

Abstract
Transformation promotes genome plasticity in bacteria via RecA-driven homologous recombination. In the Gram-positive human pathogen Streptococcus pneumoniae, the transformasome a multiprotein complex, internalizes, protects, and processes transforming DNA to generate chromosomal recombinants. Double-stranded DNA is internalized as single strands, onto which the transformation-dedicated DNA processing protein A (DprA) ensures the loading of RecA to form presynaptic filaments. We report that the structure of DprA consists of the association of a sterile alpha motif domain and a Rossmann fold and that DprA forms tail-to-tail dimers. The isolation of DprA self-interaction mutants revealed that dimerization is crucial for the formation of nucleocomplexes in vitro and for genetic transformation. Residues important for DprA–RecA interaction also were identified and mutated, establishing this interaction as equally important for transformation. Positioning of key interaction residues on the DprA structure revealed an overlap of DprA–DprA and DprA–RecA interaction surfaces. We propose a model in which RecA interaction promotes rearrangement or disruption of the DprA dimer, enabling the subsequent nucleation of RecA and its polymerization onto ssDNA.
Keywords: bacterial transformation, genetic exchange, recombinase loader, recombination mediator protein, horizontal gene transfer
Homologous recombination (HR) plays essential roles in genome maintenance, the generation of genetic diversity, and proper chromosome segregation in all living organisms. HR is universally catalyzed by recombinases, i.e., RecA in bacteria, Rad51 in eukaryotes, and RadA in archaea (1, 2). HR recombinases promote the exchange of homologous DNA strands, a prerequisite for which is the formation of a complex between ssDNA and the recombinase called the “presynaptic filament.” Nucleation of the recombinase [i.e., binding of its first subunit(s) to DNA] is the rate-limiting step, hence the need for cofactors. A specialized class of cofactors interacting with ssDNA is represented by the recombination mediator proteins (RMPs), which have evolved to ensure recombinase loading on ssDNA bound by ssDNA-binding proteins (SSBs) (3). Thus, the well-characterized RMPs UvsY of bacteriophage T4, Rec(F)OR of Escherichia coli, and yeast Rad52 alleviate the SSB barrier and promote the loading of UvsX, RecA, and Rad51 on Gp32-, SSB-, and RP-A–precoated ssDNA, respectively (3). The E. coli RecBCD complex represents another category of cofactors that process linear dsDNA into ssDNA onto which they simultaneously ensure the loading of the recombinase (4).
Neither the RecBCD equivalent RexAB (5) nor RecFOR is required for genetic transformation of the Gram-positive bacterium Streptococcus pneumoniae (6). However, we recently identified the widespread bacterial protein, DNA processing protein A (DprA), as a transformation-dedicated RecA loader (7). Transformation is believed to play a major role in the genetic plasticity of this human commensal and major pathogen (8, 9). For example, transformation allows variation of the polysaccharide capsule (the primary determinant of pneumococcal virulence) through access to a species gene pool almost equivalent to the size of an individual genome (10).
Pneumococcal transformation requires transient differentiation to competence, during which expression of a specific set of genes (11) allows assembly of the transformasome (6). This dynamic machine involves both membrane and cytosolic proteins (including DprA and RecA), and promotes internalization, protection, and processing of transforming DNA into recombinants. Competence (or X-state) (11) is induced in exponentially growing cultures by a peptide pheromone, competence-stimulating peptide (CSP) (12), which ultimately activates the synthesis of an alternative sigma factor specific for competence (σX) (13). Induction of competence by some antibiotics and DNA-damaging agents (14) supports the view that CSP functions as an alarmone and that competence/X-state constitutes an SOS substitute for S. pneumoniae (11).
During the course of transformation, DprA may interact with and facilitate the loading of RecA onto two types of substrates, naked and SSB-coated ssDNA. The former could occur close to the membrane immediately after initiation of exogenous dsDNA uptake, which results in the internalization of ssDNA with 3′ → 5′ polarity (6), and may lead to the formation of mixed DprA-RecA nucleofilaments. Such mixed filaments were documented previously in vitro and were shown to be proficient in the catalysis of homology-dependent synapsis (7). Internalized ssDNA also could be complexed with the transformation-dedicated SSB protein, SsbB. Such complexes were detected in vivo (15) and were proposed to represent a reservoir of transforming ssDNA (7); recently, evidence was provided favoring a reservoir-maintenance role for SsbB (16). In vitro data revealed the capacity of DprA to promote the loading of RecA on SSB–ssDNA complexes (7).
DprA possesses all the distinctive properties of RMPs, including the ability to bind ssDNA and interact with RecA, leading us to propose that it is a member of the RMP family dedicated to natural bacterial transformation (7). However, no evidence was provided that both the ssDNA-binding and RecA-interaction properties of DprA are required for transformation. In addition, the mechanism of the DprA-facilitated loading of RecA onto ssDNA remained elusive. Here, we report that the structure of DprA consists of the association of a sterile alpha motif (SAM) domain and a Rossmann fold (RF). We show that DprA forms tail-to-tail dimers and that dimerization is crucial for formation of nucleocomplexes in vitro. Taking advantage of the previous detection of DprA–DprA and DprA–RecA interactions in yeast two-hybrid assays (Y2H) (7), we isolated DprA mutants deficient in either of these interactions. This identification of key DprA residues allowed us to demonstrate that dimerization and interaction with RecA are equally important for transformation. Structural comparison between S. pneumoniae DprA and DprA from Rhodopseudomonas palustris [recently deposited in the Protein Data Bank (PDB); ID 3MAJ] and with 3D models of DprA from E. coli led us to conclude that (i) the widespread DprA family is structurally defined by the association of a SAM and an RF, and (ii) DprA dimerization potential and interface residues are evolutionarily conserved. On the other hand, the finding that DprA dimerization and RecA interaction residues partially overlap, together with the puzzling observation that RecA interaction residues appear to be evolutionarily less conserved, led us to postulate a much larger overlap of the two interaction surfaces. Site-directed mutagenesis of the most conserved DprA dimerization residues then established that these residues also are important for interaction with RecA. We conclude that there is a large overlap between the dimerization and RecA interaction interfaces, suggesting that, upon interaction, RecA promotes rearrangement or disruption of the DprA dimer, enabling subsequent nucleation and loading of RecA onto transforming ssDNA fragments.
Results
Overall Structure of S. pneumoniae DprA.
The structure of S. pneumoniae DprA was solved at 2.7-Å resolution by single-wavelength anomalous dispersion using seleno-methionine–labeled protein. Refinement statistics are presented in Table S1 and show good stereochemistry for the final model. Three copies of DprA (chains A, B, and C) are present per asymmetric unit in the crystal. The final model is complete, containing all residues (1–282) for chains A and C with the addition of one histidine from the His-tag for chain A. Chain B lacks the last two residues, E281 and F282. A sulfate ion is visible on the surface of each chain, linked via hydrogen bonds with R115, S230, and G229.
