

Abstract
Measles virus (MeV) is the poster child for acute infection followed by lifelong immunity. However, recent work shows the presence of MeV RNA in multiple sites for up to 3 mo after infection in a proportion of infected children. Here, we use experimental infection of rhesus macaques to show that prolonged RNA presence is characteristic of primary infection. We found that viral RNA persisted in the blood, respiratory tract, or lymph nodes four to five times longer than the infectious virus and that the clearance of MeV RNA from blood happened in three phases: rapid decline coincident with clearance of infectious virus, a rebound phase with increases up to 10-fold, and a phase of slow decrease to undetectable levels. To examine the effect of individual host immune factors on MeV load dynamics further, we developed a mathematical model that expressed viral replication and elimination in terms of the strength of MeV-specific T-cell responses, antibody responses, target cell limitations, and immunosuppressive activity of regulatory T cells. Based on the model, we demonstrate that viral dynamics, although initially regulated by T cells, require antibody to eliminate viral RNA. These results have profound consequences for our view of acute viral infections, the development of prolonged immunity, and, potentially, viral evolution.
Keywords: immune responses, within-host modeling, virus clearance
Measles is a highly contagious human disease that affects millions of children each year. Although vaccination has controlled measles in many countries, the disease remains an important cause of child mortality (1). Epidemiological studies conducted by Panum (2) during an 1846 measles epidemic in the Faroe Islands established its remarkable infectiousness and ability to confer lifelong immunity. The acute, strongly immunizing nature of measles virus (MeV) infection is underlined by a long history of epidemic modeling, coupled to analyses of surveillance data (3–5). Early studies also established measles as an immunosuppressive disease (6) with a prolonged increase in susceptibility to secondary infections that leads to most measles-related deaths (7, 8).
MeV has been studied as a paradigm for acute viral infections. Infection is initiated in the respiratory tract, and virus spreads to local lymphoid tissue and then to multiple organs, including the skin (9). In uncomplicated measles, clinical recovery occurs soon after the onset of the rash 10–14 d after infection and coincides with rapid clearance of infectious virus from the blood (10, 11). Because of the short period of infectiousness and solid immunity against retransmission after recovery, large populations are required to maintain MeV transmission (12), especially in regions with highly seasonal population aggregation (13).
Recent studies of children with measles showed that MeV RNA persists at multiple sites for many months after resolution of the rash and apparent recovery. At 1 mo after hospital discharge, MeV RNA was more frequently detected in HIV-infected children, suggesting slowed clearance when cellular immune responses are compromised (14, 15). Furthermore, during studies of rhesus macaques given partially protective experimental measles vaccines, reactivation of MeV-specific T-cell responses 3–4 mo after WT MeV challenge coincided with detection of MeV RNA in peripheral blood mononuclear cells (PBMCs) (16, 17). These data suggest that clearance of MeV after acute primary infection is more complicated and prolonged than previously recognized. However, the mechanisms and timing of normal MeV clearance are not known.
Understanding MeV clearance is crucial for advancing our knowledge of measles pathogenesis, lifelong immunity, complications associated with immune suppression, persistent infection, and transmission dynamics (18). To dissect the relationship between the within-host dynamics of MeV and the immune responses to infection, data with frequent and continuous measurements that describe the virological and immunological changes in MeV-infected hosts over a long period are needed. Rhesus macaques are susceptible to measles and develop a disease that faithfully mimics human measles (19). Therefore, we have synthesized experimental infections and dynamic models to perform a detailed investigation of MeV and immune kinetics in individual macaques infected with a WT strain of MeV.
Results
Eight MeV-naive juvenile rhesus macaques (Table S1) were infected with the Bilthoven strain of WT MeV (20) by the respiratory route. After infection, all macaques developed a transient lymphopenia followed by a prolonged period of leukocytosis (Fig. S1). Seven macaques developed a viremia detected by cocultivation of PBMCs with MeV-susceptible B95-8 or Vero/hSLAM cells (21) and a maculopapular rash (Fig. 1A). The rash resolved ∼14 d after infection, and soon thereafter, infectious virus was no longer detectable. Therefore, seven of the eight macaques developed measles typical of the human disease and were used for further evaluation.
Fig. 1.

Prolonged presence of MeV RNA in multiple sites after infection. (A) Infectious virus in the blood detected by cocultivation of PBMCs with susceptible cells. Results converted to tissue culture 50% infectious doses (TCID50) are shown as number of infected cells per 106 PBMCs (red lines). The MeV RNA load was measured by N-specific qRT-PCR using RNA extracted from 2 × 106 PBMCs. Points with undetectable MeV RNA are plotted with open circles. Lines mark 14 and 24 d after infection. Macaques are identified at the top of individual graphs. (B) Presence of MeV RNA in the respiratory tract determined by N-specific RT-PCR on RNA extracted from nasal swab cell pellets. RT-PCR products run on gels were read as positive or negative (Table S2). Results are plotted as the percentage of animals with detection of MeV RNA. (C) MeV RNA loads in lymph nodes (LN) were determined by N-specific qRT-PCR. Inguinal lymph nodes from three macaques were biopsied 71 d after infection. n.d., not detected.
