

Abstract
We inoculated BALB/c mice deficient in STAT6 (STAT6−/−) and their wild-type (wt) littermates (STAT6+/+) with the natural mouse pathogen, ectromelia virus (EV). STAT6−/− mice exhibited increased resistance to generalized infection with EV when compared with STAT6+/+ mice. In the spleens and lymph nodes of STAT6−/− mice, T helper 1 (Th1) cytokines were induced at earlier time points and at higher levels postinfection when compared with those in STAT6+/+ mice. Elevated levels of NO were evident in plasma and splenocyte cultures of EV-infected STAT6−/− mice in comparison with STAT6+/+ mice. The induction of high levels of Th1 cytokines in the mutant mice correlated with a strong natural killer cell response. We demonstrate in genetically susceptible BALB/c mice that the STAT6 locus is critical for progression of EV infection. Furthermore, in the absence of this transcription factor, the immune system defaults toward a protective Th1-like response, conferring pronounced resistance to EV infection and disease progression.
Ectromelia virus (EV) infection of mice causes a disease known as mousepox. EV, a cytopathic virus, is closely related to variola virus (the pathogen that causes smallpox in humans) and vaccinia virus (a live-virus vaccine used to eradicate smallpox). The mousepox model has been used successfully to investigate the pathogenesis of generalized viral infections, experimental epidemiology, cell-mediated immunity to viral infections, and mechanisms of genetic resistance to infection and disease expression (1–4). Importantly, these investigations have shown that various strains of mice are resistant or susceptible to mousepox infection (4–6). For instance, breeds of mice most sensitive to infection are Swiss and inbred strains such as A and A/J (both H-2a), BALB/c and DBA (both H-2d), and C3H (H-2k) (6, 7). By contrast, the AKR (H-2k) strains exhibits mild sensitivity, whereas C57BL/6, C57BL/10, and 129/Sv (all H-2b) are resistant to infection (6, 7).
Both the humoral and cell-mediated arms of the immune system have been examined for their relative importance in the recovery of mice from EV infection. Pioneering research by Fenner (8) showed that systemic administration of anti-EV antibody reduced the clinical severity of disease, but had no effect on the multiplication of virus in the inoculated foot. This work was extended by Blanden (9, 10), who used cell transfer and graft vs. host experiments to provide compelling evidence that mononuclear phagocytes and cytotoxic T cells were most important for recovery from infection and disease. Further work by Karupiah et al. (1, 11) showed that interferons (IFNs) and CD4+ and CD8+ T cells are important in the recovery process from mousepox infection. It also has been shown that virus-specific cytotoxic T cells appear earlier in regional lymph nodes (LNs) in resistant (C57BL/6 mice) in comparison with susceptible (BALB/c) strains (12). Thus, anti-EV defense mechanisms come into effect much earlier in the initial phase of infection in resistant strains of mice.
The differences between the BALB/c and C57BL/6 strains, with respect to generation of immune responses, have been well characterized in Leishmania major infection. Studies have indicated that control of lesion growth induced by L. major in genetically resistant mice (C57BL/6) is associated with the expansion of CD4+ T helper 1 (Th1) cells and the production of cytokines such as IL-12, IFN-γ, and IL-2 (13, 14). By contrast, nonhealing responses in susceptible mice (BALB/c) have been related to the expansion of CD4+ Th2 cells and the production of cytokines such as IL-4 and IL-10 (13, 14). Generation of the Th2 immune response does not provide protection, and often a progressive and fatal disease in BALB/c mice infected with L. major is observed (13–15). The disease-exacerbating role of IL-4 has been shown to be due to its ability to down-regulate the development of a Th1-like response in response to L. major infection while providing a growth and differentiation signal for Th2 cells (16, 17). The pathway responsible for IL-4-mediated differentiation events involves phosphorylation of Janus kinase 1 (JAK1) and JAK3 and subsequent activation of STAT6 (signal transducer and activator of transcription 6) (18, 19). STAT6 is essential for IL-4 signaling and the subsequent development of the Th2 subset (20, 21). Furthermore, IL-13, a cytokine closely related to IL-4 in biologic function, has been shown to share receptor components with IL-4 and also signal through the STAT6 pathway (22). Recent studies have shown that STAT6-deficient mice exhibit enhanced Th1-type responses and are resistant to infection with Trypanosoma cruzi and Leishmania mexicana (23, 24).
Because STAT6 is essential for the development of the Th2 subset, it was of interest to investigate the role of the STAT6-signaling pathway on the replication of EV in susceptible BALB/c mice. In this investigation, we show that the lack of STAT6 strongly promotes the generation of a Th1 response after infection with EV. By contrast, EV infection did not promote Th1 responses in STAT6+/+ mice but enhanced Th2 responses that were characterized by increases in IL-4 production. These investigations show that the STAT6-signaling pathway plays a pivotal role in determining susceptibility to EV by promoting Th2 immunity. These investigations also highlight the potential differential role of Th subsets in the fundamental mechanism regulating the resolution of viral infections.