DprA consists of two domains (Fig. 1A). The N-terminal domain (residue M1-F71) is composed of five helices (Fig. 1 A and C) and presents considerable structural similarity to the SAM domain of PA4738, a protein of unknown function from Pseudomonas aeruginosa (PDB code 1YWW; Z score 2.1; rmsd of 2.97 Å). SAM domains frequently are involved in various types of protein interaction (17).
Fig. 1.

3D structure of DprA from S. pneumoniae. (A) Ribbon schematic presentation of the DprA 3D structure. The N and C termini and secondary structure elements are indicated. Color code: red, SAM domain; blue-green, RF; green, eRF extension. S, SAM; e, RF extension; RF, Rossmann fold. (B) Linear representation of DprA with limits of Pfam02481 and structural domains (color code as in A). The Pfam02481 domain is shown in blue. (C) Topological representation of DprA (color code as in A). (D) Ribbon schematic presentation of the 3D structure of AB′ or CC′ DprA dimers. (Inset) The C-terminal dimerization zone is enlarged to show some of the residues (represented as yellow sticks) involved in the interaction (see also Fig. 2 A and B and Table S2). Hydrogen bonds are represented by dashed lines, and distances between residues are indicated in Å.
The C-terminal region of DprA, starting approximately from residue P107, adopts an RF-like topology with a typical three-layer (αβα) sandwich (hereafter called “RF”) (Fig. 1 A and C). The short region connecting the SAM and the RF is composed of two antiparallel β-strands, each followed by α-helices. The β-strands of the connecting region are associated by main-chain hydrogen bonds to the main β-sheet of the RF and therefore can be considered as an extension of the RF. Furthermore, the limits of Pfam02481, which characterizes the DprA protein family, almost exactly match the extension plus the RF (Fig. 1B). We refer to this entity hereafter as “extended RF” (eRF). The SAM packs closely onto the eRF, and the whole DprA structure forms a bean-shaped, globular structure.
DprA Dimers in the Crystal.
Analysis of crystal packing revealed two distinct types of DprA dimers involving RF–RF and SAM–SAM interactions, respectively. RF–RF interactions result in the formation of two nearly identical tail-to-tail (hereafter “C/C”) and antiparallel dimers [rmsd of 0.59 Å and a template-modeling (TM) score of 0.99118 (TM-align server, http://zhanglab.ccmb.med.umich.edu/TM-align/)] (Fig. 1D) (18). Both the first C/C dimer, composed of molecules A and its symmetric B′, and the second dimer, composed of molecules C and C′, are generated by a crystallographic twofold-symmetry axis. An angle of 130–150° is observed between the two subunits of C/C dimers. Their interfaces bury very similar surface areas: 1,496 Å2 and 1,430 Å2 for the AB′ and CC′ dimers, respectively [calculated by addition of the surfaces of the two monomers in contact; European Bioinformatics Institute Protein Interactions Surfaces and Assemblies (PISA) service; http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html]. Residues that contribute to C/C dimerization in the crystal are listed in Table S2, together with an estimate of accessibility of individual residues in the dimer versus the monomer. Interface residues are located on α6, the loop between β8 and α11, α11, β9, and α12 (Figs. 1D and 2A and Table S2). The interface is mainly hydrophobic, with the addition of hydrogen bonds between Q264 of one monomer and L269–D275 from the opposite monomer (Fig. 1D).
Fig. 2.

Location of DprA residues involved in the dimerization interface and in interaction with RecA. (A) Ribbon view with DprA residues involved in C/C interactions shown in yellow. Green residues indicate C/C interaction residues mutated to generate the DprAAK and DprAAR monomeric mutants. (B) Linear representation of DprA with DprA interaction residues and mutants. C/C dimerization interface residues are shown immediately above the linear map (see also Table S2). DprA mutants unable to self-interact but retaining full capacity to interact with RecA are shown in yellow within a gray rectangle above the linear map. Mutations affecting interaction with RecA but not dimerization are shown in purple below the linear map. For each mutation, the wild-type residue is indicated first, followed by its position, and then the mutant residue is shown. (C) Ribbon view with DprA residues involved in interaction with RecA shown in purple. Color code is as in Fig. 1A.
The solvent content of DprA crystals is high (70%), and the crystals are characterized by remarkable major and minor channels in the lattice, with diameters of 130 and 100 Å, respectively (Fig. S1A). DprA forms filament-like packing along these solvent channels. These twisted filaments are composed of C/C dimers interacting through their N termini, i.e., via SAM/SAM interaction, to form multimers (Fig. S1B). [SAM/SAM interactions (Fig. S1C) are referred to as “N/N” interactions hereafter.]
Y2H Screen for DprA-Interaction Mutants.
We first sought to establish which C/C or N/N interactions are required for dimerization in vivo by taking advantage of Y2H, which previously revealed DprA self-interaction (7). Therefore a Y2H screen for DprA mutants that are unable to self-interact but retain full capacity to interact with RecA was launched (Materials and Methods) with the aim of identifying DprA residues important for self-interaction in vivo. This genetic screen isolated three double mutants, C234R-F245L, I251V-H260R, and D257G-L269S (Fig. 2B). Interestingly, these mutations affected only C-terminal residues, three of which (I251, H260, and L269) are involved directly in the C/C dimerization interface in the crystal. On the other hand, no variant supporting the N/N interfacing was isolated, allowing us to conclude tentatively that in vivo DprA self-interaction primarily involves C/C interactions.
Purified DprA Forms C/C Dimers in Solution.
To investigate whether DprA also forms dimers in solution and to characterize the interaction interface, we carried out gel filtration and small-angle X-ray scattering (SAXS) experiments with purified proteins. We compared wild-type DprA with the Y2H C-terminal interaction mutant DprAI251V-H260R and with the DprAH260A-L269K and DprAH260A-L269R double mutants (hereafter referred to as DprAVR, DprAAK, and DprAAR, respectively). The latter two were designed to combine mutations of residues first identified in the screen for self-interaction mutants and also engaged in the C/C dimer interface in the crystal (Fig. 2B and Table S2).
Gel filtration experiments indicated that both DprA and DrpAAR behave as a single homogeneous species, but DprA eluted from the sizing column with an apparent mass between a monomer and a dimer, whereas DprAAR eluted with an apparent mass about half that observed for DprA (Fig. S2). Although the apparent molecular mass of DprAAR was lower than that predicted for DprA (31.885 kDa including the His-tag), these results were consistent with the DprAAR mutant protein being monomeric. Analysis of DprA SAXS data (Fig. 3A) then allowed two independent estimates of molecular mass, establishing unequivocally that DprA is a dimer in solution (SI Materials and Methods, SAXS Data Analysis). Then model reconstruction from the DprA experimental curve was carried out and revealed quite satisfactory superimposition of the C/C crystallographic dimer on the reconstructed volume (Fig. 3C). Moreover the calculated curve (red line in Fig. 3A) from the C/C crystal structure was in good agreement with the experimental curve (SI Materials and Methods, SAXS Data Analysis). A similar analysis performed using the N/N dimer indicated a weaker fit between the calculation from the N/N dimer and the experimental data (Fig. S3 B–E). Together, these results established unambiguously that purified DprA self-assembles as a dimer in solution, more likely as a C/C than an N/N dimer. It is of note that the envelope of the C/C dimer obtained from SAXS data appeared somewhat more closed than the crystal structure (Fig. S3C), suggesting some flexibility between the two monomers (Fig. S3E).