Levels of MeV RNA in PBMCs were measured with a quantitative reverse transcriptase-PCR (qRT-PCR) assay specific for the MeV nucleoprotein (N) gene (16) (Fig. 1A). In contrast to clearance of infectious virus, clearance of MeV RNA from PBMCs occurred over 5–10 wk. MeV RNA was detected in the blood for a median of 50 d (range: 24–67 d) and declined in three phases: a rapid decline coincident with clearance of infectious virus (days 10–14) and a rebound phase in which viral load increased up to 10-fold (days 14–24), followed by a slow decrease to undetectable levels (day >24). MeV RNA was also detected in nasal swab samples by RT-PCR for a median of 17 d (Fig. 1B and Table S2) and in inguinal lymph nodes by qRT-PCR 71 d after infection (Fig. 1C), indicating the prolonged presence of MeV RNA in multiple sites with persistence in lymphoid tissue after clearance from PBMCs.
To identify the host immune factors involved in MeV clearance, we analyzed both cellular and humoral responses to MeV. CD4+ and CD8+ T cells specific for MeV hemagglutinin (H), fusion (F), and N proteins producing IFN-γ and TNF were detectable by 10 d after infection (Fig. 2A and Fig. S2). MeV-specific T cells increased as infectious virus and RNA loads decreased between days 10 and 14. Conversely, as T cells decreased, MeV RNA load stabilized or increased between days 14 and 24. To follow the induction of regulatory T cells (Tregs) (22, 23), we measured the expression of FoxP3, a Treg-specific transcription factor, using qRT-PCR (24–26). Six of seven monkeys had sustained up-regulation of Foxp3 gene expression in PBMCs after clearance of infectious virus (Fig. 2D). In four of seven animals, there was a transient spike of FoxP3 expression 3 d after infection. This suggests that some Treg populations, presumably natural Tregs (27), enter the circulation before the systemic spread of virus.
Fig. 2.

MeV-specific cellular and humoral immune responses during the course of infection. (A) MeV-specific T-cell responses were assessed by IFN-γ ELISpot assays. Numbers of specific spot-forming cells (SFCs) were calculated by subtracting nonspecific (NS) responses. Data are plotted as the means of the duplicates from each condition. Macaques are identified at the top of individual graphs. (B) Reciprocal titers of neutralizing antibody in plasma were determined by PRN titer (PRNT). Results were calculated from assay triplicates. Avidity of MeV-specific IgG was assessed by disruption of antibody binding with 0–3.5 M NH4SCN and calculation of avidity index 75%, indicating the concentration needed to remove 75% of bound antibody. (C) MeV-specific IgG, IgM, and IgA in the plasma were measured by EIAs using plates coated with a lysate from MeV-infected Vero cells. H-specific IgG was measured using plates coated with a lysate from MeV H-expressing L cells. Antibody titers were determined based on the OD values of serially diluted plasma and assay-specific background. (D) FoxP3 expression as an indicator of the induction of Tregs was determined by qRT-PCR using RNA extracted from PBMCs.
MeV neutralizing antibody was detected in plasma as early as 10 d after infection (Fig. 2B), which reflected MeV-specific IgM and low levels of IgA and IgG (Fig. 2C). IgM declined rapidly after infectious virus was cleared, whereas the titer and avidity of MeV-specific IgG continued to increase over 2–4 mo (Fig. 2 B and C). A month after infection, most of the MeV-specific IgG in the plasma was H-specific IgG but varied in the time of appearance (Fig. 2C).
To dissect the effects of individual host immune factors on MeV load dynamics further, we developed mathematical models (Fig. 3A) for the process of within-host clearance of MeV RNA and fitted these models to the quantitative measurements acquired from the macaque experiments (28–30). We considered three nested models, which take different components of the immune system into account. The T cell-AB-FoxP3 model explains virus clearance by taking T cells, antibodies, and FoxP3 into account; it is given by the following differential equation:
Fig. 3.

