

Abstract
The circadian clock controls many physiological parameters including immune response to infectious agents, which is mediated by activation of the transcription factor NF-κB. It is widely accepted that circadian regulation is based on periodic changes in gene expression that are triggered by transcriptional activity of the CLOCK/BMAL1 complex. Through the use of a mouse model system we show that daily variations in the intensity of the NF-κB response to a variety of immunomodulators are mediated by core circadian protein CLOCK, which can up-regulate NF-κB–mediated transcription in the absence of BMAL1; moreover, BMAL1 counteracts the CLOCK-dependent increase in the activation of NF-κB–responsive genes. Consistent with its regulatory function, CLOCK is found in protein complexes with the p65 subunit of NF-κB, and its overexpression correlates with an increase in specific phosphorylated and acetylated transcriptionally active forms of p65. In addition, activation of NF-κB in response to immunostimuli in mouse embryonic fibroblasts and primary hepatocytes isolated from Clock-deficient mice is significantly reduced compared with WT cells, whereas Clock-Δ19 mutation, which reduces the transactivation capacity of CLOCK on E-box–containing circadian promoters, has no effect on the ability of CLOCK to up-regulate NF-κB–responsive promoters. These findings establish a molecular link between two essential determinants of the circadian and immune mechanisms, the transcription factors CLOCK and NF-κB, respectively.
Virtually all aspects of an animal’s biochemical, physiological, and behavioral functions are linked to circadian regulation. These functions include such fundamental biological processes as cell-cycle regulation, cellular response to genotoxic stress, and regulation of components of the immune system. Circadian oscillations in the symptom intensity of infectious diseases have been described and linked to variations in immune response. Many immune parameters exhibit daily variations, including the number of specific immune cells in circulation and plasma levels of cytokines (1). Consistent with this variation, daily fluctuations in susceptibility to a powerful challenge to the immune system [i.e., to the administration of a bacterial lipopolysaccharide (LPS)] (2) were correlated with daily variation in the induction of proinflammatory cytokines (3). A growing body of evidence shows that peripheral immune cells harbor functional molecular clocks. Thus, rhythmic expression of clock genes has been described in mouse peritoneal macrophages (4), jejunum (5), and spleen and lymph nodes (6). It also is becoming evident that disruption of daily rhythms affects immune response (7). However, despite much evidence for the existence of cross-talk between the circadian and immune systems, the molecular mechanism that links these two pathways remains largely unknown.
The first line of an animal’s protection against various pathogens is implemented by the components of the innate immune system, which provides immediate defense by inducing a strong inflammatory reaction mediated by activation of the transcription factor NF-κB (8). In mammals, transcriptionally active NF-κB is comprised of homo- or heterodimers of different subunits belonging to the Rel family of transcription factors. In most cell types, NF-κB is represented mainly by the p65/p50 heterodimeric complex. In a nonactive form this complex is retained in the cytoplasm through interaction with the inhibitory protein IκB, the main negative regulator of NF-κB, which undergoes phosphorylation and degradation after immunostimulating treatment. NF-κB activation is triggered by a variety of microbial and viral factors via pattern-recognition receptors, such as Toll-like receptors (TLR), Retinoic acid-inducible gene 1–like receptors, and NOD-like receptors (9, 10), or immunomodulators, such as TNF-α or other cytokines acting via their specific receptors. Ligand recognition leads to the recruitment of various adapter proteins, triggering the activation of the IκB kinase (IKK) complex followed by IκBα phosphorylation and subsequent degradation. The activated NF-κB complex enters the nucleus, binds to its consensus sites in promoters of specific genes (such as cytokines and various regulators of cellular survival and proliferation), and activates their expression (11). Transcription of IκBα is tightly controlled by NF-κB and provides an autoregulatory negative feedback loop for this signaling pathway. Although NF-κB is a trigger of all immune responses and is critical for antimicrobial defense, its excessive or deregulated activation can lead to the development of pathological inflammation that may be a cause of acute and chronic diseases. Hence, NF-κB is considered a plausible target for both therapeutic activation and repression.
The mammalian circadian clock comprises a network of transcriptional and translational feedback loops that drive 24-h–based oscillations in RNA and protein abundance of key clock components (12). At the core of the major circadian loop are two basic helix-loop-helix (bHLH)-PAS domain transcription factors, CLOCK and BMAL1, that form a heterodimer to drive rhythmic expression of genes harboring E-box elements in their promoter region. Numerous studies involving global temporal gene-expression profiling revealed that about 10% of the mammalian transcriptome displays 24-h periodicity in steady-state mRNA levels, suggesting that these genes are controlled by circadian transcription regulators (13). Importantly, the list of clock-controlled genes includes many key regulators of cell cycle, DNA repair, genotoxic stress response, and immune function, and circadian oscillations in their concentration and/or activity would be expected to underlie daily variations in corresponding physiological processes.
In the current study, we describe a direct molecular link between the circadian and NF-κB pathways that is independent of circadian transcriptional regulation by the CLOCK/BMAL1 transactivator complex. We show that the two pathways are coupled through the CLOCK protein, which functions as a positive regulator of NF-κB–responsive promoters even in the absence of BMAL1. These data extend the function of CLOCK beyond the confines of traditional circadian transcriptional control and identify CLOCK as a modulator of the key regulator of immune response, transcription factor NF-κB.
Results
Daily Variations in Acute Response to Inflammatory Agents Correlate with Variations in Transcriptional Activity of NF-κB.
To test whether daily variations in susceptibility to inflammatory challenge correlates with NF-κB activation, we first compared the acute response of BALB/c mice to the TLR4 agonist LPS administered at two time points: at the middle of the light period of the daily cycle at Zeitgeber time ZT6 (in Zeitgeber time, ZT0 corresponds to the beginning of the lights-on period of the daily cycle) or at the middle of the dark period (ZT18). Systemic administration of LPS is highly toxic because the strong induction of proinflammatory cytokines results in septic shock (14). Consistent with previous reports, mice challenged at ZT18 (during the active phase in nocturnal animals) tolerated LPS-induced acute inflammation much better than mice challenged in the middle of their rest period (ZT6) (Fig. 1A).
Fig. 1.

Daily variations in response to immune challenge correlate with variations in the activation of NF-κB transcription factor. (A) LPS-induced toxicity depends on the time of immunostimulation. Kaplan–Meier survival curves of BALB/c mice maintained on a 12:12-h LD cycle and challenged with 20 mg/kg LPS at ZT6 (red line) or ZT18 (blue line) (n = 10 per group). Animals treated in the middle of the active phase of their daily cycle (ZT18) showed higher tolerance (P = 0.018, log-rank test). (B) The activation of the NF-κB–responsive promoter in vivo depends on the time of immunostimulation. Six BALB/C-Tg(IκBα-Luc) reporter mice received 1 μg of CBLB502 at ZT6 or ZT18. Animals were injected i.p. with d-luciferin 1, 2, and 4 h later, and the luciferase signal was monitored in live animals using the Xenogen IVIS 50 system. (C) Quantitation of the luciferase signal shown in B. Closed squares represent animals injected at ZT6; open squares represent animals injected at ZT18. Values are mean of three animals ± SD. (D) Induction of IL-1α mRNA in livers of IkB-Luc reporter mice 20, 40, and 60 min after CBLB502 administration at ZT6 (closed squares) or ZT18 (open squares). (E) Daily variations in NF-κB activation. Eighteen BALB/C-Tg(IκBα-Luc) male mice that were maintained on a 12:12-h LD cycle were injected s.c. with 1 ug CBLB502 at different times of the day. Three hours later animals were injected i.p. with d-luciferin, and luciferase signal was monitored in live animals using the Xenogen IVIS 50 system. Shown are representative images for each time group. Maximum IκB promoter activation is detected in animals treated at ZT6. (F) Quantitative analysis of in vivo images. Values are mean of three animals ± SD. There is a statistically significant difference in IκB-Luc activation in animals treated at ZT6 and animals treated between ZT10–ZT18 (P < 0.001; one-way ANOVA with post hoc Tukey’s test).
