

Abstract
Werner syndrome (WS) is a disorder characterized by features of premature aging and increased cancer that is caused by loss of the RecQ helicase WRN. Telomeres consisting of duplex TTAGGG repeats in humans protect chromosome ends and sustain cellular proliferation. WRN prevents the loss of telomeres replicated from the G-rich strand, which can form secondary G-quadruplex (G4) structures. Here, we dissected WRN roles in the replication of telomeric sequences by examining factors inherent to telomeric repeats, such as G4 DNA, independently from other factors at chromosome ends that can also impede replication. For this we used the supF shuttle vector (SV) mutagenesis assay. We demonstrate that SVs with [TTAGGG]6 sequences are stably replicated in human cells, and that the repeats suppress the frequency of large deletions despite G4 folding potential. WRN depletion increased the supF mutant frequency for both the telomeric and non-telomeric SVs, compared with the control cells, but this increase was much greater (27-fold) for telomeric SVs. The higher SV mutant frequencies in WRN-deficient cells were primarily due to an increase in large sequence deletions and rearrangements. However, WRN depletion caused a more dramatic increase in deletions and rearrangements arising within the telomeric SV (70-fold), compared with non-telomeric SV (8-fold). Our results indicate that WRN prevents large deletions and rearrangements during replication, and that this role is particularly important in templates with telomeric sequence. This provides a possible explanation for increased telomere loss in WS cells.
Keywords: DNA replication, G-quadruplex, WRN helicase, mutagenesis, telomere
Introduction
Werner syndrome (WS) is a rare autosomal recessive disorder characterized by features of premature aging and a predisposition to cancer1. WS is caused by loss of the WRN helicase/exonuclease, which is a member of the highly conserved RecQ helicase family.2 Cells from WS patients undergo premature replicative senescence, exhibit an extended S-phase and show increased levels of genomic and telomere instability.3,4 Evidence indicates that WRN protein has a critical role in responding to DNA replication stress, and functions in the prevention or restoration of stalled and broken DNA replication forks.5-7 WRN-deficient cells exhibit reduced rates of replication fork elongation after exposures to agents that induce replication stress by either generating DNA damage or by depleting nucleotide pools.3 Consistent with this, WRN is required to prevent replication-associated breaks at fragile sites in vivo and to prevent DNA polymerase δ from stalling at fragile site sequences in vitro.8,9 Together the data indicate that WRN has roles in facilitating DNA replication, which are critical for genome maintenance and cancer prevention.
Telomeres are protein-DNA structures that protect chromosome ends and are critical for sustaining cell proliferation and genome stability. Telomere dysfunction contributes to aging-related pathologies and carcinogenesis.10,11 Human telomeres are comprised of 5–15 kb of duplex TTAGGG repeats followed by a single stranded G-rich 3' overhang and are bound by the 6-member shelterin protein complex.12 The 3' overhang folds back and invades the duplex telomeric DNA to form a displacement loop (D-loop) that stabilizes the lasso-like t-loop end structure.13 Telomere shortening results from the inability to completely replicate chromosome ends and from defects in DNA repair or replication at telomeres.12 The loss of telomeric repeats is compensated for by telomerase, but most human somatic cells lack telomerase activity.14,15 Telomere dysfunction results when telomeres reach a critical length, or when shelterin proteins are defective.16,17 This activates a DNA damage response resulting in apoptosis or senescence, or chromosome end fusions and aberrations if checkpoint proteins are defective.12
Accumulating evidence indicates that WRN is required for proper telomere replication. WRN-defective cells show (1) an increase in stochastic loss of telomeres that were replicated from the G-rich lagging strand template, and (2) elevated sister chromatid exchanges in telomeric regions.4,11,18-21 Premature senescence, genomic instability and stochastic telomere loss in WS cells can be rescued with either WRN or telomerase, indicating that telomerase can compensate for WRN at telomeric ends.18 We and others showed that WRN is required to prevent telomere loss resulting from endogenous and environmental effectors of DNA replication stress.18,22 However, the precise role of WRN in preventing telomere loss is not well defined.
Recent data indicate that telomeres resemble common fragile sites that are prone to breakage during DNA replication stress. Fragile sites are known hotspots for DNA deletions and rearrangements.23,24 Cells lacking shelterin TRF1 or exposed to a DNA polymerase inhibitor all exhibit aberrant chromosomal telomeres thought to arise from fragmentation.25 Several obstacles to telomere replication have been described, which offer various scenarios for replication fork stalling and WRN roles at telomeres. (1) WRN disrupts the telomeric end D-loops that may impede replication fork progression.26-28 (2) Single-stranded DNA at the telomeric 3' overhang, in the D-loop and in Okazaki fragments during telomere replication are G-rich and can fold into G-quadruplex structures (G4 DNA).29 G4 DNA is a non B-DNA structure that blocks DNA synthesis and is unwound by WRN and BLM helicases in vitro.30 Telomere fragility is suppressed by BLM and other helicases but increased with G4 stabilizing ligands,25,31,32 suggesting that G4 folds interfere with telomere replication. (3) Misalignment of DNA primers and templates in repetitive sequences may provoke replication fork stalling.33 (4) Telomeres are transcribed into non-coding RNAs, which can impede telomere replication.34 Thus, obstacles specific to chromosome ends and/or inherent to the telomeric repeats themselves may impede DNA replication and cause telomere loss.
In this study, we investigated more directly whether WRN is required to replicate telomeric sequences by using supF mutation reporter vectors harboring telomeric repeats. This system eliminates other potential replication obstacles that exist at chromosome ends, such as t-loop/D-loops or telomerase. Previously we showed that human TTAGGG repeats are replicated accurately in human cells despite their ability to form G4 DNA, unlike other sequences that can form non-B DNA structures.35-37 Here we report that WRN depletion elevated the supF mutation frequency for vectors with control non-telomeric or telomeric sequences, but the increase was significantly higher for the telomeric vector and was primarily due to a dramatic increase in sequence deletion events. Our results establish that WRN is required to accurately replicate human telomeric sequences and provide a mechanism to explain the stochastic loss of telomeres in WRN-deficient cells.4
Results
Development of telomeric supF mutagenesis assay
To determine whether WRN is required to replicate telomeric sequences in human cells, we constructed a shuttle vector containing six telomeric repeats upstream to and within the mutation reporter supF gene. This approach offers several critical advantages. (1) Deletions of telomeric DNA within the SV will not affect cell survival. However, loss of telomeric repeats at chromosome ends can cause apoptosis or senescence,12 and short telomeres and defects in repair proteins (i.e., WRN) may synergistically decrease cell survival. (2) Episomal vectors allow for direct comparison in different genetic backgrounds. Since the vector is not integrated in the genome, there are no confounding effects of different integration points. (3) The vector has sufficient telomeric repeats to form G4 DNA, but does not form complex telomeric t/D-loops and lacks the substrate for telomerase. Thus, the addition of telomerase inhibitors, as done previously,4 is not required to unmask WRN roles in preserving telomeric sequence. This approach allows us to examine WRN roles in modulating factors inherent in telomeric sequence, independently from potential confounding effects of complex end structures, telomerase activity and telomere transcription. The new supF vector integrates the last repeat of the inserted [TTAGGG]6 sequence in the acceptor stem of the supF tRNA (Fig. 1). The control vector contains a 36 bp scrambled sequence of identical nucleotide composition as the [TTAGGG]6 sequence (Table S1). These vectors have very low background mutant frequencies in E. coli. For the scrambled control vector no mutants were detected in 14,694 colonies, yielding an estimated mutant frequency of < 6.8 × 10−5. The telomeric vector provided a background mutant frequency of 3.9 × 10−5 (1 mutant/25,549 total colonies) indicating that both vectors are highly stable upon replication in the indicator E. coli strain.