Fig. 3.

Both DprA dimerization in solution and oligomerization in pneumococcal cells rely on C/C interactions. (A) SAXS analysis of purified DprA protein. Adjustment of the curve calculated from crystal structure of DprA C/C dimer (red line) to the experimental scattering curve (black dots) of DprA (SI Materials and Methods). Arrows indicate the theoretical scattering values calculated from the sequence-derived molecular mass (including the His-tag) for the monomer and for the dimer, 31,885 and 63,770 respectively. (B) SAXS analysis of purified DprAAR mutant. Experimental scattering curve of DprAAR (black dots) superimposed on the curve calculated from crystal structure of DprA monomer (mauve line). (C) Crystal structure of the DprA C/C dimer superimposed on a typical envelope of the protein deduced from the SAXS experimental curve of DprA in A using the program GASBOR (SI Materials and Methods). The lower envelope is rotated 90° on the horizontal axis compared with top envelope. (D) Crystal structure of the monomer superimposed on a typical envelope of the protein deduced from the SAXS experimental curve of DprAAR in B. The lower envelope is rotated 90° on the horizontal axis compared with top envelope. (E) Copurification of DprA and SPA-tagged DprAs documenting the existence of C/C interactions in pneumococcal cells. Total extracts from dprA+ competent cells ectopically expressing DprA-SPA or DprAAR-SPA fusions were immunoprecipitated using anti-Flag antibodies, affinity purified, and eluted by proteolytic cleavage (SI Materials and Methods). Load, i.e., soluble whole-cell extract (L), flow through, i.e., fraction of immunoprecipitate not retained on the affinity column (FT1), and capture, i.e., eluate from the first affinity column (C1) were immunoblot analyzed using anti-DprA antibodies (α-DprA). The asterisk indicates tagged product released by proteolysis. All three capture experiments shown in E and in Fig. S3G were carried out in parallel and were analyzed on the same gel and membrane. (F) Control experiment with a GFP-SPA fusion.
In contrast, the DprAAR and DprAAK mutants appeared fully monomeric (Fig. 3B and Fig. S3A), whereas SAXS experiments suggested equilibrium between monomer and dimer for the DprAVR mutant (Fig. S3F). For DprAAR and DprAAK, the crystal structure of the monomer is nicely accommodated within the envelope deduced from the scattered-intensity curve I(q) curve (see Fig. 3D for DprAAR; identical results were obtained for DprAAK). Moreover the curves calculated from the crystal structure are in good agreement with the experimental ones (see Fig. 3B for DprAAR and Fig. S3A for DprAAK). These results establish that the DprAAR and DprAAK mutant proteins are monomeric in solution, providing evidence that DprA dimerization relies primarily on C/C interactions.
DprA Oligomerization in Pneumococcal Cells Relies on C/C Interactions.
To investigate DprA oligomerization status in S. pneumoniae competent cells, we tested whether the selective purification of a tagged DprA protein could mediate the copurification of a wild-type (i.e., untagged) DprA expressed in the same cell. To this end, sequential peptide affinity (SPA)-tagged DprA [i.e., harboring a C-terminal Flag epitope separated from a calmodulin-binding domain by a cleavage site for the TEV protease (19)] was ectopically expressed under the control of a competence-inducible promoter (Materials and Methods). The resulting ectopic construct was found to complement fully the transformation defect of a dprA-null mutant strain, demonstrating that the DprA-SPA fusion is functional. Capture on beads coated by monoclonal anti-Flag antibodies proved to be quantitative, because most, if not all, DprA-SPA molecules were removed from soluble protein extract (Fig. 3E, lane FT1). Interestingly, substantial amounts of DprA molecules were copurified (Fig. 3E, lane C1), whereas only trace amounts of DprA were detected in a control experiment with SPA-tagged GFP expressed under the control of a competence-inducible promoter (Material and Methods and Fig. 3F). These results demonstrated that DprA is dragged along specifically and efficiently by DprA-SPA during the Flag purification step and provided evidence for DprA oligomerization in pneumococcal cells, i.e., under physiological conditions.
To check whether the copurification of DprA with DprA-SPA relied on interaction via the C-terminal dimerization interface, we then duplicated this experiment with pneumococcal cells expressing the DprAAR-SPA fusion. Remarkably, although DprAAR-SPA was captured efficiently on anti-Flag beads, only trace amounts of DprA were copurified (Fig. 3E, lane C1), providing direct evidence that DprA dimerizes via its C terminal in S. pneumoniae. The same conclusion was attained through capture of a DprAVR-SPA fusion, which similarly led to the copurification of only trace amounts of DprA (Fig. S3G). We conclude that DprA oligomerization, presumably dimerization, occurs readily in S. pneumoniae competent cells and relies on C/C interactions.
DprA Oligomerization Is Crucial for Genetic Transformation.
Next, the H260A-L269R pair of mutations was introduced at the dprA chromosomal locus to investigate its effects on pneumococcal transformation. The resulting dprAAR strain was found to express wild-type protein levels (Fig. 4A, compare lanes L). Interestingly, this mutant presented a more than 2-log decrease in transformation efficiency compared with wild type (Fig. 4B), pinpointing the importance of DprA C/C dimerization for genetic transformation. The DprAVR mutant displayed a similar transformation defect (Fig. 4B). Because DprAVR is only partially dimeric in solution (Fig. S3F), we concluded that stable DprA dimerization is crucial for chromosomal transformation.
Fig. 4.

DprA dimerization is required for the formation of NPC and for genetic transformation but not for interaction with RecA. (A) DprA dimerization is not required for interaction with RecA in S. pneumoniae. Cocaptures of DprA (Left) and DprAAR (Right) using RecA-SPA expressed ectopically in recA+ dprA+ or recA+ dprAAR competent cells. RecA and DprA proteins were revealed using α-RecA and α-DprA antibodies. See legend of Fig. 3E for details. (B) Comparison of chromosomal transformation deficiency in the DprAAR and DprAVR dimerization mutants and in wild-type DprA cells. (C) Equilibrium binding of DprA and DprAAR to fluorescein-labeled dT20. Fluorescence anisotropy variation with DprA (black circles) and DprAAR (open circles) concentrations fit to a single-ligand–binding model with SigmaPlot-calculated (apparent) Kd of 220 nM and 567 nM for DprA and DprAAR, respectively. (D) EMSA of DprA and DprAAR binding to a 32P-dT100 probe. Increasing amounts of purified proteins were incubated with a 0.1-nM probe as described in SI Materials and Methods. Apparent Kd calculated for DprA, 28 nM.
Although the DprAVR change disrupts DprA dimerization in Y2H without affecting interaction with RecA, the effect of DprAAR with respect to the latter had not been documented. We took advantage of pull-down experiments with tagged proteins to establish whether the DprAAR mutations affect the interaction with RecA in pneumococcal cells. Using a functional RecA-SPA fusion expressed ectopically and specifically in competent cells (Materials and Methods), similar amounts of DprA and DprAAR were captured (Fig. 4A, compare lanes C1). This experiment established that the DprAAR monomeric mutant is not affected by interaction with RecA and ruled out the possibility that its transformation deficiency was caused by an inability to interact with RecA. This conclusion was confirmed by Y2H showing that DprAAR fails to self-interact but interacts normally with RecA (Table 1).