Mathematical models for the process of within-host clearance of MeV RNA. (A) Diagram of the linear model. The virus load was directly modulated by different immune components. V(t) denotes the virus load at time t, R(t) the abundance of target cells (lymphocyte concentration), T(t) the abundance of MeV-specific IFN-γ–producing T cells, A(t) the abundance of MeV-specific antibodies (PRN titer), and S(t) the immunosuppressive activity (FoxP3 mRNA). (B) Models were fitted by maximum likelihood to the virus load data (see Materials and Methods). Colors indicate different versions of the model. The T cell-AB-FoxP3 model (Eq. 1; blue line) takes T cells, antibodies, and FoxP3 into account; the T cell-AB model (green line) takes only T cells and antibodies into account; and the T-cell model (red line) takes only T cells into account. The models are fitted for each animal individually (fits in which all parameters except 1 are common between animals are provided in Fig. S9). Log-likelihood from the different models is shown in right upper quadrants with colors corresponding to lines. The observed MeV RNA load is shown in open circles. Gray vertical lines mark the estimated lowest detection level of the qRT-PCR assay at given time points. Macaques are identified at the top of individual graphs.
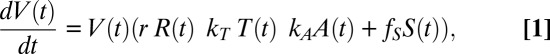 |
in which V(t) denotes the virus load at time t, R(t) the abundance of target cells, T(t) the abundance of MeV-specific T cells, A(t) the abundance of MeV-specific antibodies, and S(t) the suppressive activity of Tregs. Here, r denotes the growth rate of the virus, KT the inhibitory effect of T cells on MeV, KA the inhibitory effect of antibodies on MeV and fS the immunosuppressive effect of Tregs. The dynamics of the immune variables are obtained directly from the data as the linear interpolation of the measurements (29, 30). To assess the importance of the different immune components, we also derived two submodels from Eq. 1. The T cell-AB model captures only the effects of T cells and antibodies and is derived from Eq. 1 by setting fS = 0. The T-cell model captures only the effect of T cells and is derived by setting fS = 0 and kA = 0. These three models were then fitted separately to the virus load data from each animal by maximum likelihood (Figs. S3–S7), using the observed immune kinetics for each animal.
Conventional wisdom holds that T cells should be the major force in regulating primary infection (31). In contrast, we find that taking only viral clearance by T cells into account leads to poor model fits (Fig. 3B). In particular, the T-cell model cannot explain the decline and clearance of virus in the late phase of the infection, even when we allow for saturation of the T-cell response (Fig. S8). The model fits improve strongly and significantly (P < 10−5, likelihood ratio test) once antibodies are also taken into account. Including FoxP3 as well leads to a weaker but overall significant improvement of the model fits, although the level of significance varied among animals (see Figs. S3–S7). These results indicate that both MeV-specific T cells and antibodies contribute to controlling MeV replication in naive animals (Fig. 4) and that suppression of the immune system may slow viral clearance, and thereby contribute to the secondary peaks in virus load.
Fig. 4.

Rash, viral dynamics, and T-cell and antibody dynamics. Results of viremia, viral RNA, MeV-specific IFN-γ production, and PRN titer are plotted as the average from the study macaques + SEM. T-cell response data (green) are calculated as the sum of the H-, F-, and N-specific IFN-γ responses and plotted on a linear scale of 0–800 spot-forming cells per 106 PBMCs. Antibody response data from PRNT (orange) are plotted on a linear scale of 0–8,000 reciprocal titer. Viremia (cyan) and viral RNA (gray) are plotted in a logarithmic scale with an axis of 0–5 log tissue culture 50% infectious dose per 106 PBMCs or 0–105 MeV/GAPDH × 5,000. Based on the mathematical models, we show that viral dynamics, although initially regulated by T cells, require antibody to eliminate viral RNA.
Discussion
Here, we demonstrate that prolonged presence of viral RNA is characteristic of primary MeV infection. In the current study, we measured MeV RNA primarily in the blood, because blood is the most accessible tissue for longitudinal studies and has the least impact on the health of the animals. However, to define further when MeV RNA is actually cleared, future studies of samples collected from other tissues are necessary. The role of lymphoid tissue in MeV persistence is particularly intriguing, because we recovered MeV RNA from lymph node biopsies even after MeV RNA was no longer detectable in blood. Viral genome sequence information at different phases of infection is also of interest, but we think it is unlikely that virus mutation is the primary mechanism of virus persistence. In our study of children with naturally acquired measles, N gene sequences for five children 96–109 d after rash onset were the same as the genotype D2 MeV in circulation at the time, and for one child, the N and H sequences from PBMCs 118 d after rash onset were identical to those of the virus collected during the acute illness (14). Therefore, the prolonged presence of MeV RNA is likely attributable to persistence rather than immune evasion.