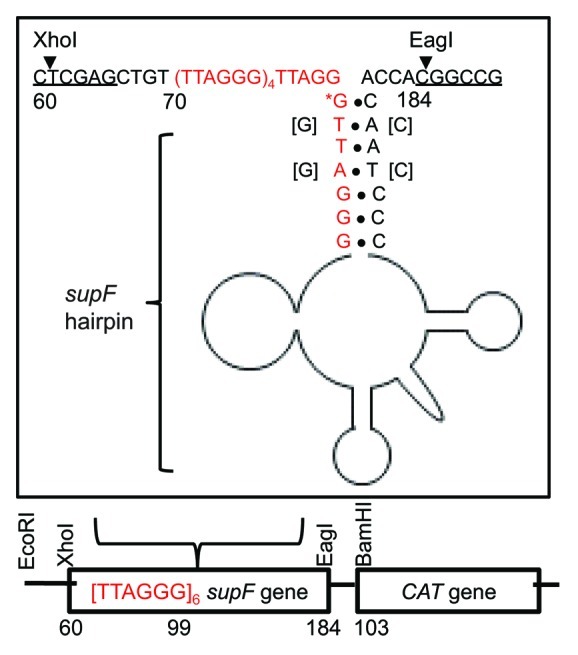
Figure 1. Structure of shuttle vector containing telomeric DNA. Six telomeric repeats were inserted upstream and within the supF gene of the pSP189 shuttle vector as shown. Four nucleotides of the supF gene were mutated as indicated so the last repeat was located in the stem of the supF tRNA hairpin structure. A scrambled telomeric sequence of identical size and nucleotide context was introduced to generate the control shuttle vector (see Table S1). The chloramphenicol aceytltransferase gene was introduced at the BamHI site. The 5' end of the mature tRNA (*G) is marked as position 99 following the traditional nomenclature.
WRN depletion increases the mutant frequency of the telomeric vector
The control and telomeric SVs were transfected into human U2OS cells stably expressing either a control shRNA (shCTRL) or an shRNA targeted against WRN (shWRN)38 (Fig. 2). WRN expression was decreased to 24% of the control cells (Fig. 2B). SVs were replicated for 48 h, isolated and subjected to DpnI digestion to select for replicated vectors, which were then transformed into the E.coli reporter strain and subjected to blue/white screening for supF mutants. The mean mutant frequencies for the scrambled and telomeric vectors after replication in shCTRL U2OS cells were very similar at 5.2 × 10−4 and 5.6 × 10−4, respectively (Fig. 2A). Thus, human telomeric repeats are not mutagenic and are stably replicated in normal human cells in agreement with our previous results using a different shuttle vector mutagenesis system.35 In contrast, replication of the telomeric vector in WRN deficient U2OS cells yielded a significantly higher (6-fold) mutant frequency compared with the scrambled vector (150 × 10−4 vs. 25 × 10−4, p-value = 0.0076). Thus, vectors with telomeric repeats are more mutagenic in the absence of WRN protein. Consistent with this, WRN depletion increased the scrambled vector mutant frequency 4.8-fold but significantly elevated the telomeric vector mutant frequency 27-fold (p-value = 0.0064), compared with control cells (Fig. 2A). The vectors with telomeric repeats exhibit an increased dependence on WRN protein to suppress mutagenic events during replication compared with non-telomeric vectors.
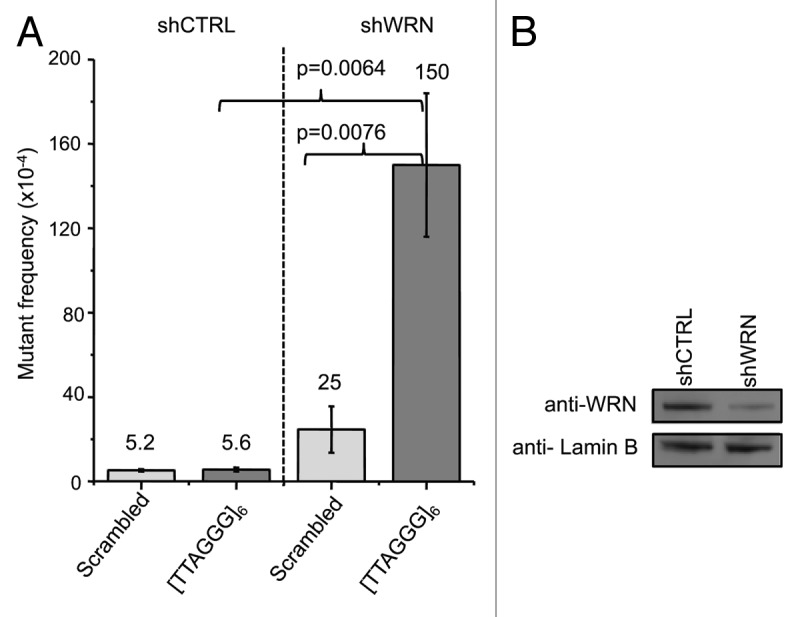
Figure 2. WRN depletion significantly increases the supF mutant frequencies for the telomeric vector. (A) supF mutant frequencies for scrambled control and telomeric vectors after replication in WRN-proficient and -deficient cells. Shuttle vectors were replicated in U2OS cells expressing control or WRN shRNAs for 48 h, isolated and transformed into the reporter E.coli strain to screen for supF mutants. Values are the mean and error bars indicate SEM from at least three independent experiments. (B) Western blot shows WRN protein levels in U2OS cells stably expressing control or WRN shRNAs. Quantification revealed a 76% knockdown in WRN expression.