Methods
Mice.
Adult (8- to 12-week-old) female STAT6−/− mice (20) (crossed to BALB/c for 6 generations, N6) and their wild-type littermates (STAT6+/+) were obtained from the specific-pathogen-free facility John Curtin School of Medical Research, Australian National University. Nullizygous STAT6−/− mice were confirmed by PCR. Experiments were performed according to institutional guidelines for animal care and use.
Virus Infection and Enumeration.
Virulent EV (Moscow strain) propagated in BS-C-1 cells (ATCC CCL 26) was purified on sucrose density gradients (1). Mice were inoculated in the right and left hind footpad with 103 plaque-forming units (pfu) of virus in 20 μl of PBS. At selected times postinfection (p.i.), organs were removed aseptically and homogenized, and serial dilutions were assayed for formation of plaques on BS-C-1 cell monolayers (1).
Reverse Transcriptase–PCR (RT-PCR) Analysis.
Total RNA was isolated from spleens by standard methods with RNAzol B (Biotech Laboratories, Houston, TX). RT-PCR was performed to determine relative quantities of mRNA for various cytokines (25). The primers and probes for all genes were purchased from GIBCO/BRL. Primer and probe sequences for IFN-γ, IFN-α, and hypoxanthine phosphoribosyltransferase (HPRT) have been described (26), and primer and probe sequences for IL-4, tumor necrosis factor α (TNF-α), and IL-12(p40) are as follows. IL-4: GAATGTACCAGGAGCCATATC (sense), CTCAGTACTACGAGTAATCCA (antisense), AGGGCTTCCAAGGTGCTTCGCA (probe); IL-12(p40): CGTGCTCATGGCTGGTGCAAAG (sense), CTTCATCTGCAAGTTCTTGGGC (antisense), TCTGTCTGCAGAGAAGGTCACA (probe); and TNF-α: GATCTCAAAGACAACCAACTAGTG (sense), CTCCAGCTGGAAGACTCCTCCCAG (antisense), CCCGACTACGTGCTCCTCACC (probe). The cycle numbers used for amplification of each gene product are as follows: IFN-γ and IFN-α, 27 cycles; IL-12 (p40) and TNF-α, 30 cycles; IL-4, 34 cycles; and HPRT, 23 cycles. After the appropriate number of PCR cycles, the amplified DNA was analyzed by gel electrophoresis and Southern blotting and detected by using the enhanced chemiluminescence (ECL) detection system as recommended by the manufacturer (Amersham Pharmacia). PCR amplification with the HPRT reference gene was performed to assess variations in cDNA or total RNA loading between samples. Mean relative transcript levels per group were determined from cDNA panels as described previously (24). Briefly, values were derived by dividing the mean of the triplicate values measured for the transcript of interest by the mean of triplicate HPRT values for the sample.
Cytokine ELISA.
IFN-γ and IL-4 production by Con A (4 μg/ml)-stimulated cells (splenocytes, 2.5 × 106) and nonstimulated cells from EV-infected STAT6+/+ and STAT6−/− mice was measured by capture ELISA (26, 27). Recombinant murine IFN-γ (Genzyme) or IL-4 (Endogen, Cambridge, MA) served as the standard.
Plasma and Splenocyte Reactive Nitrogen Intermediates (RNI) Assay.
Measurement of RNI in splenocyte culture supernatants or plasma provides an indication of the amount of NO that has been produced (28). Briefly, nitrite was measured by the addition of 100 μl of Griess reagent to 30 μl of test sample. Protein was removed by the addition of 100 μl of trichloroacetic acid followed by centrifugation, and the optical density of the samples was read at 540 nm with a reference at 650 nm by using a microplate reader (Molecular Devices). For nitrate measurements, the nitrate first was converted to nitrite by incubation with nitrate reductase and NADPH (Boehringer Mannheim) for 20 min. The results were quantified by reading against appropriate nitrite and nitrate standard curves. Both nitrite and nitrate were measured in culture supernatants, whereas only nitrate was measured in plasma because almost all of the RNI in plasma are in this form.
Cytotoxicity Assays.
Standard 51Cr-release assays were used for the measurement of antiviral natural killer (NK) activity (29).
Statistical Analysis.
Unpaired Student's t tests and one-way ANOVA at 95% confidence levels were performed by using instat 2.00 software (GraphPad, San Diego). Data represent mean ± SEM (significant if P < 0.05).
Results
Mortality, Virus Titers, and Organ Pathology in STAT6−/− Mice.