To evaluate the extent of NF-κB activation in response to immune challenge and its correlation with daily variations in toxicity, we used BALB/C-Tg(IκBα-Luc) reporter mice, a model that allows direct monitoring of NF-κB activation in live animals. To avoid the high toxicity associated with LPS administration, we chose to use the TLR5 agonist CBLB502, an optimized, highly efficient, and nontoxic derivative of bacterial flagellin (15). Both LPS and bacterial flagellin/CBLB502 induce NF-κB activation; however, because of different tissue specificities and the different spectra of cytokines produced in response to these ligands, the TLR5 agonist does not create a self-amplifying acute inflammation cascade.
Animals received a single injection of CBLB502 at either ZT6 or ZT18, and the level of NF-κB activation in liver was evaluated by in vivo luciferase imaging performed 1, 2, or 4 h later. Results show that TLR5-mediated activation of the NF-κB–responsive promoter was significantly higher in animals that were treated during their rest period (ZT6) (Fig. 1 B and C). Consistent with results of in vivo imaging of κB-luciferase (κB-Luc) reporter activation, animals treated with CBLB502 at ZT6 also showed a higher level of induction of IL-1α mRNA, another direct target of the NF-κB transcription factor (Fig. 1D). To confirm that the times at which we administered CBLB502 do in fact represent the daily peak and trough of NF-κB activation, animals were treated with the ligand at six different times of day and were monitored for IκB-Luc activation 3 h later. As shown in Fig. 1E, NF-κB activation displayed prominent daily variations with a maximum at ZT6. These data suggest that the severity of inflammatory response correlates with the intensity of NF-κB activation and that NF-κB activation is, in fact, under circadian control.
CLOCK Enhances NF-κB–Mediated Transcriptional Activation Independent of BMAL1.
Numerous experimental data suggest that circadian control of various pathways depends on periodic transcriptional activation/repression of key components by the circadian CLOCK/BMAL1 transcriptional complex. However, activation of the NF-κB pathway does not involve transcriptional steps and is regulated largely at the level of phosphorylation-induced degradation of the inhibitory IκB subunit followed by translocation of the p65/p50 dimer into the nucleus. Therefore we hypothesized that circadian modulation of NF-κB activity may occur through a transcription-independent mechanism.
To gain insight into potential mechanisms underlying daily variations in NF-κB activity, we assessed the effect of the CLOCK/BMAL1 transcriptional complex on κB-responsive promoters by transcriptional assays. HEK-293T cells were transfected with the κB-Luc reporter plasmid along with different combinations of p65-, CLOCK- and BMAL1-expressing plasmids. The coexpression of CLOCK/BMAL1 alone had no effect on κB-Luc reporter activity (Fig. 2A). Overexpression of ectopic p65 resulted in strong promoter activation that was increased modestly by coexpression with CLOCK/BMAL1. Surprisingly, when CLOCK and BMAL1 each were expressed separately with p65, they demonstrated very different effects on κB-Luc reporter activation, so that CLOCK alone was responsible for the up-regulated p65-mediated transcription in a concentration-dependent manner. Furthermore, the promoter response to CLOCK/BMAL1/p65 coexpression was slightly less than to CLOCK/p65, indicating that BMAL1 may counteract CLOCK coactivity with p65 (Fig. 2A). To demonstrate that the CLOCK-mediated up-regulation of the reporter does not require BMAL1, we performed a similar experiment in mouse embryonic fibroblasts (MEFs) isolated from Bma1l−/− mice. As shown in Fig. 2B, ectopic CLOCK increased p65-mediated activation of the reporter even in the absence of BMAL1. A similar effect of CLOCK was observed when NF-κB activation was induced by treatment with TNF-α (Fig. 2C). Importantly, the CLOCK-mediated increase in κB promoter activation correlated with a dose-dependent increase in a specifically phosphorylated (Ser536) form of p65, which is characteristic of the active state of p65 (Fig. 2 D and E) (reviewed in ref. 16). The effect of CLOCK was not restricted to the kB-Luc reporter containing consensus elements from the IκB promoter; other NF-κB–responsive promoters (including a 1.2-kb fragment of human TNF-α and E-selectin genes) were coactivated by CLOCK in a similar way (Fig. S1). Together, these data indicate that CLOCK increases NF-κB–mediated transcriptional activation of responsive promoters independent of its circadian partner BMAL1.
Fig. 2.

CLOCK enhances NF-κB activation. (A) Increasing CLOCK expression correlates with NF-κB activation. HEK-293T cells were transfected with 10 ng κB-Luc reporter plasmid, various combinations of p65-, MYC-Clock–, and HA-Bmal1–expressing plasmids, and pcDNA empty vector to equalize the total amount of DNA used in transfection. Bars represent relative luciferase signal normalized for efficiency of transfection using the β-gal assay. Experiments were performed at least three times in duplicate. Values are mean ± SD. The expression of 25 ng of p65-expressing plasmid was used as a positive control to show IκB-Luc reporter gene activation mimicking activation of the NF-κB pathway by stimuli. The effect of higher CLOCK expression on the activation of the reporter was determined to be statistically greater than the effect of p65 expression alone (P = 0.01; Student’s t test). Overexpression of BMAL1 blocks CLOCK-mediated up-regulation of the NF-κB–responsive promoter (P = 0.05). (B) CLOCK-mediated up-regulation of NF-κB does not require BMAL1. MEFs isolated from Bmal1−/− mice were transfected with 10 ng of κB-Luc reporter, 25 ng of p65-expressing plasmid, and either 100 ng MYC-CLOCK– or 40 ng HA-BMAL1–expressing plasmid. Ectopic CLOCK significantly up-regulates the κB-Luc reporter (P = 0.02). Bars represent mean values ± SD. (C) CLOCK enhances NF-κB activation in response to TNF-α. HEK-293T cells were transfected with the κB-Luc reporter gene with either pcDNA- or MYC-CLOCK–expressing plasmid. The following morning cells were treated with 2 ng/mL of TNF-α for 6 h. Ectopic CLOCK significantly enhances TNF-α–mediated activation of the κB-Luc reporter (P < 0.01; Student’s t test). (D) CLOCK-dependent up-regulation of κB-Luc reporter in response to TNF-α correlates with an increase in the active phosphorylated form of p65. HEK-293T cells were transfected with increasing concentrations of CLOCK-expressing plasmid; 24 h posttransfection cells were treated with 2 ng/mL TNF-α for 5 h, and transcriptionally active forms of p65 were visualized by Western blot with antibodies against CLOCK, total p65, pSer536-p65, and Actin for loading control. (E) Quantitative analysis of Western blot presented in D. The increase in the ratio of pSer536-p65 to total p65 correlates with an increase in CLOCK abundance. Experiments were repeated three times with similar results.
CLOCK Is Found in a Protein Complex with p65.
Our transcriptional data showing a functional interaction between CLOCK and NF-κB suggest that CLOCK may exist in a regulatory complex with p65. To test this assumption, coimmunoprecipitation assays were performed. Expression plasmids for CLOCK and BMAL1 were transfected into HEK-293T cells, and whole-cell lysates were resolved in SDS/PAGE along with anti-p65 (Fig. 3A) or anti-CLOCK (Fig. 3B) immunoprecipitations. Western blot results show that in both cases CLOCK is coimmunoprecipitated with p65. CLOCK–p65 interaction also was confirmed by coimmunoprecipitation assays performed in L929 cells with endogenous proteins (Fig. 3C). Consistent with the results of our luciferase assays, CLOCK interaction with p65 was independent of BMAL1, because it still could be precipitated with anti-p65 antibody when ectopically expressed in Bmal1-deficient MEFs (Fig. 3D). Interestingly, less CLOCK was found in complex with p65 when BMAL1 was overexpressed (Fig. 3 A and D). This phenomenon may be attributed to BMAL1–induced phosphorylation and proteolytic degradation of CLOCK (17, 18), which may underlie the observed repression of the modulation function of CLOCK on κB-responsive promoters in the presence of BMAL1 (Fig. 2A).
