

Abstract
Cytosolic bacterial pathogens must evade intracellular innate immune recognition and clearance systems such as autophagy to ensure their survival and proliferation. The intracellular cycle of the bacterium Francisella tularensis is characterized by rapid phagosomal escape followed by extensive proliferation in the macrophage cytoplasm. Cytosolic replication, but not phagosomal escape, requires the locus FTT0369c, which encodes the dipA gene (deficient in intracellular replication A). Here, we show that a replication-deficient, ∆dipA mutant of the prototypical SchuS4 strain is eventually captured from the cytosol of murine and human macrophages into double-membrane vacuoles displaying the late endosomal marker, LAMP1, and the autophagy-associated protein, LC3, coinciding with a reduction in viable intracellular bacteria. Capture of SchuS4ΔdipA was not dipA-specific as other replication-deficient bacteria, such as chloramphenicol-treated SchuS4 and a purine auxotroph mutant SchuS4ΔpurMCD, were similarly targeted to autophagic vacuoles. Vacuoles containing replication-deficient bacteria were labeled with ubiquitin and the autophagy receptors SQSTM1/p62 and NBR1, and their formation was decreased in macrophages from either ATG5-, LC3B- or SQSTM1-deficient mice, indicating recognition by the ubiquitin-SQSTM1-LC3 pathway. While a fraction of both the wild-type and the replication-impaired strains were ubiquitinated and recruited SQSTM1, only the replication-defective strains progressed to autophagic capture, suggesting that wild-type Francisella interferes with the autophagic cascade. Survival of replication-deficient strains was not restored in autophagy-deficient macrophages, as these bacteria died in the cytosol prior to autophagic capture. Collectively, our results demonstrate that replication-impaired strains of Francisella are cleared by autophagy, while replication-competent bacteria seem to interfere with autophagic recognition, therefore ensuring survival and proliferation.
Keywords: Francisella, LC3, SQSTM1/p62, autophagy, clearance, cytosol, pathogenesis, ubiquitin
Introduction
Bacterial pathogens possess strategies to avoid elimination by microbicidal mechanisms, while exploiting other cellular processes, in order to survive the inhospitable intracellular environment. Some intracellular pathogens modify the phagosome into a permissive niche while others physically escape from this degradative compartment to establish residence in the cytoplasm.1 Phagosome maturation, whereby the pathogen-containing phagosome is progressively transformed into a degradative compartment via sequential interactions with the endosomal pathway, has long been recognized to be important in combatting infection.2 Eukaryotic cells also utilize autophagy, initially described as a cytosolic process to dispose of defunct and/or surplus organelles and protein aggregates, as a defense mechanism against vacuolar and cytosolic bacterial pathogens including Mycobacterium tuberculosis,3 Salmonella enterica serovar Typhimurium,4,5 Listeria monocytogenes,6,7 Shigella flexneri8,9 and Group A Streptococcus pyogenes.10 While autophagy can be a nondiscriminatory process when induced as a homeostatic response, e.g., by starvation, it is becoming evident that specific cargo can be selectively targeted to autophagosomes for degradation.11-14 Recent studies have demonstrated the involvement of ubiquitin and autophagy adaptor proteins, such as SQSTM1/p62 (sequestosome 1), CALCOCO2/NDP52 (nuclear dot protein 52), OPTN/optineurin and NBR1 (neighbor of BRCA1 gene 1) in selectively targeting bacterial pathogens to the autophagy pathway.15-22 Some pathogens, such as S. flexneri8 and L. monocytogenes,21,23 employ strategies to avoid autophagic recognition, whereas others, like Coxiella burnetii,24 Legionella pneumophila,25 Staphylococcus aureus,26 and Brucella abortus27 have evolved mechanisms to subvert the autophagy pathway in establishing a replicative compartment, and in the case of B. abortus, to promote cell-to-cell dissemination.28
Francisella tularensis is a highly infectious, Gram negative, bacterial pathogen with broad host and cell tropism range.29 Macrophages, however, are an important target for infection and the bacterium’s virulence is linked to its ability to survive and proliferate within these cells.29-31 Upon internalization, Francisella initially resides within a phagosome which it rapidly disrupts to reach the cytosol.32-36 Following extensive replication in the cytosol, bacterial release occurs through macrophage apoptosis37,38 and pyroptosis.39 Additionally, a subset of cytosolic bacteria can re-enter LAMP1-positive vacuoles, designated Francisella-containing vacuoles (FCVs), in murine bone marrow-derived macrophages (BMMs),33 but not in human blood monocyte-derived macrophages,40,41 via an autophagy-mediated process. Thus, Francisella has developed multilayered mechanisms to successfully adapt to the different compartments it encounters during its intracellular cycle.
Two critical aspects of Francisella intracellular pathogenesis are (1) phagosomal escape and (2) cytosolic replication. Mutants unable to reach the cytosol, such as those with mutations in the Francisella pathogenicity island genes (e.g., ΔiglC), are defective for intracellular proliferation and survival.42-47 Mutants that are specifically impaired for cytosolic replication, such as those with mutations in purine biosynthesis genes (e.g., ΔpurMCD), are similarly affected for intracellular survival.39,42,48-50 Recently, we identified FTT0369c, a locus that was transcriptionally upregulated during the cytosolic stage of F. tularensis subsp tularensis strain SchuS4 infection of BMMs.42 Deletion of FTT0369c in SchuS4 did not affect phagosomal escape but impaired cytosolic replication and intracellular survival.42 Thus, we have re-named this locus dipA for deficient in intracellular proliferation A. Here, we report that the SchuS4ΔdipA mutant and other replication-deficient bacteria are redirected into autophagic vacuoles via the ubiquitin-SQSTM1-LC3 pathway, as a result of their compromised viability in the macrophage cytosol. By contrast, wild-type bacteria avoid autophagic recognition and capture, suggesting that Francisella interferes with the autophagic pathway during cytosolic proliferation.
Results
Enclosure of cytoplasmic SchuS4ΔdipA bacteria into endocytic vacuoles
Our previous work has identified dipA as required for intracellular proliferation and survival of SchuS4 in BMMs.42 In this study, we sought to further analyze the intracellular fate of a ΔdipA in-frame deletion mutant (SchuS4∆dipA). Compared with the wild-type SchuS4 strain, which displayed intracellular growth of 2.5-Log over a 24 h period in BMMs, a SchuS4ΔdipA mutant did not show intracellular replication (Fig. 1A), as described previously.42 The lack of replication of the dipA mutant was also associated with subsequent intracellular killing, since the number of viable intracellular mutants decreased by 2-Log between 10 and 24 h pi (Fig. 1A). Next, we monitored the vacuolar or cytosolic localization of the mutant bacteria over time. Compared with a phagosomal escape-deficient mutant (SchuS4ΔfevR)42 of which no more than 10% was detected as cytoplasmic over a 24 h period (Fig. 1B), SchuS4ΔdipA escaped from its original phagosome efficiently by 1 h p.i. (84 ± 2.7%; Fig. 1B) and remained cytoplasmic until 10 h p.i. (94 ± 4.6%; Fig. 1B) similarly to wild-type organisms (98 ± 1.9%; Fig. 1B). Thereafter, the number of cytoplasmic mutant bacteria decreased with time (72 ± 4.4% at 16 h p.i and 58 ± 1.5% at 24 h p.i.) indicating that a subpopulation of the ΔdipA mutant became enclosed within a vacuolar compartment (Fig. 1B). Immunostaining of infected BMMs for the late endosomal/lysosomal marker LAMP1 at 20 h p.i. showed that a fraction of SchuS4ΔdipA bacteria were surrounded by LAMP1-positive membranes, indicating enclosure within the endocytic compartment (Fig. 1C–E).

Figure 1. Capture of cytosolic Francisella into endosomal vacuoles in BMMs. (A) Intracellular growth of SchuS4, SchuS4 (in 1% FBS-medium), SchuS4 treated with chloramphenicol at 6 h p.i., and its isogenic ΔpurMCD and ΔdipA mutants in BMMs. Intracellular bacteria were enumerated from CFUs at various times p.i. Data are means ± SD from a representative experiment performed in triplicate out of two independent repeats. (B) Intracellular trafficking of SchuS4 and its derivatives in BMMs. At various times p.i., infected BMMs were subjected to a phagosomal integrity assay to quantify the percentage of cytoplasmic bacteria. At least 100 bacteria per experiment were scored for each condition. Data are means ± SD from three independent experiments. (C) Quantification of infected cells containing bacteria enclosed within endosomal vacuoles at 20 h p.i. Infected BMMs were scored for number of infected cells with bacteria enclosed within LAMP1-positive compartments. At least 100 BMMs per experiment were scored for each condition. Data are means ± SD from three independent experiments. (D) Quantification of bacteria enclosed within endosomal vacuoles at 20 h p.i. Infected BMMs were scored for the number of bacteria enclosed within LAMP1-positive compartments. At least 100 BMMs or bacteria per experiment were scored for each condition. Data are means ± SD from three independent experiments. Asterisks indicate statistically significant differences (**p < 0.01, 1-way ANOVA, Tukey’s post-test). (E) Representative confocal micrographs of BMMs infected for 20 h with either SchuS4 or its derivatives. Samples were processed for immunofluorescence labeling of bacteria (green) and LAMP1-positive membranes (red). Magnified insets show single channel images of the boxed areas. White arrows indicate either bacterial clusters or bacteria of interest. Scale bars: 10 or 2 μm.