We have developed a model that explains multiple aspects of in-host MeV infection and reveals a clear interaction between virus and effector immune responses. Our data highlight an important but commonly underestimated (32) role of antibody in the control and clearance of a systemic viral infection. The necessity for antibody in determining MeV load dynamics is a clear consensus of the family of mathematical models that we have developed; these include the standard model Eq. 1 and three other alternative model fits (SI Alternative Models): (i) a fit of the standard model in which all parameters except kT are shared between animals (Fig. S9), (ii) a nonlinear version of the standard model (Fig. S10), and (iii) a nonlinear T-cell saturation model that allows the magnitude of the T-cell response to saturate with increasing concentrations of T cells (Fig. S8). Antibody in previously infected hosts is well recognized to prevent the spread of infectious virus by neutralization (33); however, antibodies are generally believed to have very minimal roles in clearing primary infection of MeV (11). The current model captures viral load in terms of observed immune kinetics. An important area for future empirical and modeling work is to generate fully dynamic models, where viral load feeds back into the immune kinetics (34, 35).
In support of this finding, antibodies clear alphavirus infection from neurons (36) and control the persistence of lymphocytic choriomeningitis virus in mice (37), and there is in vitro evidence that antibodies to the MeV H protein can alter expression of MeV proteins in the cytoplasm of infected cells (38–41). Consistent with a demonstrated role of CD8+ T cells in controlling viral infection (10, 34), our model showed that the activation of the MeV-specific T-cell response correlated with the control of infectious virus and the initial decline in levels of MeV RNA (Fig. 3 and Fig. S8). However, the T-cell response waned before MeV had been controlled. Collectively, our data suggest that host control and clearance of MeV were achieved by temporal involvement of different arms of the effector immune responses; the cellular response is critical for initial virus control and the humoral response is important for the subsequent clearance of viral RNA and suppression of recurrent production of infectious virus (Fig. 4).
Studies of acute infection have brought valuable insights for defining viral virulence and host immunity (42). MeV has long been recognized as a paradigm for acute viral infection, but our data provide clear evidence that the virus persists in the host for months longer than generally recognized. These findings have a profound impact on our existing view of acute viral infections, the development of prolonged immunity, and viral transmission. Localized persistence of viral RNA has been identified in animal models of acute viral hepatitis (43) and encephalitis (36), but the prevalence and consequences of the prolonged presence of viral RNA in humans after clinical recovery from acute infection with RNA viruses remain largely unknown. A critical question for measles is the causal relationship between persistence of MeV RNA and the induction of prolonged immune suppression and lifelong immunity. Considering that low levels of viral antigen are important for continued stimulation of CD8+ T cells after acute infections with influenza and vesicular stomatitis viruses (44, 45), and that MeV RNA persists in the blood and lymphoid tissue for more than 2 mo after infection, we speculate that lifelong protective immunity after primary MeV infection could be a consequence of prolonged antigen stimulation.
Because we predict that recrudescence of infectious MeV might occur in individuals with impaired humoral immune responses, another key area for future work is to assess implications for measles transmission and persistence at the population level. For instance, could prolonged persistence of MeV in an individual prevent extinction of the virus during epidemic troughs? Although preventing extinction is probably not central for maintaining MeV transmission in humans because the human population largely exceeds the critical community size of measles, this mechanism could be critical for the transmission of related morbilliviruses in wildlife populations.
Materials and Methods
Animals.
Eight 2- to 3-y-old male rhesus macaques (Macaca mulatta; 15U, 46U, 55U, 67U, 40V, 43V, 55V, and 86U) (Table S1) negative for herpes B virus, tuberculosis, and antibody to MeV were obtained and housed at the Johns Hopkins University primate facility. All studies were performed in accordance with experimental protocols approved by the Johns Hopkins University Animal Care and Use Committee.
MeV Infection and Sample Collection.
Macaques were sedated with ketamine and infected intratracheally with 104 tissue culture 50% infectious doses of the Bilthoven strain of MeV (a gift from Albert Osterhaus, Erasmus University, Rotterdam, The Netherlands) in four batches (Table S1). After infection, monkeys were shaved and monitored for development of a rash. Nasal swabs and venous blood were collected at regular intervals to monitor the virus load and immune responses. Secretion and epithelial cells from the nasal cavity were collected using sterile cotton swabs. Swabs were dipped into 1 mL of PBS to release the cells and fluid. Cell pellets and fluid were separated by centrifugation. A complete blood cell count and differential count were determined on heparinized blood by means of an automated cell counter. Plasma and PBMCs were separated by density gradient centrifugation using Lympholyte-Mammal (Cedarlane Laboratories). Seventy-one days after infection, surgery was performed under general anesthesia to collect inguinal lymph nodes from three macaques (46U, 55U, and 67U).
Cells, Virus, and Virus Assays.
The stock virus for macaque infection was grown in phytohemagglutinin-stimulated human cord blood mononuclear cells and titered by syncytia formation in B95-8 cells. Viremia was quantitated by cocultivation of serially diluted PBMCs with B95-8 or Vero/hSLAM cells. Cultures were scored for cytopathic effect after 5 d, and data are reported as the number of infected cells per 106 PBMCs. The Chicago-1 strain of MeV was grown and assayed by plaque formation in Vero cells. Vero and Vero/hSLAM cells were grown in DMEM supplemented with 10% (vol/vol) FBS.
