

Abstract
Exposure to exogenous hormones during development can result in permanent health problems. In utero exposure to diethylstilbestrol (DES) is probably the most well documented case in human history. DES, an orally active synthetic estrogen, was believed to prevent adverse pregnancy outcome and thus was routinely given to selected pregnant women from the 1940s to the 1960s. It has been estimated that 5 million pregnant women worldwide were prescribed with DES during this period. In the early 1970s, vaginal clear cell adenocarcinomas (CCACs) were diagnosed in daughters whose mother took DES during pregnancy (known as DES daughters). Follow up studies demonstrated that exposure to DES in utero causes a spectrum of congenital anomalies in female reproductive tracts and CCACs. Among those, cervical and vaginal adenoses are most commonly found, which are believed to be the precursors of CCACs. Transformation related protein 63 (TRP63/p63) marks the cell fate decision of Müllerian duct epithelium (MDE) to become squamous epithelium in the cervix and vagina. DES disrupts the TRP63 expression in mice and induces adenosis lesions in the cervix and vagina. This review describes mouse models can be used to study the development of DES-induced anomalies, focusing on cervical and vaginal adenoses, and discusses its molecular pathogenesis.
Keywords: diethylstilbestrol, vaginal adenocarcinoma, adenosis, TRP63, p63
I. History of Diethylstilbestrol (DES) Administration and Resulting Epidemiology
Clinical use of DES
DES, an orally active nonsteroidal estrogen, was first synthesized in 1938 by a research team at the University of Oxford led by Sir Robert Robinson and based on a formulae by Sir Edward Charles Dodds (Dodds et al., 1939; Dodds et al., 1938) (Fig. 1). It became the first estrogenic substance to be easily synthesized and administered. Pharmaceutical companies, such as Eli Lily and Winthrop, registered DES for sale in the United States and several European countries. In September of 1941, the United States Food and Drug Administration (FDA) approved the use of DES to treat gonorrheal vaginitis, atrophic vaginitis, menopausal symptoms, and postpartum lactation suppression (Meyers, 1983). Immediately after the approval, DES use spread in the US, and physicians immediately started prescribing DES to treat conditions other than those approved, such as menstrual problems, morning sickness and infertility.
Figure 1.
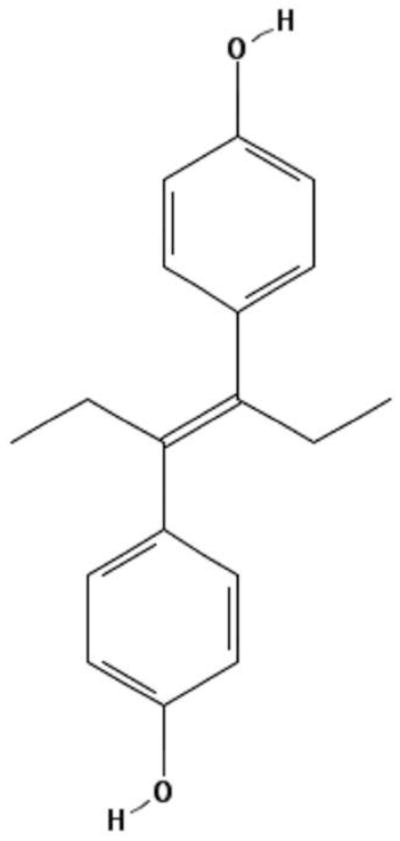
Chemical structure of diethylstilbestrol (DES).
The structure of DES (C18H20O2) is based on the PubChem Public Chemical Database, CID 448537.
A husband and wife team at Harvard, Dr. George Van Siclen Smith and Dr. Olive Watkins Smith, strongly promoted prescribing DES to a subset of pregnant women (Smith et al., 1946). They had previously discovered a correlation between pregnancy loss and decreased estrogen levels in urine (Smith et al., 1941). Thus, the Smiths believed that estrogen deficiency was the major cause of pregnancy loss. They began a clinical trial in 1943 and concluded that DES administration was beneficial in preventing premature deliveries and miscarriages. Based on their observations, the Smiths recommended administration of DES to women that were at risk for complications during pregnancy to improve their outcomes. Candidates included pregnant women with diabetes, a history of miscarriage or operations on their ovaries, uterus or cervix (Smith et al., 1946; Smith, 1948). In 1947, the FDA approved DES for pregnant women to prevent miscarriages. DES treatment of pregnant women became common practice for obstetricians (Seaman, 2003). That same year, DES was also prescribed to women in the Netherlands. However, its use to prevent complications during pregnancy was not strongly endorsed until the 1950’s. DES was later approved for use during pregnancy in France, Finland, Denmark, England, Germany, and other European countries, though the extent of its use has not been well documented (Palmlund et al., 1993a). The recommended regimen began at seven to eight weeks post last menstruation (PLM) at 5 mg/day, and was increased by 5 mg every other week up to 125 mg/day at 35 weeks PLM. The National Cooperative Diethylstilbestrol-Adenosis (DESAD) project reported that the median total dose was over 4 g among participants with complete information on dosage (Labarthe et al., 1978; O’Brien et al., 1979). However, DES doses given to pregnant women varied between hospitals, and there was nearly a 10-fold difference between the median total dose at Baylor College of Medicine (1,625 mg) and Massachusetts General Hospital (10,424 mg). The latter hospital had the highest median total dosage and used the Smith and Smith regimen most frequently.
Despite its common use, the effectiveness of DES in improving the outcome of a given pregnancy was questioned by scientists, and the Smiths’ original studies were criticized for the lack of proper control groups. In 1953, the result of a large-scale double-blind clinical trial at the University of Chicago was published involving 1,646 pregnant participants. The clinical trial concluded that DES did not reduce the incidence of abortion, prematurity or postmaturity. It even suggested that DES may “favor” premature labor (Dieckmann et al., 1953). DES usage declined approximately 30 percent in the US in the 5 years following this publication. Nonetheless, the Smiths maintained their position on the benefit of DES treatment in the prevention of miscarriage, and published a paper to defend their theory (Smith and Smith, 1954). Pharmaceutical companies continued to promote DES for pregnant women, and DES remained a routine treatment for pregnant women throughout the 1960’s.
Increased risk of vaginal adenocarcinomas following DES exposure
Seven young patients between 15 and 22 years old were diagnosed with vaginal adenocarcinomas at Massachusetts General Hospital between 1966 and 1969. One case was endometrioid and the other six cases were clear cell adenocarcinomas (CCACs) (Herbst and Scully, 1970). Vaginal cancers usually occur in patients at 40 years or older, and were almost always epidermoid (squamous cell) carcinomas, rendering these cases highly unusual (Herbst et al., 1970). A retrospective case-control study based on 8 vaginal adenocarcinoma cases and 32 matched controls was published in 1971 (Herbst et al., 1971). A statistically significant association between in utero DES exposure and risk of vaginal adenocarcinoma (P < 0.00001) was discovered. This finding was supported by another case-control study published that same year, which identified five cases of vaginal adenocarcinoma in young women who were born between 1951 and 1953 (Greenwald et al., 1971). All five mothers took synthetic estrogens (DES or Dienestrol) during their pregnancy. In response to these reports, the US FDA issued a drug bulletin in late 1971 warning physicians that prescribing DES to a pregnant woman could harm the unborn baby (FDA, 1972). In 1972, a study further strengthened the link between developmental DES exposure and vaginal adenocarcinoma by identifying 7 more cases of vaginal CCACs in girls, ages 7 to 19 years old, whose mother took DES during the first trimester of pregnancy. Although administration of DES to one mother was not confirmed, she took an unknown hormone for vaginal bleeding during pregnancy (Noller et al., 1972).