Reduced recovery of vectors from WRN-depleted U2OS cells
Next, we examined whether insertion of the telomeric repeats altered the efficiency of recovering replicated vectors from the control and WRN-depleted human cells. For this the chloramphenicol (chlor) resistant scrambled or telomeric vectors were co-transfected with the kanamycin (kan) resistant vector pEYFP-C1, which served as an internal standard, and were isolated 48 h after culturing. The mean recovery efficiency (see Methods for calculation) of the replicated scrambled and telomeric vectors from the shCTRL cells was 53 for both, indicating the telomeric repeats did not interfere with SV replication or recovery in WRN proficient cells (Fig. 3). The mean recovery efficiency for the scrambled vector from shWRN cells was 27, which was slightly higher than for the telomeric vector at 20. WRN depletion resulted in a near 2-fold decrease in recovery for both the scrambled and telomeric vectors compared with WRN proficient cells. This suggests WRN is required for efficient SV replication and/or to prevent large deletions that impact the chlor resistant gene or the mammalian/bacterial origins of replication on the vector.
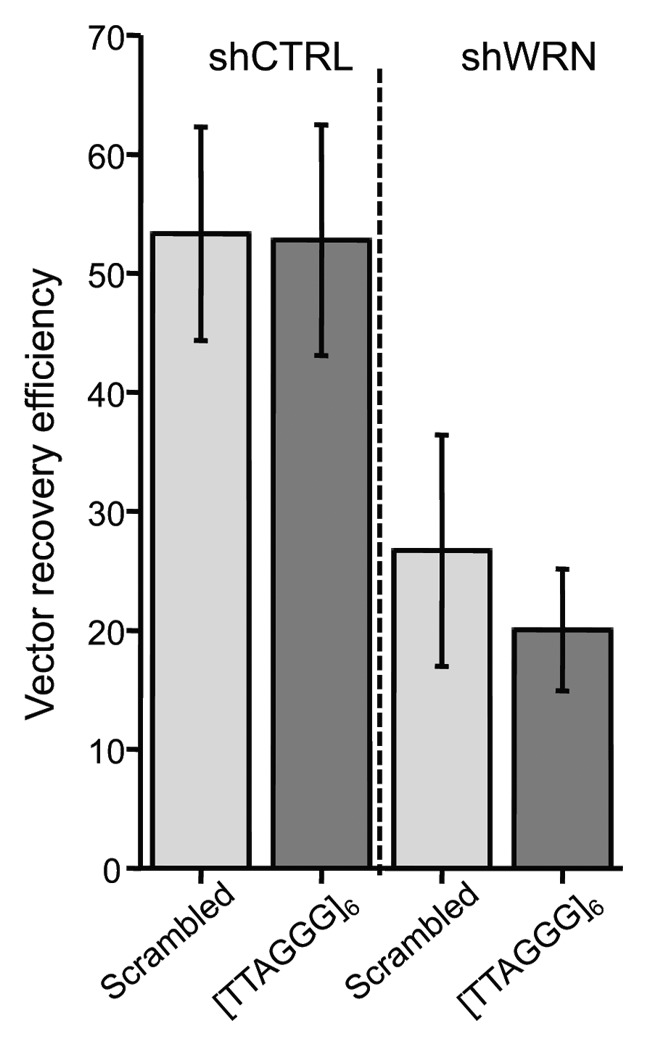
Figure 3. The pSP189 vector recovery efficiency is decreased in WRN-deficient cells. The graph shows vector recovery efficiencies for the scrambled control and telomeric vectors after replication in control and shWRN U2OS cells. The kan-resistant vector pEYFP-C1 was co-transfected with scrambled or telomeric pSP189 vectors into U2OS cells. Vectors were isolated after 48 h, electroporated into E. coli and plated on selective media. The recovery efficiency was calculated as the ratio of chlor- to kan-resistant colonies. Values are the mean and error bars indicate SEM from two to three independent experiments.
WRN depletion causes a large increase in deletion formation in SVs with telomeric repeats
To determine the mechanism(s) for the significant increase in supF mutant frequency for the telomeric vector upon WRN depletion, we investigated the mutations arising from vector replication in the human cells. SVs replicated in U2OS cells were transformed into the reporter E. coli strain. Plasmids were isolated from independent white mutant bacteria colonies and subjected to double digestion with XhoI and EagI to determine whether the telomeric or scrambled sequences and the supF gene were present (see Methods and Fig. 1). The majority of mutant telomeric vectors from the WRN-deficient cells exhibited abnormal digest patterns (Fig. S1) indicating that the supF gene was likely deleted. Therefore, vectors were sequenced using a primer that binds at the 5'end of the CAT gene rather than previously used primers.36 Mutations sorted into two categories (1) point mutations (PM) within the supF gene or (2) deletions and unknown rearrangements (del/rrg). All the PM detected were base substitutions with an obvious bias at G•C base pairs (94%), compared with A•T base pairs (3%), arising primarily at GA•CT dinucleotide sites (Fig. S2), consistent with previous reports for these cells.39 Neither the presence of telomeric repeats or WRN depletion altered this bias. The majority of del/rrg were well defined deletions greater than 500 bp and approximately 30–40% exhibited 1–3 bp of microhomology (Tables S2–5). Again, neither the presence of telomeric repeats or WRN altered the deletion sizes in an obvious way.
Insertion of the telomeric repeats did not significantly alter the overall mutant frequency, but rather altered the types of mutations generated after SV replication in shCTRL U2OS cells (Table 1). While the PM and del/rrg frequencies were nearly identical for the scrambled vector (2.7 × 10−4 and 2.5 × 10−4, respectively), the PM frequency was 2.2-fold higher than the del/rrg frequency (3.9 × 10−4 and 1.7 × 10−4, respectively) for the telomeric vector. This agrees well with our previous result using the HSV-tk shuttle vector that the insertion of human telomeric repeats stabilizes the vectors and suppresses the occurrence of large deletions and rearrangements.35
Table 1. Sequenced mutations arising in the supF SV after replication in human cells.