Fig. 3.

CLOCK is detected in a protein complex with p65. (A) Endogenous p65 pulls down ectopic CLOCK. HEK-293T cells were transfected with expression plasmids for CLOCK and BMAL1 either together or separately. Cells lysates were immunoprecipitated with anti-p65 antibody (IP p65) and analyzed for CLOCK by Western blot. (B) HEK-293T cells were transfected with CLOCK-expressing plasmid. The cell lysates were divided into two equal portions, and anti-CLOCK and nonspecific GAL4 antibody (ns-Ab) were used to pull down immunoprecipitates which were analyzed by Western blot with anti-p65 antibody. (C) Endogenous CLOCK–p65 interaction. L929 cells, which are high in CLOCK protein, were harvested in lysis buffer and immunoprecipitated with control nonimmune serum or with specific antibodies directed against either p65 or CLOCK. The immunoprecipitates were resolved in SDS/PAGE and analyzed for p65 expression by Western blot. (D) CLOCK interacts with p65 independently of BMAL1. Bmal1-deficient MEFs were transfected with expression plasmids of CLOCK and BMAL1 as indicated. The whole-cell extracts of the transfected cells and the corresponding anti-p65 immunoprecipitates were analyzed for ectopic CLOCK and BMAL1 proteins by anti-HA/MYC Western blot. WCE, whole-cell extract.
NF-κB Activation Is Impaired in Clock-Deficient Cells and Livers of Clock−/− Mice.
If CLOCK is, in fact, involved in the up-regulation of p65-dependent transcription, one might predict that activation of NF-κB target genes would be reduced in Clock-deficient cells. To test this prediction, we generated WT and Clock−/− MEFs stably expressing the κB-Luc reporter and compared their activation in response to TNF-α treatment. As shown in Fig. 4A, activation of the κB-Luc reporter was impaired in Clock-deficient MEFs compared with WT. Consistent with reduced response, nuclear accumulation of p65 in Clock−/− MEFs and its phospho-activation also were decreased significantly (Fig. 4 B and C). Importantly, total p65 levels were comparable in WT and Clock-deficient MEFs under both basal and induced conditions (Fig. 4D). These data were confirmed by immunostaining with anti-p65 antibody, showing reduced levels of nuclear p65 in Clock-deficient MEFs following TNF-α treatment (Fig. S2A). An EMSA assay performed in the same cell lysates also detected a reduction in p65 binding to DNA oligonucleotides carrying kB-binding sites in Clock−/− MEFs (Fig. 4 E and F). Most likely, this reduction reflects an overall decrease in the abundance of the active NF-κB complex in the absence of CLOCK rather than its specific effect on p65 affinity for DNA, because CLOCK has the capacity to exert strong up-regulation of the activity of a GAL4-p65 fusion protein on GAL4 promoter (Fig. 4G). Together, these results suggest that CLOCK is required for full-scale activation of NF-κB and that CLOCK promotes an increase in nuclear transcriptionally active phospho-p65.
Fig. 4.

NF-κB activation is reduced in Clock-deficient MEFs. (A) WT (black bars) and Clock-deficient (gray bars) MEFs stably expressing κB-Luc reporter were treated with 2 ng/mL TNF-α for 5 h, and luciferase activity was measured in cell lysates. (B) Clock-deficient cells show reduced levels of nuclear phospho-active p65 after TNF-α–mediated NF-κB activation. WT and Clock-deficient MEF cells were treated with 2 ng/mL of TNF-α for the indicated times and were used to prepare nuclear extracts, which were analyzed by Western blots with antibodies against total p65 and its pSer536 phosphorylated form. (C) Quantitative analysis of the Western blot shown in B. ACTIN was used as a loading control. Black bars, WT; gray bars, Clock-deficient MEFs. (D) Deficiency in Clock has no effect on total levels of p65. WT and Clock-deficient MEFs were treated with 2 ng/mL of TNF-α for the indicated times. Total cell lysates were analyzed by Western blot as in B. (E) Impaired induction of NF-κB DNA binding in Clock−/− MEF cells treated with TNF-α. WT and Clock-deficient MEFs were treated with 1 ng/mL TNF-α for the indicated times. (F) Quantitative analysis of intensity of the band corresponding to DNA-bound p65/p50 dimer. The amount of DNA-bound complex in Clock-deficient MEFs (gray bars) is reduced compared with WT MEFs (black bars); the decrease is particularly pronounced after 45 min of TNF-α treatment, suggesting that the overall amount of active NF-κB is diminished in Clock-deficient MEFs. (G) CLOCK has no effect on the DNA-binding properties of p65. HEK-293T cells were cotransfected with a luciferase reporter gene encoding a GAL4-binding element and plasmid encoding a p65 fusion of the GAL4 DNA-binding domain. The CLOCK- and BMAL1-expressing plasmids were transfected as indicated to show CLOCK’s dose-dependent effect on p65 activation and BMAL1’s capacity to counter this effect (Left). The GAL4 luciferase reporter gene also was transfected with the CLOCK-expressing plasmid to show that CLOCK did not drive expression of the reporter gene (Right).
Because our in vivo data showed strong daily variations in NF-κB activation in response to the TLR5 agonist CBLB502, we sought to test whether CLOCK is involved in this regulation. To do so, we generated a genetic cross between BALB/C-Tg(IκBα-Luc) and Clock−/− mice. The progeny that were heterozygous for Clock and hemizygous for the presence of the IκB-Luc reporter (IκB-Luc+/−; Clock+/−) were treated with CBLB502 at ZT06, and NF-κB activation in liver was monitored by in vivo imaging 2 h later. As shown in Fig. 5 A and B, in heterozygous animals a reduction in Clock gene dosage resulted in significant down-regulation of CBLB502-mediated activation of the κB-Luc reporter in liver. Consistent with this result, 2 h after CBLB502 treatment at ZT6 the plasma levels of IL-6 (one of the major NF-κB targets activated by CBLB502 administration) were much lower in Clock−/−mice than in WT mice and were comparable with IL-6 levels induced in WT mice treated at ZT18 (Fig. 5C). To confirm these differences further, we isolated and cultured primary hepatocytes from WT and Clock−/− mice and treated them with CBLB502 in vitro. As shown in Fig. 5 D and E, the amount of DNA-bound p65 was impaired in hepatocytes of Clock−/− mice, and less nuclear p65 was detected by immunocytochemical staining (Fig. S2B). Thus, deficiency in CLOCK in two different cell types (MEFs and primary hepatocytes) treated with two different immunomodulators (TNF-α and CBLB502) that trigger the NF-κB pathway via distinct reporter classes (TNF receptor and TLR5) reduces the ability of NF-κB to activate responsive promoters. These data suggest that CLOCK up-regulates NF-κB–mediated transcription in several different cell types. Thus, in addition to its role in the transcriptional regulation of circadian promoters in complex with BMAL1, CLOCK can enhance the activity of other key transcriptional regulators in a BMAL1-independent manner.
Fig. 5.