In response to these reports, the registry of women exposed to DES in utero (DES daughters) was founded in 1974, and the DESAD project was initiated to locate and monitor the health of DES daughters over time (Labarthe et al., 1978). The Registry for Research on Hormonal Transplacental Carcinogenesis was developed by Dr. Arthur Herbst and colleagues at Massachusetts General Hospital to track cases of cervical and vaginal CCAC world-wide and is now located at the University of Chicago. As of December 2007, the registry has accessioned approximately 760 cases of CCAC, two-thirds of which were exposed to DES in utero. Other organizations, such as DES Action, created in the Netherlands in 1981, and Association Info-DES of France in 1992, were formed to help provide information and support to DES daughters in their respective countries (Palmlund et al., 1993b). Data collected through the DESAD project have established correlations between the schedule of treatment (timing and dose) and symptoms. Decades of follow up studies enabled scientists to determine the long-term effects of in utero DES exposure.
II. Cervical and Vaginal Adenosis
Definition
Two types of epithelia line the human cervix; columnar epithelium lines the endocervix, as in the uterus, and squamous epithelium lines the ectocervix, as in the vagina. The location of the boundary between these two cell types is called the squamocolumnar junction (SCJ). Cervical and vaginal adenoses, which are defined as the development of columnar epithelium in the ectocervix or vagina, are the most common reproductive anomalies in DES daughters. This congenital anomaly has been observed in fetuses and newborns, in addition to young girls and adults, who were exposed to DES in utero (Johnson et al., 1979). The columnar epithelium may cover the surface (surface adenosis), or form glands within the stromal wall of the cervix and vagina (occult adenosis) (Johnson et al., 1979) (Fig. 2). Although the location of the SCJ varies depending on age, menstrual status, pregnancy and contraceptive use, it is usually found within the cervical canal. However, in DES daughters the SCJ can be found within the vagina, instead of the cervix, forming surface adenosis (Hart et al., 1976).
Figure 2.

Adult mice exposed neonatally to DES develop occult and surface adenosis.
Keratin 14 (A & C) or TRP63 (B & D) were detected in the vaginae of C57BL6 adult (P60) mice that were injected subcutaneously with 2 μg DES from P0 – P4. The neonatal mice received oil or DES every 24 hours, were ovariectomised at P35 and tissue was harvested at P60. Examples of both occult adenosis (A & B) and surface adenosis (C & D) are shown.
Incidence
Since cervical and vaginal adenosis is benign and can be asymptomatic, the presence of this disease is not always brought to the attention of a physician. In addition, sensitive detection of vaginal adenosis requires histological analysis of biopsy samples. A routine cytology screening detected vaginal adenosis in only 20 – 40% of patients in which biopsy histology identified the presence of a lesion (Robboy et al., 1976). Although an accurate prevalence rate for this disease is not known, vaginal adenosis has been estimated to affect between 34 – 91% of DES-exposed women (Antonioli and Burke, 1975; Bibbo et al., 1975; Herbst et al., 1975; Ng et al., 1975; Kaufman et al., 1977; O’Brien et al., 1979; Johnson et al., 1979) and less than 4% of unexposed women (Bibbo et al., 1975; Herbst et al., 1975; Johnson et al., 1979). The significantly varied prevalence among DES-daughters appeared to reflect the sensitivity of detection methods used, but may also be due to the point in gestation at which DES treatment began (Sherman et al., 1974; Stafl et al., 1974). The presence of adenosis increased with earlier administration and higher dose of DES treatment. In particular, exposure prior to the 18th week of gestation was strongly associated with a higher incidence of adenosis (O’Brien et al., 1979). The incidence of adenosis was also correlated with the incidence of CCAC. The presence of adenosis lesions has been detected in almost all of the DES daughters with cervical or vaginal CCAC. Therefore, adenosis is widely accepted to be the precursor of adenocarcinoma in DES daughters, though it has not been directly proven. In fact, adenosis lesions are benign, and the progression of adenosis to CCAC should require multiple events. In this regard, the atypical adenosis has been suggested to be the immediate precursor of CCACs (Robboy et al., 1984). CCACs of the cervix and vagina will be discussed in a later section.
Development of the vagina
A clear understanding of the biology of the developing vagina is essential to understanding the molecular pathogenesis of vaginal adenosis. The cellular origin of vaginal epithelium and the contributions of Müllerian duct, Wolffian duct and urogenital sinus cells had been debated for decades. The recent cell-lineage tracing experiment in mice finally determined that Müllerian duct epithelium (MDE) is the only source of adult vaginal epithelium (Kurita, 2010). The Müllerian duct, urogenital sinus and residual Wolffian duct epithelium (WDE) were detected in the lower genital tract of female mice at birth (P0). As females developed, the sinus vagina retreated posteriorly as the Müllerian duct elongated caudally. The Müllerian vagina developed with a lumen, unlike the sinus vagina, whose epithelium remained solid throughout development (Kurita, 2010, 2011). In adult mice, the entire vagina is lined with epithelial cells of Müllerian duct origin, while urogenital sinus epithelial cells were found in the vulva (Fig. 3).
Figure 3.

Müllerian duct epithelium (MDE) is the solo source of the adult vaginal epithelium.
The contributions of MDE, Wolffian duct epithelium (WDE) and urogenital sinus epithelium (UGE) were examined by the presence of enhanced green fluorescent protein (EGFP)-positive cells throughout the surface of the entire vagina in 4-week-old females containing the transgene for Rosa-tdTomato-EGFP and one of the following: Pax2-Cre (EGFP is expressed in the MDE and WDE, A), Osr1-Cre (EGFP is expressed in the UGE, B), and Hoxb7-Cre (EGFP is expressed in WDE, C). The cranial portion of the vagina is visible at the top of each panel. EGFP-positive cells were present in the vaginal epithelium of Pax2-Cre, but not Osr1-Cre or Hoxb7-Cre reporter mice. Bars = 50 μm (Kurita, 2010).
The MDE is developmentally plastic and is induced to differentiate as either squamous or columnar epithelium by paracrine factors produced in the adjacent mesenchyme. Neonatal vaginal epithelium recombined with uterine mesenchyme differentiated into columnar cells that expressed appropriate markers for uterine epithelium. Likewise, neonatal uterine epithelium recombined with vaginal mesenchyme differentiated into vaginal epithelium (Cunha, 1976; Boutin et al., 1991; Kurita et al., 2001). However, adult uterine and vaginal epithelia have been terminally differentiated into their respective cell types and do not dedifferentiate in response to mesenchymal induction (Kurita et al., 2001). These observations suggest that vaginal and cervical adenoses are a failure of squamous differentiation in the MDE during organogenesis of the lower female reproductive tract.
Mouse models of DES exposure and pathogenesis
Following the discovery of the link between in utero DES exposure and vaginal adenocarcinoma, Forsberg reported formation of vaginal adenosis in mice treated neonatally with estrogen (Forsberg, 1972). When newborn female mice were given 5 μg per day of DES or 17β-estradiol for 5 days after birth, extensive adenosis lesions were identified in the anterior part of the vagina at 10 days of age. Susceptibility to DES-induced adenosis is strain dependent. Nonetheless, formations of adenosis lesions in mice exposed neonatally to DES have been confirmed in several mouse strains (Newbold and McLachlan, 1982; Plapinger and Bern, 1979).
One critical problem with this mouse model is that developmental exposure to DES has astounding effects on the physiology of female mice, including on the neuroendocrine system (Halling, 1992; Lopez et al., 1984; 1986; Dalterio et al., 1985; Halling and Forsberg, 1990). Since these organs do not appear to be the primary targets of fetal DES exposure in humans, the effects on these systems are not likely to be involved in the pathogenesis of cervical and vaginal adenoses. A disrupted endocrine system can affect the development and physiology of pubertal and mature mice, and thus alterations in the gene expression in mature urogenital organs could be secondary to these changes. Therefore, analyses of mice at pubertal or adult ages, that had been treated with DES neonatally, would not elucidate the molecular mechanism underlying the pathogenesis of cervical and vaginal adenoses. In addition, as we discuss below, DES induces vaginal adenosis by changing the epithelial cell fate of undifferentiated MDE. The cell fate of MDE is established during the first five days of postnatal life in mice (Cunha, 1976; Kurita et al., 2001). To make a clear distinction between the cause and effect of DES-induced vaginal adenosis, the effects of DES action must be studied before epithelial cell fate is established.