Table 1.
Overview of results regarding DprA structure–function relationships
| Interaction with DprA |
ssDNA binding* | Interaction with RecA |
Transformation† | |||||
| DprA protein | Crystal | In vitro‡ | Y2H | In vivo§ | Y2H | In vivo¶ | ||
| Wild type | Yes | Yes | Yes | Yes | Yes | Yes | Yes | + |
| 234R-245L | ‖ | No | ↓ ∼1 log | |||||
| 257G-269S | No | ↓ ∼1 log | ||||||
| VR (251V-260R) | Mix** | No | No | Yes | Yes | ↓ 183 ± 23-fold | ||
| AR (260A-269R) | No†† | No | No | No | Yes | Yes | ↓ 176 ± 23-fold | |
| 235G, 243G, 264R or 265G‡‡ | Yes | No | + | |||||
| QN (235Q-243N) | Yes | Yes | ±§§ | ↓ 7.9 ± 0.9-fold | ||||
| QNQ (235Q-243N-265Q) | Yes | Yes | Yes | No | ↓ 154 ± 18-fold | |||
*Formation of NPC based on EMSA; note that FAT revealed interaction of the DprAAR protein with a 20-mer oligonucleotide (Fig. 4C).
†+, wild-type transformation frequency; fold reduction is indicated compared with wild type.
‡Based on SAXS analysis (Fig. 3A): yes, dimer; no, monomer.
§Revealed by cocapture from S. pneumoniae competent cells using DprA-SPA.
¶Revealed by cocapture from S. pneumoniae competent cells using RecA-SPA.
‖Empty box, not determined.
**SAXS indicated equilibrium between monomer and dimer (Fig. S3F).
††Beside SAXS results, in gel filtration experiments DprAAR eluted from the sizing column as a single homogeneous species with an apparent mass about half that observed for DprA and DprAQNQ (Fig. S2).
‡‡All individual mutants behave similarly.
§§Reduced cocapture of DprAQN with RecA-SPA (Fig. 5A).
DprA Dimerization Is Required for Formation of Stable Nucleocomplexes.
In search of an explanation for the transformation defect of the DprAAR monomeric mutant, we then investigated its ssDNA-binding activity in vitro using two types of assay. We first used fluorescence anisotropy titration (FAT), which showed that both purified DprA and DprAAR proteins could interact efficiently with a 5′ fluorescein-labeled dT20 oligonucleotide in solution, with the latter possibly exhibiting a slightly lower affinity (Fig. 4C). However, the difference in apparent Kd was only twofold (Fig. 4C), probably too mild to account for DprAAR transformation defect. In any case, these results suggested the presence of an ssDNA binding site on the monomer.
We then carried out EMSA, which were used previously to document the binding of DprA to dT90 oligonucleotide (7). EMSA had revealed that DprA produces nucleoprotein complexes (NPC) that barely enter the polyacrylamide gel (7); ssDNA trapped by DprA in the wells could be released by the addition of excess cold competitor ssDNA, demonstrating that NPC are formed reversibly and do not represent dead-end reaction products. Further studies by transmission electron microscopy established that NPC consist of a network of several ssDNA molecules bridged by DprA molecules (7). Fig. 4D shows that although DprA readily formed NPC with a labeled dT100 oligonucleotide, the DprAAR monomeric mutant failed to produce any complex. This striking observation strongly suggested that C/C dimerization of DprA is crucial for NPC formation. To understand NPC formation by DprA dimer better, the oligonucleotide size limit for NPC detection by EMSA was investigated. Examination of wild-type DprA binding to dT50, dT40, dT30, and dT20 revealed that NPC formed readily only with dT50 (Fig. S4). A significant increase in apparent Kd was observed with dT40, and NPC formation was severely affected with dT30, whereas no NPC could be detected with dT20. This lack of NPC formation with dT20 contrasted with the detection of DprA binding through FAT. This difference is inherent to the two assays; FAT involves equilibrium binding in solution, whereas EMSA requires nucleocomplexes stable enough to resist electrophoresis. We conclude from these data that primary ssDNA binding (including binding with the DprAAR monomeric mutant) occurs readily with a 20-nt-long substrate, but that formation of stable NPC requires a C/C dimer of DprA and ssDNA molecules longer than 30 nt, and becomes optimal with 50 nt. We propose that the transformation deficiency of monomeric mutants of DprA results from the failure to form stable NPC with incoming ssDNA.
DprA Residues Required for Interaction with RecA in Vivo.
Although previous data provided evidence for direct interactions between DprA and RecA (7), the residues involved remain unknown. Therefore we used the Y2H screen for DprA interaction mutants to isolate mutants that are disrupted in their interaction with RecA but retain a full capacity to self-interact. Seven single and three double mutants were isolated. All mutated residues map in the RF, several on the surface of DprA and, interestingly, some are very close to the C-terminal dimerization interface (Fig. 2 B and C). To investigate the effect of such mutations in pneumococcal cells, we retained four point mutations located at the C terminal of DprA, namely, E235G, D243G, Q264R, and E265G. When introduced individually at the dprA chromosomal locus, none affected chromosomal transformation (Table 1).
Several explanations could account for the differential effect of single-residue mutations in S. pneumoniae and yeast. First, DprA–RecA interaction may not be required for transformation. Second, the nature of the physical interaction required for proper functioning in the two systems may differ. A slight variant of the latter explanation would be that in pneumococcal cells, DprA–RecA interaction is robust and can be abolished only through the simultaneous mutation of several residues. To investigate the latter possibility, we attempted to combine mutations of residues important for interaction with RecA in yeast, in the hope of generating a stronger effect in S. pneumoniae. We targeted three acidic residues (E235, D243, and E265) that are clustered on the surface of DprA (Fig. 2C; see also Fig. 7A). To minimize structural alterations resulting from the accumulation of mutations, we changed these residues into Q, N, and Q, respectively. The triple mutant, “DprAQNQ” hereafter, was characterized first in vitro. Gel filtration (Fig. S2 B and C) and SAXS analysis (Fig. S5A) indicated that purified DprAQNQ is a homogeneous dimer in solution. DprAQNQ also was found to be indistinguishable from wild-type DprA with respect to both ssDNA binding activity (Fig. S5B) and ability to form NPC (Fig. S5C).
Fig. 7.