| Mutation class | scrambled shCTRL |
[TTAGGG]6 shCTRL |
scrambled shWRN |
[TTAGGG]6 shWRN |
|
|---|---|---|---|---|---|
|
Overall mean mutant frequency |
5.2x10−4 |
5.6x10−4 |
25x10−4 |
150x10−4 |
|
|
Point Mutations | |||||
| |
mean mutation frequency |
2.7x10−4 |
3.9x10−4 |
5.2x10−4 |
33x10−4 |
| number |
11 |
16 |
4 |
5 |
|
| proportion of total |
0.52 |
0.70 |
0.21 |
0.21 |
|
|
Deletions and Rearrangementsa | |||||
| |
mean mutation Frequency |
2.5x10−4 |
1.7x10−4 |
20x10−4 |
120x10−4 |
| number of mutations |
10 |
7b |
15c |
19d |
|
| proportion of total |
0.48 |
0.30 |
0.79 |
0.79 |
|
| | |||||
| Total mutations (mutants) | 21 (21) | 23 (17) | 19 (19) | 24 (21) | |
a Identical deletions or rearrangements from the same SV transfection experiment in human cells were scored once to avoid the inclusion of potential SV siblings (progeny from a single mutagenic event). bIncludes a mutation in which one (AGGGTT) repeat was inserted into the [TTAGGG]6 sequence. cIncludes one unknown rearrangement and 14 defined deletions. dIncludes four unknown rearrangements, 14 defined deletions, and a mutation in which one (AGGGTT) repeat was deleted from the [TTAGGG]6 sequence.
WRN is required to suppress both PM and del/rrg arising within the telomeric SV upon replication in human cells. WRN depletion increased the PM frequency slightly for the scrambled SV and more noticeably (8.5-fold) for the telomeric SV compared with the control cells (Table 1). However, WRN deficiency caused a much greater effect on del/rrg events. The del/rrg frequency for the scrambled vector was moderately increased 8-fold to 20 × 10−4, but was dramatically elevated 70-fold to 120 × 10−4 for the telomeric SV, compared with shCTRL cells. Next we compared the proportions of mutation types. U2OS cells exhibited a higher proportion of PM relative to del/rrg; however, WRN depletion shifted the bias toward del/rrg for both vectors (Table 1). The shWRN-mediated increase in the proportion of del/rrg was statistically significant for the telomeric vector (p = 0.0012) but not for the non-telomeric vector (p = 0.055), relative to shCTRL cell (Fisher’s exact test). Several of the del/rrg events exhibited endpoints within the supF gene and within or near the telomeric repeats, although endpoints also existed within the scrambled sequence of the control vector (Fig. 4). In summary, the data indicate that WRN has a greater role in suppressing deletions and rearrangements, compared with point mutations, and that this role is more critical in templates with telomeric repeats.

Figure 4. Deletions and rearrangements with endpoints in the supF gene and telomeric repeats. The supF gene with borders marked by “|,” telomeric or scrambled sequence insert and the XhoI and EagI restriction sites are shown 5' to 3'. Bases marked in grey are alterations in the tRNA acceptor stem. End points of deletions or rearrangements are marked by an open arrow head (shCTRL) cells or a solid arrow head (shWRN) cells. Deleted or altered sequences are all 3' to arrowheads unless otherwise noted with an arrow. The 5' end of the mature tRNA (*G) is marked as position 99 following the traditional nomenclature.
G4 DNA structures form in single-stranded [TTAGGG]6 sequences
One potential mechanism for the increase in deletions arising within the telomeric SV upon WRN depletion is unresolved G4 DNA structures that interfered with replication. Therefore, we asked whether the telomeric repeats fold into G4 DNA in the context of the supF gene flanking sequences. We and others established atomic force microscopy (AFM) as a useful tool for detecting G4 folds in ssDNA, and showed G4 DNA generates structures with distinct peaks at heights close to 1 nm.40,41 AFM images were collected of the DNA oligonucleotides used to construct the SVs with [TTAGGG]6 repeats or the scrambled sequence (Table S1). The mean peak-height of structures in AFM images of the scrambled sequence oligonucleotides was 0.6 nm (± 0.1 nm) (Fig. 5A and C), which is consistent with peak-heights of ssDNA.41 On the contrary, the AFM images of the [TTAGGG]6 containing oligonucleotides exhibited structures with a mean peak-height at 0.9 (± 0.2) nm (Fig. 5B and D), which is consistent with G4 DNA folds or intermediate structures in the folding pathway.41,42 A small population of the [TTAGGG]6 molecules displayed peak-heights at 0.6 nm (Fig. 5D, black arrow), representing unfolded ssDNA. This unfolded population served as an internal standard for peak-height comparisons between structures from the scrambled sequence molecules and the telomeric molecules. These data show that the telomeric repeats can spontaneously fold into G4 DNA in the context of SV flanking sequence and ssDNA.

Figure 5. Telomeric repeats form G4 DNA in the context of the supF gene. Representative AFM images of oligonucleotides with scrambled sequence (A) or 6 telomeric repeats (B) and supF gene flanking sequence (see Table S1). The images are 400 nm × 400 nm at 2 nm Z-scale. Histogram of peak-heights of structures in the AFM images of oligonucleotides with scrambled sequence (C) or 6 telomeric repeats (D), n = 97 for each.
Supercoiled telomeric SVs do not form G4 DNA prior to replication in human cells
Next we wished to determine whether the deletions induced in the telomeric SV were associated with replication. It is well established that the high-energy state of supercoiled DNA can lead to formations of non-canonical DNA structures, such as cruciforms, Z-DNA and H-DNA.43-45 Furthermore, transitions to these structures simultaneously relax the DNA. Preformed alternate structures in supercoiled plasmids can induce deletions independently of DNA replication.37 To investigate whether telomeric sequences formed G4 structures in the supercoiled SVs prior to transfection into human cells, we visualized telomeric SVs using AFM. Previous work showed AFM can be used to detect cruciforms and H-DNA in supercoiled plasmids.43,46 A representative AFM image of the telomeric SV is shown in Figure 6. All circular DNA molecules observed (n = 80 molecules) were in a plectonemic shape, with formations of large loops between very tightly twisted segments. These images are consistent with previous AFM images of supercoiled DNA under similar sample preparation conditions.46 However, no structures with peaks near 1 nm in height were observed on the telomeric SVs except at regions where strands of duplex DNA overlapped (Fig. 6, white arrow). These regions of overlapping DNA were distinctly different from the defined peaks of a single G4 structure formed from six telomeric repeats41 (Fig. 5B). In summary, contrary to Z-DNA and H-DNA, the data indicate that G4 structures do no form on duplex telomeric DNA by introducing supercoiling.

Figure 6. Supercoiled telomeric shuttle vectors lack pre-folded G4 structures. Representative AFM image of telomeric SVs (0.5 µg/ml). The image is 1.8 μm × 1.8 μm at 2 nm Z-scale. The white arrow points to a region where strands of duplex DNA overlap.