MeV N and FoxP3 Gene Expression.
Total RNA was isolated from 2 × 106 PBMCs, nasal swab cells, and lymph nodes using an RNeasy mini kit (Qiagen). For qRT-PCR, MeV N and FoxP3 genes were amplified using a one-step RT-PCR kit with TaqMan primers and measured with gene-specific probes in an Applied Biosystems Prism 7500. Controls included GAPDH gene amplification (Applied Biosystems). Copy number was determined by construction of a standard curve of 1–106 copies of MeV RNA or 10–107 copies of FoxP3 RNA synthesized by in vitro transcription from a plasmid encoding the Edmonston N gene or rhesus FoxP3 gene. Data were normalized to the GAPDH control, and results were expressed as [(number of copies of MeV N RNA)/(number of copies of GAPDH RNA)] × 5,000. MeV in nasal swab cell pellets was detected by N-specific RT-PCR. RT-PCR products were run on gels and read as positive or negative.
Antibody Assays.
For plaque reduction neutralization (PRN), the Chicago-1 strain of MeV was mixed with serially diluted plasma or media alone and assayed for plaque formation on Vero cells. Enzyme immunoassays (EIAs) were used to measure MeV-specific IgM, IgG, and IgA. Maxisorp 96-well plates (Nalge Nunc International) were coated with a lysate from MeV-infected Vero cells (1.16 μg of protein per well; Advanced Biotechnologies) or with a lysate from MeV H-expressing L cells. Twofold serially diluted plasma (1:100–1:204,800 for MeV-specific IgG and 1:50–1:12,800 for IgA) and pooled plasma (1:100) from MeV-seronegative monkeys were added and incubated overnight at 4 °C. HRP-conjugated goat antibody to monkey IgG or IgA (Nordic) or alkaline phosphatase-conjugated goat antibody to monkey IgM (Nordic) was used as a secondary antibody, and 3,39,5,59-tetramethylbenzidine (Sigma) or p-nitrophenyl phosphate (Sigma) was used as the substrate. Plates were read at 405 nm or 450 nm using a Multiskan MCC microplate reader (Fisher Scientific). To decrease the interference of MeV-specific IgG with measurement of IgA, plasma was incubated with a 50% protein G Sepharose slurry (Thermo Scientific) for 2 h at room temperature and collected using microspin columns. The EIA titer was the dilution at which the OD value for the sample intersected with the MeV-seronegative cutoff (mean + 2 SD). To assess antibody avidity, MeV lysate-coated plates were incubated with twofold serially diluted plasma overnight at 4 °C, washed, and treated with increasing concentrations (0.5–3.5 M) of ammonium thiocyanate (NH4SCN) for 15 min to disrupt the MeV-antibody interaction. OD values of serially diluted plasma samples under different NH4SCN treatment were plotted, and the avidity index 75%, defined as the concentration of NH4SCN to elute 75% of the bound antibody, was calculated by interpolation.
T-Cell Assays.
Enzyme-linked immunospot (ELISpot) assays were used to measure IFN-γ–producing T cells. Multiscreen HTS HA Opaque ELISpot plates (Millipore) were coated with antibody to human IFN-γ (BD Biosciences). After plates were washed and blocked with Roswell Park Memorial Institute (RPMI)-10 medium, 1–5 × 105 fresh PBMCs were added, along with 1 μg/mL pooled MeV peptides (20-mer overlapping by 11 amino acids) from the H, F, or N protein or 5 μg/mL Con A. After 40 h of incubation, plates were washed and incubated with biotinylated antibody to IFN-γ (Mabtech), followed by HRP-conjugated avidin (Vector). Assays were developed with stable diaminobenzidine (Invitrogen) solution and read on an ImmunoSpot plate reader (Cellular Technology). Data were analyzed using ImmunoSpot version 3.0 software (Cellular Technology). Results are presented as the number of spot-forming cells per 106 PBMCs.
Multiparameter Flow Cytometry.
To assess CD4+ and CD8+ T-cell activation and cytokine production during MeV infection, eight-color flow cytometry with surface and intracellular cytokine staining was used. Before staining, 106 fresh PBMCs were stimulated with staphylococcal enterotoxin B (Sigma–Aldrich), media alone, or pooled MeV H or N peptide in the presence of anti-human CD107a (BD Biosciences), brefeldin A, (Sigma–Aldrich), and Golgistop (BD Biosciences) for 12 h. Cells were then stained with ViViD live- dead discriminator (Invitrogen) and antibodies to CD3, CD4, CD8, CD14, and CD20. Cells were permeabilized using the Fixation/Permeabilization solution kit (BD Cytofix/Cytoperm) and stained for intracellular IFN-γ, TNF, and IL-2. All fluorescent-conjugated antibodies were purchased from BD Biosciences, except anti-CD20 and TNF (eBioscience). Cells were read on a FACSCanto II flow cytometer (BD Biosciences). A total of 400,000 events were collected per sample. Analysis was performed using FlowJo (Tree Star, version 8.8.6). After eliminating artifacts from acquisition noise, doublets, and dead cells, cytokine-producing cells were gated from CD3+CD4+ and CD3+CD8+ cells.
