

Abstract
Cell-selective metabolic labeling of proteins with non-canonical amino acids enables the study of proteomic changes in specified subpopulations of complex multi-cellular systems. For example, azidonorleucine (Anl) and 2-aminooctynoic acid, both of which are activated by an engineered methionyl-tRNA synthetase (designated NLL-MetRS), are excluded from proteins made in wild-type cells but incorporated readily into proteins made in cells that carry NLL-MetRS. To expand the set of tools available for cell-selective metabolic labeling, we sought a MetRS variant capable of activating propargylglycine (Pra). Pra was chosen as the target amino acid because its alkynyl side chain can be selectively and efficiently conjugated to azide-functionalized fluorescence probes and affinity tags. Directed evolution, using active-site randomization and error-prone PCR, yielded a MetRS variant (designated PraRS) capable of incorporating Pra at near-quantitative levels into proteins made in a Met-auxotrophic strain of Escherichia coli cultured in Met-depleted media. Proteins made in E. coli strains expressing PraRS were labeled with Pra in Met-supplemented media as shown by in-gel fluorescence after conjugation to Cy5-azide. The combined use of NLL-MetRS and PraRS enabled differential, cell-selective labeling of marker proteins derived from two bacterial strains co-cultured in media supplemented with Met, Anl, and Pra. Treatment of the mixed marker proteins by sequential strain-promoted and copper (I)-catalyzed cycloadditions allowed straightforward identification of the cellular origin of each protein.
Introduction
The methionyl-tRNA synthetase (MetRS) of E. coli is promiscuous with respect to activation of natural and artificial amino acids under certain conditions.1,2 For example, homopropargylglycine (Hpg) and azidohomoalanine (Aha) can be charged to tRNAMet by the wild-type MetRS and incorporated into cellular proteins; replacement of methionine (Met, 1) occurs in statistical fashion throughout the proteome. The reactive side chains of Hpg and Aha provide sites for selective attachment of affinity tags or fluorescent probes. 3,4
Bio-orthogonal non-canonical amino acid tagging (BONCAT) uses pulse labeling with reactive amino acids to separate newly synthesized cellular proteins from the preexisting proteome.5,6 Recent BONCAT experiments have enabled determination of the kinetics of nucleosome turnover,7 analysis of localized synthesis of proteins critical to axonal maintenance,8 visualization of localized protein synthesis regulated by transmembrane receptors,9 in situ fluorescence imaging of new protein synthesis in rat hippocampal neurons,10 and labeling of newly synthesized proteins in multicellular organisms.11
Engineering of the MetRS binding pocket has allowed further expansion of the set of Met analogs that can be incorporated into proteins.12–15 Labeling of cellular proteins with azidonorleucine (Anl, 2) requires expression of the L13N, Y260L, H301L variant of MetRS (NLL-MetRS), which activates Anl faster than Met.16 Ngo and coworkers recently reported the use of NLL-MetRS and Anl to effect selective, proteome-wide labeling of bacterial proteins made in mixed cultures of E. coli cells and mammalian macrophages.16 Hang and coworkers have reported similar studies with NLL-MetRS and 2-aminooctynoic acid.15
In an effort to expand the set of tools available for cell-specific metabolic labeling of proteins, we chose an alkynyl amino acid, propargylglycine (Pra, 3), an amino acid smaller than methionine (supporting information), which is activated slowly or not at all by the wild-type E. coli MetRS.17 Here we describe the engineering of a new variant of the E. coli MetRS (designated PraRS) that enables near-quantitative replacement of Met by Pra in bacterial proteins. We also show that Pra can be used as a cell-specific metabolic label in conjunction with PraRS. Finally, we report a new method that uses Anl and Pra to effect simultaneous, two-strain, cell-specific labeling of bacterial proteins (Figure 1).
Figure 1.

