sc-34150); Cytochrome c, 0.5 μg/ml (Invitrogen, 33-8500); COX4/COX IV, 0.25 μg/ml (Abcam, ab14744); and SOD2/MnSOD, 0.1 μg/ml (BD Biosciences, 611580).
Supplementary Material
Acknowledgments
The authors thank Stephane Pelletier for assistance with the immunofluorescent localization of FLAG-BNIP3L. The authors also thank the members of the Flow Cytometry Laboratory and the Animal Resource Center of St. Jude Children’s Research Hospital. Bax and Bak mutant mice were a gift of the Korsmeyer laboratory and Joseph Opferman. BclXfl/fl and Tg(MMTV-Cre) mutant mice were a gift of Lothar Henighausen. This work was supported by a grant from the National Institutes of Health, R21 DK074519 (P.A.N.), by the New York Blood Center, and by the American, Lebanese and Syrian Associated Charities.
Glossary
Abbreviations:
- NIX
NIP-like protein X
- BNIP3
adenovirus E1B interacting protein 3
- MER
minimal essential region
- LIR
LC3-interacting region
- ID
internal deletion
- MTR
MitoTracker Red
- 5-FU
5-fluorouracil
- WT
wild type
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/20764
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/20764
References
- 1.Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem. 2012;287:4434–40. doi: 10.1074/jbc.R111.271999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–46. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–8. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 5.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–60. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 6.Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–6. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 7.Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–61. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 8.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–31. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–80. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104:19500–5. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–5. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Chen Y, Lewis W, Diwan A, Cheng EH, Matkovich SJ, Dorn GW., 2nd Dual autonomous mitochondrial cell death pathways are activated by Nix/BNip3L and induce cardiomyopathy. Proc Natl Acad Sci U S A. 2010;107:9035–42. doi: 10.1073/pnas.0914013107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 17.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–39. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570–81. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang J, Randall MS, Loyd MR, Dorsey FC, Kundu M, Cleveland JL, et al. Mitochondrial clearance is regulated by Atg7-dependent and -independent mechanisms during reticulocyte maturation. Blood. 2009;114:157–64. doi: 10.1182/blood-2008-04-151639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mortensen M, Ferguson DJ, Edelmann M, Kessler B, Morten KJ, Komatsu M, et al. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci U S A. 2010;107:832–7. doi: 10.1073/pnas.0913170107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, et al. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112:1493–502. doi: 10.1182/blood-2008-02-137398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aerbajinai W, Giattina M, Lee YT, Raffeld M, Miller JL. The proapoptotic factor Nix is coexpressed with Bcl-xL during terminal erythroid differentiation. Blood. 2003;102:712–7. doi: 10.1182/blood-2002-11-3324. [DOI] [PubMed] [Google Scholar]
- 23.Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwarten M, Mohrlüder J, Ma P, Stoldt M, Thielmann Y, Stangler T, et al. Nix directly binds to GABARAP: a possible crosstalk between apoptosis and autophagy. Autophagy. 2009;5:690–8. doi: 10.4161/auto.5.5.8494. [DOI] [PubMed] [Google Scholar]
- 25.Bocharov EV, Pustovalova YE, Pavlov KV, Volynsky PE, Goncharuk MV, Ermolyuk YS, et al. Unique dimeric structure of BNip3 transmembrane domain suggests membrane permeabilization as a cell death trigger. J Biol Chem. 2007;282:16256–66. doi: 10.1074/jbc.M701745200. [DOI] [PubMed] [Google Scholar]