Previously, we have shown that a subset of wild-type SchuS4 bacteria re-enter LAMP1-positive vacuoles (FCVs) in a replication-dependent manner in BMMs.33 To compare FCV formation by SchuS4 with the endocytic capture of the SchuS4ΔdipA mutant, we quantified the numbers of infected BMMs harboring bacteria enclosed within LAMP1-positive compartments. Indeed, we observed that 37 ± 1.7% of SchuS4-infected BMMs contained clusters of bacteria in LAMP1-positive vacuoles at 20 h p.i. (Fig. 1C and 1E), reflecting FCV formation. Comparably, 38 ± 3.0% of SchuS4ΔdipA-infected BMMs harbored bacteria in LAMP1-positive vacuoles, suggesting the two vacuolar structures originate from similar events. Yet, FCV formation requires bacterial replication,33 whereas vacuolar enclosure of the dipA mutant was associated with a lack of proliferation (Fig. 1A). To determine whether the endosomal vacuoles enclosing the dipA mutant are distinct from FCVs, we examined whether SchuS4ΔdipA bacteria re-enter the endocytic compartment in human blood monocyte-derived macrophages (MDMs), which do not support FCV formation by SchuS4.40 In MDMs, SchuS4 grew exponentially over 24 h whereas the ΔdipA mutant did not proliferate nor survive after 10 h p.i. (Fig. 2A), similar to phenotypes observed in BMMs (Fig. 1A). First, we quantified the percentage of MDMs containing bacteria enclosed within LAMP1-positive structures at 20 h p.i. (Fig. 2B). Few MDMs harbored SchuS4 in LAMP1-positive vacuoles (5 ± 1.5%), verifying that FCVs do not develop in MDMs (Fig. 2B and D). In contrast, 42 ± 1.6% MDMs contained SchuS4ΔdipA bacteria in LAMP1-positive vacuoles, consistent with the observations in BMMs, indicating that, unlike FCVs, enclosure of the dipA mutant within endosomal vacuoles occurs in MDMs. To refine this analysis, we assessed the numbers of bacteria that were sequestered in LAMP1 vacuoles and found that only 1 ± 1.2% of wild-type, but 34 ± 2.5% of mutant bacteria were contained within LAMP1-positive vacuoles (Fig. 2C). Collectively, these findings suggest that enclosure of the replication-deficient ΔdipA mutant within endocytic vacuoles is a process distinct from FCV formation.

Figure 2. Intracellular replication-deficient Francisella are targeted into endosomal vacuoles in human MDMs. (A) Intracellular growth of SchuS4, SchuS4 (in 1% FBS-medium), SchuS4 treated with chloramphenicol at 6 h p.i., and its isogenic ΔpurMCD and ΔdipA mutants in MDMs. Intracellular bacteria were enumerated from CFUs at various times p.i. Data are means ± SD from a representative experiment performed in triplicate out of two independent repeats. (B) Quantification of infected cells containing bacteria enclosed within endosomal vacuoles at 20 h p.i. Infected MDMs were scored for number of infected cells with bacteria enclosed within LAMP1-positive compartments. At least 100 MDMs per experiment were scored for each condition. Data are means ± SD from three independent experiments. Asterisks indicate statistically significant differences (**p < 0.01, 1-way ANOVA, Tukey’s post-test). (C) Quantification of bacteria enclosed within endosomal vacuoles at 20 h p.i. Infected MDMs were scored for the number of bacteria enclosed within LAMP1-positive compartments. At least 100 bacteria per experiment were scored for each condition. Data are means ± SD from three independent experiments. Asterisks indicate statistically significant differences (**p < 0.01, 1-way ANOVA, Tukey’s post-test). (D) Representative confocal micrographs of MDMs infected for 20 h with either SchuS4 or its derivatives. Samples were processed for immunofluorescence labeling of bacteria (green) and LAMP1-positive membranes (red). Magnified insets show single channel images of the boxed areas. White arrows indicate bacteria of interest. Scale bars: 10 or 2 μm.
Vacuolar capture of F. tularensis correlates with replication deficiency
To assess whether capture of SchuS4ΔdipA into vacuoles resulted from the specific loss of the dipA function or the consequent inability to replicate in the macrophage cytosol, we examined in BMMs (Fig. 1A and B) and MDMs (Fig. 2A) the intracellular fate of other Francisella strains displaying similar cytosolic replication defects, such as SchuS4 treated with chloramphenicol [SchuS4 (Cm)] and SchuS4ΔpurMCD, a purine auxotroph mutant capable of phagosome escape but defective in cytosolic replication.49 Addition of chloramphenicol to wild-type SchuS4 at 6 h p.i. inhibited bacterial replication (Figs. 1A and 2A) at a time point where the majority of SchuS4 bacteria had escaped into the cytosol (Fig. 1B) and mimicked the intracellular replication defect of SchuS4ΔdipA. Both chloramphenicol-treated SchuS4 and SchuS4ΔpurMCD bacteria remained cytoplasmic until 10 h p.i. (Fig. 1B), after which a fraction of these replication-impaired cytoplasmic bacteria became re-enclosed within LAMP1-positive vacuoles, similarly to SchuS4ΔdipA bacteria (Figs. 1C–E and 2B–D). The parallel intracellular trafficking among all three replication-deficient SchuS4 strains suggests that this phenomenon arises from limited bacterial proliferation within the macrophage cytosol, and not mutation in dipA only.
Vacuoles sequestering replication-impaired bacteria display features of autophagosomes
Autophagy is a homeostatic process that directly sequesters cytoplasmic components for degradation and is also appreciated to function as a defense mechanism against invading intracellular pathogens.51 Antibacterial autophagy has been demonstrated to target both vacuolar and cytosolic pathogens for degradation.51 To determine whether vacuolar capture of replication-deficient SchuS4 is mediated by autophagy, we first examined LAMP1-positive vacuoles containing replication-impaired bacteria for the presence of the autophagy marker LC3, in BMMs from GFP-LC3-transgenic mice. At 20 h p.i., 11 ± 4.0% and 13 ± 4.5% of LAMP1-positive vacuoles containing chloramphenicol-treated SchuS4 and SchuS4ΔpurMCD were GFP-LC3-positive, respectively, whereas a higher proportion of SchuS4ΔdipA bacteria within LAMP1-positive vacuoles were GFP-LC3-positive (30 ± 5.6%; Fig. 3A). At 24 h p.i., the levels of GFP-LC3 enriched LAMP1-positive vacuoles containing replication-impaired bacteria were similar among the three strains (19 ± 5.4% SchuS4 (Cm), 17 ± 2.0% SchuS4ΔpurMCD, 18 ± 4.2% SchuS4ΔdipA; Fig. 3A). The varying levels of GFP-LC3 recruitment to LAMP1-positive vacuoles observed among the different replication-deficient SchuS4 strains suggests differing vacuole maturation kinetics along the autophagic pathway. Ultrastructural analysis by TEM at 20 h p.i. in BMMs revealed individual or groups of two or three replication-deficient bacteria within double-membrane structures resembling autophagosomes (Fig. 3B), consistent with the observations made by fluorescence microscopy. Hence, Francisella replication-deficient strains are captured within vacuoles displaying autophagic features.