We first examined the ability of genetically deficient, susceptible BALB/c mice to resolve infection with EV. Mice were infected with 103 pfu of EV in each of the right and left footpads. All STAT6+/+ mice succumbed to infection and died between days 6 and 10 p.i. (mean, 7.4 ± 0.4 days; n = 10), whereas there was a significant (P < 0.005) delay in mortality observed in the STAT6−/− mice (mean, 19 ± 0.9 days; n = 10) (Fig. 1a). To investigate the mechanism(s) underlying the delay in mortality in STAT6−/− mice, the kinetics of EV replication in organs of mice was determined. At various days, groups of mice were killed and spleens and livers were removed for determination of virus titers. In the livers of STAT6−/− mice, virus was first detected on the fourth day p.i. Virus was recovered at ≈3.1 log10 pfu on day 4 and reached 7.3 log10 pfu by day 18 p.i. (Fig. 1b). In STAT6+/+ mice, mean virus titers of 2.2 log10 pfu were recovered on day 3 p.i. and a peak titer of 7.1 log10 pfu was recorded on day 6 p.i. The kinetics of virus replication in the spleen of STAT6−/− mice was similar to that in the liver. Virus was below the limit of detection in spleens of STAT6−/− mice at day 3 after infection (Fig. 1b). By day 4 p.i., 2.6 log10 pfu of virus was recovered (Fig. 1b). However, by day 18 p.i., viral titers reached ≈7.8 log10 pfu. By contrast, virus was detected in spleens of STAT6+/+ mice as early as day 3 p.i. By day 6 p.i., this had increased to more than 8 log10 pfu. Histological examination of liver and spleen from STAT6−/− mice on day 6 p.i. revealed a few necrotic foci whereas there was pronounced necrosis in these organs from STAT6+/+ mice (data not shown).
Figure 1.

Absence of STAT6 confers pronounced resistance to primary EV infection. (a) Mortality in female STAT6+/+ or STAT6−/− mice (n = 10) inoculated with 103 pfu EV and monitored for 25 days. Data are representative of two independent experiments. (b) Viral titers in the livers and spleens of STAT6+/+ mice (n = 4) and STAT6−/− mice (n = 4) given 103 pfu EV determined at various days. Data are representative of two experiments and are expressed as log10 virus titers/gram tissue ± SEM. The sensitivity of the assay is 2 log10 pfu and is indicated by the dotted line.
Enhanced Levels of Th1 Cytokine Production in Spleens of STAT6−/− Mice.
Results from the preceding section clearly demonstrate that STAT6−/− mice exhibit significant resistance to a generalized infection with EV whereas STAT6+/+ mice rapidly succumb to infection. This is apparent from the differential levels of virus replication in both groups of mice (Fig. 1b) as well as mean time to death (Fig. 1a). These differences could be attributed to the development of a more effective immune response in the STAT6−/− mice. Therefore, it was of interest to first establish the nature of the cytokine response that may have contributed to the outcome of infection and subsequent disease progression in both groups of mice. Compared with STAT6+/+ mice, spleens from STAT6−/− mice 3 days p.i. had relative higher transcript levels of TNF-α (5-fold), IFN-γ (10-fold), IFN-α (20-fold), and IL-12 (10-fold) (Fig. 2). Notably, levels of IL-4 mRNA transcripts were 3-fold higher in STAT6+/+ mice compared with STAT6−/− mice (Fig. 2). A bias toward Th1 immunity in STAT6−/− mice after infection also was observed in popliteal LNs on days 1 and 3 p.i. in which a similar cytokine profile was observed (data not shown). In addition, time course analysis revealed that IFN-γ, IFN-α, and IL-12 mRNA transcripts in splenocytes of EV-infected STAT6−/− mice were present as early as day 1 p.i. (data not shown). In contrast, IFN-γ, IFN-α, and IL-12 mRNA transcripts were detected only at low levels by day 3 p.i. in splenocytes from control mice (data not shown). IL-4 mRNA transcripts were detected as early as day 1 p.i. in both groups of mice, with higher expression levels seen in STAT6+/+ mice at all time points tested (data not shown). To address whether this observation was consistent at the protein level, splenocytes from EV-infected mice from both groups obtained on days 0, 3, and 6 p.i. were cultured in vitro in the presence of Con A (4 μg/ml) for 48 h. Supernatants then were analyzed for IFN-γ and IL-4 by ELISA. Indeed, splenocytes taken from virus-infected STAT6−/− mice cultured with Con A produced higher levels (1.5- to 2-fold) of IFN-γ in comparison with splenocytes from STAT6+/+ mice (Fig. 3a). Levels of plasma IFN-γ also were elevated (1.5- to 1.8-fold) in STAT6−/− mice compared with STAT6+/+ mice (Fig. 3b). By contrast, splenocytes of virus-infected STAT6+/+ mice produced higher levels (1.7- to 2-fold) of IL-4 when compared with STAT6−/− mice (Fig. 3c). Collectively, these cytokine profiles are suggestive of a shift from a Th2 response in STAT6−/− mice toward a greater capacity to produce Th1-related cytokines, which directly correlates with increased resistance to EV.
Figure 2.