- 26.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–71. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 27.Rost B, Yachdav G, Liu J. The PredictProtein server. Nucleic Acids Res. 2004;32(Web Server issue):W321-6. doi: 10.1093/nar/gkh377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::AID-PROT50>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 29.Yasuda M, D’Sa-Eipper C, Gong XL, Chinnadurai G. Regulation of apoptosis by a Caenorhabditis elegans BNIP3 homolog. Oncogene. 1998;17:2525–30. doi: 10.1038/sj.onc.1202467. [DOI] [PubMed] [Google Scholar]
- 30.Cizeau J, Ray R, Chen G, Gietz RD, Greenberg AH. The C. elegans orthologue ceBNIP3 interacts with CED-9 and CED-3 but kills through a BH3- and caspase-independent mechanism. Oncogene. 2000;19:5453–63. doi: 10.1038/sj.onc.1203929. [DOI] [PubMed] [Google Scholar]
- 31.Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–85. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 32.Diwan A, Koesters AG, Odley AM, Pushkaran S, Baines CP, Spike BT, et al. Unrestrained erythroblast development in Nix-/- mice reveals a mechanism for apoptotic modulation of erythropoiesis. Proc Natl Acad Sci U S A. 2007;104:6794–9. doi: 10.1073/pnas.0610666104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simpson CF, Kling JM. The mechanism of mitochondrial extrusion from phenylhydrazine-induced reticulocytes in the circulating blood. J Cell Biol. 1968;36:103–9. doi: 10.1083/jcb.36.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gronowicz G, Swift H, Steck TL. Maturation of the reticulocyte in vitro. J Cell Sci. 1984;71:177–97. doi: 10.1242/jcs.71.1.177. [DOI] [PubMed] [Google Scholar]
- 35.Heynen MJ, Tricot G, Verwilghen RL. Autophagy of mitochondria in rat bone marrow erythroid cells. Relation to nuclear extrusion. Cell Tissue Res. 1985;239:235–9. doi: 10.1007/BF00214924. [DOI] [PubMed] [Google Scholar]
- 36.Strappazzon F, Vietri-Rudan M, Campello S, Nazio F, Florenzano F, Fimia GM, et al. Mitochondrial BCL-2 inhibits AMBRA1-induced autophagy. EMBO J. 2011;30:1195–208. doi: 10.1038/emboj.2011.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–9. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 38.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654–8. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- 39.Davey NE, Van Roey K, Weatheritt RJ, Toedt G, Uyar B, Altenberg B, et al. Attributes of short linear motifs. Mol Biosyst. 2012;8:268–81. doi: 10.1039/c1mb05231d. [DOI] [PubMed] [Google Scholar]
- 40.Davey NE, Travé G, Gibson TJ. How viruses hijack cell regulation. Trends Biochem Sci. 2011;36:159–69. doi: 10.1016/j.tibs.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 41.Nair U, Jotwani A, Geng J, Gammoh N, Richerson D, Yen WL, et al. SNARE proteins are required for macroautophagy. Cell. 2011;146:290–302. doi: 10.1016/j.cell.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bodemann BO, Orvedahl A, Cheng T, Ram RR, Ou YH, Formstecher E, et al. RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell. 2011;144:253–67. doi: 10.1016/j.cell.2010.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–98. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Persons DA, Hargrove PW, Allay ER, Hanawa H, Nienhuis AW. The degree of phenotypic correction of murine beta -thalassemia intermedia following lentiviral-mediated transfer of a human gamma-globin gene is influenced by chromosomal position effects and vector copy number. Blood. 2003;101:2175–83. doi: 10.1182/blood-2002-07-2211. [DOI] [PubMed] [Google Scholar]
- 45.Zhang J, Kundu M, Ney PA. Mitophagy in mammalian cells: the reticulocyte model. Methods Enzymol. 2009;452:227–45. doi: 10.1016/S0076-6879(08)03615-X. [DOI] [PubMed] [Google Scholar]
- 46.Koury MJ, Sawyer ST, Bondurant MC. Splenic erythroblasts in anemia-inducing Friend disease: a source of cells for studies of erythropoietin-mediated differentiation. J Cell Physiol. 1984;121:526–32. doi: 10.1002/jcp.1041210311. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.