NF-κB activation in response to CBLB502 is reduced in liver and primary hepatocytes of Clock-deficient mice. (A) BALB/C-Tg(IκBα-Luc) mice were crossed to C57BL/6J WT or Clock-deficient mice (C57BL/6J background). The progeny (WT or Clock+/−) received 1 ug of CBLB502 at ZT06 and were monitored for luciferase expression in vivo 2 h later. Representative images for each genotype are shown. (B) Quantitative analysis of luciferase expression in livers of WT (n = 3) and Clock+/− (n = 3) mice measured 2 h post CBLB502 administration. CBLB502-mediated NF-κB activation is reduced in Clock+/− mice (P < 0.05). UT, untreated (C) CBLB502-mediated induction of IL-6 correlates with the scale of NF-κB activation. Two WT and two Clock−/− mice received a single injection of CBLB502 at the times indicated. Blood was collected 2 h later, and the plasma concentration of IL-6 was measured by ELISA. (D) Impaired induction of NF-κB DNA binding in primary hepatocytes of Clock−/− mice treated with CBLB502 in vitro. Primary hepatocytes isolated from age-matched WT and Clock−/− mice were treated with 100 ng/mL of CBLB502 for the indicated times. (E) Quantitative analysis of the intensity of the band corresponding to DNA-bound p65/p50 dimer. The amount of DNA-bound complex in Clock-deficient hepatocytes (gray bars) is reduced significantly compared with WT hepatocytes (black bars).
CLOCK-Mediated Up-Regulation of NF-κB–Responsive Promoters Is Distinct from Its Function on E-Box Promoters of CLOCK/BMAL1 Target Genes.
The fact that CLOCK-dependent up-regulation of NF-κB–responsive genes does not require its dimerization with BMAL1 was our first indication that this function of CLOCK is distinct from its ability to activate E-box–containing promoters of circadian-regulated genes (Fig. 2). To characterize CLOCK function on NF-κB–responsive promoters further, we tested the ability of mutant CLOCK protein (CLOCK-Δ19) to up-regulate the κB-Luc reporter in a manner similar to WT CLOCK. The Clock-Δ19 mutation was identified originally in an N-ethyl-N-nitrosourea mutagenesis screen for mutations affecting circadian behavior in mice (19). The mutation results in deletion of 51 amino acids in the transactivation domain of the CLOCK protein (20). CLOCK-Δ19 can bind DNA and dimerize with BMAL1; however, the complex is deficient in transactivation (21). As a result, Clock-Δ19 mutant mice show reduced levels of expression of many clock and clock-controlled genes in various tissues (22). Results of luciferase assays in HEK-293T cells treated with TNF-α showed that both WT CLOCK and CLOCK-Δ19 effectively coactivated κB-Luc reporter (Fig. 6A). Consistent with this finding, the amount of DNA-bound p65/p50 complex in primary hepatocytes isolated from livers of Clock-Δ19 mice and treated with CBLB502 was indistinguishable from that of WT animals (Fig. 6 B and C). Also, staining for nuclear p65 did not reveal any differences between the two genotypes (Fig. S2C). Moreover, unlike cryptochrome (CRY)-mediated repression of E-box promoters, the regulatory function of CLOCK on NF-κB–responsive promoters was not affected by overexpression of CRY1 (Fig. 6D). Together, these results suggest that CLOCK’s function as a modulator of the transcriptional activation of NF-κB–responsive promoters is distinct from its transactivator function in complex with BMAL1 on E-box–responsive promoters.
Fig. 6.

CLOCK-dependent modulation of NF-κB activation is distinct from its transactivation function on circadian promoters. (A) The CLOCK-Δ19 mutant modulates NF-κB–responsive promoters as well as WT CLOCK. HEK-293T cells were cotransfected with the κB-Luc reporter in combination with Clock- or Clock-Δ19 –expressing plasmids. Twenty-four hours posttransfection cells were treated with 2 ng/mL of TNF-α for 5 h. Both constructs modulate NF-κB (P < 0.05). (B) The Clock-Δ19 mutation has no effect on the amount of DNA-bound p65/p50 complex. Primary hepatocytes obtained from WT and Clock-Δ19–mutant mice were treated with CBLB502 (100 ng/mL) for the times indicated. (C) Quantitative analysis of intensity of the band corresponding to DNA-bound p65/p50 dimer shows no difference in the strength and kinetics of CBLB502-induced NF-κB DNA binding in Clock-Δ19 (gray bars) and WT (black bars) cells. Values are the average of two samples run on the same gel ± SD. (D) The circadian repressor CRY1 has no effect on CLOCK’s ability to up-regulate NF-κB–responsive promoters. HEK-293T cells were transfected with 10 ng of Per1-Luc or kB-Luc reporters and various expression plasmids of clock proteins. CRY1 represses CLOCK/BMAL1-dependent transactivation on the Per1 promoter but has no effect on CLOCK-dependent up-regulation of the κB promoter in response to TNF-α.
CLOCK Cooperates with NF-κB Coactivators.
CLOCK has been shown to exhibit intrinsic histone acetyltransferase (HAT) activity and to transacetylate histones (23), its transcriptional partner BMAL1 (24), and the glucocorticoid receptor (25). Acetylation of p65 is an important posttranslational modification that is involved in the up-regulation of NF-κB target genes. To test whether CLOCK exerts its effects on NF-κB–responsive promoters via p65 acetylation, we first tested the effects of CLOCK overexpression on the abundance of specifically acetylated Lys-310, a form of p65 that is required for its full activation potential (26). The results of a Western blot assay showed more p65 acetylation in the presence of CLOCK (Fig. 7A). To test whether the intrinsic transacetylase activity of CLOCK is required for the up-regulation of the κB-Luc reporter, we generated a CLOCK mutant lacking HAT activity (CLOCK-ΔHAT). 293T cells were transfected with the kB-Luc reporter gene and plasmids expressing full-length CLOCK or CLOCK-ΔHAT and were treated with TNF-α. The results show that the ability of CLOCK-ΔHAT to up-regulate the NF-κB–responsive promoter was similar to that of WT CLOCK (Fig. 7B). These data indicate that the intrinsic HAT activity of CLOCK is not important for mediating the increased acetylation of p65 suggesting that CLOCK may recruit other, more potent HATs, such as p300 and/or CREB-binding protein (CBP) to NF-κB-responsive promoters. To address this hypothesis, a κB-Luc reporter assay was performed with expression plasmids of CLOCK, CBP, and BMAL1. As shown in Fig. 7C, both CBP and CLOCK up-regulated NF-κB–dependent transcription and, when expressed in combination, clearly demonstrate a cooperative effect. Consistent with previous data, BMAL1 negated CLOCK-mediated up-regulation of NF-κB in this assay.
Fig. 7.
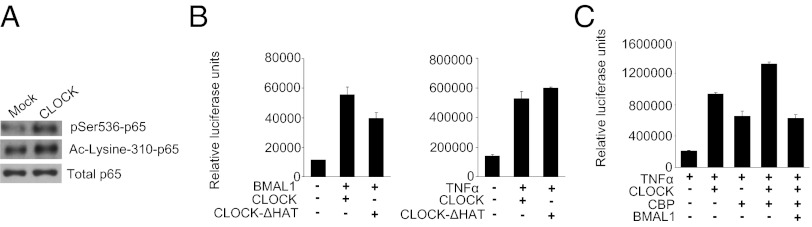
CLOCK cooperates with NF-κB coactivators. (A) Overexpression of CLOCK facilitates acetylation of p65 at Lys-310. HEK-293T cells were transfected with either the control pcDNA vector or CLOCK-expressing plasmid. Cells were harvested in Nonidet P-40 buffer 24 h posttransfection, and lysates were immunoprecipitated with anti-p65. The precipitates were analyzed by Western blot using p65-specific Ac-Lysine 310 antibody. An anti-p65 Western blot was used to normalize for loading. (B) Intrinsic HAT activity is not important for CLOCK-dependent up-regulation of NF-κB–responsive promoters. HEK-293T cells were cotransfected with a luciferase reporter gene driven by either the Per1 promoter (Left) or the NF-κB–responsive promoter (Right) and the combination of the BMAL1-expressing plasmid and a plasmid expressing either WT CLOCK or HAT-deficient CLOCK as indicated. TNF-α was added at 2 ng/mL 24 h posttransfection. (C) CLOCK cooperates with CBP in activating the κB-responsive promoter. 293T cells were cotransfected with the kB-Luc reporter and combinations of plasmids expressing CLOCK, BMAL1, and CBP. TNF-α was added at 2 ng/mL 24 h posttransfection. The coexpression of CLOCK and CBP results in higher NF-κB activation (P < 0.05).