Inhibition of TRP63 by DES in the developing cervix and vagina
The transcription factor transformation related protein 63 (TRP63/p63) (Yang et al., 1998) was highly expressed in the squamous epithelia of the cervix and vagina, but not in the columnar epithelium of the uterus (Kurita and Cunha, 2001; Kurita et al., 2004). In mice, TRP63 was undetectable in the vagina and cervix until just before birth, suggesting that TRP63 may be involved in squamous differentiation of the MDE (Kurita et al., 2004; Kurita et al., 2005). Since Trp63-null mice died at birth due to defects in skin development, their female reproductive tracts could not be directly studied. Therefore, anlagen of uterus, cervix and vagina were developed into mature organs by grafting them under the kidney capsule of mature female mice (Kurita and Cunha, 2001; Kurita et al., 2004). In such grafts, the Trp63-null uterine epithelium appeared normal, but the entire Trp63-null vagina and cervix were lined with columnar epithelium and contained gland-like structures instead of normal squamous epithelium, demonstrating that the developmental loss of Trp63 is the cause of adenosis. When wild-type newborn mice were exposed to DES, expression of Trp63 in MDE was inhibited and TRP63-negative MDE differentiated into uterine-like glandular epithelium within the cervix and vagina, as seen in Trp63-null mice (Fig 4) (Kurita and Cunha, 2001; Kurita et al., 2004). However, once MDE cells became positive for TRP63, expression was not affected by DES exposure, suggesting that DES inhibited induction, but not maintenance of Trp63 expression in MDE (Kurita and Cunha, 2001; Kurita et al., 2004). Hence, the timing of exposure was critical for the pathogenesis of DES-induced adenosis as described below.
Figure 4.

DES disrupts expression of TRP63 in the vaginae of neonatal wild-type mice.
TRP63 was detected in C57BL6 neonatal mice that were injected subcutaneously with 30 μl corn oil (A) or 2 μg DES in 30 μl corn oil (B) from P0 – P4. The neonatal mice received oil or DES every 12 hours. Tissue was harvested at P5. Pictures show the fornix of the vagina. Scale bar: 50 μm.
The expression pattern of tumor protein 63 (TP63/p63), the ortholog of mouse TRP63, has been studied in the female reproductive tracts of human fetuses (Kurita et al., 2005). TP63 expression was greater in the caudal and lesser or absent in the cranial cells of the MDE in the human fetus at PLM week 12 – 13, which is equivalent to the TRP63 expression in mice at embryonic day (E) 16 - postnatal day (P) 1. The human specimens at PLM week 16 – 18 contained both TP63-positive and negative cells within the stratified epithelium of ectocervical or vaginal anlagen, and the concentration of TP63-positive cells increased caudally to cranially as the fetus developed. The expression pattern of TP63 in the vagina of PLM 18 week fetuses appeared to be comparable to the expression in P2 – 3 mice. While most epithelial cells were positive for TRP63 in the caudal (lower vaginal) regions, the cranial (upper vaginal and cervical) regions contained substantial TRP63-negative epithelial cells. Like P2 – 3 is the last point of greatest susceptibility to DES-induced adenosis in mice, exposure to DES in humans within the first 18 weeks of pregnancy resulted in a higher incidence of vaginal epithelial changes (O’Brien et al., 1979). This suggests that DES induces vaginal or cervical adenosis in human fetuses by blocking the expression of TP63 in MDE, as in mice.
The Trp63 gene is transcribed into two alternative splice variants producing isoforms with and without the transactivation domain (TA). The isoforms containing or missing a TA domain are called the TA and ΔN forms, respectively, and each of them has three alternative splice variants of the carboxy terminus, identified as α, β and γ forms. In mice, the ΔNα form (ΔNp63α), lacking the transactivating domain and containing the sterile alpha motif domain, is the dominant form of TRP63 expressed in the neonatal and adult cervix and vagina (Kurita et al., 2005). This is also true for the vaginal epithelium of the human fetus (Kurita et al., 2005). These commonalities confirm the rationale for utilization of mouse models to study the molecular pathogenesis of cervical and vaginal adenoses in DES daughters.
ESR1-mediated DES action in vaginal epithelium
Estrogen receptor alpha (ERα) knockout mice (Esr1-null) exhibit resistance to neonatal DES exposure, indicating that ERα is essential for DES-induced urogenital abnormalities (Couse et al., 2001). However, whether ERα mediated DES action in the pathogenesis of vaginal adenosis was not fully established due to a low prevalence of DES-induced adenosis in the wild-type mice. The essential role of ERα in DES-induced vaginal adenosis was later established by analysis of TRP63 expression in DES-treated Esr1-null mice (Kurita et al., 2004). In the study, neonatal DES treatment inhibited TRP63 expression in the vaginal epithelium of wild-type but not Esr1-null mice. In developing vaginae, ERα is expressed in both epithelium and mesenchyme (Kurita et al., 2001). Therefore, the role of epithelial versus mesenchymal ERα in DES-induced vaginal adenosis was further clarified through tissue-recombination experiments. In this study, MDE from Esr1-null (ERα negative) and wild-type (ERα positive) mice was combined with Esr1-null or wild-type vaginal mesenchyme. While DES failed to induce adenosis in Esr1-null MDE, DES completely blocked TRP63 expression in wild-type MDE (Kurita et al., 2004; Kurita, 2011). Expression of ERα in MDE was the single determinant if DES inhibited expression of TRP63, and the presence or absence of mesenchymal ERα did not affect the result. Therefore, DES acts through ERα within MDE and induces vaginal adenosis by blocking expression of TRP63.
A recent study suggested that Activin signaling from the stroma is the major factor regulating proliferation and differentiation of vaginal epithelial cells (Nakajima et al., 2011). The study further proposed that DES induces vaginal adenosis through disruption of this signaling, because DES reduced Acvr2b and Inhba mRNA by 20% and 40%, respective, and upregulated Inhbb mRNA 20 fold, as normalized to the level of Ppia mRNA. However, Ppia is upregulated by estrogen in mouse uterus (Schroder et al., 2009), thus the analysis may have been skewed by the effect of DES on the Ppia level. The authors also demonstrated reduced Activin signaling by immunofluorescence analysis of phospho-SMAD2. Although the overall signal level for phospho-SMAD2 was reduced in the entire vagina of neonatally DES-treated mice, the epithelial tissue that contained reduced phosphorylated-SMAD2 was fully differentiated as squamous stratified epithelium, indicating that reduced Activin A signaling is not necessarily associated with adenosis (Nakajima et al., 2011). While Activin A signaling may play a role in vaginal epithelial differentiation or proliferation, it has been shown that the underlying pathway through which DES causes adenosis is mediated by intrinsic epithelial ERα signaling (Kurita, 2011). Therefore, alterations in pathways through the stroma, such as down-regulation of Activin ligand, would not play a role in the pathogenesis. How DES disrupts expression of TRP63 through ERα within epithelial cells has yet to be elucidated. Recently, an essential role of androgen receptor (AR) binding to a regulatory element of Trp63 gene was demonstrated in developing mouse epididymal epithelium (Murashima et al., 2011). Likewise, ERα may directly bind to the regulatory elements of Trp63 to block expression (Fig. 5).
Figure 5.

DES inhibits activation of TP63 and induces adenosis through ERα within vaginal or cervical epithelial cells.
Additional studies of DES-induced abnormalities
Though mouse models for DES-induced pathology have existed for decades, little advancement has been made toward our understanding of the mechanism for cervical or vaginal pathology following DES treatment. Most studies have analyzed the effect of DES on gene expression at a point in which epithelial cell fate is irreversibly altered. In addition, cell fate is altered only in a small subpopulation of MDEs in DES-induced adenosis, thus, global analysis may not detect a gene expression change that is critical for the pathogenesis of vaginal adenosis. These are the challenges for future studies.