Expression of cytokine mRNA transcripts in spleens of STAT6−/− and control mice during infection with EV. Total RNA was prepared from spleens of mice infected for 3 days. Specific mRNA species were amplified by RT-PCR. The amplification products were blotted onto nylon membranes and hybridized to fluorescein-labeled oligonucleotide probes specific for the PCR product. Groups of four mice were used for each time point. The reference gene, HPRT, was used to assess variation between RNA and cDNA loading. Results for IFN-γ, IL-12 p40, IFN-α, TNF-α, and IL-4 are shown. Representative blots for each gene also are shown. The intensities of the bands were quantified by using a scanner. The bar graph shows normalized values for each gene in STAT6+/+ and STAT6−/− mice derived by taking the ratio of the mean of triplicate values for each animal. Data are expressed as the mean ± SEM (*, P < 0.005; **, P < 0.05).
Figure 3.

Production of IFN-γ [splenocyte (a) and plasma (b)] and IL-4 [splenocyte (c)] proteins in STAT6−/− and STAT6+/+ mice. Splenocytes from EV-infected STAT6−/− mice (n = 5) and control mice (n = 5) were taken on various days and left untreated (control) or cultured in vitro with Con A for 48 h. Data are presented as means of triplicate cultures + SEM and are representative of two experiments. Asterisks indicate statistically significant differences between groups (P < 0.05).
Elevated NO Production in STAT6−/− Mice.
It has been shown previously that NO is antiviral against EV (30).
Thus, we determined the effects of elevated IFN-γ production in
STAT6−/− mice on NO production. On days 0, 3,
and 6 p.i., plasma NO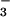 and splenocyte
NO
and splenocyte
NO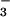 and NO
and NO (RNI) levels were
determined. Plasma NO
(RNI) levels were
determined. Plasma NO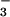 levels were elevated (1.7- to
1.9-fold) in mutant mice on days 3 and 6 p.i. compared with levels
in STAT6+/+ mice (Fig.
4a). RNI levels were measured
in supernatants of spleen cell cultures from EV-infected mice on
various days p.i. Spleen cells from EV-infected mice were cultured with
or without Con A stimulation. After 72 h, the concentration of RNI
in the supernatant was measured. Elevated levels of RNI (1.5- to
2-fold) were produced in Con A-stimulated spleen cultures from
EV-infected STAT6−/− mice when compared with
STAT6+/+ mice (Fig. 4b).
levels were elevated (1.7- to
1.9-fold) in mutant mice on days 3 and 6 p.i. compared with levels
in STAT6+/+ mice (Fig.
4a). RNI levels were measured
in supernatants of spleen cell cultures from EV-infected mice on
various days p.i. Spleen cells from EV-infected mice were cultured with
or without Con A stimulation. After 72 h, the concentration of RNI
in the supernatant was measured. Elevated levels of RNI (1.5- to
2-fold) were produced in Con A-stimulated spleen cultures from
EV-infected STAT6−/− mice when compared with
STAT6+/+ mice (Fig. 4b).
Figure 4.

Levels of reactive nitrogen intermediates in plasma
(NO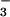 ) (a) and splenocyte cultures
(NO
) (a) and splenocyte cultures
(NO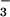 and NO
and NO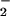 ) (b) of
STAT6+/+ mice (n = 4) and
STAT6−/− mice (n = 4). Splenocyte
cultures were obtained from mice on various days p.i. and were left
untreated (control) or cultured in vitro with Con A for
72 h before measurement of reactive nitrogen intermediates. Data
represent individual samples assayed in triplicate. Asterisks indicate
statistically significant differences between groups
(P < 0.05).
) (b) of
STAT6+/+ mice (n = 4) and
STAT6−/− mice (n = 4). Splenocyte
cultures were obtained from mice on various days p.i. and were left
untreated (control) or cultured in vitro with Con A for
72 h before measurement of reactive nitrogen intermediates. Data
represent individual samples assayed in triplicate. Asterisks indicate
statistically significant differences between groups
(P < 0.05).
Enhanced NK Cytotoxic Activity in STAT6−/− Mice.
Previous work has shown that production of IFN-γ and IL-12 contributes to NK cell activation and augmented cytolytic activity (31, 32). Because NK cells are important for the control of virus replication during the early stages of infection, we measured their cytolytic activity in both groups of mice. On days 1 and 3 p.i. with EV, significant lysis of the NK-sensitive cell line YAC-1 was observed with splenocytes from STAT6−/− mice, but less killing was observed by splenocytes from STAT6+/+ mice (Fig. 5). NK cytotoxic activity was 3-fold higher in STAT6−/− mice compared with STAT6+/+ mice. Cells from uninfected STAT6+/+ mice or mutant mice displayed no measurable lytic activity (Fig. 5). The anti-EV cytotoxic T lymphocyte (CTL) responses in spleens from both groups of mice on day 6 p.i., the time at which peak cytolytic effector function normally is observed, showed similar levels of killing (data not shown).
Figure 5.

Splenic NK cell activity (day 3 p.i.) in STAT6+/+ mice and STAT6−/− mice infected with 103 pfu EV. Spleen cells from uninfected STAT6−/− mice were used as controls. Data shown are means of lysis values from four individual mice for each group. SEM was less than 5% and has been omitted for clarity.