Discussion
Circadian oscillations of physiological functions and immune response to infectious agents are two major adaptive control mechanisms that are regulated through the nuclear abundance of their corresponding multi-subunit transcription factors. Phenomenological indications of their functional interconnection are well documented in circadian studies and are likely to reflect adaptive benefits to the organism, although the rationale remains to be explained. Thus, previous studies have demonstrated that an animal’s susceptibility to various infectious agents is time dependent and often is correlated with differential induction of pro- and anti-inflammatory cytokines (3). Our data support this phenomenon by demonstrating dramatic differences in mouse sensitivity to LPS at different circadian phases (Fig. 1A). Moreover, it has been shown recently that deficiency in CRY proteins, core components of the molecular clock, increases susceptibility of cells to TNF-α–induced apoptosis resulting from impaired activation of NF-κB (27). However, the exact molecular mechanisms underlying cross-talk between circadian and NF-κB pathways have not been understood. Here we provide evidence that the two pathways are linked through the activity of the core circadian protein, CLOCK.
CLOCK always has been viewed as the essential component of the major circadian transactivation complex that dimerizes with BMAL1 to drive rhythmic expression from E-box–containing promoters. We now demonstrate that CLOCK also functions as an enhancer of the transactivation potential of NF-κB and that this modulatory function of CLOCK does not require BMAL1. Consistent with this finding, CLOCK is found in protein complexes with the p65 subunit of NF-κB, although we do not know whether this physical interaction is direct or if it is required for their functional cooperation. Furthermore, the NF-κB response is reduced significantly in Clock-deficient cells and in the tissues of Clock-knockout mice. We also provide evidence that the role of CLOCK as an enhancer of NF-κB signaling is independent of specific transactivator functions that are required of CLOCK in BMAL1-dependent regulation of circadian genes. Together, these findings demonstrate a mechanistic link between key regulators of immune response and circadian function.
Although our work defines CLOCK as a modulator of the transcriptional activity of NF-κB, the exact molecular mechanism(s) of CLOCK/NF-κB interaction and subsequent up-regulation of NF-κB–responsive promoters requires more in-depth studies. Transcriptional activation is a dynamic process involving gene-specific activators binding to their response elements, chromatin remodeling, and recruitment of a series of coactivators that generate protein bridges between transcription factors and basal transcription machinery. Generally speaking, in its role as an enhancer of the functional activity of other transcription factors, CLOCK resembles the best-studied transcriptional coactivator, steroid receptor coactivator 3 (SRC-3), with which it shares sequence and functional similarities. CLOCK possesses intrinsic HAT activity; a nuclear receptor-interacting domain; and potential CBP/p300- and methyltransferase-interacting domains (28), suggesting that it may acetylate p65 directly or recruit other transcriptional regulators to NF-κB–responsive promoters. Our transcriptional assays show that full-length and HAT-deficient CLOCKs are equally effective in up-regulating the NF-κB–responsive promoter, a finding that agues in favor of the latter option. This result also is consistent with our transcriptional assay showing functional cooperation of CLOCK and CBP in activating the κB-Luc reporter and with previously published data demonstrating the interaction of both CLOCK and its homolog NPAS2 with chromatin-modifying enzymes on E-box–containing promoters (29, 30). It is noteworthy that recently CLOCK was identified as one of only three proteins that are recruited to sites of dsDNA breaks (31). Although the detailed mechanism and functional significance of this finding have not been investigated, it represents another example of the BMAL1-independent function of CLOCK and suggests that CLOCK may be involved in DNA repair by recruiting chromatin-modifying or DNA repair enzymes to the sites of DNA lesions by a mechanism similar to NF-κB coactivation.
The fact that CLOCK, which is an integral component of circadian machinery, can cooperate with a key regulator of immune response in the absence of BMAL1 uncovers a regulatory level of circadian control of cellular responses. Our in vivo data demonstrate that NF-κB activation in response to bacterial flagellin/CBLB502 displays prominent daily variations, with a peak corresponding to the middle of the daily rest period, ZT6 (Fig. 1E), and that these variations are determined by CLOCK. This conclusion may seem paradoxical, because it has been shown that during the circadian cycle the expression of CLOCK is nearly constitutive at both the mRNA and protein levels (32). At the same time, many functional characteristics of CLOCK are regulated by BMAL1, which is subject to strong circadian regulation at multiple levels. Thus, BMAL1 mediates the nuclear translocation (17, 33) and site-specific phosphorylation/degradation of CLOCK (18), both of which are tightly linked to the activation of responsive circadian promoters. These findings allow us to speculate that the sharp peak in CLOCK-mediated up-regulation of NF-κB–responsive promoters observed at ZT6 may coincide with the daily peak in BMAL1-dependent shuttling of CLOCK into the nucleus, thereby generating an abundant pool of “active nuclear CLOCK” available for cooperation with other transcriptional regulators on noncircadian promoters. After being recruited to E-box promoters, CLOCK availability is decreased dramatically by transactivation-dependent degradation (33), possibly leading to a decrease in the transactivation potential of NF-κB. Although this hypothesis has yet to be tested, it is supported by the timing of the effect of CLOCK on the NF-κB response, by BMAL1-dependent repression of CLOCK modulation of the NF-κB response, and by several reports showing less nuclear CLOCK at times of higher CLOCK/BMAL1-dependent transcriptional activation of circadian promoters (17, 32, 33). Although at this time it cannot be concluded that the previously observed impairment of NF-κB activation in the absence of CRYs occurs through the mechanism that we have described, the reversal of this phenotype by the down-regulation of BMAL1 (27) and the demonstration of the formation of a ternary complex involving CLOCK, BMAL1, and CRY1 (34) are consistent with our hypothetical model.
Other important questions that remain to be answered include the broader applications of CLOCK modulator functions, whether CLOCK cooperation with NF-κB is ligand- and/or tissue-specific, and whether CLOCK can cooperate with other transcriptional regulators. Our data show that CLOCK up-regulates several different NF-κB–responsive promoters (synthetic and endogenous) in two different types of tissues (MEFs and primary hepatocytes in liver) in response to two stimuli (TNF-α and TLR5 agonist). Because all immunostimulators induce acute activation of NF-κB, it seems reasonable that CLOCK-mediated up-regulation of NF-κB–dependent transcription may represent a general mechanism that is independent of the type of immune challenge. At the same time, it is possible that some mediators of the immune response may be under direct circadian transcriptional control. A recent report suggests that the circadian clock controls the expression and function of another type of TLR, TLR9, which is activated in response to bacterial and viral DNA and leads to acute activation of NF-κB (35). This control was attributed to the direct regulation of Tlr9 mRNA expression by the CLOCK/BMAL1 complex through putative E-box elements in its promoter (36). In our study, Tlr5 mRNA profiling did not reveal a circadian pattern in its expression (Fig. S3); furthermore, the fast kinetics of NF-κB activation in response to CBLB502 argues in favor of a nontranscriptional regulatory mechanism.