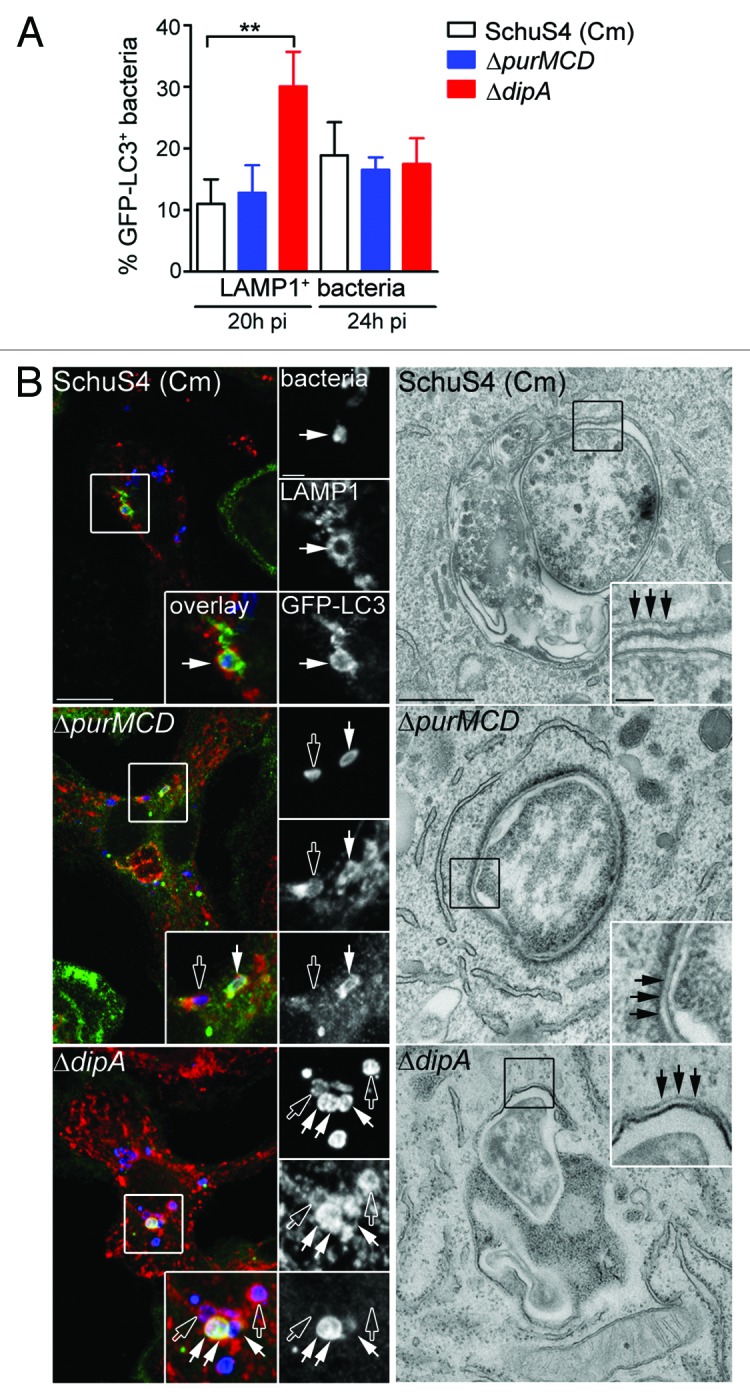
Figure 3. Endosomal vacuoles containing replication-deficient Francisella display features of autophagosomes. (A) Quantification of GFP-LC3 recruitment to LAMP1-positive vacuoles containing replication-deficient Francisella at 20 h and 24 h p.i. BMMs expressing GFP-LC3 were infected with either SchuS4 treated with chloramphenicol at 6 h p.i. or its isogenic ΔpurMCD or ΔdipA mutants, and processed for immunofluorescence labeling of bacteria, LAMP1-positive membranes and GFP. At least 30 LAMP1-positive bacteria per experiment were scored for LC3 recruitment in each condition. Data are means ± SD from three independent experiments. Asterisks indicate statistically significant differences (** p < 0.01, 1-way ANOVA, Tukey’s post-test). (B) Representative confocal (left panels) and transmission electron (right panels) micrographs of BMMs infected for 20 h with strains described in (A). Left panels, BMMs expressing GFP-LC3 were infected and processed for immunofluorescence labeling of bacteria (blue), LAMP1-positive membranes (red) and GFP (green). Magnified insets show single channel images of the boxed areas. Empty white arrows indicate bacteria enclosed within LAMP1-positive, LC3-negative vacuoles; solid white arrows indicate bacteria enclosed within LAMP1-positive and LC3-positive vacuoles. Right panels, BMMs were infected and processed for TEM as described in Materials and Methods. Insets show a magnification of the boxed areas. Black arrows indicate double membranes surrounding intracellular bacteria. Scale bars: 10 or 2 μm (confocal images) and 500 or 100 nm (TEM images).
Cytosolic detection of F. tularensis by ubiquitin, SQSTM1 and NBR1
To further elucidate the mechanisms of capture of replication-deficient Francisella, we next examined how these bacteria are recognized and targeted to autophagosomes. Ubiquitination is a well-characterized signal for antibacterial autophagy.5,17,52,53 Ubiquitin-coated bacteria are delivered to autophagosomes through recruitment of autophagy adaptor proteins which bind to both ubiquitin and LC3 directly, thus bridging the two systems.9,15,18,19,21,54 We first examined whether the replication-deficient strains contained within LC3-positive vacuoles were labeled with ubiquitin and found that 52 ± 6.1% of SchuS4(Cm), 71 ± 20.5% of ΔpurMCD and 48 ± 6.9% of ΔdipA bacteria were ubiquitin-coated at 16 h p.i. (Fig. 4A and B). Moreover, 76 ± 1.8% of SchuS4(Cm), 81 ± 8.0% of ΔpurMCD and 82 ± 5.3% of ΔdipA bacteria enclosed within LC3-positive vacuoles were also positive for SQSTM1, indicating a role for this adaptor in the autophagic capture of replication-deficient SchuS4 (Fig. 4C and D). A fraction of the LC3-positive bacteria did not colocalize with SQSTM1, suggesting the involvement of additional adaptors. Unlike the replication-deficient strains, very few wild-type SchuS4 were observed within LC3-positive compartments (n < 15 per experiment), so we could not significantly evaluate the association of SQSTM1 or ubiquitin with such a small population. Nonetheless, most LC3-positive SchuS4 observed were also labeled with ubiquitin and SQSTM1 (data not shown), suggesting a similar recognition process. Taken together, our results indicate that replication-deficient F. tularensis captured within autophagosomes are recognized by the ubiquitin system and the autophagy receptor SQSTM1.

Figure 4. Autophagy-targeted Francisella are tagged with ubiquitin and recruit the autophagy receptor SQSTM1. (A) Quantification of LC3-positive bacteria colocalizing with ubiquitin (Ub) at 16 h p.i. in BMMs expressing GFP-LC3. At least 30 LC3-positive bacteria per experiment were scored for ubiquitin colocalization in each condition. Data are means ± SD from three independent experiments. ND, not determined. (B) Representative confocal images of BMMs expressing GFP-LC3 infected for 16 h with either SchuS4 treated with chloramphenicol at 6 h p.i. or its isogenic ΔpurMCD or ΔdipA mutants. Samples were processed for immunofluorescence labeling of GFP (green) and ubiquitin (red), and stained with DAPI to label DNA and intracellular bacteria (blue). Magnified insets show single channel images of the boxed areas. White arrows indicate bacteria of interest. Scale bars: 10 or 2 μm. (C) Quantification of LC3-positive bacteria displaying SQSTM1 recruitment at 16 h p.i. in BMMs. At least 30 LC3-positive bacteria per experiment were scored for SQSTM1 colocalization in each condition. Data are means ± SD from three independent experiments. ND, not determined. (D) Representative confocal images of BMMs infected for 16 h with either SchuS4 treated with chloramphenicol at 6 h p.i. or its isogenic ΔpurMCD or ΔdipA mutants. Samples were processed for immunofluorescence labeling of LC3 (red) and SQSTM1 (green), and stained with DAPI to label DNA and intracellular bacteria (blue) at 16 h p.i. Magnified insets show single channel images of the boxed areas. White arrows indicate bacteria of interest. Scale bars: 10 or 2 μm.
To evaluate Francisella detection by ubiquitin and SQSTM1, we examined the accumulation of these proteins on intracellular bacteria, regardless of their colocalization with LC3-positive structures. Few wild-type and replication-deficient SchuS4 were decorated with either ubiquitin or SQSTM1 at 16 h p.i. (< 10%; Fig. 5A and B), indicating that most cytosolic Francisella are not detected via ubiquitination. Yet ~2-fold more replication-deficient bacteria than wild-type SchuS4 were associated with SQSTM1 (Fig. 5B), suggesting an enhanced autophagic recognition of replication-deficient bacteria. Despite these low numbers, the majority of ubiquitin-coated bacteria, whether replication-competent or not, colocalized with SQSTM1 (Fig. 5C and D), suggesting that ubiquitinated Francisella mostly proceed toward recognition by SQSTM1. Due to the low level recognition by SQSTM1 and because NBR1 possesses functional similarity to SQSTM1,12,13 we also examined the recruitment of NBR1 to intracellular Francisella and found similarly few numbers of NBR1-labeled bacteria (Fig. 5E). Yet, the majority of SQSTM1-positive bacteria were also labeled with NBR1 (Fig. 5F and G), indicating that both adaptors recognize the same population of bacteria, consistent with their cooperative function in promoting the capture of ubiquitin-tagged cargo.12 Hence, we used SQSTM1 as a marker of this bacterial population in the remainder of our studies. Interestingly, while the majority of SQSTM1-positive replication-deficient bacteria also recruited LC3, consistent with their targeting to autophagic capture, this was not the case of SQSTM1-positive wild-type SchuS4 bacteria, the majority of which did not recruit LC3 (Fig. 5E and F). Consistent with the near absence of LC3-positive SchuS4 (Fig. 4), this indicates that wild-type Francisella interfere with the autophagic cascade when targeted for autophagic capture via ubiquitination and SQSTM1 recruitment.