Studies conducted on the effects of DES on developmental genes have focused on changes in the uterus more than the cervix and vagina. A number of mouse studies have shown anomalies within the uterus following exposure to DES, which include disorganized myometrium (Iguchi and Takasugi, 1987; Brody and Cunha, 1989), reduced glands (Iguchi and Takasugi, 1987) and squamous metaplasia (Forsberg and Kalland, 1981). In mice, neonatal DES-exposure disrupts expression of several genes that are essential for normal development of the uterus. For example, DES down-regulated Wnt7a in neonatal uteri, and Wnt7a-null mice exhibit similar uterine defects to neonatally DES-exposed mice including under-differentiation of myometrium, lack of uterine glands and squamous metaplasia (Miller and Sassoon, 1998). This suggests that DES elicits its teratogenic effects via down-regulation of Wnt7a. However, null mutations in Wnt7a altered uterine expression of other developmental regulators including Hoxa10 and Hoxa11, which are also down-regulated in the uterus of neonatally DES-exposed mice (Gendron et al., 1997; Ma et al., 1998). Therefore, the molecular mechanism of DES-induced uterine abnormalities appears to be more than the disruption of Wnt7a expression. Since these uterine abnormalities are not observed in DES daughters, they are out of the scope of this review and are not discussed further. For the potential molecular mechanisms underlying DES-induced uterine abnormalities, please read a recent review by Ma (Ma, 2009).
III. Cervical and Vaginal Adenocarcinoma
There is an association between prenatal DES exposure and increased risk of vaginal and cervical CCAC. It has been hypothesized that CCACs develop from cervical and vaginal adenoses because adenoses have been identified in nearly all CCAC cases (Herbst et al., 1972; Herbst et al., 1974; Robboy et al., 1984). The Combined Cohort Follow-up Study, which follows more than 4,600 women exposed to DES, estimated the incidence of CCAC from birth to age 39 is 1.6 in 1,000 exposed women (Hoover et al., 2011; Troisi et al., 2007). The significant divergence between the incidence rates of adenosis (approximately 90%) versus CCACs (approximately 0.1 %) indicates that the progression of adenosis to CCAC is an extremely rare event, even among DES daughters. The observed to expected ratio of vaginal and cervical CCAC was 39.0 (95% confidence interval 15–104) among exposed women (Troisi et al., 2007; Hoover et al., 2011). As discussed in the previous section, prevalence of cervical and vaginal adenoses is approximately 30 times higher in DES daughters compared to women without a history of DES exposure (4 versus 90 %). Thus, in utero DES-exposure increased the risk of adenosis and CCACs to a similar degree (30 versus 40 times), suggesting that the high incidence of CCACs in DES daughters reflects a high incidence of benign precursor lesions. Accordingly, the risk for malignant transformation of adenosis to adenocarcinoma may not significantly differ between DES daughters and the general population.
Because the prevalence of adenosis and CCACs differs greatly, the malignant transformation of DES-induced adenosis to CCACs must require additional factors. However, no postnatal factors, including pregnancy and oral contraceptive use, have been identified to influence the risk of CCAC incidence in DES daughters (Palmer et al., 2000). Several genes commonly mutated in cancers have been investigated within CCAC samples. However, there have been no mutations identified to be associated with CCACs of the cervix and vagina. In a study of 24 CCAC cases (16 were associated with prenatal DES-exposure), there were no mutations identified in the Wilms’ tumor (WT1) tumor suppressor gene, the ESR1 gene or KRAS and HRAS proto-oncogenes (Boyd et al., 1996). Samples from 14 of 21 CCAC cases had nuclear accumulation of a tumor suppressor tumor protein 53 (TP53), which is often indicative of a loss of function mutation. However, no mutations in the TP53 gene were detected by single-stranded conformational polymorphism analysis (Waggoner et al., 1996). The same authors speculated that the high BCL2 expression observed in these cancer cells may have contributed to the absence of apoptosis, despite the nuclear accumulation of wild-type TP53 (Waggoner et al., 1998). Nonetheless, causal mutations of CCACs have yet to be identified. Unlike squamous cell carcinomas and non-clear cell adenocarcinomas of the cervix (Naucler et al., 2011), CCACs appear to have minimal or no association with the infection of high-risk types of the human papillomavirus (HPV) (Pirog et al., 2000; Goto et al., 2005; Stewart et al., 2006; Liebrich et al., 2009; Waggoner et al., 1994). Loss of function mutations in PTEN (phosphatase and tensin homolog deleted on chromosome 10) have been detected in HPV-negative endometrioid (2/5) and mucinous (2/6) cervical adenocarcinomas (Minaguchi et al., 2004; Hashiguchi et al., 2006) as well as CCACs of the ovary (15/40 and 6/22) (Sato et al., 2000). However, to this date, there have been no reports on PTEN mutations in CCACs of the cervix or vagina. The study of CCACs etiology in the cervix and vagina is hindered by its low incidence rate. In this regard, the molecular mechanism underlying the progression of adenosis to CCACs may be studied using mouse genetics. For example, simultaneous loss of Trp63 and a tumor suppressor gene in vaginal epithelium may induce vaginal adenocarcinoma in a mouse genetic model.
IV. Endocrine disruptors
There have been cases of cervical and vaginal adenoses and CCACs reported in women who have no history of DES exposure. In fact, the presence of glands in vaginae were described in women who were born before DES was clinically used, or after the administration of DES to pregnant women was stopped (Plaut, 1940; Sandberg et al., 1965; Sandberg, 1967; Blaikley et al., 1971; Ruffolo et al., 1971; Kurman and Scully, 1974; Robboy et al., 1986; Chattopadhyay et al., 2001; Paczos et al., 2010; Cebesoy et al., 2007; Accetta et al., 2001; Charlton et al., 2001). In utero exposure to other hormones or environmental chemicals may be the cause of non-DES associated adenosis in the cervix and vagina. We have demonstrated that DES induces vaginal and cervical adenoses in mice by disrupting induction of Trp63 expression in MDE through epithelial ERα (Kurita et al., 2004; Kurita, 2011). Thus, compounds that can modulate ERα signaling are candidates for causal factors of non-DES associated adenosis. In fact, estrogens or estrogen receptor modulators other than DES have been shown to induce cervical or vaginal adenosis in humans (Sherman et al., 1974) and animal models. In rodent models, 17β-estradiol (Forsberg, 1972), 17α-estradiol (Forsberg and Kalland, 1981), estradiol benzoate (Plapinger and Bern, 1979), dienestrol (Forsberg and Kalland, 1981), clomiphene citrate (Gorwill et al., 1982), tamoxifen (Taguchi and Nishizuka, 1985), nafoxidine (Iguchi et al., 1986), coumestrol (Burroughs et al., 1990) and bisphenol A (BPA) (Newbold et al., 2009) have been shown to induce vaginal adenosis. Among these compounds, BPA is particularly important because there is a high rate of exposure to this compound in the industrialized world (Vandenberg et al., 2010).
BPA is present in our environment as an essential component of polycarbonate plastic and epoxy resins. Commercial production of BPA began in 1957 in the United States and over 6 billion pounds is produced each year (Vandenberg et al., 2009). Though it has been suggested that BPA is promptly metabolized in the liver and intestines and excreted (Volkel et al., 2002), a review of over 80 biomonitoring studies revealed that unconjugated BPA is detected in blood, urine and other fluids of individuals of all ages from around the world (Vandenberg et al., 2010). Additionally, studies in the United States, Germany and Japan have shown detectable levels of BPA in the circulation of both pregnant women and their fetuses (Yamada et al., 2002; Vandenberg et al., 2009; Ikezuki et al., 2002; Schonfelder et al., 2002; Padmanabhan et al., 2008). Several other environmental chemicals possess estrogen-like activities in some assays (Sonnenschein and Soto, 1998; Roy et al., 2009; Vassalli et al., 1994; Korach et al., 1988). These xenoestrogens are detected in many products, including pesticides (Soto et al., 1994), industrial detergents (White et al., 1994) and flame-retardants (Legler and Brouwer, 2003). Exposure to a combination of these compounds may cause non-DES associated adenosis.
