

Abstract
Background
Activation of innate immunity through polyinosinic:polycytidylic acid (poly(I:C)) causes acute salivary gland hypofunction. Since a major consequence of poly(I:C) treatment is type I interferon (IFN) production, this study was undertaken to investigate their role in salivary gland dysfunction.
Methods
Different strains of mice, deficient either in interferon alpha receptor (IFNAR1−/−), or IL-6−/−, or IL-10−/−, or EBI3−/− were treated with poly(I:C). Salivary gland function was determined by measuring pilocarpine induced saliva volume. Gene expression levels were measured by real time PCR. Ca2+ mobilization studies were done using ex-vivo acinar cells.
Results
A single injection of poly(I:C) rapidly induced salivary gland hypofunction in wild type B6 mice (41% drop in saliva volumes compared to PBS treated mice). In contrast, the loss of function in poly(I:C) treated IFNAR−/− mice was only 9.6%. Gene expression analysis showed reduced levels of Il-6, Il-10 and Il-27 in submandibular glands of poly(I:C) treated IFNAR−/− mice. While salivary gland dysfunction in poly(I:C) treated IL-10−/− and EBI3−/− mice was comparable to wild type mice, the IL-6−/− mice were more resistant, with only a 21 % drop in function. Pilocarpine induced Ca2+ flux was significantly suppressed in acinar cells obtained from poly(I:C) treated wild type mice.
Conclusions
Our data demonstrates that a combined action of type I IFNs and IL-6 contributes towards salivary gland hypofunction. This happens through interference with Ca2+ mobilization within acinar cells. Thus, in acute viral infections and diseases like Sjögren’s syndrome, elevated levels of type I IFNs and IL-6 can directly affect glandular function.
Keywords: Xerostomia, Sjogren’s syndrome, Innate Immunity, Interferon
Introduction
Salivary gland hypofunction and xerostomia can be caused by medications, radiation therapy, acute viral infections and autoimmune disorders such as Sjögren’s syndrome (1). Salivary gland destruction by severe lymphocytic infiltration is considered an important mechanism for functional loss in Sjögren’s syndrome. However, other mechanisms also seem to play a role. Evidence for this comes from both human and mouse studies. Several patients show dissociation between the severity of sialoadenitis and unstimulated or stimulated salivary flow rates (2–4). Data from experimental mouse model systems demonstrates that glandular hypofunction can precede the formation of lymphocytic foci within the tissue (5). In addition, salivary gland hypofunction induced by viral infections such as by Hepatitis C virus also suggest that mechanisms not dependent on glandular destruction are important in this process (6).
Recently we demonstrated that activation of innate immunity by toll-like receptor 3 (TLR3) agonist poly(I:C) causes a rapid loss in salivary gland function in mice (7). This acute functional loss was not associated with sialoadenitis or autoantibody production and was reversible; as removal of innate stimuli caused a complete recovery of salivary gland function. Following poly(I:C) injection, there was a considerable spike in the expression levels of type I IFNs, both systemically and locally within the submandibular glands. Collectively these data suggested that exposure of salivary glands to type I IFNs and proinflammatory cytokines, interferes with their ability to make saliva. Clearly, this scenario can be envisioned not only in viral infections but also in Sjögren’s syndrome, wherein type I IFNs are considered an important pathogenic factor (8). This study was undertaken to investigate the role of type I IFNs in salivary gland hypofunction.
Type I IFNs are pleiotropic cytokines affecting multiple cell types (9). A major consequence of viral infections is activation of innate immunity through TLR3, TLR7 and TLR9, and production of large amounts of type I IFNs. In Sjögren’s syndrome, plasmacytoid dendritic cells infiltrating the salivary glands are considered a major source of type I IFNs (10, 11). In addition, salivary gland epithelial cells activated by different TLRs can also contribute towards local production of type I IFNs (12). Currently, in the context of Sjögren’s syndrome, type I IFNs are only considered to amplify an autoimmune response (13). Whether they can directly affect glandular function is not known. The poly(I:C) induced model for acute xerostomia allows us to address this issue. Thus, in the current study we asked the question, in absence of type I IFNs, can poly(I:C) still induce glandular dysfunction? The type I IFN family comprises several Ifn- α genes, Ifn-β, Ifn-ε, Ifn-κ and Ifn-ω genes (9). These IFNs signal through a common receptor and in mice lacking Ifnar1, all signaling through type I IFNs is abrogated (14). We treated the Ifnar1 deficient (IFNAR−/−) mice with poly(I:C) and investigated salivary gland function. Our data shows that mice lacking IFNAR, are significantly more resistant to loss of glandular function compared to wild type mice (IFNAR+/+). An investigation into other cytokines elevated in poly(I:C) treated B6 mice shows that in addition to type I IFNs, IL-6 can also directly affect salivary gland function. Our results have significant implications for understanding the pathogenesis of Sjögren’s syndrome as well as xerostomia induced by viral infections.