Figure 5. Detection of intracellular Francisella by the macrophage ubiquitin system, SQSTM1 and NBR1. (A and B) Quantification of (A) ubiquitin- and (B) SQSTM1-decorated bacteria at 16 h p.i. in BMMs. At least 100 bacteria per experiment were scored for colocalization with each marker in each condition. Data are means ± SD from three independent experiments. (*p < 0.05, 1-way ANOVA, Tukey’s post-test). (C) Quantification of ubiquitinated bacteria colocalizing with SQSTM1. BMMs were infected for 16 h and processed for immunofluorescence labeling of ubiquitin and SQSTM1, followed by DAPI staining. At least 30 ubiquitin-positive bacteria per experiment were scored for SQSTM1 recruitment in each condition. Data are means ± SD from three independent experiments. (D) Representative confocal images of BMMs infected for 16 h with either SchuS4, or its derivatives. Samples were processed for immunofluorescence labeling of ubiquitinated proteins (red), SQSTM1 (green) and stained with DAPI to label DNA and intracellular bacteria (blue). Magnified insets show single channel images of the boxed areas. White arrows indicate ubiquitin-positive, p62-positive bacteria. Scale bars: 10 or 2 μm. (E) Quantification of NBR1-decorated bacteria at 16 h p.i. in BMMs. At least 100 bacteria per experiment were scored for NBR1 recruitment in each condition. Data are means ± SD from three independent experiments. (*p < 0.05, 1-way ANOVA, Tukey’s post-test). (F) Quantification of SQSTM1-positive bacteria that recruit NBR1. BMMs were infected for 16 h and processed for immunofluorescence labeling of SQSTM1 and NBR1, followed by DAPI staining. At least 30 ubiquitin-positive bacteria per experiment were scored for SQSTM1 recruitment in each condition. Data are means ± SD from three independent experiments. (G) Representative confocal images of BMMs infected for 16 h with either SchuS4, or its derivatives. Samples were processed for immunofluorescence labeling of SQSTM1 (green), NBR1 (red) and stained with DAPI to label DNA and intracellular bacteria (blue). Magnified insets show single channel images of the boxed areas. White arrows indicate SQSTM1-positive, NBR1-positive bacteria. Scale bars: 10 or 2 μm. (H) Quantification of LC3-recruitment to SQSTM1-positive bacteria at 16 h p.i. in BMMs. At least 30 SQSTM1-positive bacteria per experiment were scored for LC3 recruitment in each condition. Data are means ± SD from three independent experiments. Asterisks indicate statistically significant differences (***p < 0.001, 1-way ANOVA, Tukey’s post-test). (I) Representative confocal images of BMMs infected for 16 h with SchuS4 and processed for immunofluorescence labeling of SQSTM1 (green) and LC3 (red), and stained with DAPI to label DNA and intracellular bacteria (blue). Magnified insets show single channel images of the boxed areas. Empty white arrows indicate SQSTM1-positive and LC3-negative bacteria; solid white arrows indicate SQSTM1-positive bacteria enclosed within LC3-positive vacuoles. Scale bars: 10 or 2 μm.
Capture of replication-impaired bacteria requires ATG5, LC3B and SQSTM1
Given the autophagic features of the capture process of replication-deficient Francisella, we examined the functional requirements of the autophagy-associated proteins SQSTM1, ATG5 and LC3B in the formation of these vacuoles. BMMs lacking either SQSTM1, ATG5 or LC3B were infected with either the wild-type or replication-deficient strains and the percentages of bacteria sequestered within LAMP1-positive vacuoles were quantified at 16 h p.i. Few to no wild-type SchuS4 bacteria were found within LAMP1-positive vacuoles in either Sqstm1−/−, Atg5f/f-Lyz-Cre, Lc3b−/− or their respective control BMMs (Fig. 6A–C). In Atg5f/f-Lyz-Cre BMMs, vacuolar recapture of replication-deficient bacteria was considerably diminished compared with control Atg5f/f BMMs (Fig. 6A), indicating that this process is dependent upon the autophagy protein ATG5. A significant reduction in the numbers of replication-deficient bacteria targeted into LAMP1 vacuoles was also observed in both Lc3b−/− and Sqstm1−/− BMMs compared with control C57BL/6 BMMs (Fig. 6B and C), consistent with the recruitment of these proteins to replication deficient bacteria (Figs. 3 and 4). Hence, vacuolar capture of replication-deficient Francisella requires the canonical autophagy-associated proteins ATG5, LC3B and the autophagy adaptor SQSTM1.

Figure 6. Vacuolar capture, but not intracellular killing, of replication-deficient Francisella is dependent on Atg5, Lc3b and Sqstm1. BMMs were infected with either SchuS4 or its derivatives, left untreated or treated with 100 nM BAF at 10 h p.i., and processed for immunofluorescence microscopy at 16 h p.i. (A–D) or enumeration of viable intracellular bacteria at 24 h p.i. (E–H). (A–D) Quantification of bacteria within endosomal vacuoles in (A) Atg5flox/flox and Atg5flox/flox-Lyz-Cre, (B) C57BL/6 and Lc3b−/−, (C) C57BL/6 and Sqstm1−/−, or (D) untreated and bafilomycin A1 (BAF)-treated C57BL/6 BMMs. Infection and data analysis of Lc3b−/− (B) and Sqstm1−/− (C) BMMs share the same set of C57BL/6 controls because these experiments were done simultaneously. Samples were processed for immunofluorescence labeling of bacteria and LAMP1-positive membranes, and the numbers of bacteria enclosed within LAMP1-positive compartments were scored. At least 100 bacteria per experiment were scored for each condition. Data are means ± SD from three (A and D) or four (B and C) independent experiments. Asterisks indicate statistically significant differences (*p < 0.05, **p < 0.01, ***p < 0.001, two-tailed unpaired Student’s t-test). (E–H) Intracellular survival of SchuS4 or its derivatives in (E) Atg5flox/flox and Atg5flox/flox-Lyz-Cre, (F) C57BL/6 and Lc3b−/−, (G) C57BL/6 and Sqstm1−/− or (H) untreated and BAF-treated C57BL/6 BMMs. Intracellular bacteria were enumerated from CFUs at 24 h p.i. Data are means ± SD from a representative experiment performed in triplicate out of two independent repeats.
Given that autophagy is a degradative process, we sought to examine whether the observed loss of bacterial viability (Figs. 1A and 2A) concomitant with autophagic capture of replication-deficient strains requires autophagy proteins. To our surprise, the absence of ATG5 in BMMs did not restore viability of the replication-deficient strains, and had little effect on growth of wild-type bacteria (Fig. 6E; Fig. S1). The absence of LC3B or SQSTM1 in BMMs had similarly little effect on the viability of intracellular bacteria at 24 h p.i. (Fig. 6F and G). Altogether, these results suggest that formation of the autophagic vacuoles does not account for bacterial killing. Consistently, treatment of infected BMMs with bafilomycin A1, which blocks autophagosome maturation,55 significantly reduced the numbers of replication-deficient bacteria targeted into LAMP1 vacuoles but did not restore viability of replication-deficient bacteria at 24 h p.i. (Fig. 6D and H), indicating that autophagosome maturation into degradative autolysosomes does not constitute a significant bactericidal step in the decreased viability of replication-deficient strains.
Autophagic clearance of replication-deficient Francisella dying in the macrophage cytosol
To account for the lack of restored viability of replication-deficient Francisella when autophagosome formation is decreased (Fig. 6E–G) or autophagosome maturation is inhibited (Fig. 6H), we hypothesized that a significant fraction of these bacteria die in the cytosol prior to autophagic capture. To test this hypothesis, we designed a fluorescence microscopy assay based on the membrane-impermeant nucleic acid dye propidium iodide (PI) that specifically labels either cytosolic or all intracellular bacteria with compromised membranes as an indicator of viability (see Experimental Procedures). Intracellular killing of 99.9% of cytosolic SchuS4 with tetracycline (Tc, 50 µg/ml for 6 h; data not shown) led to the detection of 54 ± 1.6% of PI-positive bacteria at 8 h p.i. (Fig. 7A), indicating that about half of Tc-killed bacteria had detectable damaged membranes under our experimental conditions. Comparatively, ~20% of wild-type and replication-deficient strains were PI-positive at 8 h p.i. (Fig. 7A), indicating that most cytosolic bacteria have intact membranes at this stage, yet some membrane damage occur in a small fraction of cytosolic bacteria, regardless of their replication competency. At 16 h p.i., while only 13 ± 5.3% of cytosolic SchuS4 were PI-positive, the percentages of PI-positive replication-deficient bacteria ranged from 33 ± 4.0% [SchuS4(Cm)] to 47 ± 6.4% (∆dipA), indicating high numbers of damaged cytosolic bacteria that are replication deficient (Fig. 7B). This intracellular viability assay may however underrepresent the percentage of non-viable bacteria, since the fraction of PI-negative, captured bacteria could also include nonviable organisms whose membranes have not been sufficiently compromised to be detected by PI staining. Nonetheless, these results clearly demonstrate that a large fraction of replication-deficient strains die in the macrophage cytosol between 8 and 16 h p.i.