Amino acids and fluorescent probes used in this study. 1: Met; 2: Anl; 3: Pra; 4: TAMRA-azide; 5: Cy5-azide; 6: DIBO-Alexa Fluor 488
Results and Discussion
Although Pra bears an alkyne group suitable for copper (I)-catalyzed azide-alkyne [3+2] cycloaddition, we chose a cell-based screening method that does not require exposure of host cells to the copper catalyst. The screening method used a previously evolved variant of the green fluorescent protein (GFPrm_AM) that is nearly insensitive to global replacement of Met by non-canonical amino acids.13 Because expression of GFPrm_AM in Met-auxotrophic host cells in Met-depleted medium is limited by charging of tRNAMet, fluorescence-activated cell sorting (FACS) can be used to identify MetRS variants that activate alternative substrates.
Our initial attempts to evolve MetRS variants for activation of Pra focused on the amino acid binding pocket, in which five positions (L13, A256, P257, Y260, and H301) were randomized using NNK codons. Thirteen rounds of positive and negative FACS screening yielded a MetRS variant (MO2b_13-2; L13P/A256G/P257T/Y260Q/H301F; Figure 2) capable of activating Pra. MO2b_13-2 was also found to misincorporate an unidentified amino acid at Met codons (data not shown). A new library (MO9) based on MO2b_13-2 was generated by error-prone PCR to improve specificity for Pra. Highly active mutants obtained from screening MO9 shared a common trait: amino acid changes were localized around the KMSKS motif, a highly conserved sequence that plays an important role in stabilizing the aminoacyl adenylate intermediate in the charging of tRNA.18,19 In addition, a truncation (E548Δ; Δ is the TGA codon) at the end of the catalytic core of MetRS was found to be reversed in some of the most active MetRS variants.20 The C-terminal domain of MetRS has been shown to play a role in dimerization of the synthetase and contributes to increased tRNA affinity.21 Mutations identified by screening the MO9 library were recombined to generate a MetRS variant that we designated PraRS (L13P/A256G/P257T/Y260Q/H301F/A331V/Δ548E; Figure 2).
Figure 2.

FACS histograms of E. coli variants obtained from evolution of MetRS for Pra activity. Addition of Pra to E. coli cultures in M9 medium supplemented with 19 amino acids (-Met) did not change the GFPrm_AM fluorescence of cells expressing endogenous levels of wild-type MetRS (compare pREP4-19aa and pREP4-Pra; pREP4 is the vector lacking the MetRS cassette). Variant MO2b_13-2 was isolated from the library prepared by randomizing the amino acid binding pocket. MO9c_4-16 was isolated from the error-prone PCR library generated from MO2b_13-2, and carries an additional mutation (A331V). Reverting the stop codon in MO9c_4-16 to Glu yielded PraRS, which shows the highest fluorescence signal (PraRS-Pra) for cells expressed in media supplemented with Pra.
Lysates derived from E. coli strain TYJV2, which carries a plasmid-borne copy of the gene encoding PraRS, were treated with tetramethylrhodamine-azide (TAMRA-azide, 4) to confirm incorporation of Pra into cellular proteins (Figure 3a). Protein labeling was detected by in-gel fluorescence within 5 min of addition of 4 mM Pra to the culture medium. Although labeling was detected in lysates treated with lower concentrations (e.g., 500 µM) of Pra, supplementation at higher concentrations increased the incorporation level, as shown by increases in the apparent mass of GFPrm_AM due to multi-site conjugation of TAMRA-azide (Figure 3b). Matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) of trypsinized GFPrm_AM expressed in Met-depleted M9 minimal medium supplemented with 4 mM Pra confirmed near-quantitative incorporation of Pra at positions encoding Met (Figure 4). Lower levels of Met replacement were accomplished without depletion of the natural amino acid. For example, expression of GFPrm_AM in M9 medium supplemented with 269 µM Met and 4 mM Pra yielded a protein containing approximately a 10:1 ratio of Met to Pra (supporting information).
Figure 3.