DprA dimerization, formation of NPC, and interaction with RecA in the processing of transforming DNA. (A) Overlap between DprA dimerization and RecA interaction interfaces. Color code: green, surface residues required for both DprA dimerization and interaction with RecA; mauve, surface residues affecting interaction with RecA but not DprA dimerization; yellow, surface residues involved only in DprA dimerization. (B) Schematic representation of the production of NPC in vitro. Orange, DprA C-terminal dimerization interface; blue, ssDNA-binding domain. Black and red lines represent different ssDNA molecules. The possible contribution of N/N interactions (Fig. S1C) to the formation of NPC via the bridging of C/C dimers is illustrated. The previously demonstrated cooperative binding of DprA to ssDNA (7) is attributed tentatively to a conformational change in the dimer, triggered upon ssDNA binding, leading to protein–protein interactions (red open arrow). This possibility is supported indirectly by the observation in the crystal of monomer piles relying on interaction between oppositely charged central regions (M/M interaction) from two monomers (1,940.6-Å2 surface area buried in the interface). (C) Interplay of DprA and RecA in the early processing of incoming ssDNA. *Despite the ability of both DprA monomers and dimers to bind ssDNA, only the latter can form NPC, which is crucial for transformation. **Interaction of RecA with DprA is illustrated as triggering dimer dissociation. Alternatively, RecA may induce only a large conformational change facilitating interaction with residues whose accessibility is limited in the dimer (see D). ***Nucleation of RecA is drawn arbitrarily as involving three monomers. (D) Accessibility of RecA interaction residues in DprA dimer versus monomer. Residues in mauve remain fully accessible to RecA in a C/C dimer of DprA. We suggest they are targeted first by RecA and belong to a primary interaction site (“1st”). A subsequent conformational change in the dimer or its complete dissociation could provide full access to residues buried in the dimer and allow establishment of stronger DprA–RecA interactions (“2nd”).
These three mutations then were introduced at the dprA chromosomal locus. The strategy used led to the isolation of a double mutant (mutated for E235 and D243 residues; hereafter “DprAQN”) in addition to the triple mutant. Expression levels and solubility of DprAQN and DprAQNQ in S. pneumoniae cells were found to be identical to those of DprA (Fig. S5D, lanes L). Pull-down experiments revealed that both DprAQN and DprAQNQ interacted with RecA less efficiently than wild-type DprA (Fig. 5A). Quantification of the amount of DprA cocaptured by the RecA-SPA fusion indicated a 1.6-fold and fivefold decrease in DprAQN and DprAQNQ recovery, respectively. Taken together, these results led to the identification of three DprA surface residues that collectively are important for stable physical association with RecA in competent cells of S. pneumoniae. It is of note that DprA–RecA complexes formed in vivo are quite stable, being maintained through two consecutive stages of purification (SI Materials and Methods; and Fig. S5D, lane C2).
Fig. 5.

DprA–RecA interaction is required for genetic transformation. (A) DprAQN and DprAQNQ display reduced stability of complexes with RecA in vivo. Cocaptures of DprA (Left), DprAQN (Center), and DprAQNQ (Right) using RecA-SPA expressed ectopically in recA+ cells harboring the dprA+, dprAQN, or dprAQNQ alleles were conducted as described in the legend of Fig. 3E but with an additional purification step before elution (SI Materials and Methods). Only C2 final eluates are shown in this figure, but note that all three capture experiments were carried out in parallel and were analyzed on the same membrane (Fig. S5D). The amount of mutant DprAs captured is indicated relative to that of wild-type DprA. Signals were quantified using a Fujifilm LAS-4000 luminescent image analyzer. (B) Chromosomal transformation deficiency of the RecA-interaction mutants DprAQN and DprAQNQ compared with wild-type DprA.
DprA–RecA Interaction Is Required for Genetic Transformation.
Both DprAQN and DprAQNQ mutants exhibited a significant reduction in the frequency of chromosomal transformation (Fig. 5B), although the triple mutant was clearly more affected (154- versus eightfold reduction). Because DprAQNQ also was more affected in its capacity to form complexes with RecA (Fig. 5A), we conclude both that DprA interaction with RecA is required for pneumococcal transformation and that the efficiency of transformation correlates directly with the ability of DprA to interact and form stable complexes with RecA in competent cells.
Evolutionary Conservation of DprA Structural Features.
To determine whether the main structural features established for S. pneumoniae DprA (SpDprA) could be extended to other members of the family, we took advantage of the recent availability of the structure of DprA from R. palustris (RpDprA; PDB ID 3MAJ) to launch a comparative analysis. Examination of the RpDprA structure confirmed the presence of an eRF (Fig. S6A) that exhibited excellent structural overlap with that of SpDprA (Fig. S6B), as expected given the conservation of Pfam02481. In addition, a C-terminal extradomain folded independently of the eRF in RpDprA (yellow domain in Fig. S6A).
Interestingly, dimers were present in the RpDprA crystal, and, despite the presence of the C-terminal extradomain, they also involved RF–RF interactions (Fig. S6C, C/C dimer). Comparison of residues involved in SpDprA and RpDprA dimerization surfaces revealed good sequence conservation (Table S2). Nine of 15 RpDprA interface residues were identical to those in SpDprA. Stabilization of SpDprA and RpDprA dimers involves predominantly hydrophobic interactions, together with hydrogen bonds for the former and a combination of hydrogen bonds and salt bridges for the latter (Table S2). Three dimerization interface residues identified in SpDprA (P248, G249, and I263) (Fig. 6A) were identical in RpDprA, E. coli DprA (EcDprA), and Bacillus subtilis DprA (BsDprA) (Fig. S7). More generally, these residues appeared to be the best-conserved surface residues among 60 selected Pfam02481 sequences (Fig. 6B), arguing in favor of the evolutionary conservation of the DprA dimerization potential and interfacing residues.
Fig. 6.

Evolutionary conservation of DprA structural features. (A) Location of residues involved in dimerization on the DprA surface. Residues in mauve rectangles also are involved in interaction with RecA. Residues labeled in green are amino acids mutated to generate the DprAAK and DprAAR monomeric mutants. (B) Conservation of residues of the dimerization surface among 60 representative DprA proteins. The intensity of the green gradient indicates the degree of conservation among 60 selected Pfam02481 sequences. The conservation gradient shown on the SpDprA monomer surface was generated using ESPript 2.2 (32). (C) DprA surface residues important for interaction with RecA are shown in bright pink. The two residues labeled in green, which correspond to amino acids mutated to generate DprA monomeric mutants, are shown to facilitate comparison with A. (D) Nonconservation of SpDprA surface residues important for interaction with RecA among 60 representative DprA proteins (see legend of B).
In contrast, only 3 of 13 SpDprA residues involved in interaction with RecA (Fig. 6C) appeared strictly conserved in RpDprA, BsDprA, and EcDprA (Table S3). All three correspond to buried residues suggesting they are structurally important rather than directly engaged in DprA–RecA interaction. This observation suggested a rather limited evolutionary conservation, which was confirmed by examination of 60 selected Pfam02481 sequences (Fig. 6D).
Overlap Between DprA Dimerization and RecA Interaction Interfaces.
The apparent lack of conservation of RecA interaction residues in the DprA family appeared paradoxical for a protein involved primarily in the loading of RecA onto ssDNA. We first considered the possibility that DprA–RecA interaction could involve surface residues prone to species-specific evolution to adapt to their corresponding cytosolic environment. In support of this idea was the finding that DprAE265Q substitution (Table 1) affected interaction with RecA in pneumococcal cells, whereas a Q residue was present at that same position in three wild-type proteins, RpDprA, BsDprA, and EcDprA (Table S3). On the other hand, the previous observation that SpDprA interacted with BsRecA in Y2H and promoted the loading of EcRecA onto ssDNA (7) provided evidence for the maintenance of functional interactions between phylogenetically distant proteins, which is difficult to reconcile with the idea of a species-specific evolution of DprA surface residues involved in RecA interaction.