Discussion
EV is an excellent model of virus infection to elucidate pathogenic mechanisms not only associated with mousepox but also human infectious diseases, such as molluscum contagiosum (which is caused by a poxvirus and is a frequent infection in HIV-infected AIDS patients), herpetic (herpes simplex virus type 1) skin lesions, and smallpox. Thus, understanding the mechanism of EV infection has important implications in understanding viral pathogenesis in humans.
Three primary components of host immunity are required to eliminate EV. These are IFNs (11, 33), class I MHC-restricted CD8+ cytotoxic T cells (1, 12), and nitric oxide synthase 2 (NOS2) (30). The roles of NK cells (innate immune response) or class II MHC-restricted CD4+ helper T cells (acquired immune response) also contribute but to a lesser extent (1, 33). For instance, cytokines produced by the CD4+ Th1 cells such as IFN-γ and IL-12 are believed to be involved in the generation of optimal CD8+ CTL responses (1). The major finding in this study is that the STAT6 pathway, a critical regulator of Th2 immunity, plays a pivotal role in the susceptibility to EV infection. STAT6−/− mice infected with EV displayed lower infectivity in the liver and spleen, minimal lesions, and substantially delayed mortality, which correlated with a suppressed Th2 immunity. In STAT6+/+ mice, infection was disseminated throughout the liver and spleen. Furthermore, severe necrosis was evident in these tissues and was linked to high viral load and earlier mortality. Differences in infection and disease progression in STAT6−/− mice correlated with the development of a more pronounced Th1 immune response in these mutant mice.
Spleen and LN cells provide the environment for the generation of a specific immune response after antigen exposure. In the present study, spleens and LNs (data not shown) of STAT6−/− mice produced significantly higher levels of IL-12, IFN-γ, IFN-α, and TNF-α mRNA transcripts than that of STAT6+/+ mice. By contrast, levels of IL-4 mRNA transcript were significantly higher in STAT6+/+ mice. The high levels of IFN-γ and IL-4 mRNA in mutant and control mice, respectively, correlated with increased protein levels of these cytokines. The higher levels of IL-4 in STAT6+/+ mice may be attributed to the reduced IL-12 and IFN-γ levels in STAT6+/+ mice that can inhibit IL-4 production by T cells (34). Furthermore, the absence or reduced levels of IL-12, IFN-γ, and TNF-α in STAT6+/+ mice most likely are due to the ability of IL-4 to down-regulate IL-12 and IFN-γ production and inhibit the production of TNF-α by macrophages (35–37). In particular, the absence or reduced levels of IL-12 may be the key factor that regulates susceptibility of STAT6+/+ mice to EV. IL-12 is critical in the induction of IFN-γ production from T and NK cells and initiates the development of cell-mediated immunity by promoting the differentiation of Th1 cells from naïve T cells (38). It is likely that the increased Th1-like responses in EV-infected STAT6−/− mice are due to the absence of IL-4-mediated suppression of IL-12 production and, subsequently, IFN-γ. More recently, it was reported that genetically resistant mice infected with a recombinant EV expressing IL-4 developed symptoms of acute mousepox accompanied by high mortality (39). The IL-4-encoding EV suppressed cytolytic responses of NK and CTL and the expression of IFN-γ (39). The importance of IFN-γ in the development of a Th1-like response and resistance to EV has been shown in investigations by using neutralizing mAb for IFN-γ. Treatment of genetically resistant C57BL/6 mice with IFN-γ mAb converts infection from a self-limiting disease to fulminant mousepox (11). The importance of TNF-α in the control of EV infection also was reported recently, with partial protection to infection conferred by this cytokine via signaling predominantly through the TNF receptor 2 (40). Additionally, EV infection was shown to elicit the production of a TNF homolog that binds to TNF-α to neutralize its effects, suggesting a role for this molecule in establishing viral infectivity (41). Interestingly, this homolog not only binds mouse TNF-α but also human and rat TNF (41). However, there is no evidence showing that EV infects rats or humans. Together, these results demonstrate that, in the absence of STAT6, a rapid Th1 immune response to EV develops, whereas in STAT6+/+, genetically susceptible BALB/c mice infection predominantly elicits a Th2 response with delayed Th1 immunity.
IFN-γ activates macrophages to kill viruses by (among other methods)
production of NO (30). In addition, we have shown previously that
NOS2−/− mice inoculated with increasing doses
of EV directly correlated with the development of fulminant mousepox
and rate of death (42). Thus, NOS2 is a resistance loci that
is considered important for recovery from EV infection and disease
(42). Here, we show that STAT6−/− mice exhibit
high-plasma NO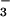 and splenocyte NO
and splenocyte NO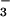 and NO
and NO levels. It is clear that the reduced IFN-γ
in STAT6+/+ mice subsequently results in low NO
production. Furthermore, the prominent production of IL-4 in
STAT6+/+ mice may further inhibit NO production
via the STAT6 pathway. Interestingly, a binding site for STAT6 has been
identified in the promoter region of the NOS2 gene (43). In this
regard, our finding is supported by investigations that showed that the
IL-4-mediated suppression of IFN-γ-induced NOS2 gene expression was
abolished in peritoneal macrophages from
STAT6−/− mice (43). Thus, STAT6, by regulating
NOS2 gene expression, links these two key susceptibility and resistance
loci.