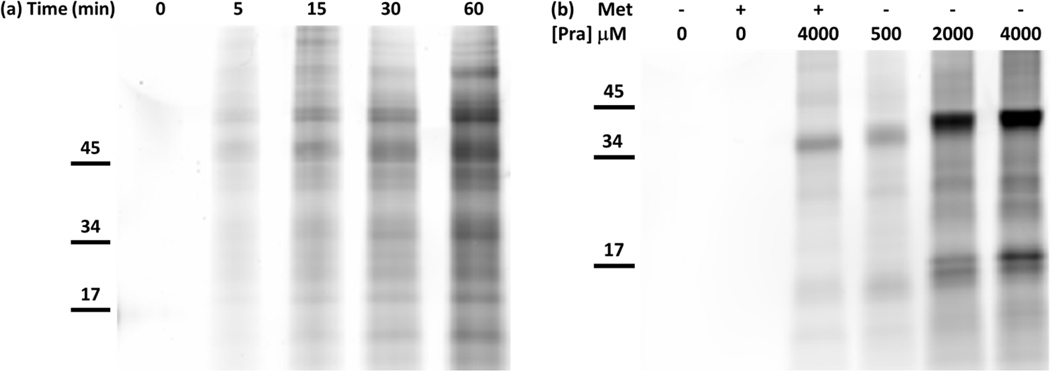
In-gel fluorescence detection of TAMRA-azide after labeling of lysates from Met-auxotrophic E. coli cells. a) Time-dependent, proteomic labeling after addition of Pra. At time = 0, 4 mM Pra was added to the medium without depletion of Met (269 µM). Aliquots were removed at regular time intervals and chloramphenicol was added to prevent further protein synthesis. Incorporation of Pra into cellular proteins was detected by labeling with TAMRA-azide within 5 min of addition of Pra. b) The extent of proteomic labeling was dependent on Pra concentration. Expression of GFPrm_AM (top band) was induced after Met depletion and resuspension in medium with and without 269 µM Met and various concentrations of Pra. TAMRA-azide was detected in all lanes where Pra was added to the culture medium. The apparent molecular weight of GFPrm_AM increased with increasing amounts of Pra due to higher levels of labeling with TAMRA-azide.
Figure 4.

MALDI-TOF spectrum of tryptic digest of purified GFPrm_AM. His-tagged protein was expressed in a Met-auxotrophic strain of E. coli harboring PraRS in medium supplemented with 19 aa (-Met) plus 4 mM Pra. Replacement of Met by Pra in the peptide HNVMDGSVQLADHYQQNTPIGDGDPVR results in a decrease in mass of 36 Da.
The use of PraRS for cell-selective proteomic labeling does not require the use of auxotrophic bacterial strains. Prototrophic DH10B E. coli strains harboring plasmid-borne, constitutively expressed genes encoding wild-type MetRS, NLL-MetRS, or PraRS were grown to mid-log phase and then treated with 1 mM Pra and 269 µM Met for 1 h. Cells were lysed and lysates were subjected to copper (I)-catalyzed ligation to a Cy5-azide probe (5). Labeling was detected only in lysates derived from cells bearing PraRS (Figure 5). Strains that over-expressed either wild-type MetRS or NLL-MetRS showed insignificant levels of proteomic labeling. Encouraged by the fact that Pra is not a substrate for the NLL-MetRS, we sought to develop a method for independently labeling the proteomes of two intermixed populations of cells (Figure 6a). His-tagged marker proteins DHFR and GFPm were co-expressed with NLL-MetRS and PraRS, respectively, in prototrophic DH10B cells. The two strains were grown separately in M9 medium supplemented with all 20 canonical amino acids until OD600 ~ 1 and then mixed together in equal amounts. The mixed culture was then supplemented with 1 mM Anl, 4 mM Pra, and 269 µM Met. Expression of the marker proteins was induced by addition of IPTG. After 2 h, His-tagged proteins were purified from the mixed culture. To prevent cross-coupling of marker proteins derived from different strains, tagging reactions were run sequentially (Figure 6). The mixture of DHFR and GFPm was first treated with dibenzocyclooctyne-Alexa Fluor 488 (DIBO-Alexa Fluor 488, 6) to tag Anl side chains. Without purification, the mixture was then treated with Cy5-azide under the copper-catalyzed conditions required to convert the unstrained alkyne side chains of Pra. In-gel fluorescence scanning showed that Alexa Fluor emission was confined to the marker derived from the NLL-MetRS strain, while Cy5 labeling was apparent only for the PraRS marker (Figure 7). Thus the use of Anl and Pra as metabolic labels, combined with the sequential labeling strategy described here, permits confident identification of the cellular origins of proteins derived from mixed cell populations. Extension of this approach to cell-selective proteomic analysis via high-throughput mass spectrometry is straightforward.5,15
Figure 5.