These considerations prompted us to consider the alternative hypothesis of an overlap between DprA dimerization and RecA interaction interfaces. This hypothesis would be consistent with the identification of S124 and Q264 as potentially involved in both types of interaction (Figs. 2B and 6 A and C). According to this hypothesis, our Y2H screen for DprA mutants selectively affected in their interaction with RecA but retaining full capacity to self-interact (i.e., to dimerize) could have failed to identify most RecA interaction residues. To check this possibility, six residues belonging to the dimerization interface of SpDprA and well-conserved in the family—G249, S250, I263, L269, T271, and D275—were selected for site-directed mutagenesis (SI Matherials and Methods). Twenty-four individual residue changes were obtained with one or more substitutions at each site affecting the potential of DprA interaction in Y2H (Table 2). Although five changes had no effect, six substitutions significantly reduced and 13 abolished the DprA–RecA interaction. On the other hand, only one substitution, the L269W change, abolished DprA self-interaction. We tentatively attribute the last observation to the fact that the DprA dimerization assay involved a combination of DprA mutant and wild-type proteins. Most importantly, for each of the selected dimerization residues mutagenized, at least one substitution resulted in a significant reduction or the complete abolition of the interaction with RecA. We conclude that there is a large overlap of the two interaction surfaces in Y2H, suggesting that in S. pneumoniae the two interactions could be mutually exclusive (i.e., that interaction with RecA could promote the discruption of the DprA C/C dimer) (Fig. 7C).
Table 2.
Y2H-based identification of additional RecA interaction residues by site-directed mutagenesis of DprA dimerization residues
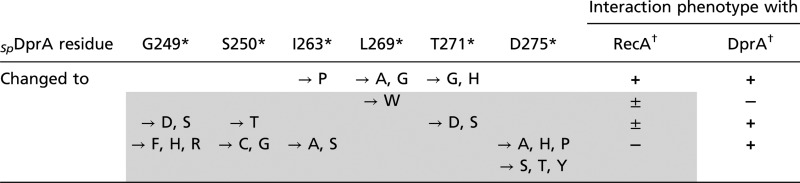 |
The gray-shaded area identifies changes in conserved dimerization residue affecting the interaction of DprA with RecA
*Residue and position in wild-type SpDprA. Their involvement in dimerization and conservation in other DprA proteins are documented in Table S2.
†From + (wild-type–like Y2H interaction) to − (no interaction), with ± indicating slow growth of yeast diploid cells (i.e., significantly reduced interaction).
Discussion
Structural Definition of the DprA Family.
The resolution of the full-length structure of SpDprA, the transformation-dedicated RecA loader of S. pneumoniae, revealed that it consists of the association of an SAM and an eRF (Fig. 1A). Although an eRF also was present in the structure of the phylogenetically distant RpDprA, its N terminal did not exhibit a SAM fold (Fig. S6A). Nevertheless, the intriguing structural similarity between SpDprA and RpDprA N terminals prompted us to deepen our analysis. Firstly, we observed that the SAM of SpDprA could not be predicted through 3D modeling using RpDprA coordinates (Fig. S6D), suggesting that constraints in the RpDprA crystal prohibited SAM folding. Second, in both cases 3D modeling of the N terminal of RpDprA and EcDprA using SpDprA coordinates predicted a bona fide SAM fold (Fig. S6E). Taken together, these observations led us to propose that all members of the DprA family harbor a SAM fold at their N terminals. The DprA family, previously defined at the primary sequence level by the Pfam02481 (7), now can be regarded at the structural level as a larger entity comprising an eRF, which entirely overlaps Pfam02481, preceded by a SAM fold. The SAM-eRF structural association is totally unrelated to the structure of any other recombinase loaders, i.e., RecBCD (20), RecFOR (ref.21 and references therein), Rad52 (22, 23), and BRCA2 (24, 25).
Previous primary sequence analysis indicated that some DprA orthologs, including RpDprA (Fig. S6A) and EcDprA, contain a C-terminal extradomain next to Pfam02481. This extradomain could modulate properties common to all DprAs or confer distinct additional properties. With respect to the latter possibility, it might be relevant that the C-terminal extradomain of RpDprA exhibited an excellent structural match with the Z-DNA–binding tumor-associated protein DML-1 (26). This extradomain does not prevent the formation of RF-RF dimers (Fig. S6C), strongly suggesting that dimerization, which is crucial for transformation of S. pneumoniae, is an evolutionarily conserved property of DprA proteins. This conclusion is fully consistent with a previous report that BsDprA also displays self-interaction in Y2H (7).
Dimerization/NPC Formation and RecA-Interaction Properties of SpDprA Are Crucial for Transformation.
Previous studies provided evidence for DprA–DprA and DprA–RecA interaction in yeast, as well as for DprA–RecA interaction in pneumococcal cell extracts (7). Here, our detailed phenotypic characterization of strains harboring point mutations in dprA established that these interactions are functionally important for genetic transformation. Both the DprA monomeric mutant, DprAAR, and the RecA-interaction mutant, DprAQNQ, display a 2-log transformation defect (Table 1), demonstrating that each interaction is crucial for this process.
Although the importance of DprA–RecA interactions for pneumococcal transformation was not unexpected in the light of previous data demonstrating that SpDprA mediated the loading of EcRecA onto ssDNA (7), the biological role of DprA self-interactions (if any) remained obscure. Investigation of the ssDNA binding capacity of the monomeric mutant DprAAR indicated that this mutant was still capable of interacting with ssDNA in solution as deduced from FAT (Fig. 4C) but, in contrast to DprA and DprAQNQ, failed to form stable NPC in EMSA (Fig. 4D). We propose that dimerization plays a key role in the stabilization of DprA onto ssDNA and the formation of stable NPC (Fig. 7B). The transformation defect of the DprAAR monomeric mutant thus would result directly from its inability to form such complexes with internalized ssDNA.
Working Model of DprA–RecA Interplay in Genetic Transformation.