The identification of CLOCK as an enhancer of NF-κB–mediated transcription, whose activity is distinct from the transactivation of circadian genes, has important translational applications. Although the induction of the NF-κB response is critical for antimicrobial defense, the response must be well-balanced, because its excessive activation results in both acute toxicity and chronic diseases. It also is assumed that the most effective immune response correlates with the active period of an organism’s daily cycle, because this is the time when the risk of infection from a variety of routes (food, wounding, sexual behavior, among others) is higher. Interestingly, our data demonstrate that the daily peak in NF-κB activation in response to bacterial flagellin/CBLB502 occurs at the middle of the rest period (ZT6 in nocturnal mice) and that this time coincides with the peak of LPS-induced toxicity. This finding suggests that the excess of nuclear CLOCK may promote NF-κB activation, leading to overproduction of inflammatory cytokines and systemic toxicity, whereas BMAL1 is important for reducing the scale of inflammatory response by downregulating the CLOCK-dependent modulation of NF-κB activation. Thus, CLOCK and BMAL1 seem to affect the inflammatory response in different but interdependent ways. In line with this hypothesis is our initial observation of a high basal level of active NF-κB in MEFs of Bmal1−/− mice (Fig. S4), suggesting that BMAL1 deficiency may result in chronic inflammation. Chronic inflammation often is linked to pathologies such as arthritis, asthma, septic shock, lung fibrosis, glomerulonephritis, atherosclerosis, and premature aging (37). In previous work we demonstrated that Bmal1−/− mice develop a syndrome of premature aging, which was attributed to excessive production of reactive oxygen species (ROS) (38). Our current work suggests that BMAL1 deficiency may cause an misbalance in ROS generation/neutralization not only through the deregulation of downstream transcriptional anti- and pro-oxidant CLOCK/BMAL1 targets but also by promoting chronic inflammation via an NF-κB–dependent mechanism (39). Because NF-κB is considered a plausible target for both therapeutic activation and repression, detailed mechanistic understanding of the relative roles of CLOCK and BMAL1 in regulating NF-κB activity may result in the development of novel therapeutic tools and strategies in treatment of immune disorders.
Materials and Methods
Animals.
BALB/C-Tg(IκBα-Luc) reporter mice were obtained from Xenogen Corp. Clock-knockout mice were obtained from David Weaver (University of Massachusetts Medical School, Worcester, MA) and were transferred to the C57BL/6J background for 10 backcross generations. Clock-Δ19 mice were obtained originally from J. Takahashi (University of Texas Southwestern Medical Center, Dallas) and were backcrossed to the C57BL/6J background for 20 generations. WT C57BL/6J mice were purchased from Jackson Laboratory. All animals were housed at a 12:12-h light/dark (LD) cycle. All manipulations during the dark phase of the cycle were performed under infrared light. Escherichia coli LPS (005:B5) was injected i.p. at 20 mg/kg; CBLB502 was injected s.c. at a dose of 1ug/mouse. Animal husbandry is in compliance with NIH, USDA, and New York State Standards. All animal experiments were approved by the Institutional Animal Care and Use Committee of Roswell Park Cancer Institute.
In Vivo Bioluminescence Imaging.
Bioluminescence imaging was performed using the IVIS 50 imaging system (Caliper Life Sciences Xenogen Corp.). One to three hours after immunostimulation with CBLB502, mice were injected i.p. with firefly d-Luciferin (150 mg/kg) (Caliper Life Sciences), anesthetized with isoflurane, and imaged 15 min later using an integration time of 10 s and medium binning. Data were quantified as the sum of photon flux within the region of interest using Living Image software (Xenogen Corp.).
Plasmids.
Expression constructs encoding HA-tagged BMAL1, CLOCK, and CRY1 proteins were described previously (17). TNF-nonresponsive expression plasmids encoding MYC-CLOCK were generously provided by Kunho Lee (Seoul National University, Seoul, Korea). Untagged-p65, GAL4-p65, and GAL4-Luc reporter are described in ref. 40. The mPer1 reporter construct containing 1.8 kb of mPer1 promoter is described in ref. 41. The κB-Luc reporter containing three tandem copies of the two adjacent NF-κB sites from the HIV enhancer is described in ref. 42. The hTNF-Luc reporter was generously provided by Dmitry Kuprash (Engelhardt Institute of Molecular Biology, Moscow) and is described in ref. 43. The CLOCK-ΔHAT construct was generated as described in ref. 23. The CLOCK-Δ19 expression plasmid was provided by J. Takahashi. The pRc/RSV-mCBP-HA expression plasmid was acquired from Addgene.
Cells.
HEK-293T and L929 cells were obtained from ATCC. MEFs were isolated from WT, Bmal1−/−, and Clock−/− embryos at embryonic day 13 and were immortalized by a retrovirus-mediated expression of GSE56 (a fragment of p53 encoding for a peptide working as a dominant-negative inhibitor of p53) as described (44). Cells were maintained in DMEM supplemented with 10% (vol/vol) FCS.
Transient Transfection, Luciferase Reporter Assay, Cellular Fractionation, and Western Blotting.
HEK-293T cells were seeded in 24-well plates and transfected with various proteins encoding plasmids using Fugene 6 (Roche Diagnostics) according to the manufacturer’s protocol. The amount of total DNA was equalized with pcDNA3 empty vector (Invitrogen). A β-gal expression plasmid driven by the TNF-nonresponsive RSV promoter was used to normalize transfection efficiency. When cells were treated with TNF-α (Sigma-Aldrich), it was added 18–24 h posttransfection for 5–6 h. Cells were harvested in Promega Reporter Lysis buffer and analyzed for luciferase and β-gal activity as previously described (18). Results are representative of at least three experiments performed in duplicate. For Western blot analysis, whole-cell lysates were prepared 24 h posttransfection by boiling in 2% SDS. Nuclear extraction was performed using the Active Motif nuclear extraction kit following the manufacturer’s specifications. Proteins were resolved by SDS/PAGE, transferred to nitrocellulose membrane, and probed with specific antibodies. HA-tagged proteins were detected with mouse anti-HA.11 (Covance Research Products). Anti-CLOCK and anti-BMAL1 were raised in guinea pig as previously described (18). Antibodies against pSer535-p65 and Ac310-p65 were purchased from Cell Signaling. Monoclonal anti-Myc antibody 9E10, polyclonal anti-p65, and HRP-conjugated secondary antibodies were purchased from Santa Cruz Biotechnologies. Transferred proteins were visualized with the ECL detection kit (Jackson Research Laboratories); the intensities of the autoradiographic signals were quantitated by scanning with an imaging densitometer, followed by integration of the signals using ImageJ software. Intensities of corresponding ACTIN bands were used to normalize for protein loading.
Coimmunoprecipitation.
HEK-293T and MEF cells transfected with plasmids encoding various combinations of CLOCK, BMAL1, and p65 proteins were lysed in buffer [100 mM NaCl, 50 mM Tris⋅HCL (pH 7.5), 2 mM EDTA, 1% Nonidet P-40, 0.1% SDS] supplemented with proteinase inhibitors mixture (Roche Diagnostics) and phenylmethylsulfonyl fluoride (Sigma). The lysate of equal amounts of protein was incubated at 4 °C with primary antibody for 2 h and then with protein A/G beads for an additional 2 h, after which beads were washed with TBS washing buffer [50 mM Tris⋅HCl (pH 7.4), and 150 mM NaCl] supplemented with 0.1% Tween. Bound proteins were solubilized in SDS sample buffer for Western blot analysis.
EMSA and Electrophoretic Mobility Supershift Assay.