Materials and Methods
Mice
All experiments involving mice were approved by the University of Virginia, Animal Care and Use Committee. C57BL6/J (B6 or IFNAR+/+), B6.129S2-Il6tm1Kopf/J (IL-6−/−), B6.129P2-Il10tm1Cgn/J (IL-10−/−) and B6.129X1-Ebi3tm1Rsb/J (Ebi3−/−) mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). The Ebi3−/− mice were used to investigate the effects of IL-27. EBI3 is a common chain for IL-27 and IL-35 and the Ebi3−/− mice lack both IL-27 and IL-35. B6.Ifnar1−/− mice were kindly provided by Dr Ann Marshak-Rothstein, University of Massachusetts Medical School, USA. The IFNAR−/− mice were bred and maintained in specific pathogen free conditions in the University of Virginia, Vivarium. All mice were used at an age of 12–14 weeks, unless noted otherwise. Female mice were injected with 50μg of poly(I:C) (Invivogen, San Diego, CA, USA) by intra peritoneal route. Control mice were injected with PBS. One day after treatment, pilocarpine induced saliva was measured as described before (7).
Real Time PCR
For gene expression analysis, mice were injected either with poly(I:C) or PBS. Three and half hours later mice were euthanized and submandibular glands harvested. Expression of different genes within the submandibular glands was done by employing Taqman Gene Expression assays (Applied Biosystems, Carlsbad, CA, USA) as described before (7).
Ca2+ mobilization studies
Mice were injected 2 times with 50 μg of poly(I:C) by intraperitoneal route on alternate days. Control mice were similarly injected with saline. Submandibular glands were harvested on day 3 and acini were isolated using a method described by Kondo et al (15). Submandibular glands were finely minced in Krebs- Henseleit-Ringer solution supplemented with 1mg/ml BSA and digested with a mixture of Collagenase and Hyaluronidase (Worthington Biochemical Corp., Lakewood, NJ, USA) for 30 min at 37°C. Following two washes, the tissue was suspended in 1 ml of buffer. Larger clusters of cells were allowed to settle and 0.1 ml of cell suspension was used for measurement of intracellular calcium as described previously (16, 17). Briefly, cells from both untreated and poly(I:C)-treated mice were loaded with 1 uM fura-2 AM (Life Technologies, Grand Island, NY, USA) for 30 min in separate wells. During incubation with fura-2 AM, one set of cells was additionally given 0.2 uM Cell Tracker Red CMTPX (Life Technologies, Grand Island, NY, USA), a membrane-penetrating fluorescent probe, in order to distinguish cells from untreated versus treated mice (16). Cell clusters were then transferred to a recording chamber, and calcium ratios were recorded every 5 seconds with a Hamamatsu ORCA-ER camera (Hamamatsu Photonics, Japan) attached to an Olympus BX51WI fluorescence microscope (Olympus, Tokyo, Japan) using 340 and 380 nm excitation light and 510 nm emission as described (17). The ratio of emitted light from 340 nm and 380 nm stimulation was used to calculate the calcium ratio (340/380 nm) for each 5-sec image pair. Basal calcium readings were taken for 2-min, and then cell cluster were exposed to 5 uM pilocarpine for 3-min followed by a 5-min wash in PBS. Data were recorded and analyzed with IP Lab software Version 4.0 (Scanalytics, Rockville, MD, USA)
Statistical Analysis
Unpaired t test was used to determine statistical significance. A p<0.05 at 95% confidence interval was considered significant. For samples not passing normality test, statistical significance was determined using the Mann-Whitney test. All analysis was performed using GraphPad PRISM software (GaphPad Software, La Jolla, CA, USA).