We propose a rational mechanistic model of the interplay of DprA and RecA in the processing of internalized transforming DNA (Fig. 7C). The proposal is based on the main features presented in this study regarding the importance of DprA dimerization/NPC formation and interaction with RecA capacities for genetic transformation. We suggest that the binding of a DprA C/C dimer is the first step in the processing of incoming ssDNA (Fig. 7C, steps 1 and 2). Presumably, it occurs close to the entry pore (6). We interpret the minimal size requirement for NPC formation (Fig. S4) as reflecting the ssDNA length necessary for the simultaneous interaction with the two binding sites present in the dimer. It is postulated that a conformational change in the dimer, possibly its opening, then occurs and accounts for the previously documented cooperativity of DprA binding to ssDNA (Fig. 7B). Whatever the underlying mechanism may be, NPC formation increases DprA concentration locally and presumably facilitates the subsequent binding of RecA to DprA–ssDNA complexes (Fig. 7C, steps 2–4). Because residues important for interaction with RecA are partially masked in the DprA dimer (Fig. 7D, Left), interaction with RecA presumably triggers major rearrangement (i.e., wide opening to expose dimerization interface residues) or disruption of the dimer, exposing residues important for DprA–RecA association (Fig. 7D, Right). Because of the large overlap between DprA dimerization and RecA interaction interfaces (Fig. 7A), we favor the latter hypothesis. In any case, the flexibility between two interacting DprA monomers suggested by the comparison between the C/C crystal structure and SAXS data (Fig. S3E) could facilitate RecA-triggered conformational changes. In the proposed scenario, contacts of RecA with α11 residues Q265 and E264 (50% accessibility in SpDprA dimer; 40% accessibility for K283, its counterpart in RpDprA dimer; Table S2), would be of particular importance for initiating RecA–DprA association. DprA α11 thus could act as a pivot controlling DprA dimerization and RecA interaction. The maintenance of interactions between RecA and DprA once RecA is loaded onto ssDNA (Fig. 7C, steps 4 and 5) is based on the previously documented formation of DprA-RecA mixed nucleofilaments (7). The finding that the DprAAR monomeric mutant retained a full capacity to form stable complexes with RecA (Fig. 4A) is consistent with this drawing. Finally, nucleation of RecA is drawn arbitrarily as involving three monomers, although this step was not investigated in our study (Fig. 7B, step 4). However, previous studies on E. coli RecA filament formation imply nucleation by a species larger than a monomer (27, 28).
To summarize, this study presents the structure of pneumococcal DprA, consisting of the association of the SAM and eRF domains, and establishes that the capacity of DprA to form RF-RF dimers is evolutionarily conserved, as are the residues involved in dimerization and RecA interaction. In addition, this study provides genetic evidence that RecA and DprA compete for the same DprA-binding surface and demonstrates that the abilities of SpDprA to interact stably with ssDNA (as a dimer) and with RecA, two key functional properties of RMPs, are absolutely required for full transformation proficiency.
Materials and Methods
Bacterial Strains, Plasmids, Primers, Pneumococcal Transformation, and Y2H.
S. pneumoniae strains and plasmids used are listed in Table S4; primers used are listed in Table S5. Pneumococcal transformation was performed as previously described (29). Random mutagenesis and selection for DprA interaction-defective mutants using Y2H were performed as previously described (30). Details of the Y2H screen and plasmid constructions are described in SI Materials and Methods.
Protein Purification and Western Blot Analysis.
His-tagged wild-type and mutant DprA proteins were purified from E. coli by a two-step procedure, i.e., an Ni-NTA column followed by gel filtration. SPA-tagged proteins were purified from pneumococcal cells using a protocol developed for B. subtilis (31). Details regarding these protocols and the immunodetection of DprA and RecA by Western blotting are given in SI Materials and Methods.
Crystallization, Structure Determination, Comparison, and 3D Model Building.
Native and Selenomethionine-labeled protein crystals were grown in hanging drops by mixing protein and reservoir solution in a 1:1 ratio. For crystallization conditions, see SI Materials and Methods. Sorbitol cryoprotected crystals were flash frozen in liquid nitrogen and exposed on the Proxima-1 beamline at the SOLEIL synchrotron, St-Aubin, France. Diffraction data and refinement statistics are given in Table S1. The structure was determined by the SAD method using SeM-labeled protein data. The refinement was done as described in SI Materials and Methods.
Exploration of 3D structures was performed using the following tools: Dali server, I-TASSER, and SWISS-MODEL servers, and the PyMOL Molecular Graphics System (SI Materials and Methods).
SAXS Measurements and Data Analysis.
SAXS experiments with wild-type DprA and the DprAQNQ mutant were performed using the Nanostar instrument at the Institut de Biochimie et Biophysique Moléculaire et Cellulaire (Orsay, France). SAXS experiments on DprAAR and DprAVR were performed on the SWING beamline at the SOLEIL synchrotron. For each sample, data were collected at two or three protein concentrations: about 1, 2, and sometimes 5 mg mL−1, in 50 mM MES (pH 6.5), 2 M NaCl. Data analysis and shape reconstruction for the scattering object are described in SI Materials and Methods.
DNA-Binding Assays.
EMSA were performed exactly as described previously (7) with a 32P dT100 as ssDNA substrate. After electrophoresis, the gel was dried, revealed with a FLA-3000 series fluorescent image analyzer (Fuji), and signal was quantified with MultiGauge software V 3.0 (FujiFilm). FAT was measured in a Fluoromax-4 (Horiba Scientific) (Fig. 4C) or a CARY Eclipse (Varian) spectrofluorometer (Fig. S5B), at 20 °C, in a final reaction volume of 200 μL buffered with 25 mM NaCl, 20 mM Tris (pH 7.5), 2.5 mM MgCl2, 2.5% (vol/vol) glycerol, and supplemented with 25 nM (Fig. S5B) or 100 nM (Fig. 4C) of a dT20 5′-labeled with fluoresceine (GeneCust). The excitation wavelength was set at 490 nm, and emission was observed at 525 nm (10-nm bandwidth). Protein injections were 0.25–1 μL from a 10-mg/mL stock solution. Each titration curve was made in triplicate. The data were treated with SigmaPlot 12.0 (Systat Software).
Supplementary Material
Acknowledgments
We thank Dr. Andrew Thompson and Pierre Legrand for assistance with beamline Proxima-1; Dr. Javier Pérez for assistance with SWING beamline at the SOLEIL synchrotron facility; Dr. François Lecointe for helping us adapt the protocol of purification of SPA-tagged proteins for S. pneumoniae; Jérome Cicolari for help with crystallization; Ines Li de la Sierra-Gallay for the last refinement steps of the structure; Maud Hertzog for performing a control fluorescence anisotropy titration experiment; and Calum Johnston for critical reading of the manuscript. The European Synchrotron Radiation Facility and the SOLEIL synchrotron provided synchrotron radiation facilities. This work was supported in part by a grant from the Agence Nationale de la Recherche (projet n_ BLAN06-3_141806 to J.-P. C. and P.P.), by Grant INE20080311761 from the Fondation pour la Recherche Médicale (to P.P.), and by the Programme Incitatifs à Mobilité d'Équipe from the Centre National de la Recherche Scientifique (to P.P.). N.M. was the recipient of PhD Thesis Fellowship 10/2006-09/2007 from the Association pour la Recherche sur le Cancer.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: Crystallography, atomic coordinates, and structure factors reported in this work for the structure of SpDprA have been deposited in the Protein Data Bank (PDB), www.pdb.org (PDB ID code 3UQZ).
See Author Summary on page 14738 (volume 109, number 37).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1205638109/-/DCSupplemental.