Mathematical Modeling.
The aim of our model is to fit the virus load dynamics (measured as the concentration of viral RNA) by using immune variables as explanatory variables. The dynamics of the explanatory variables [T(t), R(t), A(t), S(t)] were taken directly from the data as the linear interpolation of the measurements. The explanatory variables relate as follows to the immunological measurements. Because lymphocytes are one of the main target cells, we use their concentration as a proxy for R(t). As a proxy for cellular immunity, we use the concentration of MeV-specific IFN-γ–producing T cells in the blood (measured by IFN-γ ELISpot). As a proxy for the action of antibodies, we use the PRN titer value that measures the capacity of serum to neutralize MeV in cell culture. FoxP3 is a transcription factor mainly expressed in Tregs, which suppress activation of the immune system, and the concentration of FoxP3-RNA is used as a proxy for the immunosuppressive activity of measles.
The values of the parameters (r, kT, kA, fS) of the model are determined by maximizing the likelihood of the model. For a given set of parameters, we compute the solution V(t) of Eq. 1 and then compute the likelihood of observing the virus load measurements M(ti) given this solution as
where 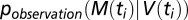 captures the uncertainty associated with the observation/measurement process; that is,
captures the uncertainty associated with the observation/measurement process; that is, 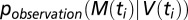 is the probability of observing M(ti) if the real underlying virus load is V(ti). Specifically, we assume that this observation process is given by the negative binomial distribution (which we chose for its ability to capture overdispersion) with mean V(ti) and overdispersion parameter k. We could determine k directly from the virus load measurements because two viral load measurements were available for each time point (Fig. S11). The thus computed likelihood was then maximized using the routine optim() in R.
is the probability of observing M(ti) if the real underlying virus load is V(ti). Specifically, we assume that this observation process is given by the negative binomial distribution (which we chose for its ability to capture overdispersion) with mean V(ti) and overdispersion parameter k. We could determine k directly from the virus load measurements because two viral load measurements were available for each time point (Fig. S11). The thus computed likelihood was then maximized using the routine optim() in R.
Given the large variability between immune responses, it is not possible to fit all animals with the same parameters (i.e., the resulting fits do not qualitatively match the virus load trajectories). Therefore, we fit each animal individually and computed the total log-likelihood as the sum of the log-likelihoods for the individual animal fits. Although it is not possible to fit all animals together, we find the same qualitative results if we assume that that the different animals share all parameters except kTC (the rate with which T cells clear the virus), which we allow to vary between animals (Fig. S9).
Model 1 explains viral clearance on the basis of T cells, antibodies, and FoxP3 (henceforth, we will refer to it as the T cell-AB-FoxP3 model). To assess the importance of the different immune components, we also derive two submodels. The T cell-AB model considers only the effect of T cells and antibodies on viral clearance:
Finally, the T-cell model only considers the effect of T cells:
Estimating the overdispersion parameter.
For each time point ti, two virus load measurements were available, v1,ti and v2,ti. These data were fitted with the negative binomial distribution under the assumption that the two measurements at each time point represent samples from the same mean (μti) and that all samples share the same k. Thus, the likelihood reads:
In fact, the maximum likelihood estimate for μti is given by the means mti = (v1,ti + v2,ti)/2, such that we can obtain the maximum likelihood estimate for k by maximizing the one-dimensional function:
Supplementary Material
Acknowledgments
We thank Dr. David I. Watkins (University of Wisconsin) for macaque MHC typing; Dr. Sebastien Ballesteros (Princeton University) and Dr. Chien-Hsiung Pan and Dr. William Moss (Johns Hopkins University) for helpful discussions; and Debra Hauer and Brandyn Lau (Johns Hopkins University) for technical assistance. This work was supported by grants from the National Institutes of Health (to D.E.G.); the Bill and Melinda Gates Foundation (to D.E.G. and B.T.G.); a Marjorie Gilbert Scholarship (to W.-H.W.L.); the Research and Policy for Infectious Disease Dynamics Program of the Science and Technology Directorate, Department of Homeland Security, and the Fogarty International Center, National Institutes of Health (to B.T.G.); and the Swiss National Science Foundation Grant PA00P3_131498 (to R.D.K).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1211138109/-/DCSupplemental.
See Commentary on page 14724.