In-gel fluorescence detection of Cy5-azide (top) after labeling of E. coli lysates. Colloidal blue staining (bottom) confirms protein loading in all lanes. Conjugation of the fluorescent probe to cellular proteins requires both expression of PraRS and addition of 1 mM Pra to the medium (lane 7). Over-expression of the wild-type E. coli MetRS (lane 3) confirms the lack of activity toward Pra previously reported by Kiick et al.17 NLL-MetRS also shows little or no evidence of activation of Pra (lane 5). Incorporation of Pra into cellular proteins in the presence of Met is confirmed by MALDI-MS (supporting information).
Figure 6.

Schematic representation of simultaneous, cell-selective metabolic labeling of two proteomes in a mixed bacterial culture.
Figure 7.

In-gel fluorescence detection of DIBO-Alexa Fluor 488 and Cy5-azide after sequential labeling of mixed cell lysates. Dihydrofolate reductase (DHFR) metabolically labeled with Anl was conjugated to DIBO-Alexa Fluor 488 via copper-free, strain-promoted cycloaddition. GFPm metabolically labeled with Pra was conjugated to Cy5-azide by Cu(I)-catalyzed cycloaddition.
Conclusions
A MetRS engineered to use Pra, a small non-canonical amino acid with a reactive side chain, was developed to enable differential, cell-selective proteome-wide labeling of two intermixed bacterial populations. The mutations that differentiate PraRS from wild-type MetRS highlight the importance of error-prone PCR in the evolution of synthetase variants that activate non-canonical amino acid substrates. Previous efforts in synthetase engineering have focused for the most part on the amino acid binding pocket of the enzyme.13,14,22–24 Here we found that mutations adjacent to the KMSKS region of MetRS can enhance charging of Pra; these mutations would not have been identified without the use of error-prone PCR.
We and others have shown previously that the use of appropriately-designed amino acids and mutant synthetases can enable time-resolved, cell-selective metabolic labeling of proteins.15,16 By using the Anl/NLL-MetRS system in conjunction with Pra and PraRS, investigators can now interrogate the proteomic responses of two intermixed bacterial strains without crosstalk. Important cellular phenomena that would be amenable to study via dual cell-selective labeling would include bacterial quorum sensing, the development of biofilms, and interactions among commensal bacterial.25–32
Materials and Methods
Materials
Restriction enzymes and T4 DNA ligase were purchased from New England Biolabs (Beverly, MA). PfuUltra II Fusion and Mutazyme II were purchased from Agilent Technologies (Santa Clara, CA). DNA oligomers were synthesized by Integrated DNA Technologies (Coralville, IA). L-Anl was prepared by previously reported methods.33 Canonical amino acids and L-Pra were purchased from Sigma-Aldrich (St. Louis, MO). His-tag purifications were carried out using nickel-nitrilotriacetic acid resin purchased from Qiagen (Valencia, CA). Sequencing grade trypsin was purchased from Promega (Madison, WI).
Construction of MetRS Libraries
Plasmid pMTY11, which contains a MetRS expression cassette, was described previously.13 Active site randomization of the MetRS gene was carried out at the L13, A256, P257, Y260, and H301 positions by site-overlap extension using primers with NNK codons. PCR products were purified on 1% agarose gels. Fragments containing the randomized positions were assembled and amplified using PfuUltra II Fusion DNA polymerase to construct the MetRS library with 5 randomized positions. The library was re-ligated into pMTY11 using BamHI/NotI restriction sites. The ligation product was transformed into E. coli strain TYJV2 bearing the previously described plasmid pQE80L/GFPrm_AM via electroporation.13
Error-prone PCR mutagenesis of MO2b_13-2 was performed with Mutazyme II using primers Lib_Fwd (5’-TTCCGACAGCTACGTCGCGGAAC-3’) and Lib_Rev (5’-GAAGGACCGTAGAAGGTCCTTTAGAG-3’), which flank the MetRS gene. The target mutation rate was two to four mutations per MetRS gene in the library. The PCR product was purified on a 1% agarose gel and subsequently cloned into pREP4/MO2b_13-2 using the BamHI/NotI restriction sites. The ligation product was transformed into E. coli strain TYJV2 bearing pQE80L/GFPrm_AM via electroporation.