References
- 1.Cox MM. Motoring along with the bacterial RecA protein. Nat Rev Mol Cell Biol. 2007;8:127–138. doi: 10.1038/nrm2099. [DOI] [PubMed] [Google Scholar]
- 2.Haldenby S, White MF, Allers T. RecA family proteins in archaea: RadA and its cousins. Biochem Soc Trans. 2009;37:102–107. doi: 10.1042/BST0370102. [DOI] [PubMed] [Google Scholar]
- 3.Beernink HT, Morrical SW. RMPs: Recombination/replication mediator proteins. Trends Biochem Sci. 1999;24:385–389. doi: 10.1016/s0968-0004(99)01451-6. [DOI] [PubMed] [Google Scholar]
- 4.Dillingham MS, Kowalczykowski SC. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol Mol Biol Rev. 2008;72:642–671. doi: 10.1128/MMBR.00020-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halpern D, Gruss A, Claverys JP, El-Karoui M. rexAB mutants in Streptococcus pneumoniae. Microbiology. 2004;150:2409–2414. doi: 10.1099/mic.0.27106-0. [DOI] [PubMed] [Google Scholar]
- 6.Claverys JP, Martin B, Polard P. The genetic transformation machinery: Composition, localization, and mechanism. FEMS Microbiol Rev. 2009;33:643–656. doi: 10.1111/j.1574-6976.2009.00164.x. [DOI] [PubMed] [Google Scholar]
- 7.Mortier-Barrière I, et al. A key presynaptic role in transformation for a widespread bacterial protein: DprA conveys incoming ssDNA to RecA. Cell. 2007;130:824–836. doi: 10.1016/j.cell.2007.07.038. [DOI] [PubMed] [Google Scholar]
- 8.Hiller NL, et al. Generation of genic diversity among Streptococcus pneumoniae strains via horizontal gene transfer during a chronic polyclonal pediatric infection. PLoS Pathog. 2010;6:e1001108. doi: 10.1371/journal.ppat.1001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Croucher NJ, et al. Rapid pneumococcal evolution in response to clinical interventions. Science. 2011;331:430–434. doi: 10.1126/science.1198545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bentley SD, et al. Genetic analysis of the capsular biosynthetic locus from all 90 pneumococcal serotypes. PLoS Genet. 2006;2:e31. doi: 10.1371/journal.pgen.0020031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Claverys JP, Prudhomme M, Martin B. Induction of competence regulons as a general response to stress in gram-positive bacteria. Annu Rev Microbiol. 2006;60:451–475. doi: 10.1146/annurev.micro.60.080805.142139. [DOI] [PubMed] [Google Scholar]
- 12.Håvarstein LS, Coomaraswamy G, Morrison DA. An unmodified heptadecapeptide pheromone induces competence for genetic transformation in Streptococcus pneumoniae. Proc Natl Acad Sci USA. 1995;92:11140–11144. doi: 10.1073/pnas.92.24.11140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee MS, Morrison DA. Identification of a new regulator in Streptococcus pneumoniae linking quorum sensing to competence for genetic transformation. J Bacteriol. 1999;181:5004–5016. doi: 10.1128/jb.181.16.5004-5016.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prudhomme M, Attaiech L, Sanchez G, Martin B, Claverys JP. Antibiotic stress induces genetic transformability in the human pathogen Streptococcus pneumoniae. Science. 2006;313:89–92. doi: 10.1126/science.1127912. [DOI] [PubMed] [Google Scholar]
- 15.Morrison DA, Mortier-Barrière I, Attaiech L, Claverys JP. Identification of the major protein component of the pneumococcal eclipse complex. J Bacteriol. 2007;189:6497–6500. doi: 10.1128/JB.00687-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Attaiech L, et al. Role of the single-stranded DNA-binding protein SsbB in pneumococcal transformation: Maintenance of a reservoir for genetic plasticity. PLoS Genet. 2011;7:e1002156. doi: 10.1371/journal.pgen.1002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim CA, Gingery M, Pilpa RM, Bowie JU. The SAM domain of polyhomeotic forms a helical polymer. Nat Struct Biol. 2002;9:453–457. doi: 10.1038/nsb802. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Skolnick J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeghouf M, et al. Sequential peptide affinity (SPA) system for the identification of mammalian and bacterial protein complexes. J Proteome Res. 2004;3:463–468. doi: 10.1021/pr034084x. [DOI] [PubMed] [Google Scholar]
- 20.Singleton MR, Dillingham MS, Gaudier M, Kowalczykowski SC, Wigley DB. Crystal structure of RecBCD enzyme reveals a machine for processing DNA breaks. Nature. 2004;432:187–193. doi: 10.1038/nature02988. [DOI] [PubMed] [Google Scholar]
- 21.Timmins J, Leiros I, McSweeney S. Crystal structure and mutational study of RecOR provide insight into its mode of DNA binding. EMBO J. 2007;26:3260–3271. doi: 10.1038/sj.emboj.7601760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kagawa W, et al. Crystal structure of the homologous-pairing domain from the human Rad52 recombinase in the undecameric form. Mol Cell. 2002;10:359–371. doi: 10.1016/s1097-2765(02)00587-7. [DOI] [PubMed] [Google Scholar]
- 23.Singleton MR, Wentzell LM, Liu Y, West SC, Wigley DB. Structure of the single-strand annealing domain of human RAD52 protein. Proc Natl Acad Sci USA. 2002;99:13492–13497. doi: 10.1073/pnas.212449899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pellegrini L, et al. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature. 2002;420:287–293. doi: 10.1038/nature01230. [DOI] [PubMed] [Google Scholar]
- 25.Yang H, et al. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002;297:1837–1848. doi: 10.1126/science.297.5588.1837. [DOI] [PubMed] [Google Scholar]
- 26.Schwartz T, Behlke J, Lowenhaupt K, Heinemann U, Rich A. Structure of the DLM-1-Z-DNA complex reveals a conserved family of Z-DNA-binding proteins. Nat Struct Biol. 2001;8:761–765. doi: 10.1038/nsb0901-761. [DOI] [PubMed] [Google Scholar]
- 27.Spies M, Kowalczykowski SC. The RecA binding locus of RecBCD is a general domain for recruitment of DNA strand exchange proteins. Mol Cell. 2006;21:573–580. doi: 10.1016/j.molcel.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 28.Joo C, et al. Real-time observation of RecA filament dynamics with single monomer resolution. Cell. 2006;126:515–527. doi: 10.1016/j.cell.2006.06.042. [DOI] [PubMed] [Google Scholar]
- 29.Martin B, Prudhomme M, Alloing G, Granadel C, Claverys JP. Cross-regulation of competence pheromone production and export in the early control of transformation in Streptococcus pneumoniae. Mol Microbiol. 2000;38:867–878. doi: 10.1046/j.1365-2958.2000.02187.x. [DOI] [PubMed] [Google Scholar]
- 30.Noirot-Gros MF, et al. Functional dissection of YabA, a negative regulator of DNA replication initiation in Bacillus subtilis. Proc Natl Acad Sci USA. 2006;103:2368–2373. doi: 10.1073/pnas.0506914103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lecointe F, et al. Anticipating chromosomal replication fork arrest: SSB targets repair DNA helicases to active forks. EMBO J. 2007;26:4239–4251. doi: 10.1038/sj.emboj.7601848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gouet P, Courcelle E, Stuart DI, Métoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