Murine normal primary hepatocytes or MEF cells (2–2.5 × 106 for the each time point, 80% cell density) were treated with 100 ng/mL of CBLB502, a derivative of bacterial flagellin (Cleveland BioLabs, Inc.) or10 ng/mL of rhTNF-α (R&D Systems), respectively, for the indicated times. Before collection cells were washed twice with ice-cold 1× PBS buffer, collected by a cell lifter on ice, and centrifuged (500 × g at 4 °C for 10 min). Cell pellets were snap frozen in liquid nitrogen and stored at −70 °C until protein isolation. Nuclear protein extracts were prepared as described in ref. 45 with some modifications (46). The binding reaction for EMSA contained 10 mM Hepes (pH 7.5), 80 mM KCl, 1 mM EDTA, 1 mM EGTA, 6% glycerol, 0.5 μg poly(dI-dC), 0.5 μg sonicated salmon sperm DNA, [γ-32P]-labeled (2–3 × 105 cpm) double-stranded κB-consensus oligonucleotide (Promega), and 10 μg of nuclear protein extract. DNA-binding reaction was performed at room temperature for 45 min in a final volume of 20 μL. For the electrophoretic mobility supershift assay, antibodies against p65 or p50 (Santa Cruz) were added to the reaction before the addition of labeled oligonucleotide. DNA–protein complexes were analyzed on 6% PAGE with × 0.25 Tris/borate/EDTA buffer. Dried gels were subjected to radiography. The intensities of the autoradiographic signals were quantitated by scanning with an imaging densitometer (Canon 4400F), followed by integration of the signals using ImageJ software. Integrated intensities of the autoradiographic signals of the complex (arbitrary numbers) are presented as a percentage of the most abundant band that was set for 100%.
ELISA.
WT and Clock−/− male mice received a single injection of 1 ug of CBLB502. Two hours later blood samples were collected through retroorbital bleeding and plasma were separated using Microtainer Plasma Separation Tubes with Lithium Heparin (Becton Dickinson). The levels of IL-6 were measured using the Mouse IL-6 ELISA kit (Invitrogen) per the manufacturer’s protocol.
Isolation and Cultivation of Adult Mouse Hepatocytes.
Mouse hepatocytes were isolated from adult male C57BL/6 wild-type and Clock−/− mice after two-step perfusion as described previously (47) with some modifications. Liver perfusion was performed on deeply anesthetized mice through the inferior vena cava using peristaltic pump at a rate of 3–4 mL/min. The liver was perfused with EGTA (0.5 mM EGTA in PBS without calcium and magnesium) following 0.02% collagenase type IV (Sigma) in DMEM. After perfusion, the liver was dissected, disrupted in DMEM, and filtered through 40-μm cell strainers. Cells were allowed to sediment for 20 min at +4 °C, after which the pellet was dissolved in 20 mL of DMEM and overlaid on the top of a two-step Percoll gradient [50% and 25% Percoll (wt/vol) in PBS]. Purified hepatocytes were collected from the bottom of the tube after centrifugation at 1,750 × g for 20 min and were resuspended in DMEM supplemented with 10% (vol/vol) FCS. Cells were plated at a density of 70 × 103 nuclei/cm2 and were allowed to attach for 1–1.5 h. Then the medium was replaced with William’s E medium (Invitrogen) supplemented with 10% FCS, penicillin/streptomycin, 2 mM glutamine, 10 mM nicotinamide, ITS (insulin, transferrin, sodium selenit), 50 ng/mL EGF, and 10−7M dexamethasone (48). The typical yield was 40–50 × 106 hepatocyte nuclei per mouse liver with 90–93% viability.
Hepatocyte Treatment with CBLB502 and Immunostaining.
After 24 h hepatocyte cultures were treated with CBLB502 at 100 ng/mL for 30–120 min. Cultures were fixed with 4% formaldehyde in PBS for 10 min at room temperature, washed with PBS, and blocked from unspecific antibody binding by incubation for 10 min with 2% glycine in PBS with 0.2% Triton X-100 and 0.2% Tween-20. Primary antibody (rabbit antibody against p65; ChiP grade, 1:200 dilution) (Abcam) was mixed with Alexa Fluor 594- or Alexa Fluor 647-conjugated phalloidin (1:100 dilution) (Invitrogen) and incubated with cultures for 30–45 min at room temperature followed by washing with PBS and incubation with secondary donkey anti-rabbit DyLight488 antibody (1:500 dilution) (Jackson ImmunoResearch) for 30 min at room temperature. Primary and secondary antibodies were diluted in blocking solution (5% normal donkey serum, PBS, 0.2% Triton X-100, 0.2% Tween-20). After washing with PBS cultures were mounted in ProLong antifade reagent (Invitrogen) with DAPI (DNA counterstain) and were studied under an AxioImager Z1 fluorescent microscope (Carl Zeiss Inc.). Images were captured by an AxioCam MRm digital camera (Carl Zeiss Inc.) using AxioVision software (rel.3.4.6; Carl Zeiss Inc.).
Statistical Analyses.
All experiments were performed in triplicate unless stated, all values are presented as mean ± SD. Statistical analyses were performed with SigmaStat 3.5 (Systat Software) using log-rank analysis for survival curves, Student’s t test for two-group comparisons, and ANOVA for comparisons of more than two groups with post hoc Tukey’s test. P values <0.05 were considered significant.
Supplementary Material
Acknowledgments
We thank Drs. Joseph Takahashi for providing Clock-Δ19 mutant mice, David Weaver for providing Clock−/− mice, Dmitry Kuprash for the hTNF-α–κB-Luc reporter, Kunho Lee for the MYC-CLOCK expression construct, T. Gilmore for p65-expressing constructs, and Roman Kondratov for critical comments on the manuscript. This work was supported by National Institutes of Health Grants GM095847 (to M.P.A.) and AI080446 and AI087616 (to A. V. Gudkov).
Footnotes
Conflict of interest statement: A. V. Gudkov is a co-founder and a shareholder of Cleveland Biolabs, Inc., the company that is developing CBLB502 for medical and biodefense applications.
This article is a PNAS Direct Submission.
See Author Summary on page 14736 (volume 109, number 37).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1206274109/-/DCSupplemental.