Screening of MetRS libraries via FACS
Met auxotrophic TYJV2 cells containing pQE80L/GFPrm_AM and the MetRS libraries were grown at 37°C to mid-log phase (OD600 = 0.8–1.0) in M9 minimal medium (M9 salts, 0.2% glucose, 1 mM MgSO4, 0.1 mM CaCl2, 35 mg/L Vitamin B) supplemented with all 20 canonical amino acids (each at 40 mg/L), ampicillin (200 mg/L) and kanamycin (35 mg/L). Upon reaching mid-log phase, cells were washed twice with cold 0.9% NaCl and resuspended in M9 minimal media supplemented either with 19 amino acids (minus Met) at 40 mg/L or with 19 amino acids plus 4 mM Pra. After 30 min, expression of GFPrm_AM was induced by addition of 1 mM IPTG. After 2 h, cells were washed and resuspended in phosphate buffered saline (pH 7.4) for FACS screening.
FACS screening was performed on a MoFLO XDP cell sorter (Beckman Coulter, Brea, CA) using an argon laser (488 nm) and 530/40 nm bandpass filter. Side scatter was utilized for event triggering; forward and side scatter gating was used to remove non-E. coli events. Event rate was maintained between 20000 and 30000 events per second. Positive sorting to enrich MetRS variants specific for Pra involved expression of GFPrm_AM in Pra-supplemented media and collecting the 0.5–1% of cells characterized by the highest levels of fluorescence. Negative sorting to remove MetRS variants that mischarge canonical amino acids was accomplished by collecting non-fluorescent cells when GFP was expressed in Met-depleted media. Cells were sorted into SOC medium (2 mL), rescued at 37°C for 1 h, and further diluted 1:10 with LB medium for overnight growth. Sorted populations were stored at −80°C in 25% glycerol.
Pulse Labeling
Met auxotrophic DH10B cells containing a constitutively expressed PraRS cassette in the pREP4 plasmid (pREP4/PraRS) were grown to mid-log phase in M9 minimal medium containing all 20 canonical amino acids. Addition of 4 mM Pra to the growth medium was performed without Met depletion. At selected time points, aliquots of cells were removed and chloramphenicol (170 mg/L) was added to inhibit further protein synthesis. Cells were pelleted, lysed, and subjected to copper (I)-catalyzed cycloaddition with 25 µM TAMRA-azide, 100 µM CuSO4, 500 µM tris(3-hydroxypropyltriazolylmethyl)amine (THPTA), and 5000 µM sodium ascorbate as the reducing agent.34 After 30 min, each sample was run on a 12% SDS-PAGE gel without purification. Labeled proteins were visualized using a Typhoon Trio (GE Healthcare, Piscataway, NJ) with a 532 nm laser and 580/30 nm bandpass filter.
Mass Spectrometry
Expression of GFPrm_AM was carried out in TYJV2 cells transformed with pQE80L/GFPrm_AM and pREP4/PraRS. Cells were grown to mid-log phase and then washed twice with cold 0.9% NaCl. Cells were subsequently resuspended in M9 minimal medium supplemented with 19 amino acids (minus Met), 20 amino acids, or 19 amino acids plus 4 mM Pra. After 30 min, IPTG was added to a final concentration of 1 mM to induce expression of GFPrm_AM. Utilizing the N-terminal 6 × histidine tag of GFPrm_AM, the protein was purified under denaturing conditions according to the manufacturer’s protocol. Tryptic digests of purified GFPrm_AM were subjected to MALDI time of flight mass spectrometry with α-cyano-4-hydroxycinnamic acid as matrix. An Applied Biosystems Voyager DE-PRO equipped with a 20-Hz nitrogen laser was used for all peptide analyses.