References
- 1.Simons E, et al. Assessment of the 2010 global measles mortality reduction goal: Results from a model of surveillance data. Lancet. 2012;379:2173–2178. doi: 10.1016/S0140-6736(12)60522-4. [DOI] [PubMed] [Google Scholar]
- 2.Panum P. Observations made during the epidemic of measles on the Faroe Islands in the year 1846. Med Classics. 1938;3:829–886. [Google Scholar]
- 3.Anderson RM, May RM. Infectious Diseases of Humans: Dynamics and Control. Oxford: Oxford Science Publications; 1992. [Google Scholar]
- 4.Bjornstad ON. Dynamics of measles epidemics: Estimating scaling of transmission rates using a time series SIR model. Ecol Monogr. 2002;72(2):169–184. [Google Scholar]
- 5.Grenfell BT. Dynamics of measles epidemics: Scaling noise, determinism, and predictability with the TSIR model. Ecol Monogr. 2002;72(2):185–202. [Google Scholar]
- 6.von Pirquet C. Das verhalten der kutanen tuberkulin-reaktion warend der masern. Duet Med Wochenshr. 1908;34:1297–1300. [Google Scholar]
- 7.Beckford AP, Kaschula RO, Stephen C. Factors associated with fatal cases of measles. A retrospective autopsy study. S Afr Med J. 1985;68:858–863. [PubMed] [Google Scholar]
- 8.Greenberg BL, et al. Measles-associated diarrhea in hospitalized children in Lima, Peru: Pathogenic agents and impact on growth. J Infect Dis. 1991;163:495–502. doi: 10.1093/infdis/163.3.495. [DOI] [PubMed] [Google Scholar]
- 9.Moench TR, Griffin DE, Obriecht CR, Vaisberg AJ, Johnson RT. Acute measles in patients with and without neurological involvement: Distribution of measles virus antigen and RNA. J Infect Dis. 1988;158:433–442. doi: 10.1093/infdis/158.2.433. [DOI] [PubMed] [Google Scholar]
- 10.Permar SR, et al. Role of CD8(+) lymphocytes in control and clearance of measles virus infection of rhesus monkeys. J Virol. 2003;77:4396–4400. doi: 10.1128/JVI.77.7.4396-4400.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Permar SR, et al. Limited contribution of humoral immunity to the clearance of measles viremia in rhesus monkeys. J Infect Dis. 2004;190:998–1005. doi: 10.1086/422846. [DOI] [PubMed] [Google Scholar]
- 12.Keeling MJ, Grenfell BT. Disease extinction and community size: Modeling the persistence of measles. Science. 1997;275:65–67. doi: 10.1126/science.275.5296.65. [DOI] [PubMed] [Google Scholar]
- 13.Ferrari MJ, et al. The dynamics of measles in sub-Saharan Africa. Nature. 2008;451:679–684. doi: 10.1038/nature06509. [DOI] [PubMed] [Google Scholar]
- 14.Riddell MA, Moss WJ, Hauer D, Monze M, Griffin DE. Slow clearance of measles virus RNA after acute infection. J Clin Virol. 2007;39:312–317. doi: 10.1016/j.jcv.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 15.Permar SR, et al. Prolonged measles virus shedding in human immunodeficiency virus-infected children, detected by reverse transcriptase-polymerase chain reaction. J Infect Dis. 2001;183:532–538. doi: 10.1086/318533. [DOI] [PubMed] [Google Scholar]
- 16.Pan CH, et al. Modulation of disease, T cell responses, and measles virus clearance in monkeys vaccinated with H-encoding alphavirus replicon particles. Proc Natl Acad Sci USA. 2005;102:11581–11588. doi: 10.1073/pnas.0504592102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan CH, et al. A chimeric alphavirus replicon particle vaccine expressing the hemagglutinin and fusion proteins protects juvenile and infant rhesus macaques from measles. J Virol. 2010;84:3798–3807. doi: 10.1128/JVI.01566-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Griffin DE, Lin WH, Pan CH. Measles virus, immune control, and persistence. FEMS Microbiol Rev. 2012;36:649–662. doi: 10.1111/j.1574-6976.2012.00330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Auwaerter PG, et al. Measles virus infection in rhesus macaques: Altered immune responses and comparison of the virulence of six different virus strains. J Infect Dis. 1999;180:950–958. doi: 10.1086/314993. [DOI] [PubMed] [Google Scholar]
- 20.van Binnendijk RS, van der Heijden RW, van Amerongen G, UytdeHaag FG, Osterhaus AD. Viral replication and development of specific immunity in macaques after infection with different measles virus strains. J Infect Dis. 1994;170:443–448. doi: 10.1093/infdis/170.2.443. [DOI] [PubMed] [Google Scholar]
- 21.Ono N, et al. Measles viruses on throat swabs from measles patients use signaling lymphocytic activation molecule (CDw150) but not CD46 as a cellular receptor. J Virol. 2001;75:4399–4401. doi: 10.1128/JVI.75.9.4399-4401.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu XL, et al. Measles virus infection in adults induces production of IL-10 and is associated with increased CD4+ CD25+ regulatory T cells. J Immunol. 2008;181:7356–7366. doi: 10.4049/jimmunol.181.10.7356. [DOI] [PubMed] [Google Scholar]