levels. It is clear that the reduced IFN-γ
in STAT6+/+ mice subsequently results in low NO
production. Furthermore, the prominent production of IL-4 in
STAT6+/+ mice may further inhibit NO production
via the STAT6 pathway. Interestingly, a binding site for STAT6 has been
identified in the promoter region of the NOS2 gene (43). In this
regard, our finding is supported by investigations that showed that the
IL-4-mediated suppression of IFN-γ-induced NOS2 gene expression was
abolished in peritoneal macrophages from
STAT6−/− mice (43). Thus, STAT6, by regulating
NOS2 gene expression, links these two key susceptibility and resistance
loci.
NK cells are cytotoxic lymphocytes that are capable of lysing a variety of target cells and form the first line of defense against microbial pathogens. In the case of primary viral infection, NK cell responses are induced early and are temporally distinct from those of T cells. The early response to viral infection is associated with increases in both systemic and local IFN production, peaking at 3–5 days after infection. IFN production closely mirrors the time course of NK cell activation and the augmented cytolytic activity that is seen in viral infection (44). One explanation for our findings of suppressed NK cytolytic activity in STAT6+/+ mice is that reduced production of IFN-γ and IL-12 in these mice leads to the insufficient activation of NK cells. IL-12 first was identified as an inducer of NK cell-mediated cytotoxicity (32) presumably through the induction of genes involved in target cell lysis such as perforin or granzymes (45). The importance of both perforin and granzymes in the control of EV infection was established recently (46, 47). Another important function of IL-12 is the induction of IFN-γ production in resting and activated NK cells, a key step in the innate response to acute infection. Indeed, IL-12-deficient mice exhibit a defect in both NK cell cytolytic activity and IFN-γ production (48). Furthermore, in STAT4-deficient mice, IL-12-mediated increases in IFN-γ production, cellular proliferation, and NK cell cytotoxic activity of lymphocytes were impaired (49). It therefore is reasonable to speculate that STAT4−/− mice may be highly susceptible to EV infection.
IL-12 also is known to be important for the optimal generation of CTL responses. CD8+ CTL are important for recovery from primary mousepox infection, and the absence of this cell subset in genetically resistant C57BL/6 mice renders mice highly susceptible to EV infection (1). However, EV-specific CTL activity was no different in STAT6−/− or STAT6+/+ mice. Although the reasons are not clear, it is possible that the STAT6−/− mice eventually succumb to EV infection because of a lack of a potent CTL response despite the augmented NK cell and IFN-γ responses.
The proposed mechanisms underlying the development of nonhealing lesions in STAT6+/+ mice after EV infection have been linked to (i) the presence of an IL-4-driven Th2 response, (ii) failure to produce IL-12 and IFN-γ and mount a subsequent Th1 response, (iii) reduced levels of NO production, and (iv) reduced NK and CTL cytolytic activity. Despite enhanced Th1 cytokine production, STAT6−/− mice eventually succumb to infection and die. This finding may be related to the fact that genetic resistance to EV is complex and at least three resistance loci are considered important for recovery from EV infection and disease: H-2Db [termed Rmp-3, Resistance to mousepox, on chromosome 17 (4, 50)], the C5 genes (Rmp-2, on chromosome 2), and Rmp-1 (51), recently localized to a region on chromosome 6 that encodes the NK cell receptor NKR-P1 alloantigens (52). More recently, NOS2 also was shown to be an important resistant locus (42). Thus, susceptibility mechanisms that lead to susceptibility are multifactorial, and there are distinct spatial and temporal requirements for individual molecular and cellular components in the immune network that lead to infectivity and disease. We speculate here that although the absence of STAT6 alone is insufficient for BALB/c mice to express the full complement of genetic resistance to EV, this transcription factor plays a pivotal role in the immune component system that underlies genetic susceptibility to this pathogen.
Acknowledgments
We thank Mr. W. Damcevski for the provision of backcrossed mice. This work was supported by a grant from the Human Frontiers Science Program (to P.S.F.). S.M. is a recipient of the Australian National Health and Medical Research Council Peter Doherty Fellowship. G.K. is a Medical Foundation Fellow of the University of Sydney and an International Research Scholar of the Howard Hughes Medical Institute.