References
- 1.Haus E, Smolensky MH. Biologic rhythms in the immune system. Chronobiol Int. 1999;16:581–622. doi: 10.3109/07420529908998730. [DOI] [PubMed] [Google Scholar]
- 2.Halberg F, Johnson EA, Brown BW, Bittner JJ. Susceptibility rhythm to E. coli endotoxin and bioassay. Proc Soc Exp Biol Med. 1960;103:142–144. doi: 10.3181/00379727-103-25439. [DOI] [PubMed] [Google Scholar]
- 3.Marpegan L, et al. Diurnal variation in endotoxin-induced mortality in mice: Correlation with proinflammatory factors. Chronobiol Int. 2009;26:1430–1442. doi: 10.3109/07420520903408358. [DOI] [PubMed] [Google Scholar]
- 4.Hayashi M, Shimba S, Tezuka M. Characterization of the molecular clock in mouse peritoneal macrophages. Biol Pharm Bull. 2007;30:621–626. doi: 10.1248/bpb.30.621. [DOI] [PubMed] [Google Scholar]
- 5.Froy O, Chapnik N. Circadian oscillation of innate immunity components in mouse small intestine. Mol Immunol. 2007;44:1954–1960. doi: 10.1016/j.molimm.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 6.Keller M, et al. A circadian clock in macrophages controls inflammatory immune responses. Proc Natl Acad Sci USA. 2009;106:21407–21412. doi: 10.1073/pnas.0906361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Logan RW, Sarkar DK. Circadian nature of immune function. Mol Cell Endocrinol. 2012;349:82–90. doi: 10.1016/j.mce.2011.06.039. [DOI] [PubMed] [Google Scholar]
- 8.Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun. 2009;388:621–625. doi: 10.1016/j.bbrc.2009.08.062. [DOI] [PubMed] [Google Scholar]
- 9.Beutler BA. TLRs and innate immunity. Blood. 2009;113:1399–1407. doi: 10.1182/blood-2008-07-019307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 11.Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006;25:6758–6780. doi: 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]
- 12.Lowrey PL, Takahashi JS. Genetics of circadian rhythms in mammalian model organisms. In: Brody S, editor. Advances in Genetics. Vol. 74. San Diego, CA: Academic Press; 2011. pp. 175–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Panda S, et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109:307–320. doi: 10.1016/s0092-8674(02)00722-5. [DOI] [PubMed] [Google Scholar]
- 14.Mencin A, Kluwe J, Schwabe RF. Toll-like receptors as targets in chronic liver diseases. Gut. 2009;58:704–720. doi: 10.1136/gut.2008.156307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burdelya LG, et al. An agonist of toll-like receptor 5 has radioprotective activity in mouse and primate models. Science. 2008;320:226–230. doi: 10.1126/science.1154986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang B, Yang XD, Lamb A, Chen LF. Posttranslational modifications of NF-kappaB: Another layer of regulation for NF-kappaB signaling pathway. Cell Signal. 2010;22:1282–1290. doi: 10.1016/j.cellsig.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kondratov RV, et al. BMAL1-dependent circadian oscillation of nuclear CLOCK: Posttranslational events induced by dimerization of transcriptional activators of the mammalian clock system. Genes Dev. 2003;17:1921–1932. doi: 10.1101/gad.1099503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spengler ML, Kuropatwinski KK, Schumer M, Antoch MP. A serine cluster mediates BMAL1-dependent CLOCK phosphorylation and degradation. Cell Cycle. 2009;8:4138–4146. doi: 10.4161/cc.8.24.10273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vitaterna MH, et al. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science. 1994;264:719–725. doi: 10.1126/science.8171325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.King DP, et al. Positional cloning of the mouse circadian clock gene. Cell. 1997;89:641–653. doi: 10.1016/s0092-8674(00)80245-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gekakis N, et al. Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998;280:1564–1569. doi: 10.1126/science.280.5369.1564. [DOI] [PubMed] [Google Scholar]
- 22.Miller BH, et al. Circadian and CLOCK-controlled regulation of the mouse transcriptome and cell proliferation. Proc Natl Acad Sci USA. 2007;104:3342–3347. doi: 10.1073/pnas.0611724104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doi M, Hirayama J, Sassone-Corsi P. Circadian regulator CLOCK is a histone acetyltransferase. Cell. 2006;125:497–508. doi: 10.1016/j.cell.2006.03.033. [DOI] [PubMed] [Google Scholar]
- 24.Hirayama J, et al. CLOCK-mediated acetylation of BMAL1 controls circadian function. Nature. 2007;450:1086–1090. doi: 10.1038/nature06394. [DOI] [PubMed] [Google Scholar]
- 25.Nader N, Chrousos GP, Kino T. Circadian rhythm transcription factor CLOCK regulates the transcriptional activity of the glucocorticoid receptor by acetylating its hinge region lysine cluster: Potential physiological implications. FASEB J. 2009;23:1572–1583. doi: 10.1096/fj.08-117697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen LF, Mu Y, Greene WC. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002;21:6539–6548. doi: 10.1093/emboj/cdf660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JH, Sancar A. Regulation of apoptosis by the circadian clock through NF-kappaB signaling. Proc Natl Acad Sci USA. 2011;108:12036–12041. doi: 10.1073/pnas.1108125108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katada S, Sassone-Corsi P. The histone methyltransferase MLL1 permits the oscillation of circadian gene expression. Nat Struct Mol Biol. 2010;17:1414–1421. doi: 10.1038/nsmb.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Etchegaray JP, Lee C, Wade PA, Reppert SM. Rhythmic histone acetylation underlies transcription in the mammalian circadian clock. Nature. 2003;421:177–182. doi: 10.1038/nature01314. [DOI] [PubMed] [Google Scholar]
- 30.Curtis AM, et al. Histone acetyltransferase-dependent chromatin remodeling and the vascular clock. J Biol Chem. 2004;279:7091–7097. doi: 10.1074/jbc.M311973200. [DOI] [PubMed] [Google Scholar]
- 31.Cotta-Ramusino C, et al. A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science. 2011;332:1313–1317. doi: 10.1126/science.1203430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 2001;107:855–867. doi: 10.1016/s0092-8674(01)00610-9. [DOI] [PubMed] [Google Scholar]
- 33.Kwon I, et al. BMAL1 shuttling controls transactivation and degradation of the CLOCK/BMAL1 heterodimer. Mol Cell Biol. 2006;26:7318–7330. doi: 10.1128/MCB.00337-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ye R, Selby CP, Ozturk N, Annayev Y, Sancar A. Biochemical analysis of the canonical model for the mammalian circadian clock. J Biol Chem. 2011;286:25891–25902. doi: 10.1074/jbc.M111.254680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med. 2003;198:513–520. doi: 10.1084/jem.20030162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silver A. C., Arjona A, Walker W. E., Fikrig E. 2012. The circadian clock controls Toll-like receptor 9-Mediated innate and adaptive immunity. Immunity.
- 37.Alonso-Fernandez P, De la Fuente M. Role of the immune system in aging and longevity. Curr Aging Sci. 2011;4:78–100. doi: 10.2174/1874609811104020078. [DOI] [PubMed] [Google Scholar]
- 38.Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core component of the circadian clock. Genes Dev. 2006;20:1868–1873. doi: 10.1101/gad.1432206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Starczynowski DT, Reynolds JG, Gilmore TD. Mutations of tumor necrosis factor alpha-responsive serine residues within the C-terminal transactivation domain of human transcription factor REL enhance its in vitro transforming ability. Oncogene. 2005;24:7355–7368. doi: 10.1038/sj.onc.1208902. [DOI] [PubMed] [Google Scholar]
- 41.Wilsbacher LD, et al. Photic and circadian expression of luciferase in mPeriod1-luc transgenic mice invivo. Proc Natl Acad Sci USA. 2002;99:489–494. doi: 10.1073/pnas.012248599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Galang CK, Der CJ, Hauser CA. Oncogenic Ras can induce transcriptional activation through a variety of promoter elements, including tandem c-Ets-2 binding sites. Oncogene. 1994;9:2913–2921. [PubMed] [Google Scholar]
- 43.Kuprash DV, et al. Similarities and differences between human and murine TNF promoters in their response to lipopolysaccharide. J Immunol. 1999;162:4045–4052. [PubMed] [Google Scholar]
- 44.Ossovskaya VS, et al. Use of genetic suppressor elements to dissect distinct biological effects of separate p53 domains. Proc Natl Acad Sci USA. 1996;93:10309–10314. doi: 10.1073/pnas.93.19.10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lyakh LA, et al. Bacterial lipopolysaccharide, TNF-alpha, and calcium ionophore under serum-free conditions promote rapid dendritic cell-like differentiation in CD14+ monocytes through distinct pathways that activate NK-kappa B. J Immunol. 2000;165:3647–3655. doi: 10.4049/jimmunol.165.7.3647. [DOI] [PubMed] [Google Scholar]
- 47.Gleiberman AS, Sharovskaya YuYu, Chailakhjan LM. “Contact inhibition” of alpha-fetoprotein synthesis and junctional communication in adult mouse hepatocyte culture. Exp Cell Res. 1989;184:228–234. doi: 10.1016/0014-4827(89)90380-7. [DOI] [PubMed] [Google Scholar]
- 48.Fougère-Deschatrette C, et al. Plasticity of hepatic cell differentiation: Bipotential adult mouse liver clonal cell lines competent to differentiate in vitro and in vivo. Stem Cells. 2006;24:2098–2109. doi: 10.1634/stemcells.2006-0009. [DOI] [PubMed] [Google Scholar]