- 23.Koga R, Ohno S, Ikegame S, Yanagi Y. Measles virus-induced immunosuppression in SLAM knock-in mice. J Virol. 2010;84:5360–5367. doi: 10.1128/JVI.02525-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 25.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. [PubMed] [Google Scholar]
- 26.Fontenot JD, et al. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 27.Sakaguchi S. Regulatory T cells: History and perspective. Methods Mol Biol. 2011;707:3–17. doi: 10.1007/978-1-61737-979-6_1. [DOI] [PubMed] [Google Scholar]
- 28.Saenz RA, et al. Dynamics of influenza virus infection and pathology. J Virol. 2010;84:3974–3983. doi: 10.1128/JVI.02078-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kouyos RD, Gordon SN, Staprans SI, Silvestri G, Regoes RR. Similar impact of CD8+ T cell responses on early virus dynamics during SIV infections of rhesus macaques and sooty mangabeys. PLOS Comput Biol. 2010;6:e1000901. doi: 10.1371/journal.pcbi.1000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Regoes RR, et al. Roles of target cells and virus-specific cellular immunity in primary simian immunodeficiency virus infection. J Virol. 2004;78:4866–4875. doi: 10.1128/JVI.78.9.4866-4875.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fahey LM, Brooks DG. Opposing positive and negative regulation of T cell activity during viral persistence. Curr Opin Immunol. 2010;22:348–354. doi: 10.1016/j.coi.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanna PP, Burton DR. Role of antibodies in controlling viral disease: Lessons from experiments of nature and gene knockouts. J Virol. 2000;74:9813–9817. doi: 10.1128/jvi.74.21.9813-9817.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen RT, et al. Measles antibody: Reevaluation of protective titers. J Infect Dis. 1990;162:1036–1042. doi: 10.1093/infdis/162.5.1036. [DOI] [PubMed] [Google Scholar]
- 34.Heffernan JM, Keeling MJ. An in-host model of acute infection: Measles as a case study. Theor Popul Biol. 2008;73(1):134–147. doi: 10.1016/j.tpb.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 35.Nowak MA, May RM. Virus Dynamics: Mathematical Principles of Immunology and Virology. New York: Oxford Univ Press; 2000. [Google Scholar]
- 36.Levine B, et al. Antibody-mediated clearance of alphavirus infection from neurons. Science. 1991;254:856–860. doi: 10.1126/science.1658936. [DOI] [PubMed] [Google Scholar]
- 37.Bergthaler A, et al. Impaired antibody response causes persistence of prototypic T cell-contained virus. PLoS Biol. 2009;7:e1000080. doi: 10.1371/journal.pbio.1000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joseph BS, Oldstone MB. Antibody-induced redistribution of measles virus antigens on the cell surface. J Immunol. 1974;113:1205–1209. [PubMed] [Google Scholar]
- 39.Fujinami RS, Oldstone MB. Antiviral antibody reacting on the plasma membrane alters measles virus expression inside the cell. Nature. 1979;279:529–530. doi: 10.1038/279529a0. [DOI] [PubMed] [Google Scholar]
- 40.Fujinami RS, Oldstone MB. Alterations in expression of measles virus polypeptides by antibody: Molecular events in antibody-induced antigenic modulation. J Immunol. 1980;125:78–85. [PubMed] [Google Scholar]
- 41.Fujinami RS, Norrby E, Oldstone MB. Antigenic modulation induced by monoclonal antibodies: Antibodies to measles virus hemagglutinin alters expression of other viral polypeptides in infected cells. J Immunol. 1984;132:2618–2621. [PubMed] [Google Scholar]
- 42.Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell. 2009;138(1):30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 43.Lanford RE, et al. Acute hepatitis A virus infection is associated with a limited type I interferon response and persistence of intrahepatic viral RNA. Proc Natl Acad Sci USA. 2011;108:11223–11228. doi: 10.1073/pnas.1101939108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turner DL, Cauley LS, Khanna KM, Lefrançois L. Persistent antigen presentation after acute vesicular stomatitis virus infection. J Virol. 2007;81:2039–2046. doi: 10.1128/JVI.02167-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zammit DJ, Turner DL, Klonowski KD, Lefrançois L, Cauley LS. Residual antigen presentation after influenza virus infection affects CD8 T cell activation and migration. Immunity. 2006;24:439–449. doi: 10.1016/j.immuni.2006.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.