Although it has not been demonstrated, the expression of TRP63 in MDE may also be disrupted by a mechanism that does not involve ERα-mediated signaling. Since the induction of TRP63, and thus cell fate determination of MDE, occurs within a narrow developmental window, the effects of environmental compounds on vaginal development should be tested in animal models. Continued investigation into the mechanisms of cell fate decisions in the MDE is necessary to understand the onset of vaginal and cervical adenosis and possible progression into CCAC by DES.
Exposure to diethylstilbestrol (DES) in utero causes cervical and vaginal adenoses.
An estimated 5 million pregnant women were prescribed DES in the 1940s to 1960s.
P63 induces Müllerian duct epithelium to become squamous in the cervix and vagina.
DES disrupts P63 expression in mice and induces adenosis lesions in cervix and vagina.
Mouse models can be used to study onset of adenoses and clear cell adenocarcinoma.
Acknowledgments
The authors thank Vanida Ann Serna and Krystle Gadrinab for technical assistance. The work is supported by NIH/NIC R01 CA154358 and R01 HD064402.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Accetta SG, Rivoire WA, Monego HI, Vettori DV, De Oliveira Freitas DM, Edelweiss MI, Capp E. Vaginal adenosis in a non-diethylstilbestrol-exposed 6-year-old patient. Gynecol Obstet Invest. 2001;51:271–273. doi: 10.1159/000058063. [DOI] [PubMed] [Google Scholar]
- Antonioli DA, Burke L. Vaginal adenosis. Analysis of 325 biopsy specimens from 100 patients. Am J Clin Pathol. 1975;64:625–638. doi: 10.1093/ajcp/64.5.625. [DOI] [PubMed] [Google Scholar]
- Bibbo M, Ali I, Al-Naqeeb M, Baccarini I, Climaco LA, Gill W, Sonek M, Wied GL. Cytologic findings in female and male offspring of DES treated mothers. Acta Cytol. 1975;19:568–572. [PubMed] [Google Scholar]
- Blaikley JB, Dewhurst CJ, Ferreira HP, Lewis TL. Vaginal adenosis: clinical and pathological features with special reference to malignant change. J Obstet Gynaecol Br Commonw. 1971;78:1115–1122. doi: 10.1111/j.1471-0528.1971.tb00234.x. [DOI] [PubMed] [Google Scholar]
- Boutin EL, Sanderson RD, Bernfield M, Cunha GR. Epithelial-mesenchymal interactions in uterus and vagina alter the expression of the cell surface proteoglycan, syndecan. Dev Biol. 1991;148:63–74. doi: 10.1016/0012-1606(91)90317-v. [DOI] [PubMed] [Google Scholar]
- Boyd J, Takahashi H, Waggoner SE, Jones LA, Hajek RA, Wharton JT, Liu FS, Fujino T, Barrett JC, McLachlan JA. Molecular genetic analysis of clear cell adenocarcinomas of the vagina and cervix associated and unassociated with diethylstilbestrol exposure in utero. Cancer. 1996;77:507–513. doi: 10.1002/(SICI)1097-0142(19960201)77:3<507::AID-CNCR12>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Brody JR, Cunha GR. Histologic, morphometric, and immunocytochemical analysis of myometrial development in rats and mice: II. Effects of DES on development. Am J Anat. 1989;186:21–42. doi: 10.1002/aja.1001860103. [DOI] [PubMed] [Google Scholar]
- Burroughs CD, Mills KT, Bern HA. Reproductive abnormalities in female mice exposed neonatally to various doses of coumestrol. Journal of toxicology and environmental health. 1990;30:105–122. doi: 10.1080/15287399009531415. [DOI] [PubMed] [Google Scholar]
- Cebesoy FB, Kutlar I, Aydin A. Vaginal adenosis successfully treated with simple unipolar cauterization. J Natl Med Assoc. 2007;99:166–167. [PMC free article] [PubMed] [Google Scholar]
- Charlton A, Kirker JA, Robertson AK, Jones RW. Vaginal adenosis with adenocarcinoma in situ in a woman with no recognised antecedent factors. Aust N Z J Obstet Gynaecol. 2001;41:97–99. doi: 10.1111/j.1479-828x.2001.tb01303.x. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay I, Cruickshan DJ, Packer M. Non diethylstilbesterol induced vaginal adenosis--a case series and review of literature. Eur J Gynaecol Oncol. 2001;22:260–262. [PubMed] [Google Scholar]
- Couse JF, Dixon D, Yates M, Moore AB, Ma L, Maas R, Korach KS. Estrogen receptor-alpha knockout mice exhibit resistance to the developmental effects of neonatal diethylstilbestrol exposure on the female reproductive tract. Dev Biol. 2001;238:224–238. doi: 10.1006/dbio.2001.0413. [DOI] [PubMed] [Google Scholar]
- Cunha GR. Stromal induction and specification of morphogenesis and cytodifferentiation of the epithelia of the Müllerian ducts and urogenital sinus during development of the uterus and vagina in mice. J Exp Zool. 1976;196:361–370. doi: 10.1002/jez.1401960310. [DOI] [PubMed] [Google Scholar]
- Dalterio S, Bartke A, Steger R, Mayfield D. Neonatal exposure to DES in BALB/c male mice: effects on pituitary-gonadal function. Pharmacology, biochemistry, and behavior. 1985;22:1019–1024. doi: 10.1016/0091-3057(85)90312-0. [DOI] [PubMed] [Google Scholar]
- Dieckmann WJ, Davis ME, Rynkiewicz LM, Pottinger RE. Does the administration of diethylstilbestrol during pregnancy have therapeutic value? Am J Obstet Gynecol. 1953;66:1062–1081. doi: 10.1016/s0002-9378(16)38617-3. [DOI] [PubMed] [Google Scholar]
- Dodds EC, Golberg L, Lawson W, Robinson R. Synthetic Oestrogenic Compounds Related to Stilbene and Diphenylethane. Part I. Proceedings of the Royal Society of London. Series B, Biological Sciences. 1939;127:140–167. doi: 10.1098/rspb.1953.0003. [DOI] [PubMed] [Google Scholar]
- Dodds EC, Goldberg L, Lawson W, Robinson R. OEstrogenic Activity of Certain Synthetic Compounds. Nature. 1938;141:247–248. [Google Scholar]
- FDA. Selected item from the FDA drug bulletin-november 1971: diethylstilbestrol contraindicated in pregnancy. California medicine. 1972;116:85–86. [PMC free article] [PubMed] [Google Scholar]
- Forsberg JG. Estrogen, vaginal cancer, and vaginal development. Am J Obstet Gynecol. 1972;113:83–87. doi: 10.1016/0002-9378(72)90456-5. [DOI] [PubMed] [Google Scholar]
- Forsberg JG, Kalland T. Neonatal estrogen treatment and epithelial abnormalities in the cervicovaginal epithelium of adult mice. Cancer Res. 1981;41:721–734. [PubMed] [Google Scholar]
- Gendron RL, Paradis H, Hsieh-Li HM, Lee DW, Potter SS, Markoff E. Abnormal uterine stromal and glandular function associated with maternal reproductive defects in Hoxa-11 null mice. Biol Reprod. 1997;56:1097–1105. doi: 10.1095/biolreprod56.5.1097. [DOI] [PubMed] [Google Scholar]
- Gorwill RH, Steele HD, Sarda IR. Heterotopic columnar epithelium and adenosis in the vagina of the mouse after neonatal treatment with clomiphene citrate. Am J Obstet Gynecol. 1982;144:529–532. doi: 10.1016/0002-9378(82)90221-6. [DOI] [PubMed] [Google Scholar]