Discussion
Previous studies showed WRN protein localizes to telomeres in S-phase and is required to prevent the loss of chromosomal telomeres replicated from the G-rich lagging strand.4,20,26 Here we dissected WRN roles in the replication of telomeric sequences using the supF shuttle vector mutagenesis assay to test factors inherent to the repeats, such as G4 DNA, independently from chromosome end structures and enzymatic processing. Consistent with our previous results,35 we demonstrated that (1) SVs with [TTAGGG]6 sequences are stably replicated in human cells and (2) insertion of the repeats suppresses the frequency of large deletions despite G4 folding potential (Fig. 2). WRN depletion increased the supF mutant frequency for both the telomeric and non-telomeric scrambled SVs, compared with the control cells, but this increase was much greater for the telomeric SVs (Fig. 2). The higher supF SV mutant frequencies in WRN-deficient cells were primarily due to an increase in large sequence deletions and rearrangements for both vectors. However, WRN depletion caused a more dramatic increase in deletions and rearrangements arising within the telomeric SV compared with the scrambled SV. Our results show that WRN prevents deleterious mutagenic events during DNA replication, and that this role is particularly important in templates with telomeric sequence.
We observed a greater role for WRN in suppressing deletions and rearrangements compared with base substitutions (Table 1). This increase in deletions is likely an underestimate, since the recovery of SVs from shWRN U2OS cells was lower compared with shCTRL cells (Fig. 3). Large deletions that compromise the SV drug resistant genes or replication origins will cause loss of the SV, excluding these events from detection. WRN-deficient cells exhibited a slightly reduced recovery of telomeric SV compared with the scrambled SV, consistent with WRN roles in replicating telomeric sequences. However, the recovery of both vectors was reduced in WRN-deficient cells, compared with control cells, suggesting that WRN has a more general role in SV maintenance. Our data suggest this role is to prevent sequence deletions. Consistent with this, previous studies of mutation rates at the HPRT chromosomal locus from SV-40 transformed WS fibroblasts revealed an increase in large deletion events.47 Importantly, these studies also confirmed that SV-40 large T-antigen helicase could not compensate for WRN roles in suppressing deletions, consistent with our results using a SV that requires SV-40 large T-antigen for replication.
A recent study by Bacolla et al. showed that WRN depletion caused a 2-fold increase in mutant frequency for both the control vector and the SV with Z-DNA forming sequences.39 This is vastly different from the 27-fold increase in supF mutant frequency obtained when the telomeric SVs were replicated in WRN-deficient cells compared with control cells (Fig. 2). Thus, while WRN can unwind both Z-DNA and G4 DNA, it only suppresses deletions induced by telomeric sequences, but not Z-DNA forming sequences.30,39 This may be related to different mechanisms by which the telomeric and Z-DNA-forming sequences induce DNA deletions. Z-DNA is pre-folded prior to replication and the deletions are not dependent on replication,37 unlike the telomeric DNA (Fig. 5). Furthermore, the mutation spectrum of the control and Z-DNA SVs in both WRN proficient and deficient cells consisted primarily of BS.39 The domination of BS was attributed to oxidative damage, which was increased in WRN-depleted U2OS cells.39 In contrast, we observed that WRN depletion resulted in a much greater increase in deletions, compared with BS (Table 1). This difference is likely explained by our culturing the U2OS cells at 5% oxygen to minimize oxidative damage. Furthermore, the design of our supF telomeric SV allowed for the detection of larger deletions compared with the supF Z-DNA SV, since we could detect deletions into the ampicillin resistance gene up to the SV 40 origin. Thus, it was possible for our system of selection to recover a greater number of mutants with large deletions.
Our data indicate that the mechanism for SV sequence deletions in WRN-depleted cells resulted from replication-induced DSBs. Replication fork stalling due to alternate structures or other factors can cause the fork to collapse into DSBs.25,48 AFM imaging of the supercoiled SVs revealed no evidence of pre-folded alternate structures, including G4 DNA, in the telomeric SV (Fig. 6). In contrast, the introduction of Z-DNA forming sequences in the supF SV caused the formation of alternate structures prior to transfection into human cells and induced large deletions independently from DNA replication.37 Thus, the deletions could result from nuclease cleavage at Z-DNA structures. In stark contrast to Z-DNA forming sequences, the introduction of telomeric G4 forming sequences in the supF SV actually suppressed the deletion frequency in control cells (Table 1). Thus, our data are consistent with the model that deletions in the telomeric SV are due to secondary structures or others factors that arise during replication, rather than nucleolytic processing of pre-formed secondary structures.
Possible mechanisms for the increase in deletion frequencies in WRN-depleted cells are (1) WRN roles in preventing DSB formation during replication, or (2) WRN roles in promoting error-free repair of replication-induced DSBs. While WRN is not required for DSB repair, WRN minimizes nucleotide end resection to prevent large deletions during non-homologous end joining (NHEJ) of DSBs.49 However, our data showed that the mean deletion sizes in the SVs were similar in the presence or absence of WRN and so was the percent of deletion endpoints that exhibited microhomology at the junction sites. Therefore, our data indicate that WRN does not suppress end resection at break points in our SV assay. Instead, we argue that the increase in deletion frequency is a result of WRN roles in preventing replication fork demise and collapse into DSBs, consistent with data that WRN promotes replication fork progression on chromosomes.3
While our results clearly show that WRN suppresses the formation of deletions during the replication of telomeric sequences, we cannot rule out the possibility that factors inherent to telomeric sequence other than G4 formation may be responsible for causing deletions. Mutating G residues in the TTAGGG repeats or other G4 forming sequences prevents G4 folding, but also alters sequence context effects which are known to influence DNA polymerase activity.50,51 G4 folding can be suppressed in vitro by altering ionic reaction conditions;52 however, this is not possible in vivo. Indeed we observed that the replicative DNA polymerase δ stalls on TTAGGG templates even under ionic conditions that suppress G4 folding (our unpublished data). Consistent with this, WRN promotes polymerase δ DNA synthesis through the FRA16D fragile site in vitro and suppresses breakage at fragile sites in vivo.8,9 Thus, there is precedent for WRN facilitating replication through difficult templates by mechanisms other than G4 unwinding.