Abbreviations
- Th
T helper
- EV
ectromelia virus
- NK
natural killer
- IFN
interferon
- LN
lymph node
- STAT6
signal transducer and activator of transcription 6
- pfu
plaque-forming unit(s)
- p.i.
postinfection
- RT-PCR
reverse transcription–PCR
- HPRT
hypoxanthine phosphoribosyltransferase
- TNF
tumor necrosis factor
- RNI
reactive nitrogen intermediates
- CTL
cytotoxic T lymphocyte
- NOS2
nitric-oxide synthase 2
References
- 1.Karupiah G, Buller R M, Van Rooijen N, Duarte C J, Chen J. J Virol. 1996;70:8301–8309. doi: 10.1128/jvi.70.12.8301-8309.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lipman N S, Nguyen H, Perkins S. Lab Anim Sci. 1999;49:229. [PubMed] [Google Scholar]
- 3.Blanden R V, Gardner I D. Cell Immunol. 1976;22:271–282. doi: 10.1016/0008-8749(76)90029-0. [DOI] [PubMed] [Google Scholar]
- 4.O'Neill H C, Blanden R V, O'Neill T J. Immunogenetics. 1983;18:255–265. doi: 10.1007/BF00952964. [DOI] [PubMed] [Google Scholar]
- 5.Brownstein D, Bhatt P N, Jacoby R O. Arch Virol. 1989;107:35–41. doi: 10.1007/BF01313876. [DOI] [PubMed] [Google Scholar]
- 6.Bhatt P N, Jacoby R O. Lab Anim Sci. 1987;37:11–15. [PubMed] [Google Scholar]
- 7.Buller R M, Potter M, Wallace G D. Curr Top Microbiol Immunol. 1986;127:319–322. doi: 10.1007/978-3-642-71304-0_38. [DOI] [PubMed] [Google Scholar]
- 8.Fenner F. J Immunol. 1949;63:341–373. [PubMed] [Google Scholar]
- 9.Blanden R V. J Exp Med. 1970;132:1035–1054. doi: 10.1084/jem.132.5.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blanden R V. J Exp Med. 1971;133:1074–1089. doi: 10.1084/jem.133.5.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karupiah G, Fredrickson T N, Holmes K L, Khairallah L H, Buller M L. J Virol. 1993;67:4214–4226. doi: 10.1128/jvi.67.7.4214-4226.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O'Neill H C, Brenan M. J Gen Virol. 1987;68:2669–2673. doi: 10.1099/0022-1317-68-10-2669. [DOI] [PubMed] [Google Scholar]
- 13.Heinzel F P, Sadick M D, Mutha S S, Locksley R M. Proc Natl Acad Sci USA. 1991;88:7011–7015. doi: 10.1073/pnas.88.16.7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scott P, Natovitz P, Coffman R L, Pearce E, Sher A. J Exp Med. 1988;168:1675–1684. doi: 10.1084/jem.168.5.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Himmelrich H, Launois P, Maillard I, Biedermann T, Tacchini-Cottier F, Locksley R M, Rocken M, Louis J A. J Immunol. 2000;164:4819–4825. doi: 10.4049/jimmunol.164.9.4819. [DOI] [PubMed] [Google Scholar]
- 16.Oswald I P, Gazzinelli R T, Sher A, James S L. J Immunol. 1992;148:3578–3582. [PubMed] [Google Scholar]
- 17.Sher A, Gazzinelli R T, Oswald I P, Clerici M, Kullberg M, Pearce E J, Berzofsky J A, Mosmann T R, James S L, Morse H C. Immunol Rev. 1992;127:183–204. doi: 10.1111/j.1600-065x.1992.tb01414.x. [DOI] [PubMed] [Google Scholar]
- 18.Paul W E. CIBA Found Symp. 1997;204:208–216. doi: 10.1002/9780470515280.ch14. [DOI] [PubMed] [Google Scholar]
- 19.Witthuhn B A, Silvennoinen O, Miura O, Lai K S, Cwik C, Liu E T, Ihle J N. Nature (London) 1994;370:153–157. doi: 10.1038/370153a0. [DOI] [PubMed] [Google Scholar]
- 20.Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S, Nakanishi K, Yoshida N, Kishimoto T, Akira S. Nature (London) 1996;380:627–630. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- 21.Kaplan M H, Schindler U, Smiley S T, Grusby M J. Immunity. 1996;4:313–319. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 22.Wurster A L, Tanaka T, Grusby M J. Oncogene. 2000;15:2577–2584. doi: 10.1038/sj.onc.1203485. [DOI] [PubMed] [Google Scholar]
- 23.Tarleton R L, Grusby M J, Zhang L. J Immunol. 2000;165:1520–1525. doi: 10.4049/jimmunol.165.3.1520. [DOI] [PubMed] [Google Scholar]
- 24.Stamm L M, Raisanen-Sokolowski A, Okano M, Russell M E, David J R, Satoskar A R. J Immunol. 1998;161:6180–6188. [PubMed] [Google Scholar]
- 25.Wynn T A, Eltoum I, Cheever A W, Lewis F A, Gause W C, Sher A. J Immunol. 1993;151:1430–1440. [PubMed] [Google Scholar]