- Goto K, Takeuchi Y, Yakihara A, Kotsuji F. Synchronous invasive squamous cell carcinoma and clear cell adenocarcinoma of the uterine cervix: a different human papillomavirus status. Gynecol Oncol. 2005;97:976–979. doi: 10.1016/j.ygyno.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Greenwald P, Barlow JJ, Nasca PC, Burnett WS. Vaginal cancer after maternal treatment with synthetic estrogens. N Engl J Med. 1971;285:390–392. doi: 10.1056/NEJM197108122850707. [DOI] [PubMed] [Google Scholar]
- Halling A. Alterations in hypothalamic and pituitary hormone levels induced by neonatal treatment of female mice with diethylstilbestrol. Reproductive toxicology. 1992;6:335–346. doi: 10.1016/0890-6238(92)90197-2. [DOI] [PubMed] [Google Scholar]
- Halling A, Forsberg JG. Steroid synthesis in ovarian homogenates from immature mice treated with diethylstilboestrol in neonatal life. J Reprod Fertil. 1990;88:399–404. doi: 10.1530/jrf.0.0880399. [DOI] [PubMed] [Google Scholar]
- Hart WR, Townsend DE, Aldrich JO, Henderson BE, Roy M, Benton B. Histopathologic spectrum of vaginal adenosis and related changes in stilbestrol-exposed females. Cancer. 1976;37:763–775. doi: 10.1002/1097-0142(197602)37:2<763::aid-cncr2820370224>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Hashiguchi Y, Tsuda H, Inoue T, Berkowitz RS, Mok SC. PTEN expression in clear cell adenocarcinoma of the ovary. Gynecol Oncol. 2006;101:71–75. doi: 10.1016/j.ygyno.2005.09.047. [DOI] [PubMed] [Google Scholar]
- Herbst AL, Green TH, Jr, Ulfelder H. Primary carcinoma of the vagina. An analysis of 68 cases. Am J Obstet Gynecol. 1970;106:210–218. doi: 10.1016/0002-9378(70)90265-6. [DOI] [PubMed] [Google Scholar]
- Herbst AL, Kurman RJ, Scully RE, Poskanzer DC. Clear-cell adenocarcinoma of the genital tract in young females. Registry report. N Engl J Med. 1972;287:1259–1264. doi: 10.1056/NEJM197212212872501. [DOI] [PubMed] [Google Scholar]
- Herbst AL, Poskanzer DC, Robboy SJ, Friedlander L, Scully RE. Prenatal exposure to stilbestrol. A prospective comparison of exposed female offspring with unexposed controls. N Engl J Med. 1975;292:334–339. doi: 10.1056/NEJM197502132920704. [DOI] [PubMed] [Google Scholar]
- Herbst AL, Robboy SJ, Scully RE, Poskanzer DC. Clear-cell adenocarcinoma of the vagina and cervix in girls: analysis of 170 registry cases. Am J Obstet Gynecol. 1974;119:713–724. [PubMed] [Google Scholar]
- Herbst AL, Scully RE. Adenocarcinoma of the vagina in adolescence. A report of 7 cases including 6 clear-cell carcinomas (so-called mesonephromas) Cancer. 1970;25:745–757. doi: 10.1002/1097-0142(197004)25:4<745::aid-cncr2820250402>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Herbst AL, Ulfelder H, Poskanzer DC. Adenocarcinoma of the vagina. Association of maternal stilbestrol therapy with tumor appearance in young women. N Engl J Med. 1971;284:878–881. doi: 10.1056/NEJM197104222841604. [DOI] [PubMed] [Google Scholar]
- Hoover RN, Hyer M, Pfeiffer RM, Adam E, Bond B, Cheville AL, Colton T, Hartge P, Hatch EE, Herbst AL, Karlan BY, Kaufman R, Noller KL, Palmer JR, Robboy SJ, Saal RC, Strohsnitter W, Titus-Ernstoff L, Troisi R. Adverse health outcomes in women exposed in utero to diethylstilbestrol. N Engl J Med. 2011;365:1304–1314. doi: 10.1056/NEJMoa1013961. [DOI] [PubMed] [Google Scholar]
- Iguchi T, Hirokawa M, Takasugi N. Occurrence of genital tract abnormalities and bladder hernia in female mice exposed neonatally to tamoxifen. Toxicology. 1986;42:1–11. doi: 10.1016/0300-483x(86)90087-9. [DOI] [PubMed] [Google Scholar]
- Iguchi T, Takasugi N. Postnatal development of uterine abnormalities in mice exposed to DES in utero. Biol Neonate. 1987;52:97–103. doi: 10.1159/000242690. [DOI] [PubMed] [Google Scholar]
- Ikezuki Y, Tsutsumi O, Takai Y, Kamei Y, Taketani Y. Determination of bisphenol A concentrations in human biological fluids reveals significant early prenatal exposure. Human reproduction. 2002;17:2839–2841. doi: 10.1093/humrep/17.11.2839. [DOI] [PubMed] [Google Scholar]
- Johnson LD, Driscoll SG, Hertig AT, Cole PT, Nickerson RJ. Vaginal adenosis in stillborns and neonates exposed to diethylstilbestrol and steroidal estrogens and progestins. Obstet Gynecol. 1979;53:671–679. [PubMed] [Google Scholar]
- Kaufman RH, Binder GL, Gray PM, Jr, Adam E. Upper genital tract changes associated with exposure in utero to diethylstilbestrol. Am J Obstet Gynecol. 1977;128:51–59. doi: 10.1016/0002-9378(77)90294-0. [DOI] [PubMed] [Google Scholar]
- Korach KS, Sarver P, Chae K, McLachlan JA, McKinney JD. Estrogen receptor-binding activity of polychlorinated hydroxybiphenyls: conformationally restricted structural probes. Mol Pharmacol. 1988;33:120–126. [PubMed] [Google Scholar]
- Kurita T. Developmental origin of vaginal epithelium. Differentiation. 2010;80:99–105. doi: 10.1016/j.diff.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurita T. Normal and Abnormal Epithelial Differentiation in the Female Reproductive Tract. Differentiation. 2011 doi: 10.1016/j.diff.2011.04.008. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurita T, Cooke PS, Cunha GR. Epithelial-stromal tissue interaction in paramesonephric (Müllerian) epithelial differentiation. Dev Biol. 2001;240:194–211. doi: 10.1006/dbio.2001.0458. [DOI] [PubMed] [Google Scholar]
- Kurita T, Cunha GR. Roles of p63 in differentiation of Müllerian duct epithelial cells. Ann N Y Acad Sci. 2001;948:9–12. doi: 10.1111/j.1749-6632.2001.tb03982.x. [DOI] [PubMed] [Google Scholar]
- Kurita T, Cunha GR, Robboy SJ, Mills AA, Medina RT. Differential expression of p63 isoforms in female reproductive organs. Mech Dev. 2005;122:1043–1055. doi: 10.1016/j.mod.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Kurita T, Mills AA, Cunha GR. Roles of p63 in the diethylstilbestrol-induced cervicovaginal adenosis. Development. 2004;131:1639–1649. doi: 10.1242/dev.01038. [DOI] [PubMed] [Google Scholar]
- Kurman RJ, Scully RE. The incidence and histogenesis of vaginal adenosis. An autopsy study. Hum Pathol. 1974;5:265–276. doi: 10.1016/s0046-8177(74)80111-5. [DOI] [PubMed] [Google Scholar]
- Labarthe D, Adam E, Noller KL, O’Brien PC, Robboy SJ, Tilley BC, Townsend D, Barnes AB, Kaufman RH, Decker DG, Fish CR, Herbst AL, Gundersen J, Kurland LT. Design and preliminary observations of National Cooperative Diethylstilbestrol Adenosis (DESAD) Project. Obstet Gynecol. 1978;51:453–458. doi: 10.1097/00006250-197804000-00014. [DOI] [PubMed] [Google Scholar]
- Legler J, Brouwer A. Are brominated flame retardants endocrine disruptors? Environment international. 2003;29:879–885. doi: 10.1016/S0160-4120(03)00104-1. [DOI] [PubMed] [Google Scholar]
- Liebrich C, Brummer O, Von Wasielewski R, Wegener G, Meijer C, Iftner T, Petry KU. Primary cervical cancer truly negative for high-risk human papillomavirus is a rare but distinct entity that can affect virgins and young adolescents. Eur J Gynaecol Oncol. 2009;30:45–48. [PubMed] [Google Scholar]