In summary, we demonstrate that WRN is required for suppressing deleterious DNA deletions that are induced by factors inherent to telomeric repeat sequences, independently from enzymatic processes and complex DNA conformations at chromosomal ends. Our data support the model that in WRN-deficient cells, unresolved G4 DNA or other factors in telomeric sequences induce replication fork stalling and collapse, leading to large deletions. In a cellular context, this causes stochastic telomere loss, which is a characteristic of WRN-deficient cells.4
Experimental Procedures
Cell culture and reagents
Human U2OS osteosarcoma cells were cultured in Dulbecco's modified Eagle media (DMEM) supplemented with 10% fetal bovine serum and Penicillin/Streptomycin (Invitrogen) and grown at 5% O2, 5% CO2 and 37°C. U2OS cells proficient and deficient for WRN were generated by stably expressing a scrambled shRNA (shCTRL) and shRNA against WRN (shWRN) respectively, as described in reference 38. Cells were cultured in the presence of hygromycin (200 μg/ml) (EMD Chemicals Inc.) to maintain selective pressure for shRNA expression. All the restriction enzymes were obtained from New England Biolabs. Chloramphenicol (chlor), 5-bromo-4-chloro-3-indolyl β-d-galactoside (X-Gal) and kanamycin (kan) were purchased from Sigma Chemical Co. while isopropyl β-d-thiogalactoside (IPTG) was obtained from Fisher Bioreagents. All oligonucleotides were from Integrated DNA Technologies Inc. (Table S1).
Construction of supF reporter gene shuttle vectors containing telomeric repeats
The shuttle vector (SV) pSP189 harboring the supF mutagenic reporter gene was kindly provided by Dr. Karen Vasquez (University of Texas).53 To optimize vector selection in bacteria, the chloramphenicol acetyltransferase (cat) gene was cloned into the BamHI site of pSP189. The sequence (TTAGGG)6 was inserted such that the last repeat was present in the supF acceptor stem of the suppressor tRNA molecule (Fig. 1) to generate the telomeric SV. For this the following mutations were introduced into the supF gene: G100T, G102A, C176T, and C178A. To generate the scrambled control SV the sequence [CTTGCTTACGACTTACCGTTCATCGTTGA] was inserted 3' to the supF gene and the following residues of the acceptor stem were mutated: T100A, G102A, C176T, and C178A. SV construction was as described previously.37 Briefly, oligonucleotides (Table S1) containing the telomeric or scrambled control sequences and the supF gene were annealed and cloned into the XhoI and EagI restriction sites of the pSP189 vector. Ligation reactions were transformed into the MBM7070 indicator E. coli strain (provided by Dr. Karen Vasquez, University of Texas) and plated on LB agar plates supplemented with chlor (50 µg/ml), X-gal (0.12 mg/ml) and IPTG (0.3 mg/ml). Plasmids were isolated from wild type blue colonies and sequenced (ACGT Inc.) to confirm the correct sequence.
SupF mutational analysis of telomeric and control shuttle vectors
SupF mutant frequencies of the various SVs were determined after replication in human U2OS cells as described previously.37 For transfections, 2 μg SV was mixed with 2 × 106 cells in 100 μl of nucleofector kit V solutions and electroporated using the Amaxa Nucleofection system (Lonza). After 48 h of growth in supplemented DMEM media the SVs were isolated using the PureLink Quick Plasmid Miniprep kit (Invitrogen). Vectors were digested with DpnI enzyme to remove vectors that were not replicated in U2OS cells, and were then transfected into MBM7070 bacteria, incubated for 45 min at 37°C, and plated on selective media containing 50 µg/ml chlor, 0.12 mg/ml X-gal and 0.3 mg/ml IPTG to screen for supF mutants by blue-white screening.37 All mutant white colonies were confirmed by re-plating on selective media. The supF mutant frequency was determined as the number of mutant white colonies divided by the total number of chlor-resistant colonies. To test for the recovery efficiency of the SVs after replication in human cells, we co-transfected 1 × 106 U2OS cells with 1 µg of SV harboring the chlor resistant gene and 1 µg of pEYFP-C1 (Clontech) harboring the kan-resistant gene, which served as an internal standard to control for differences in transfection efficiency. Both vectors contain an SV40 origin of replication. The vectors were electroporated into human cells as described above, isolated 48 h after culturing, and transfected into MBM7070 E. coli. The recovery efficiency was calculated as the ratio of chlor-resistant bacteria colonies to kan-resistant colonies.
Generation of supF shuttle vector mutation spectra
To obtain independent mutants for analysis, after electroporation of replicated shuttle vectors into MBM7070 bacteria, the culture was placed on ice and aliquoted into multiple tubes containing 200 µl media. After the 45 min recovery at 37°C, each aliquot was plated on selective media, and one white mutant was isolated for plasmid purification and DNA sequencing. This ensures that any mutational hotspots were not due to division of bacteria harboring supF mutant vectors during the 45 min recovery. To initially screen for mutants with large deletions or rearrangements the plasmids were digested with XhoI and EagI restriction enzymes (Fig. 1). Sequence changes and mutations within the promoter and coding region of the supF gene, as well as the telomeric inserts, were determined by dideoxy DNA sequencing (ACGT Inc.). DNA sequence analysis was done using Align-X software of Vector NTI Advance (Invitrogen). The primer used for DNA sequencing (Table S1) annealed at the 5' end of the cat gene to prime sequencing through the supF gene. Mutant SVs from the same transfection into human cells that exhibited the identical deletion or rearrangement could have resulted from either (1) that mutation occurring independently in different U2OS cells, or (2) from an early mutagenic event that was replicated multiple times during the 48 h growth. To maintain rigor and consistency, these mutants were considered siblings and were scored once. The same mutations occurring in different clones were considered independent.
Statistics
Means and standard deviations of mutant frequencies were calculated and statistical significance was determined by two-tailed t-test using Microsoft Excel. Fisher’s exact test was used to calculate statistical significance between proportions of deletions/rearrangements using GraphPad InStat version 3.10 for Windows, GraphPad Software (www.graphpad.com). Significance was determined at p < 0.05.
AFM imaging and analysis
SupF telomeric SV or olignucleotides used to construct the scrambled and telomeric SVs (Table S1) were diluted to a final concentration of 0.5 or 1 µg/ml in a buffer containing 25 mM Hepes pH 7.5, 25 mM sodium acetate, and 10 mM MgCl2. Buffers were pre-heated at 65°C for 15–30 min to dissolve small salt particles that may have accumulated during storage. Molecules were pre-incubated at 37°C for 15 min immediately prior to deposition onto a freshly cleaved mica disk (SPI Supply). All samples were washed with MilliQ water and dried under a stream of nitrogen gas. Images were collected using a MultiModeV microscope (Veeco Instruments) using E scanners in tapping mode. Pointprobe® plus noncontact/tapping mode silicon probes (PPP-NCL, Agilent) with spring constants of ~50 N/m and resonance frequencies of ~190 kHz were used. Images were captured at a scan size of 2 μm × 2 μm, a scan rate of 3 Hz, a target amplitude of 0.30 to 0.35 V and a resolution of 512 × 512 pixels.