Figure 7. Survival-impaired Francisella are recognized by SQSTM1 in the macrophage cytosol. (A and B) Quantification of cytosolic bacteria for PI labeling at 8 h (A) and 16 h (B) p.i. Infected BMMs were either left untreated or treated with tetracycline at 2 h p.i. to kill cytosolic Francisella and then subjected to an intracellular viability assay to quantify the percentage of dead cytosolic bacteria. At least 100 cytosolic bacteria per experiment were scored for PI labeling in each condition. Data are means ± SD from three independent experiments. (*p < 0.05, ***p < 0.001, two-tailed unpaired Student’s t-test). (C) Quantification of the percentage of cytosolic SQSTM1-positive bacteria at 16 h p.i. Infected BMMs were subjected to the phagosome integrity assay, followed by immunofluorescence labeling of SQSTM1. SQSTM1-positive bacteria were scored for vacuolar or cytosolic localization. At least 30 bacteria per experiment were scored for each condition. Data are means ± SD from three independent experiments. (D) Quantification of cytosolic SQSTM1-positive bacteria for PI labeling at 16 h p.i. Infected BMMs were subjected to an intracellular viability assay, followed by immunofluorescence labeling of SQSTM1. Cytosolic SQSTM1-positive bacteria were scored for PI labeling. At least 30 bacteria per experiment were scored for each condition. Data are means ± SD from three independent experiments. (E) Representative confocal images of BMMs infected with either SchuS4 or its derivatives and subjected to an intracellular viability assay at 16 h p.i. Cytosolic bacteria (green) with compromised membranes are labeled with PI (red) after digitonin permeabilization. All bacteria (blue) are detected after saponin permeabilization. Magnified insets show single channel images of the boxed areas. White arrows indicate bacteria of interest. Scale bars: 10 or 2 μm. (F) Representative confocal images of BMMs infected for 16 h with either SchuS4 or its derivatives and processed for an intracellular viability assay followed by immunofluorescence labeling of SQSTM1. SQSTM1-positive (green) cytosolic bacteria (blue) with compromised membranes are labeled with PI (red). Magnified insets show single channel images of the boxed areas. White arrows indicate bacteria of interest. Scale bars: 10 or 2 μm.
Since a large fraction of replication-deficient bacteria die in the macrophage cytosol, we sought to determine whether the macrophage autophagic machinery targeted cytosolic bacteria. Using SQSTM1 recruitment as a readout of autophagic targeting, we found that > 75% of SQSTM1-positive bacteria were cytosolic (Fig. 7C), indicating that not only is autophagy detecting cytosolic Francisella but also that SQSTM1 is recruited directly to the bacteria rather than to vacuolar membranes. Additionally, about 25% of the cytosolic, SQSTM1-positive bacteria were labeled with PI (Fig. 7D), indicating that dead cytosolic bacteria are targeted to autophagy. While the fraction of PI-positive, replication-deficient bacteria within the SQSTM1-positive population did not vary significantly from that within the cytosolic population (Fig. 7B and D), the SQSTM1-positive SchuS4 population was enriched in PI-positive bacteria, compared with the whole cytosolic population (Fig. 7B and D), indicating that damaged wild-type bacteria are preferentially targeted for autophagic capture. Consistent with the SQSTM1-positive population, 28 ± 6.1% to 46 ± 2.3% of LC3-positive, replication-deficient bacteria within autophagosomes had compromised membranes (Fig. S2) and these percentages increased further in the population within LAMP1-positive, maturing or matured autolysosomes (Fig. S2), confirming bacterial degradation along the autophagic pathway. Taken together, our results demonstrate that a large fraction of replication-deficient Francisella die within the cytosol and are targeted to autophagy for clearance, which likely accounts for the limited bactericidal action of the autophagic process.
Discussion
Like other cytosol-adapted bacterial pathogens, F. tularensis resides and proliferates for extended lengths of time within this compartment. With the exception of a belated, autophagy-mediated response only observed in murine primary macrophages,33 this bacterium dwells in the cytosol without obvious recognition and capture by the host autophagic machinery, suggesting that it interferes with this process. It has been reported that Francisella downregulates several genes required for nucleation and elongation of autophagy membranes during infection of human monocytes,56,57 yet there is so far no direct evidence that this bacterium inhibits autophagy. We have recently identified a gene, dipA, in the SchuS4 locus FTT0369c, the deletion of which specifically impairs multiplication in the cytosol.42 By further characterizing the intracellular fate of a ∆dipA mutant in an attempt to gain clues about DipA function, we have uncovered its eventual autophagic capture following a lack of replication. The possible interpretation that DipA specifically prevents autophagy was, however, ruled out by the similar autophagic capture of an auxotroph mutant (∆purMCD) or the wild-type SchuS4 strain treated with the protein synthesis inhibitor chloramphenicol. While chloramphenicol-treated SchuS4 are likely deficient in DipA expression, the ∆purMCD mutant expressed wild-type levels of this protein (data not shown), yet could not avoid autophagic capture, indicating that DipA deficiency alone is not sufficient for autophagic capture. With the common phenotypes of these strains being their inability to proliferate in the cytosol, these results instead indicate that replication deficiency leads to autophagic capture of Francisella. Consistently, previous work by Fuller et al. has reported autophagic capture of another, intracellular growth-deficient mutant in the F. tularensis live vaccine strain (LVSΔripA) at 24 h p.i.,39 lending support to the lack of gene specificity in this process.
How replication deficiency leads to autophagic capture is unclear, yet the reduced intracellular viability of all replication-deficient strains, which resulted in a large part from death in the macrophage cytosol more than from killing within autolysosomes, suggests that nonviable Francisella cannot avoid autophagic recognition and capture. While our results highlight autophagy as a clearance more than a bactericidal process, they also indicate that either the lack of DipA, a major physiological defect such as purine auxotrophy, or general inhibition of protein synthesis affects the viability of Francisella within the macrophage cytosol. Whether bacteria die due to lethal physiological defects, or via active killing by macrophage bactericidal mechanisms, remains to be clarified, although it is conceivable that bacteria with major physiological defects cannot survive within the macrophage cytosol. The ∆dipA mutant is growth-proficient in rich medium in vitro,42 but any survival defect under nutrient-limiting and stress-inducing conditions similar to those potentially encountered within the macrophage cytosol needs to be examined.
In investigating the mechanism of autophagy targeting of replication-deficient Francisella, we detected bacterial tagging with ubiquitin and recruitment of the autophagy receptors SQSTM1 and NBR1. Cargo recognition in antibacterial autophagy is not fully understood, yet it clearly involves recruitment of the autophagy adaptors SQSTM1, CALCOCO2/NDP52, OPTN/optineurin and NBR1.15,16,18,19,54 While recruitment of other adaptors to ubiquitinated Francisella remains to be examined, the decreased formation of autophagic vacuoles in SQSTM1-deficient macrophages and its recruitment to the majority of ubiquitinated bacteria indicates that SQSTM1 plays a major role in autophagic recognition of this pathogen, likely in cooperation with NBR1. Despite a clear involvement of the ubiquitin-SQSTM1-LC3 pathway in Francisella capture, a surprisingly low percentage (~5%) of all intracellular Francisella were ubiquitinated and recruited SQSTM1 or NBR1 at any given time (Fig. 5A and B and data not shown). This could reflect a transient stage of a dynamic process that rapidly delivers targeted bacteria to the lysosomal compartment, therefore favoring the detection of later compartments such as LC3- and LAMP1-positive vacuoles. Nonetheless, the role of ubiquitin- and/or SQSTM1-independent pathway cannot be ruled out. The involvement of TECPR1, which facilitates ATG12–ATG5 conjugation required for selective autophagy of bacterial pathogens, remains possible, in concert with the Ub-SQSTM1-LC3 pathway, although whether TECPR1 is involved in cargo recognition remains unclear.58 It is unlikely, however, that alternative pathways involving the lipid second messenger diacylglycerol59 and the cytosolic lectin LGALS8/galectin 860 play a role in targeting replication-deficient Francisella, since these molecules detect damaged vacuolar membranes upon entry into the cytosol whereas replication-deficient Francisella are captured many hours after phagosomal disruption.
The demonstration of autophagic targeting and capture of replication-impaired Francisella also provides clues as to how this pathogen may normally interfere with autophagy to ensure its successful proliferation in the cytosol. First, the low level of ubiquitin tagging of cytosolic bacteria, whether replication-competent or deficient, suggests that Francisella efficiently avoids such recognition mechanisms. This is supported by the fact that the SQSTM1-positive SchuS4 cytosolic population was enriched in membrane-damaged bacteria (Fig. 7D) in comparison with the cytosolic population (Fig. 7B), which indicates that autophagy preferentially targets the few damaged wild-type bacteria. Second, numbers of wild-type SchuS4 bacteria decorated with ubiquitin were low and similar to replication-deficient strains (Fig. 5A), yet fewer wild-type SchuS4 were recognized by SQSTM1 and NBR1 than replication-impaired bacteria (Fig. 5B and E) and even fewer of the SQSTM1-associated wild-type SchuS4 progressed to LC3 recruitment (Fig. 5H). Given that SQSTM1 or NBR1 directly bind to LC3 via their LC3-interacting region (LIR),11,12 SchuS4 may impair such interactions to block efficient LC3 recruitment and evade targeting to autophagic membranes. Hence, in addition to avoiding autophagic recognition in the cytosol, SchuS4 may also interfere with autophagic capture by preventing phagophore assembly or elongation, or delivery to autophagosome membranes. Further studies aimed at dissecting the molecular mechanisms of autophagy evasion by Francisella are necessary to confirm our observations, which will not only shed light on this bacterium’s pathogenic traits, but also on the molecular events involved in antibacterial autophagy.
Materials and Methods
Bacterial strains and culture conditions
The prototypic Type A virulent strain, F. tularensis subsp tularensis SchuS4 was obtained from Rick Lyons (University of New Mexico, Albuquerque, NM, USA). The SchuS4∆dipA (∆FTT0369c) and SchuS4∆fevR (∆FTT0383) mutants have been described previously.42 Bacteria were grown on modified Mueller-Hinton (MMH) agar plates [Mueller-Hinton medium supplemented with 0.1% glucose (Sigma, G8270), 0.025% ferric pyrophosphate (Sigma Aldrich, P6526) and 2% IsoVitaleX (Becton Dickinson, 211875)] for 3 d at 37°C under 7% CO2. For allelic replacement, MMH medium was supplemented with either 10 µg/ml kanamycin (Sigma, K0254) or 8% sucrose (Sigma, S9370). All manipulations of F. tularensis strain SchuS4 and its derivatives were performed in a Biosafety Level 3 facility according to standard operating procedures approved by the Rocky Mountain Laboratories Institutional Biosafety Committee.