- 26.Mahalingam S, Farber J M, Karupiah G. J Virol. 1999;73:1479–1491. doi: 10.1128/jvi.73.2.1479-1491.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hogan S P, Foster P S, Tan X, Ramsay A J. Eur J Immunol. 1998;28:413–423. doi: 10.1002/(SICI)1521-4141(199802)28:02<413::AID-IMMU413>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 28.Rockett K A, Kwiatkowski D, Bate C A, Awburn M M, Rockett E J, Clark I A. J Infect. 1996;32:187–196. doi: 10.1016/s0163-4453(96)80018-1. [DOI] [PubMed] [Google Scholar]
- 29.Karupiah G, Coupar B E, Andrew M E, Boyle D B, Phillips S M, Mullbacher A, Blanden R V, Ramshaw I A. J Immunol. 1990;144:290–298. [PubMed] [Google Scholar]
- 30.Karupiah G, Xie Q W, Buller M L, Nathan C, Duarte C, MacMicking J D. Science. 1993;261:1445–1448. doi: 10.1126/science.7690156. [DOI] [PubMed] [Google Scholar]
- 31.Welsh R M. Crit Rev Immunol. 1984;5:55–93. [PubMed] [Google Scholar]
- 32.Kobayashi M, Fitz L, Ryan M, Hewick R M, Clark S C, Chan S, Loudon R, Sherman F, Perussia B, Trinchieri G. J Exp Med. 1989;170:827–845. doi: 10.1084/jem.170.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacoby R O, Bhatt P N, Brownstein D G. Arch Virol. 1989;108:49–58. doi: 10.1007/BF01313742. [DOI] [PubMed] [Google Scholar]
- 34.Seder R A, Gazzinelli R, Sher A, Paul W E. Proc Natl Acad Sci USA. 1993;90:10188–10192. doi: 10.1073/pnas.90.21.10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peleman R, Wu J, Fargeas C, Delespesse G. J Exp Med. 1989;170:1751–1756. doi: 10.1084/jem.170.5.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanaka T, Hu-Li J, Seder R A, de St. Groth B F, Paul W E. Proc Natl Acad Sci USA. 1993;90:5914–5918. doi: 10.1073/pnas.90.13.5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hart P H, Vitti G F, Burgess D R, Whitty G A, Piccoli D S, Hamilton J A. Proc Natl Acad Sci USA. 1989;86:3803–3807. doi: 10.1073/pnas.86.10.3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trinchieri G. Annu Rev Immunol. 1995;13:251–276. doi: 10.1146/annurev.iy.13.040195.001343. [DOI] [PubMed] [Google Scholar]
- 39.Jackson R J, Ramsay A J, Christensen C D, Beaton S, Hall D F, Ramshaw I A. J Virol. 2001;75:1205–1210. doi: 10.1128/JVI.75.3.1205-1210.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruby J, Bluethmann H, Peschon J J. J Exp Med. 1997;186:1591–1596. doi: 10.1084/jem.186.9.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith V P, Alcami A. J Virol. 2000;74:8460–8471. doi: 10.1128/jvi.74.18.8460-8471.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karupiah G, Chen J H, Nathan C F, Mahalingam S, MacMicking J D. J Virol. 1998;72:7703–7706. doi: 10.1128/jvi.72.9.7703-7706.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohmori Y, Hamilton T A. J Biol Chem. 1998;273:29202–29209. doi: 10.1074/jbc.273.44.29202. [DOI] [PubMed] [Google Scholar]
- 44.Biron C A. Curr Opin Immunol. 1997;9:24–34. doi: 10.1016/s0952-7915(97)80155-0. [DOI] [PubMed] [Google Scholar]
- 45.Salcedo T W, Azzoni L, Wolf S F, Perussia B. J Immunol. 1993;151:2511–2520. [PubMed] [Google Scholar]
- 46.Mullbacher A, Ebnet K, Blanden R V, Hla R T, Stehle T, Museteanu C, Simon M M. Proc Natl Acad Sci USA. 1996;93:5783–5787. doi: 10.1073/pnas.93.12.5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mullbacher A, Waring P, Tha Hla R, Tran T, Chin S, Stehle T, Museteanu C, Simon M M. Proc Natl Acad Sci USA. 1999;96:13950–13955. doi: 10.1073/pnas.96.24.13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Magram J, Connaughton S E, Warrier R R, Carvajal D M, Wu C Y, Ferrante J, Stewart C, Sarmiento U, Faherty D A, Gately M K. Immunity. 1996;4:471–481. doi: 10.1016/s1074-7613(00)80413-6. [DOI] [PubMed] [Google Scholar]
- 49.Kaplan M H, Sun Y L, Hoey T, Grusby M J. Nature (London) 1996;382:174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- 50.O'Neill H C, Blanden R V. Infect Immunol. 1983;41:1391–1394. doi: 10.1128/iai.41.3.1391-1394.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wallace G D, Buller R M, Morse H C. J Virol. 1985;55:890–891. doi: 10.1128/jvi.55.3.890-891.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brownstein D G, Gras L. Am J Pathol. 1997;150:1407–1420. [PMC free article] [PubMed] [Google Scholar]