Supplementary Material
Acknowledgments
We thank Dr. Bennett Van Houten (University of Pittsburgh) for use of the AFM instrument in his laboratory. We are grateful to Dr. Graham Wang (University of Texas) for advice regarding the SupF mutagenesis assay. This work was funded by the Ellison Medical Foundation (P.L.O.) and NIH grants ES0515052 (P.L.O.), 1K99ES016758-01 (H.W.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
Kelly E. Knickelbein performed experiments involving the SupF mutagenesis assay, data collection and data analysis along with Fu-jun Liu and Steven Strutt. Rama Rao Damerla was involved in analysis of mutations obtained from the SupF assay, generating mutation spectra, data interpretation and wrote the manuscript. Hong Wang performed experiments involving atomic force microscopy, interpreted the data and wrote the manuscript. Patricia L. Opresko proposed and supervised the project, interpreted the data and wrote the manuscript.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/21399
References
- 1.Kudlow BA, Kennedy BK, Monnat RJ., Jr. Werner and Hutchinson-Gilford progeria syndromes: mechanistic basis of human progeroid diseases. Nat Rev Mol Cell Biol. 2007;8:394–404. doi: 10.1038/nrm2161. [DOI] [PubMed] [Google Scholar]
- 2.Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, et al. Positional cloning of the Werner’s syndrome gene. Science. 1996;272:258–62. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]
- 3.Sidorova JM, Li N, Folch A, Monnat RJ., Jr. The RecQ helicase WRN is required for normal replication fork progression after DNA damage or replication fork arrest. Cell Cycle. 2008;7:796–807. doi: 10.4161/cc.7.6.5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crabbe L, Verdun RE, Haggblom CI, Karlseder J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science. 2004;306:1951–3. doi: 10.1126/science.1103619. [DOI] [PubMed] [Google Scholar]
- 5.Cheng WH, Muftic D, Muftuoglu M, Dawut L, Morris C, Helleday T, et al. WRN is required for ATM activation and the S-phase checkpoint in response to interstrand cross-link-induced DNA double-strand breaks. Mol Biol Cell. 2008;19:3923–33. doi: 10.1091/mbc.E07-07-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pichierri P, Franchitto A, Mosesso P, Palitti F. Werner’s syndrome protein is required for correct recovery after replication arrest and DNA damage induced in S-phase of cell cycle. Mol Biol Cell. 2001;12:2412–21. doi: 10.1091/mbc.12.8.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pichierri P, Ammazzalorso F, Bignami M, Franchitto A. The Werner syndrome protein: linking the replication checkpoint response to genome stability. Aging. 2011;3:311–8. doi: 10.18632/aging.100293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pirzio LM, Pichierri P, Bignami M, Franchitto A. Werner syndrome helicase activity is essential in maintaining fragile site stability. J Cell Biol. 2008;180:305–14. doi: 10.1083/jcb.200705126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shah SN, Opresko PL, Meng X, Lee MY, Eckert KA. DNA structure and the Werner protein modulate human DNA polymerase delta-dependent replication dynamics within the common fragile site FRA16D. Nucleic Acids Res. 2010;38:1149–62. doi: 10.1093/nar/gkp1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Artandi SE, DePinho RA. Telomeres and telomerase in cancer. Carcinogenesis. 2010;31:9–18. doi: 10.1093/carcin/bgp268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blagoev KB, Goodwin EH, Bailey SM. Telomere sister chromatid exchange and the process of aging. Aging (Albany NY) 2010;2:727–30. doi: 10.18632/aging.100206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–34. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 13.Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, et al. Mammalian telomeres end in a large duplex loop. Cell. 1999;97:503–14. doi: 10.1016/S0092-8674(00)80760-6. [DOI] [PubMed] [Google Scholar]
- 14.Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu C-P, Morin GB, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–52. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 15.Greider CW, Blackburn EH. The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell. 1987;51:887–98. doi: 10.1016/0092-8674(87)90576-9. [DOI] [PubMed] [Google Scholar]
- 16.van Steensel B, Smogorzewska A, de Lange T. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998;92:401–13. doi: 10.1016/S0092-8674(00)80932-0. [DOI] [PubMed] [Google Scholar]
- 17.Hemann MT, Strong MA, Hao LY, Greider CW. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell. 2001;107:67–77. doi: 10.1016/S0092-8674(01)00504-9. [DOI] [PubMed] [Google Scholar]
- 18.Crabbe L, Jauch A, Naeger CM, Holtgreve-Grez H, Karlseder J. Telomere dysfunction as a cause of genomic instability in Werner syndrome. Proc Natl Acad Sci USA. 2007;104:2205–10. doi: 10.1073/pnas.0609410104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laud PR, Multani AS, Bailey SM, Wu L, Ma J, Kingsley C, et al. Elevated telomere-telomere recombination in WRN-deficient, telomere dysfunctional cells promotes escape from senescence and engagement of the ALT pathway. Genes Dev. 2005;19:2560–70. doi: 10.1101/gad.1321305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arnoult N, Saintome C, Ourliac-Garnier I, Riou JF, Londoño-Vallejo A. Human POT1 is required for efficient telomere C-rich strand replication in the absence of WRN. Genes Dev. 2009;23:2915–24. doi: 10.1101/gad.544009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hagelstrom RT, Blagoev KB, Niedernhofer LJ, Goodwin EH, Bailey SM. Hyper telomere recombination accelerates replicative senescence and may promote premature aging. Proc Natl Acad Sci USA. 2010;107:15768–73. doi: 10.1073/pnas.1006338107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu FJ, Barchowsky A, Opresko PL. The Werner syndrome protein suppresses telomeric instability caused by chromium (VI) induced DNA replication stress. PLoS ONE. 2010;5:e11152. doi: 10.1371/journal.pone.0011152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glover TW, Stein CK. Induction of sister chromatid exchanges at common fragile sites. Am J Hum Genet. 1987;41:882–90. [PMC free article] [PubMed] [Google Scholar]
- 24.Durkin SG, Glover TW. Chromosome fragile sites. Annu Rev Genet. 2007;41:169–92. doi: 10.1146/annurev.genet.41.042007.165900. [DOI] [PubMed] [Google Scholar]