Construction of an in-frame deletion purMCD mutant of SchuS4
A SchuS4ΔpurMCD mutant was generated using the SacB-assisted allelic replacement suicide vector pJC84, as described previously.42 Deletion of the purMCD (FTT0893-FTT0894) loci was designed to preserve the integrity of downstream genes and avoid any polar effects. To engineer an in-frame deletion of purMCD, a 5′-fragment containing 1040 bp upstream of the start codon of purM (FTT0893), its start and the first 3 codons was generated by PCR amplification using the primers JC965 and JC966 and a 3′ fragment containing 960 bp downstream of the stop codon and the last 4 codons of purCD (FTT0984) using primers JC967 and JC968 (Table 1). Both hemi-fragments were fused by overlap extension PCR using primers JC965 and JC968. The resulting 2027 bp fragment containing a residual open reading frame of the first 3 codons of purM and the last 4 codons of purCD was cloned into the BamHI and SalI sites of pJC84 using the In-Fusion® PCR Cloning System (Clontech, 639650) and was fully sequenced.
Table 1. Primers used in this study.
| Name | Sequence (5′ → 3′) |
|---|---|
|
pJC84 chromosomal detection |
|
| JC420 |
CTAGCTAGCAGGAGACATGAACGATGAACATC |
| JC427 |
GGGACGTCGGATTCACCTTTATGTTGATAAG |
| JC428 |
GGGACGTCGATTAAGCATTGGTAACTGTCAGACC |
| JC589 |
ATCAGCTCACTCAAAGGCGG |
|
purMCD deletion |
|
| JC965 |
CGGTACCCGGGGATCCGAGAGATTAGCAACTCAAGTTC |
| JC966 |
TAAGCCTGCCATTTTATTTCC |
| JC967 |
AAAATGGCAGGCTTACAGGAGCTTAAATAAATAATGTCTAAGC |
| JC968 |
TATCCATACAGTCGACTAACTGGTACACCTAATACTGGTA |
| JC972 |
CAGTGTGCTCAGGAGTTGAA |
| JC973 |
TTACTAGGCTTATCTGAGCA |
| JC974 |
GCTGGTAGTAGAATTATGGT |
| JC975 | CGCTGGCATCTGTACTATTG |
To perform allelic replacement in the chromosome of Schu S4, electrocompetent bacteria were prepared and electroporated with recombinant pJC84 plasmid DNA as previously described.42 Kanamycin-resistant merodiploid colonies were tested for integration of the allelic replacement plasmid, using colony PCR with primers JC420 and JC427 (to amplify a 1.5 kb internal fragment of sacB) or JC589 and JC428 (to amplify a 900 bp fragment of the pJC84 backbone). Independent clones were then subjected to sucrose counter selection as previously described,42 to isolate clones that have undergone allelic replacement. The presence of the deleted allele and allelic replacement within the correct chromosomal locus were verified by PCR using primers JC972 and JC973 and primers JC974 and JC975 (Table 1) for the purMCD deletion, and primers JC420 and JC427 for the loss of the sacB gene. Independent clones with the correct in-frame purMCD deletion were isolated and used for further studies.
Macrophage culture and infection
Murine bone marrow-derived macrophages (BMMs) were harvested from C57BL/6J (Jackson Laboratories, 000664), Atg5flox/flox and Atg5flox/flox-Lyz-Cre,61 Lc3b−/−62 or Sqstm1−/− 63 and differentiated as described.32 GFP-LC3 transgenic mice were provided by RIKEN BRC through the National Bio-Resource Project of the MEXT, Japan. All animal rearing, handling and experimental methods were conducted under protocols approved by the RML Institutional Animal Care and Use Committee (IACUC). Human monocyte-derived macrophages (MDMs) were generated from peripheral blood monocytes subjected to apheresis and enriched by density centrifugation using Ficoll-Paque (GE Healthcare, 17-5442-03) and by negative selection using Dynabeads Untouched Human Monocytes Kit (Life Technologies, 113.50D) according to the manufacturer’s instructions. Mononuclear cells were seeded at a cell density of 2 × 105/well (24-well plate) in RPMI medium (Life Technologies, 21870) supplemented with 10% fetal bovine serum (FBS; Life Technologies, 16000-044), 2 mM L-glutamine (Life Technologies, 25030), nonessential amino acids (Life Technologies, 11140) and 50 ng/ml recombinant human macrophage colony stimulating factor (M-CSF; PeproTech Inc., Rocky 300-25). Medium was replenished on day 3 and day 5 of culture, and MDMs were used for infections on day 6. Anonymized volunteers provided the human blood cells, after written informed consent, under protocols approved by the NIH Clinical Center Institutional Review Board.
Immediately prior to infection of macrophages, a few colonies from a freshly streaked plate were resuspended in MMH broth and the OD600nm was measured to estimate bacterial numbers. Bacteria were diluted to the appropriate multiplicity of infection (MOI) in either BMM or MDM media and 0.5 ml of bacterial suspension was added to chilled cells. Macrophage infections were performed as described32 at an applied MOI of 25 (MDM) or 50 (BMM). Infections with SchuS4ΔpurMCD and the corresponding control, SchuS4 (1% FBS), were performed in complete media containing 1% FBS. When required, SchuS4-infected BMMs were treated with 10 μg/mL chloramphenicol (Sigma, C1863) at 6 h p.i. or 50 μg/mL tetracycline (Sigma-Aldrich, 87128) at 2 h p.i. Autophagosome maturation was inhibited by treating BMMs with 100 nM bafilomycin A1 (AG Scientific, B-1183) at 10 h p.i. and maintained throughout the experiment.
Determination of bacterial intracellular growth
Intracellular growth of SchuS4 and its derivatives was monitored by determining the number of colony-forming units (CFU) recovered from lysed macrophages as described previously.32 The number of viable intracellular bacteria per well was determined in triplicate for each time point.
Fluorescence microscopy
Infected BMMs on 12 mm glass coverslips were processed for immunofluorescence labeling as described previously.32 Primary antibodies used were mouse monoclonal anti-F. tularensis LPS (US Biologicals, F6070-02), rat monoclonal anti-mouse LAMP1 (clone 1D4B, developed by J.T. August and obtained from the Developmental Studies Hybridoma Band; developed under the auspices of the NICHD and maintained by The University of Iowa, Iowa City, IA), rabbit polyclonal anti-human LAMP1 (Novus Biologicals, NB600-956), rabbit polyclonal anti-GFP (Life Technologies, A6455), mouse monoclonal anti-GFP (Life Technologies, A11120), mouse monoclonal anti- mono- and poly-ubiquitinylated conjugates (Enzo Life Sciences, PW8810), guinea pig polyclonal anti-SQSTM1 (Progen Biotechnik, GP62-C), mouse monoclonal anti-NBR1 (AbCam, ab55474) and mouse monoclonal anti-LC3 (MBL International, M152-3). Secondary antibodies used were Alexa Fluor™ 488-conjugated goat anti-mouse (Life Technologies, A11029), Alexa Fluor™ 405-conjugated goat anti-mouse (Life Technologies, A31553), cyanin-5-conjugated donkey anti-rabbit (Jackson ImmunoResearch, 711-175-152), Alexa Fluor™ 568-conjugated goat anti-rat (Life Technologies, A11077), Alexa Fluor™ 488-conjugated donkey anti-rat (Life Technologies, A21208), Alexa Fluor™ 568-conjugated goat anti-rabbit (Life Technologies, A11036), Alexa Fluor™ 488-conjugated goat anti-rabbit (Life Technologies, A11034), Alexa Fluor™ 488-conjugated goat anti-guinea pig (Life Technologies, A11073). When required, macrophage nuclei and bacteria were stained with DAPI (Life Technologies, D3571) for 10 min at room temperature after incubation with secondary antibodies. Samples were observed on a Carl Zeiss Axio Imager epi-fluorescence microscope equipped with a Plan-APOCHROMAT 63×/1.4 objective for quantitative analysis, or Carl Zeiss LSM 710 confocal laser scanning microscope for image acquisition. Confocal images of 1024 × 1024 pixels were acquired and assembled using Adobe Photoshop CS3.
Transmission electron microscopy
Infected BMMs on 12 mm Aclar coverslips were processed as described previously.32 Sections were viewed in a Hitachi H7500 transmission electron microscope at 80 kV. Images were acquired with a Hamamatsu 2 K × 2 K bottom mount AMT digital camera and assembled in Adobe Photoshop CS3.