- Lopez J, Ogren L, Talamantes F. Effects of neonatal treatment with diethylstilbestrol and 17 alpha-hydroxyprogesterone caproate on in vitro pituitary prolactin secretion. Life Sci. 1984;34:2303–2311. doi: 10.1016/0024-3205(84)90221-2. [DOI] [PubMed] [Google Scholar]
- Lopez J, Ogren L, Talamantes F. Neonatal diethylstilbestrol treatment: response of prolactin to dopamine or estradiol in adult mice. Endocrinology. 1986;119:1020–1027. doi: 10.1210/endo-119-3-1020. [DOI] [PubMed] [Google Scholar]
- Ma L. Endocrine disruptors in female reproductive tract development and carcinogenesis. Trends in endocrinology and metabolism: TEM. 2009;20:357–363. doi: 10.1016/j.tem.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Benson GV, Lim H, Dey SK, Maas RL. Abdominal B (AbdB) Hoxa genes: regulation in adult uterus by estrogen and progesterone and repression in Müllerian duct by the synthetic estrogen diethylstilbestrol (DES) Dev Biol. 1998;197:141–154. doi: 10.1006/dbio.1998.8907. [DOI] [PubMed] [Google Scholar]
- Meyers R. DES, the bitter pill. Seaview/Putnam; New York: 1983. [Google Scholar]
- Miller C, Sassoon DA. Wnt-7a maintains appropriate uterine patterning during the development of the mouse female reproductive tract. Development. 1998;125:3201–3211. doi: 10.1242/dev.125.16.3201. [DOI] [PubMed] [Google Scholar]
- Minaguchi T, Yoshikawa H, Nakagawa S, Yasugi T, Yano T, Iwase H, Mizutani K, Shiromizu K, Ohmi K, Watanabe Y, Noda K, Nishiu M, Nakamura Y, Taketani Y. Association of PTEN mutation with HPV-negative adenocarcinoma of the uterine cervix. Cancer Lett. 2004;210:57–62. doi: 10.1016/j.canlet.2004.03.017. [DOI] [PubMed] [Google Scholar]
- Murashima A, Miyagawa S, Ogino Y, Nishida-Fukuda H, Araki K, Matsumoto T, Kaneko T, Yoshinaga K, Yamamura K, Kurita T, Kato S, Moon AM, Yamada G. Essential roles of androgen signaling in wolffian duct stabilization and epididymal cell differentiation. Endocrinology. 2011;152:1640–1651. doi: 10.1210/en.2010-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima T, Iguchi T, Sato T. Involvement of activin signaling in abnormalities of mouse vagina exposed neonatally to diethylstilbestrol. Cell Tissue Res. 2011 doi: 10.1007/s00441-011-1161-2. [DOI] [PubMed] [Google Scholar]
- Naucler P, Mabota da Costa F, da Costa JL, Ljungberg O, Bugalho A, Dillner J. Human papillomavirus type-specific risk of cervical cancer in a population with high human immunodeficiency virus prevalence: case-control study. The Journal of general virology. 2011 doi: 10.1099/vir.0.034298-0. [DOI] [PubMed] [Google Scholar]
- Newbold RR, Jefferson WN, Padilla-Banks E. Prenatal exposure to bisphenol a at environmentally relevant doses adversely affects the murine female reproductive tract later in life. Environ Health Perspect. 2009;117:879–885. doi: 10.1289/ehp.0800045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbold RR, McLachlan JA. Vaginal adenosis and adenocarcinoma in mice exposed prenatally or neonatally to diethylstilbestrol. Cancer Res. 1982;42:2003–2011. [PubMed] [Google Scholar]
- Ng AB, Reagan JW, Hawliczek S, Wentz WB. Cellular detection of vaginal adenosis. Obstet Gynecol. 1975;46:323–328. [PubMed] [Google Scholar]
- Noller KL, Decker DG, Lanier AP, Kurland LT. Clear-cell adenocarcinoma of the cervix after maternal treatment with synthetic estrogens. Mayo Clinic proceedings. Mayo Clinic. 1972;47:629–630. [PubMed] [Google Scholar]
- O’Brien PC, Noller KL, Robboy SJ, Barnes AB, Kaufman RH, Tilley BC, Townsend DE. Vaginal epithelial changes in young women enrolled in the National Cooperative Diethylstilbestrol Adenosis (DESAD) project. Obstet Gynecol. 1979;53:300–308. [PubMed] [Google Scholar]
- Paczos TA, Ackers S, Odunsi K, Lele S, Mhawech-Fauceglia P. Primary vaginal adenocarcinoma arising in vaginal adenosis after CO2 laser vaporization and 5-fluorouracil therapy. Int J Gynecol Pathol. 2010;29:193–196. doi: 10.1097/PGP.0b013e3181b6a7d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan V, Siefert K, Ransom S, Johnson T, Pinkerton J, Anderson L, Tao L, Kannan K. Maternal bisphenol-A levels at delivery: a looming problem? Journal of perinatology: official journal of the California Perinatal Association. 2008;28:258–263. doi: 10.1038/sj.jp.7211913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer JR, Anderson D, Helmrich SP, Herbst AL. Risk factors for diethylstilbestrol-associated clear cell adenocarcinoma. Obstet Gynecol. 2000;95:814–820. [PubMed] [Google Scholar]
- Palmlund I, Apfel R, Buitendijk S, Cabau A, Forsberg JG. Effects of diethylstilbestrol (DES) medication during pregnancy: report from a symposium at the 10th international congress of ISPOG. Journal of psychosomatic obstetrics and gynaecology. 1993a;14:71–89. doi: 10.3109/01674829309084432. [DOI] [PubMed] [Google Scholar]
- Palmlund I, Apfel R, Buitendijk S, Cabau A, Forsberg JG. Effects of diethylstilbestrol (DES) medication during pregnancy: report from a symposium at the 10th international congress of ISPOG. J Psychosom Obstet Gynaecol. 1993b;14:71–89. doi: 10.3109/01674829309084432. [DOI] [PubMed] [Google Scholar]
- Pirog EC, Kleter B, Olgac S, Bobkiewicz P, Lindeman J, Quint WG, Richart RM, Isacson C. Prevalence of human papillomavirus DNA in different histological subtypes of cervical adenocarcinoma. Am J Pathol. 2000;157:1055–1062. doi: 10.1016/S0002-9440(10)64619-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plapinger L, Bern HA. Adenosis-like lesions and other cervicovaginal abnormalities in mice treated perinatally with estrogen. J Natl Cancer Inst. 1979;63:507–518. [PubMed] [Google Scholar]
- Plaut A, Dreyfuss ML. Adenosis of vagina and its relations to primary adenocarcinoma of vagina. Surg Gynecol Obstet. 1940;71:756–765. [Google Scholar]
- Robboy SJ, Friedlander LM, Welch WR, Keh PC, Taft PD, Barnes AB, Scully RE, Herbst AL. Cytology of 575 young women with prenatal exposure to diethylstilbestrol. Obstet Gynecol. 1976;48:511–515. [PubMed] [Google Scholar]
- Robboy SJ, Hill EC, Sandberg EC, Czernobilsky B. Vaginal adenosis in women born prior to the diethylstilbestrol era. Hum Pathol. 1986;17:488–492. doi: 10.1016/s0046-8177(86)80039-9. [DOI] [PubMed] [Google Scholar]
- Robboy SJ, Young RH, Welch WR, Truslow GY, Prat J, Herbst AL, Scully RE. Atypical vaginal adenosis and cervical ectropion. Association with clear cell adenocarcinoma in diethylstilbestrol-exposed offspring. Cancer. 1984;54:869–875. doi: 10.1002/1097-0142(19840901)54:5<869::aid-cncr2820540519>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Roy JR, Chakraborty S, Chakraborty TR. Estrogen-like endocrine disrupting chemicals affecting puberty in humans--a review. Medical science monitor: international medical journal of experimental and clinical research. 2009;15:RA137–145. [PubMed] [Google Scholar]