- 25.Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, et al. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009;138:90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Opresko PL, Otterlei M, Graakjaer J, Bruheim P, Dawut L, Kølvraa S, et al. The Werner syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2. Mol Cell. 2004;14:763–74. doi: 10.1016/j.molcel.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 27.Opresko PL, Sowd G, Wang H. The Werner syndrome helicase/exonuclease processes mobile D-loops through branch migration and degradation. PLoS ONE. 2009;4:e4825. doi: 10.1371/journal.pone.0004825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kusumoto-Matsuo R, Opresko PL, Ramsden D, Tahara H, Bohr VA. Cooperation of DNA-PKcs and WRN helicase in the maintenance of telomeric D-loops. Aging (Albany NY) 2010;2:274–84. doi: 10.18632/aging.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maizels N. Dynamic roles for G4 DNA in the biology of eukaryotic cells. Nat Struct Mol Biol. 2006;13:1055–9. doi: 10.1038/nsmb1171. [DOI] [PubMed] [Google Scholar]
- 30.Mohaghegh P, Karow JK, Brosh RM, Jr., Bohr VA, Hickson ID. The Bloom’s and Werner’s syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 2001;29:2843–9. doi: 10.1093/nar/29.13.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rizzo A, Salvati E, Porru M, D’Angelo C, Stevens MF, D’Incalci M, et al. Stabilization of quadruplex DNA perturbs telomere replication leading to the activation of an ATR-dependent ATM signaling pathway. Nucleic Acids Res. 2009;37:5353–64. doi: 10.1093/nar/gkp582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vannier JB, Pavicic-Kaltenbrunner V, Petalcorin MI, Ding H, Boulton SJ. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell. 2012;149:795–806. doi: 10.1016/j.cell.2012.03.030. [DOI] [PubMed] [Google Scholar]
- 33.Shishkin AA, Voineagu I, Matera R, Cherng N, Chernet BT, Krasilnikova MM, et al. Large-scale expansions of Friedreich’s ataxia GAA repeats in yeast. Mol Cell. 2009;35:82–92. doi: 10.1016/j.molcel.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luke B, Lingner J. TERRA: telomeric repeat-containing RNA. EMBO J. 2009;28:2503–10. doi: 10.1038/emboj.2009.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Damerla RR, Knickelbein KE, Kepchia D, Jackson A, Armitage BA, Eckert KA, et al. Telomeric repeat mutagenicity in human somatic cells is modulated by repeat orientation and G-quadruplex stability. DNA Repair (Amst) 2010;9:1119–29. doi: 10.1016/j.dnarep.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang G, Vasquez KM. Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells. Proc Natl Acad Sci USA. 2004;101:13448–53. doi: 10.1073/pnas.0405116101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang G, Christensen LA, Vasquez KM. Z-DNA-forming sequences generate large-scale deletions in mammalian cells. Proc Natl Acad Sci USA. 2006;103:2677–82. doi: 10.1073/pnas.0511084103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu FJ, Barchowsky A, Opresko PL. The Werner syndrome protein functions in repair of Cr(VI)-induced replication-associated DNA damage. Toxicol Sci. 2009;110:307–18. doi: 10.1093/toxsci/kfp104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bacolla A, Wang G, Jain A, Chuzhanova NA, Cer RZ, Collins JR, et al. Non-B DNA-forming sequences and WRN deficiency independently increase the frequency of base substitution in human cells. J Biol Chem. 2011;286:10017–26. doi: 10.1074/jbc.M110.176636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neaves KJ, Huppert JL, Henderson RM, Edwardson JM. Direct visualization of G-quadruplexes in DNA using atomic force microscopy. Nucleic Acids Res. 2009;37:6269–75. doi: 10.1093/nar/gkp679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang H, Nora GJ, Ghodke H, Opresko PL. Single molecule studies of physiologically relevant telomeric tails reveal POT1 mechanism for promoting G-quadruplex unfolding. J Biol Chem. 2011;286:7479–89. doi: 10.1074/jbc.M110.205641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mashimo T, Yagi H, Sannohe Y, Rajendran A, Sugiyama H. Folding pathways of human telomeric type-1 and type-2 G-quadruplex structures. J Am Chem Soc. 2010;132:14910–8. doi: 10.1021/ja105806u. [DOI] [PubMed] [Google Scholar]
- 43.Shlyakhtenko LS, Potaman VN, Sinden RR, Lyubchenko YL. Structure and dynamics of supercoil-stabilized DNA cruciforms. J Mol Biol. 1998;280:61–72. doi: 10.1006/jmbi.1998.1855. [DOI] [PubMed] [Google Scholar]
- 44.Mirkin SM. Discovery of alternative DNA structures: a heroic decade (1979-1989) Front Biosci. 2008;13:1064–71. doi: 10.2741/2744. [DOI] [PubMed] [Google Scholar]
- 45.Rich A, Nordheim A, Wang AHJ. The chemistry and biology of left-handed Z-DNA. Annu Rev Biochem. 1984;53:791–846. doi: 10.1146/annurev.bi.53.070184.004043. [DOI] [PubMed] [Google Scholar]
- 46.Lyubchenko YL. DNA structure and dynamics: an atomic force microscopy study. Cell Biochem Biophys. 2004;41:75–98. doi: 10.1385/CBB:41:1:075. [DOI] [PubMed] [Google Scholar]
- 47.Fukuchi K, Martin GM, Monnat RJ., Jr. Mutator phenotype of Werner syndrome is characterized by extensive deletions. Proc Natl Acad Sci USA. 1989;86:5893–7. doi: 10.1073/pnas.86.15.5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang H, Freudenreich CH. An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol Cell. 2007;27:367–79. doi: 10.1016/j.molcel.2007.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oshima J, Huang S, Pae C, Campisi J, Schiestl RH. Lack of WRN results in extensive deletion at nonhomologous joining ends. Cancer Res. 2002;62:547–51. [PubMed] [Google Scholar]
- 50.Abbotts J, Bebenek K, Kunkel TA, Wilson SH. Mechanism of HIV-1 reverse transcriptase. Termination of processive synthesis on a natural DNA template is influenced by the sequence of the template-primer stem. J Biol Chem. 1993;268:10312–23. [PubMed] [Google Scholar]
- 51.Fortune JM, Pavlov YI, Welch CM, Johansson E, Burgers PM, Kunkel TA. Saccharomyces cerevisiae DNA polymerase delta: high fidelity for base substitutions but lower fidelity for single- and multi-base deletions. J Biol Chem. 2005;280:29980–7. doi: 10.1074/jbc.M505236200. [DOI] [PubMed] [Google Scholar]
- 52.Lee JY, Okumus B, Kim DS, Ha T. Extreme conformational diversity in human telomeric DNA. Proc Natl Acad Sci USA. 2005;102:18938–43. doi: 10.1073/pnas.0506144102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parris CN, Seidman MM. A signature element distinguishes sibling and independent mutations in a shuttle vector plasmid. Gene. 1992;117:1–5. doi: 10.1016/0378-1119(92)90482-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.