Phagosome integrity assay
To assess the presence and integrity of phagosomal membranes around intracellular F. tularensis, phagosomal integrity assays were performed as described previously.32 Briefly, infected BMMs were washed three times with KHM buffer (110 mM potassium acetate, 20 mM Hepes, 2 mM MgCl2, pH 7.3), the plasma membrane was selectively permeabilized with 50 μg/ml digitonin (Sigma, D141) in KHM buffer for 1 min at room temperature, immediately washed three times with KHM buffer and incubated for 12 min at 37°C with rabbit anti-calnexin (specific for the cytoplasmic-facing C-terminal tail) and Alexa Fluor™ 488-conjugated mouse anti-F. tularensis LPS antibodies to label the endoplasmic reticulum of permeabilized cells and accessible cytosolic bacteria, respectively. BMMs were then washed with PBS, fixed with 3% paraformaldehyde for 20 min at 37°C, and incubated in 50 mM NH4Cl in PBS to quench free aldehydes. All host cell membranes were permeabilized with 0.1% saponin-10% horse serum in PBS for 30 min at room temperature. Thereafter, bound anti-calnexin antibodies were detected using cyanin 5-conjugated donkey anti-rabbit antibodies and all intracellular bacteria were labeled using Alexa Fluor™ 568-conjugated mouse anti-Francisella antibodies. To evaluate the intracellular localization of SQSTM1-positive F. tularensis, infected BMMs were treated as described above with the following exceptions: Alexa Fluor™ 568-conjugated mouse anti-F. tularensis LPS antibodies in KHM buffer were used to detect cytosolic bacteria; following fixation and permeabilization, SQSTM1 was detected using guinea pig anti-SQSTM1 and Alexa Fluor™ 488-conjugated anti-guinea pig antibodies, and all intracellular bacteria were labeled using mouse anti-F. tularensis LPS and Alexa Fluor™ 405-conjugated anti-mouse antibodies. Samples were observed on a Carl Zeiss Axio Imager epifluorescence microscope equipped with a Plan-APOCHROMAT 63×/1.4 objective for quantitative analysis.
Determination of intracellular bacterial viability
To evaluate the viability of cytosolic F. tularensis, phagosomal integrity assays were adapted to allow cytosolic delivery of the cell-impermeant nucleic acid dye PI. Infected BMMs were incubated for 12 min at 37°C with Alexa Fluor™ 488-conjugated mouse anti-F. tularensis LPS antibodies and 2.6 μM PI (Life Technologies, L7007) in KHM buffer to label accessible cytosolic bacteria and compromised bacteria, respectively, in permeabilized cells. Following fixation and permeabilization of all host cell membranes, all intracellular bacteria were labeled using mouse anti-Francisella antibodies and detected using Alexa Fluor™ 405-conjugated secondary antibodies. To evaluate the viability of cytosolic SQSTM1-positive F. tularensis, infected BMMs were incubated for 12 min at 37°C with Alexa Fluor™ 647-conjugated mouse anti-F. tularensis LPS antibodies and 2.6 μM PI in KHM buffer. Following fixation and permeabilization, SQSTM1 was detected using guinea pig anti-SQSTM1 and Alexa Fluor™ 488-conjugated anti-guinea pig antibodies. To assess the viability of vacuolar bacteria, infected BMMs were permeabilized with 100 μg/ml digitonin in KHM buffer for 1 min at room temperature, immediately washed three times with KHM buffer and incubated for 12 min at 37°C with 2.6 μM PI in KHM buffer. After fixation and quenching, all intracellular bacteria were labeled using mouse anti-Francisella antibodies and Alexa Fluor™ 405-conjugated anti-mouse secondary antibodies. The macrophage endosomal membranes were labeled with rat monoclonal anti-mouse LAMP1 antibodies and detected using Alexa Fluor™ 488-conjugated anti-rat secondary antibodies; GFP-LC3 signal was enhanced with rabbit anti-GFP and Alexa Fluor™ 488-conjugated anti-rabbit antibodies.
Statistical analysis
All data are given as mean ± SD from three independent experiments. Statistical analyses were performed using either a one-way ANOVA with Tukey post-test or an unpaired, two-tailed Student t-test. *p < 0.05, **p < 0.01 and ***p < 0.001.
Supplementary Material
Acknowledgments
We thank Preeti Malik-Kale and Olivia Steele-Mortimer for providing human peripheral blood monocytes. This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases.
Glossary
Abbreviations:
- ATG5
autophagy-related 5
- BAF
bafilomycin A1
- BMM
murine bone marrow-derived macrophages
- Cm
chloramphenicol
- FCV
Francisella-containing vacuole
- GFP
green fluorescent protein
- LAMP1
lysosomal-associated membrane protein 1
- LC3
microtubule-associated protein 1 light chain 3
- MDM
human blood monocyte-derived macrophages
- NBR1
neighbor of BRCA1 gene 1
- CALCOCO2/NDP52
nuclear dot protein 52
- PI
propidium iodide
- SQSTM1/p62
sequestosome 1
- TEM
transmission electron microscopy
- Ub
ubiquitin
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/20808
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/20808
References
- 1.Alonso A, García-del Portillo F. Hijacking of eukaryotic functions by intracellular bacterial pathogens. Int Microbiol. 2004;7:181–91. [PubMed] [Google Scholar]
- 2.Huynh KK, Grinstein S. Regulation of vacuolar pH and its modulation by some microbial species. Microbiol Mol Biol Rev. 2007;71:452–62. doi: 10.1128/MMBR.00003-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–66. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 4.Birmingham CL, Brumell JH. Autophagy recognizes intracellular Salmonella enterica serovar Typhimurium in damaged vacuoles. Autophagy. 2006;2:156–8. doi: 10.4161/auto.2825. [DOI] [PubMed] [Google Scholar]
- 5.Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J Biol Chem. 2006;281:11374–83. doi: 10.1074/jbc.M509157200. [DOI] [PubMed] [Google Scholar]
- 6.Rich KA, Burkett C, Webster P. Cytoplasmic bacteria can be targets for autophagy. Cell Microbiol. 2003;5:455–68. doi: 10.1046/j.1462-5822.2003.00292.x. [DOI] [PubMed] [Google Scholar]
- 7.Py BF, Lipinski MM, Yuan J. Autophagy limits Listeria monocytogenes intracellular growth in the early phase of primary infection. Autophagy. 2007;3:117–25. doi: 10.4161/auto.3618. [DOI] [PubMed] [Google Scholar]
- 8.Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of intracellular Shigella from autophagy. Science. 2005;307:727–31. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- 9.Dupont N, Lacas-Gervais S, Bertout J, Paz I, Freche B, Van Nhieu GT, et al. Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe. 2009;6:137–49. doi: 10.1016/j.chom.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 10.Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–40. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 11.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–45. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 12.Kirkin V, Lamark T, Johansen T, Dikic I. NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy. 2009;5:732–3. doi: 10.4161/auto.5.5.8566. [DOI] [PubMed] [Google Scholar]
- 13.Kirkin V, Lamark T, Sou YS, Bjørkøy G, Nunn JL, Bruun JA, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33:505–16. doi: 10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 14.Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci U S A. 2008;105:20567–74. doi: 10.1073/pnas.0810611105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mostowy S, Sancho-Shimizu V, Hamon MA, Simeone R, Brosch R, Johansen T, et al. p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J Biol Chem. 2011;286:26987–95. doi: 10.1074/jbc.M111.223610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–33. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perrin AJ, Jiang X, Birmingham CL, So NS, Brumell JH. Recognition of bacteria in the cytosol of Mammalian cells by the ubiquitin system. Curr Biol. 2004;14:806–11. doi: 10.1016/j.cub.2004.04.033. [DOI] [PubMed] [Google Scholar]
- 18.Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10:1215–21. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 19.Cemma M, Kim PK, Brumell JH. The ubiquitin-binding adaptor proteins p62/SQSTM1 and NDP52 are recruited independently to bacteria-associated microdomains to target Salmonella to the autophagy pathway. Autophagy. 2011;7:341–5. doi: 10.4161/auto.7.3.14046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol. 2009;183:5909–16. doi: 10.4049/jimmunol.0900441. [DOI] [PubMed] [Google Scholar]
- 21.Yoshikawa Y, Ogawa M, Hain T, Yoshida M, Fukumatsu M, Kim M, et al. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol. 2009;11:1233–40. doi: 10.1038/ncb1967. [DOI] [PubMed] [Google Scholar]
- 22.Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhães JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 23.Birmingham CL, Canadien V, Gouin E, Troy EB, Yoshimori T, Cossart P, et al. Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy. 2007;3:442–51. doi: 10.4161/auto.4450. [DOI] [PubMed] [Google Scholar]
- 24.Berón W, Gutierrez MG, Rabinovitch M, Colombo MI. Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect Immun. 2002;70:5816–21. doi: 10.1128/IAI.70.10.5816-5821.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amer AO, Swanson MS. Autophagy is an immediate macrophage response to Legionella pneumophila. Cell Microbiol. 2005;7:765–78. doi: 10.1111/j.1462-5822.2005.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schnaith A, Kashkar H, Leggio SA, Addicks K, Krönke M, Krut O. Staphylococcus aureus subvert autophagy for induction of caspase-independent host cell death. J Biol Chem. 2007;282:2695–706. doi: 10.1074/jbc.M609784200. [DOI] [PubMed] [Google Scholar]