In-gel Fluorescence Detection of Two-strain, Cell-selective Protein Labeling
Overnight M9 cultures of TYJV2 cells transformed either with pQE80L/GFPm and pREP4/PraRS or with the previously described pQE80L/DHFR/NLL-MetRS (pJTN5) and pREP4 were diluted 1:100 into fresh M9 minimal medium containing all 20 amino acids.16 Cultures were grown to mid-log phase and equal numbers of cells from each of the two strains were mixed together. After mixing, the medium was supplemented with 1 mM L-Anl, 4 mM L-Pra, and 269 µM L-Met. Expression of his-tagged DHFR and his-tagged GFPm was induced by addition of 1 mM IPTG for 2 h. DHFR and GFPm were purified on nickel-nitrilotriacetic acid resin according to the manufacturer’s protocols under denaturing conditions.
Sequential labeling of the marker protein mixture was accomplished with DIBO-Alexa Fluor 488 (Invitrogen, Carlsbad, CA), and Cy5-alkyne (Lumiprobe, Hallandale Beach, FL). The protein mixture (approximately 2 mg/ml) was first treated with 10 µM DIBO-Alexa Fluor 488 in tris-buffered saline for 1 h. Copper (I)-catalyzed cycloaddition was then performed without intermediate purification using 25 µM of Cy5-azide, 100 µM CuSO4, 500 µM THPTA, and 5000 µM sodium ascorbate as the reducing agent. After 30 min, samples were run on a 12% SDS-PAGE gel. Labeled marker proteins were visualized using a Typhoon Trio with a 488 nm laser and 526 nm shortpass filter as well as a 633 nm laser and 680/30 nm bandpass filter.
Supplementary Material
Acknowledgments
We thank John Ngo, Janek Szychowski, Caglar Tanrikulu, and James Van Deventer for helpful discussions, and Felicia Rusnak, Jie Zhou and Mona Shahgholi for help with spectroscopic measurements. This work was supported by National Institutes of Health Grant NIH R01 GM062523 and by the Institute for Collaborative Biotechnologies through grant W911NF-09-0001 from the U.S. Army Research Office.
Footnotes
Supporting Information Available: Calculated side chain dimensions of Met and Pra, comparison of best performing mutants from error-prone PCR library; additional mass spectra for proteins expressed in medium containing Pra and Met. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cowie DB, Cohen GN. Biochim. Biophys. Acta. 1957;26:252–261. doi: 10.1016/0006-3002(57)90003-3. [DOI] [PubMed] [Google Scholar]
- 2.Johnson JA, Lu YY, Van Deventer JA, Tirrell DA. Curr. Opin. Chem. Bio. 2010;14:774–780. doi: 10.1016/j.cbpa.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem. Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 4.Sletten EM, Bertozzi CR. Angew. Chem. Int. Ed. 2009;48:6974–6998. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dieterich DC, Link AJ, Graumann J, Tirrell DA, Schuman EM. Proc. Natl. Acad. Sci. U.S.A. 2006;103:9482–9487. doi: 10.1073/pnas.0601637103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ngo JT, Tirrell DA. Acc. Chem. Res. 2011;44:677–685. doi: 10.1021/ar200144y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deal RB, Henikoff JG, Henikoff S. Science. 2010;328:1161–1164. doi: 10.1126/science.1186777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoon BC, Jung H, Dwivedy A, O'Hare CM, Zivraj KH, Holt CE. Cell. 2012;148:752–764. doi: 10.1016/j.cell.2011.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tcherkezian J, Brittis PA, Thomas F, Roux PP, Flanagan JG. Cell. 2010;141:632–644. doi: 10.1016/j.cell.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dieterich DC, Hodas JJL, Gouzer G, Shadrin IY, Ngo JT, Triller A, Tirrell DA, Schuman EM. Nat. Neurosci. 2010;13:897–905. doi: 10.1038/nn.