- Ruffolo EH, Foxworthy D, Fletcher JC. Vaginal adenocarcinoma arising in vaginal adenosis. Am J Obstet Gynecol. 1971;111:167–172. doi: 10.1016/0002-9378(71)90885-4. [DOI] [PubMed] [Google Scholar]
- Sandberg EC. The incidence and distribution of occult vaginal adenosis. Trans Pac Coast Obstet Gynecol Soc. 1967;35:36–48. [PubMed] [Google Scholar]
- Sandberg EC, Danielson RW, Cauwet RW, Bonar BE. Adenosis Vaginae. Am J Obstet Gynecol. 1965;93:209–222. doi: 10.1016/0002-9378(65)90660-5. [DOI] [PubMed] [Google Scholar]
- Sato N, Tsunoda H, Nishida M, Morishita Y, Takimoto Y, Kubo T, Noguchi M. Loss of heterozygosity on 10q23.3 and mutation of the tumor suppressor gene PTEN in benign endometrial cyst of the ovary: possible sequence progression from benign endometrial cyst to endometrioid carcinoma and clear cell carcinoma of the ovary. Cancer Res. 2000;60:7052–7056. [PubMed] [Google Scholar]
- Schonfelder G, Wittfoht W, Hopp H, Talsness CE, Paul M, Chahoud I. Parent bisphenol A accumulation in the human maternal-fetal-placental unit. Environmental health perspectives. 2002;110:A703–707. doi: 10.1289/ehp.110-1241091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder AL, Pelch KE, Nagel SC. Estrogen modulates expression of putative housekeeping genes in the mouse uterus. Endocrine. 2009;35:211–219. doi: 10.1007/s12020-009-9154-6. [DOI] [PubMed] [Google Scholar]
- Seaman B. The greatest experiment ever performed on women: exploding the estrogen myth. Hyperion 2003 [Google Scholar]
- Sherman AI, Goldrath M, Berlin A, Vakhariya V, Banooni F, Michaels W, Goodman P, Brown S. Cervical-vaginal adenosis after in utero exposure to synthetic estrogens. Obstet Gynecol. 1974;44:531–545. [PubMed] [Google Scholar]
- Smith GV, Smith OW. Prophylactic hormone therapy; relation to complications of pregnancy. Obstet Gynecol. 1954;4:129–141. [PubMed] [Google Scholar]
- Smith OW. Diethylstilbestrol in the prevention and treatment of complications of pregnancy. Am J Obstet Gynecol. 1948;56:821–834. [PubMed] [Google Scholar]
- Smith OW, Smith GV, Hurwitz D. Increased excretion of pregnanediol in pregnancy from diethylstilbestrol with special reference to the prevention of late pregnancy accidents. Am J Obstet Gynecol. 1946;51:411–415. doi: 10.1016/s0002-9378(16)40020-7. [DOI] [PubMed] [Google Scholar]
- Smith OW, Smith GVS, Schiller S. ESTROGEN AND PROGESTIN METABOLISM IN PREGNANCY. I. SPONTANEOUS AND INDUCED LABOR. The Journal of Clinical Endocrinology. 1941;1:461–469. [Google Scholar]
- Sonnenschein C, Soto AM. An updated review of environmental estrogen and androgen mimics and antagonists. J Steroid Biochem Mol Biol. 1998;65:143–150. doi: 10.1016/s0960-0760(98)00027-2. [DOI] [PubMed] [Google Scholar]
- Soto AM, Chung KL, Sonnenschein C. The pesticides endosulfan, toxaphene, and dieldrin have estrogenic effects on human estrogen-sensitive cells. Environmental health perspectives. 1994;102:380–383. doi: 10.1289/ehp.94102380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafl A, Mattingly RF, Foley DV, Fetherston WC. Clinical diagnosis of vaginal adenosis. Obstet Gynecol. 1974;43:118–128. [PubMed] [Google Scholar]
- Stewart J, 3rd, Bevans-Wilkins K, Ye C, Kurtycz DF. Clear-cell endocervical adenocarcinoma in a 19-year-old woman. Diagn Cytopathol. 2006;34:839–842. doi: 10.1002/dc.20569. [DOI] [PubMed] [Google Scholar]
- Taguchi O, Nishizuka Y. Reproductive tract abnormalities in female mice treated neonatally with tamoxifen. Am J Obstet Gynecol. 1985;151:675–678. doi: 10.1016/0002-9378(85)90163-2. [DOI] [PubMed] [Google Scholar]
- Troisi R, Hatch EE, Titus-Ernstoff L, Hyer M, Palmer JR, Robboy SJ, Strohsnitter WC, Kaufman R, Herbst AL, Hoover RN. Cancer risk in women prenatally exposed to diethylstilbestrol. Int J Cancer. 2007;121:356–360. doi: 10.1002/ijc.22631. [DOI] [PubMed] [Google Scholar]
- Vandenberg LN, Chahoud I, Heindel JJ, Padmanabhan V, Paumgartten FJ, Schoenfelder G. Urinary, circulating, and tissue biomonitoring studies indicate widespread exposure to bisphenol A. Environmental health perspectives. 2010;118:1055–1070. doi: 10.1289/ehp.0901716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg LN, Maffini MV, Sonnenschein C, Rubin BS, Soto AM. Bisphenol-A and the great divide: a review of controversies in the field of endocrine disruption. Endocrine reviews. 2009;30:75–95. doi: 10.1210/er.2008-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassalli A, Matzuk MM, Gardner HA, Lee KF, Jaenisch R. Activin/inhibin beta B subunit gene disruption leads to defects in eyelid development and female reproduction. Genes Dev. 1994;8:414–427. doi: 10.1101/gad.8.4.414. [DOI] [PubMed] [Google Scholar]
- Volkel W, Colnot T, Csanady GA, Filser JG, Dekant W. Metabolism and kinetics of bisphenol a in humans at low doses following oral administration. Chemical research in toxicology. 2002;15:1281–1287. doi: 10.1021/tx025548t. [DOI] [PubMed] [Google Scholar]
- Waggoner SE, Anderson SM, Luce MC, Takahashi H, Boyd J. p53 protein expression and gene analysis in clear cell adenocarcinoma of the vagina and cervix. Gynecol Oncol. 1996;60:339–344. doi: 10.1006/gyno.1996.0052. [DOI] [PubMed] [Google Scholar]
- Waggoner SE, Anderson SM, Van Eyck S, Fuller J, Luce MC, Herbst AL. Human papillomavirus detection and p53 expression in clear-cell adenocarcinoma of the vagina and cervix. Obstet Gynecol. 1994;84:404–408. [PubMed] [Google Scholar]
- Waggoner SE, Baunoch DA, Anderson SA, Leigh F, Zagaja VG. Bcl-2 protein expression associated with resistance to apoptosis in clear cell adenocarcinomas of the vagina and cervix expressing wild-type p53. Ann Surg Oncol. 1998;5:544–547. doi: 10.1007/BF02303648. [DOI] [PubMed] [Google Scholar]
- White R, Jobling S, Hoare SA, Sumpter JP, Parker MG. Environmentally persistent alkylphenolic compounds are estrogenic. Endocrinology. 1994;135:175–182. doi: 10.1210/endo.135.1.8013351. [DOI] [PubMed] [Google Scholar]
- Yamada H, Furuta I, Kato EH, Kataoka S, Usuki Y, Kobashi G, Sata F, Kishi R, Fujimoto S. Maternal serum and amniotic fluid bisphenol A concentrations in the early second trimester. Reproductive toxicology. 2002;16:735–739. doi: 10.1016/s0890-6238(02)00051-5. [DOI] [PubMed] [Google Scholar]
- Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, Andrews NC, Caput D, McKeon F. p63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell. 1998;2:305–316. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]