- 27.Pizarro-Cerdá J, Méresse S, Parton RG, van der Goot G, Sola-Landa A, Lopez-Goñi I, et al. Brucella abortus transits through the autophagic pathway and replicates in the endoplasmic reticulum of nonprofessional phagocytes. Infect Immun. 1998;66:5711–24. doi: 10.1128/iai.66.12.5711-5724.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Starr T, Child R, Wehrly TD, Hansen B, Hwang S, López-Otin C, et al. Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe. 2012;11:33–45. doi: 10.1016/j.chom.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodriguez AR, Yu JJ, Murthy AK, Guentzel MN, Klose KE, Forsthuber TG, et al. Mast cell/IL-4 control of Francisella tularensis replication and host cell death is associated with increased ATP production and phagosomal acidification. Mucosal Immunol. 2011;4:217–26. doi: 10.1038/mi.2010.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hall JD, Woolard MD, Gunn BM, Craven RR, Taft-Benz S, Frelinger JA, et al. Infected-host-cell repertoire and cellular response in the lung following inhalation of Francisella tularensis Schu S4, LVS, or U112. Infect Immun. 2008;76:5843–52. doi: 10.1128/IAI.01176-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fortier AH, Green SJ, Polsinelli T, Jones TR, Crawford RM, Leiby DA, et al. Life and death of an intracellular pathogen: Francisella tularensis and the macrophage. Immunol Ser. 1994;60:349–61. [PubMed] [Google Scholar]
- 32.Chong A, Wehrly TD, Nair V, Fischer ER, Barker JR, Klose KE, et al. The early phagosomal stage of Francisella tularensis determines optimal phagosomal escape and Francisella pathogenicity island protein expression. Infect Immun. 2008;76:5488–99. doi: 10.1128/IAI.00682-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Checroun C, Wehrly TD, Fischer ER, Hayes SF, Celli J. Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc Natl Acad Sci U S A. 2006;103:14578–83. doi: 10.1073/pnas.0601838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clemens DL, Lee BY, Horwitz MA. Virulent and avirulent strains of Francisella tularensis prevent acidification and maturation of their phagosomes and escape into the cytoplasm in human macrophages. Infect Immun. 2004;72:3204–17. doi: 10.1128/IAI.72.6.3204-3217.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Santic M, Molmeret M, Abu Kwaik Y. Modulation of biogenesis of the Francisella tularensis subsp. novicida-containing phagosome in quiescent human macrophages and its maturation into a phagolysosome upon activation by IFN-gamma. Cell Microbiol. 2005;7:957–67. doi: 10.1111/j.1462-5822.2005.00529.x. [DOI] [PubMed] [Google Scholar]
- 36.Golovliov I, Baranov V, Krocova Z, Kovarova H, Sjöstedt A. An attenuated strain of the facultative intracellular bacterium Francisella tularensis can escape the phagosome of monocytic cells. Infect Immun. 2003;71:5940–50. doi: 10.1128/IAI.71.10.5940-5950.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang MT, Mortensen BL, Taxman DJ, Craven RR, Taft-Benz S, Kijek TM, et al. Deletion of ripA alleviates suppression of the inflammasome and MAPK by Francisella tularensis. J Immunol. 2010;185:5476–85. doi: 10.4049/jimmunol.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fuller JR, Kijek TM, Taft-Benz S, Kawula TH. Environmental and intracellular regulation of Francisella tularensis ripA. BMC Microbiol. 2009;9:216. doi: 10.1186/1471-2180-9-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fuller JR, Craven RR, Hall JD, Kijek TM, Taft-Benz S, Kawula TH. RipA, a cytoplasmic membrane protein conserved among Francisella species, is required for intracellular survival. Infect Immun. 2008;76:4934–43. doi: 10.1128/IAI.00475-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edwards JA, Rockx-Brouwer D, Nair V, Celli J. Restricted cytosolic growth of Francisella tularensis subsp. tularensis by IFN-gamma activation of macrophages. Microbiology. 2010;156:327–39. doi: 10.1099/mic.0.031716-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Akimana C, Al-Khodor S, Abu Kwaik Y. Host factors required for modulation of phagosome biogenesis and proliferation of Francisella tularensis within the cytosol. PLoS One. 2010;5:e11025. doi: 10.1371/journal.pone.0011025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wehrly TD, Chong A, Virtaneva K, Sturdevant DE, Child R, Edwards JA, et al. Intracellular biology and virulence determinants of Francisella tularensis revealed by transcriptional profiling inside macrophages. Cell Microbiol. 2009;11:1128–50. doi: 10.1111/j.1462-5822.2009.01316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmerk CL, Duplantis BN, Howard PL, Nano FE. A Francisella novicida pdpA mutant exhibits limited intracellular replication and remains associated with the lysosomal marker LAMP-1. Microbiology. 2009;155:1498–504. doi: 10.1099/mic.0.025445-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lindgren H, Golovliov I, Baranov V, Ernst RK, Telepnev M, Sjöstedt A. Factors affecting the escape of Francisella tularensis from the phagolysosome. J Med Microbiol. 2004;53:953–8. doi: 10.1099/jmm.0.45685-0. [DOI] [PubMed] [Google Scholar]
- 45.Bönquist L, Lindgren H, Golovliov I, Guina T, Sjöstedt A. MglA and Igl proteins contribute to the modulation of Francisella tularensis live vaccine strain-containing phagosomes in murine macrophages. Infect Immun. 2008;76:3502–10. doi: 10.1128/IAI.00226-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Santic M, Molmeret M, Klose KE, Jones S, Kwaik YA. The Francisella tularensis pathogenicity island protein IglC and its regulator MglA are essential for modulating phagosome biogenesis and subsequent bacterial escape into the cytoplasm. Cell Microbiol. 2005;7:969–79. doi: 10.1111/j.1462-5822.2005.00526.x. [DOI] [PubMed] [Google Scholar]
- 47.Barker JR, Chong A, Wehrly TD, Yu JJ, Rodriguez SA, Liu J, et al. The Francisella tularensis pathogenicity island encodes a secretion system that is required for phagosome escape and virulence. Mol Microbiol. 2009;74:1459–70. doi: 10.1111/j.1365-2958.2009.06947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alkhuder K, Meibom KL, Dubail I, Dupuis M, Charbit A. Glutathione provides a source of cysteine essential for intracellular multiplication of Francisella tularensis. PLoS Pathog. 2009;5:e1000284. doi: 10.1371/journal.ppat.1000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pechous R, Celli J, Penoske R, Hayes SF, Frank DW, Zahrt TC. Construction and characterization of an attenuated purine auxotroph in a Francisella tularensis live vaccine strain. Infect Immun. 2006;74:4452–61. doi: 10.1128/IAI.00666-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pechous RD, McCarthy TR, Mohapatra NP, Soni S, Penoske RM, Salzman NH, et al. A Francisella tularensis Schu S4 purine auxotroph is highly attenuated in mice but offers limited protection against homologous intranasal challenge. PLoS One. 2008;3:e2487. doi: 10.1371/journal.pone.0002487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–35. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Collins CA, De Mazière A, van Dijk S, Carlsson F, Klumperman J, Brown EJ. Atg5-independent sequestration of ubiquitinated mycobacteria. PLoS Pathog. 2009;5:e1000430. doi: 10.1371/journal.ppat.1000430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol. 2010;12:836–41. doi: 10.1038/ncb0910-836. [DOI] [PubMed] [Google Scholar]
- 54.von Muhlinen N, Thurston T, Ryzhakov G, Bloor S, Randow F. NDP52, a novel autophagy receptor for ubiquitin-decorated cytosolic bacteria. Autophagy. 2010;6:288–9. doi: 10.4161/auto.6.2.11118. [DOI] [PubMed] [Google Scholar]
- 55.Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- 56.Butchar JP, Cremer TJ, Clay CD, Gavrilin MA, Wewers MD, Marsh CB, et al. Microarray analysis of human monocytes infected with Francisella tularensis identifies new targets of host response subversion. PLoS One. 2008;3:e2924. doi: 10.1371/journal.pone.0002924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cremer TJ, Amer A, Tridandapani S, Butchar JP. Francisella tularensis regulates autophagy-related host cell signaling pathways. Autophagy. 2009;5:125–8. doi: 10.4161/auto.5.1.7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ogawa M, Yoshikawa Y, Kobayashi T, Mimuro H, Fukumatsu M, Kiga K, et al. A Tecpr1-dependent selective autophagy pathway targets bacterial pathogens. Cell Host Microbe. 2011;9:376–89. doi: 10.1016/j.chom.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 59.Shahnazari S, Yen WL, Birmingham CL, Shiu J, Namolovan A, Zheng YT, et al. A diacylglycerol-dependent signaling pathway contributes to regulation of antibacterial autophagy. Cell Host Microbe. 2010;8:137–46. doi: 10.1016/j.chom.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature. 2012;482:414–8. doi: 10.1038/nature10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao Z, Thackray LB, Miller BC, Lynn TM, Becker MM, Ward E, et al. Coronavirus replication does not require the autophagy gene ATG5. Autophagy. 2007;3:581–5. doi: 10.4161/auto.4782. [DOI] [PubMed] [Google Scholar]
- 62.Cann GM, Guignabert C, Ying L, Deshpande N, Bekker JM, Wang L, et al. Developmental expression of LC3alpha and beta: absence of fibronectin or autophagy phenotype in LC3beta knockout mice. Dev Dyn. 2008;237:187–95. doi: 10.1002/dvdy.21392. [DOI] [PubMed] [Google Scholar]
- 63.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–63. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.