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hinz FI, Dieterich DC, Tirrell DA, Schuman EM. ACS Chem. Neurosci. 2012;3:40–49. doi: 10.1021/cn2000876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Link AJ, Vink MKS, Agard NJ, Prescher JA, Bertozzi CR, Tirrell DA. Proc. Natl. Acad. Sci. U.S.A. 2006;103:10180–10185. doi: 10.1073/pnas.0601167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoo TH, Tirrell DA. Angew. Chem. Int. Ed. 2007;46:5340–5343. doi: 10.1002/anie.200700779. [DOI] [PubMed] [Google Scholar]
- 14.Tanrikulu IC, Schmitt E, Mechulam Y, Goddard WA, Tirrell DA. Proc. Natl. Acad. Sci. U.S.A. 2009;106:15285–15290. doi: 10.1073/pnas.0905735106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grammel M, Zhang MM, Hang HC. Angew. Chem. Int. Ed. 2010;49:5970–5974. doi: 10.1002/anie.201002050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ngo JT, Champion JA, Mahdavi A, Tanrikulu IC, Beatty KE, Connor RE, Yoo TH, Dieterich DC, Schuman EM, Tirrell DA. Nat. Chem. Bio. 2009;5:715–717. doi: 10.1038/nchembio.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiick KL, Weberskirch R, Tirrell DA. FEBS Lett. 2001;502:25–30. doi: 10.1016/s0014-5793(01)02657-6. [DOI] [PubMed] [Google Scholar]
- 18.Schmitt E, Meinnel T, Blanquet S, Mechulam Y. J. Mol. Bio. 1994;242:566–577. doi: 10.1006/jmbi.1994.1601. [DOI] [PubMed] [Google Scholar]
- 19.Mechulam Y, Dardel F, Le Corre D, Blanquet S, Fayat G. J. Mol. Bio. 1991;217:465–475. doi: 10.1016/0022-2836(91)90750-z. [DOI] [PubMed] [Google Scholar]
- 20.Cassio D, Waller J-P. Eur. J. Biochem. 1971;20:283–300. doi: 10.1111/j.1432-1033.1971.tb01393.x. [DOI] [PubMed] [Google Scholar]
- 21.Crepin T, Schmitt E, Blanquet S, Mechulam Y. Biochemistry. 2002;41:13003–13011. doi: 10.1021/bi026343m. [DOI] [PubMed] [Google Scholar]
- 22.Neumann H, Peak-Chew SY, Chin JW. Nat. Chem. Biol. 2008;4:232–234. doi: 10.1038/nchembio.73. [DOI] [PubMed] [Google Scholar]
- 23.Xie J, Schultz PG. Methods. 2005;36:227–238. doi: 10.1016/j.ymeth.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 24.Wu N, Deiters A, Cropp TA, King D, Schultz PG. J. Am. Chem. Soc. 2004;126:14306–14307. doi: 10.1021/ja040175z. [DOI] [PubMed] [Google Scholar]
- 25.Miller MB, Skorupski K, Lenz DH, Taylor RK, Bassler BL. Cell. 2002;110:303–314. doi: 10.1016/s0092-8674(02)00829-2. [DOI] [PubMed] [Google Scholar]
- 26.Zhu J, Miller MB, Vance RE, Dziejman M, Bassler BL, Mekalanos JJ. Proc. Natl. Acad. Sci. U.S.A. 2002;99:3129–3134. doi: 10.1073/pnas.052694299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Costerton JW, Stewart PS, Greenberg EP. Science. 1999;284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 28.Davies DG, Parsek MR, Pearson JP, Iglewski BH, Costerton JW, Greenberg EP. Science. 1998;280:295–298. doi: 10.1126/science.280.5361.295. [DOI] [PubMed] [Google Scholar]
- 29.Hall-Stoodley L, Costerton JW, Stoodley P. Nat. Rev. Microbiol. 2004;2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 30.Singh PK, Schaefer AL, Parsek MR, Moninger TO, Welsh MJ, Greenberg EP. Nature. 2000;407:762–764. doi: 10.1038/35037627. [DOI] [PubMed] [Google Scholar]
- 31.Hooper LV, Gordon JI. Science. 2001;292:1115–1118. doi: 10.1126/science.1058709. [DOI] [PubMed] [Google Scholar]
- 32.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Link AJ, Vink MKS, Tirrell DA. Nat. Prot. 2007;2:1879–1883. doi: 10.1038/nprot.2007.268. [DOI] [PubMed] [Google Scholar]
- 34.Hong V, Presolski SI, Ma C, Finn MG. Angew. Chem. Int. Ed. 2009;48:9879–9883. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.