Results
Limited loss of salivary gland function occurs in poly(I:C) treated IFNAR−/− mice
In a previous study we had demonstrated that treatment of NZB/W F1 mice with poly(I:C) caused a rapid but reversible loss of salivary gland function (7). The functional loss in response to poly(I:C) also occurs in B6 mice (Figure 1 right panel). There was a 41% loss in the mean saliva volume in poly(I:C) treated B6 mice, in comparison with PBS treated controls. These mice have a functional IFNAR. Thus, to determine the role of type I IFNs in this model of xerostomia, B6.Ifnar1−/− mice were either injected with poly(I:C) or with PBS. Figure 1 (left panel) shows that the mean saliva volume ± SEM in poly(I:C) treated IFNAR−/− mice (5.884 ± 0.074) was only 9.6% lower than the PBS treated IFNAR−/− mice (6.516 ± 0.126). This drop is significantly less (p<0.0001) than the functional loss seen in IFNAR+/+ mice. The saliva volumes between PBS treated IFNAR+/+ and IFNAR−/− mice were not significantly different, indicating that lack of type I IFN signaling by itself did not impair the salivary gland function. Identical results were obtained in an independent cohort of mice (5 mice per group per treatment). These data show that the functional loss in presence of type I IFN signaling is 4 times greater than that induced in its absence.
Figure 1. Poly(I:C) induced loss of salivary gland function is much lower in the absence of type I IFN signaling.
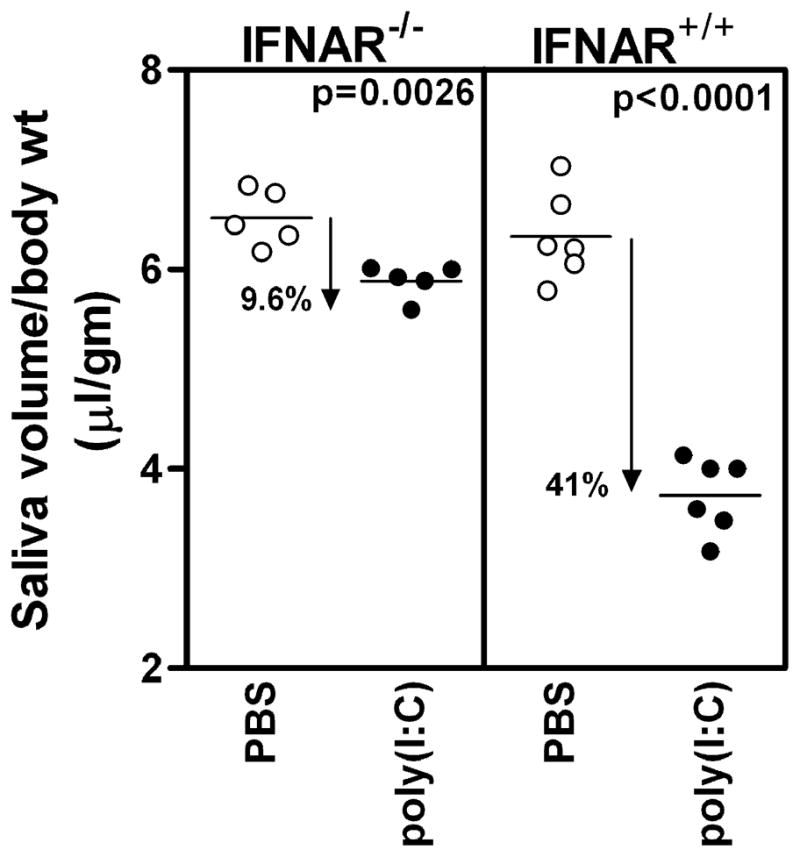
IFNAR−/− and IFNAR+/+ (wild type mice) mice were injected either with PBS or poly(I:C) and pilocarpine induced saliva was measured. The results are expressed as ratio of saliva volume (μl) to gm body weight. In the IFNAR−/−mice, the mean saliva volume ± SEM in poly(I:C) group is 5.88 ± 0.07 and in PBS group it is 6.52 ± 0.13. In wild type mice, the mean saliva volume ± SEM in poly(I:C) group is 3.73 ± 0.15 and in PBS treated group it is 6.33 ± 0.18. Thus, the loss of function in IFNAR−/− mice is 9.6% in comparison to 41% loss in IFNAR+/+ mice. Similar results were obtained in an independent cohort of mice.
Amplified cytokine gene expression in submandibular glands of poly(I:C) treated IFNAR+/+ mice
Although the IFNAR−/− mice were not as impaired in salivary gland function as the wild type mice following poly(I:C) treatment, the functional loss in IFNAR−/− mice was statistically significant. This suggests that factors other than type I IFNs also influence the salivary gland hypofunction. In addition to the production of type I IFNs through the TRIF3 pathway, poly(I:C) also induces the expression of inflammatory cytokines through the NF-kB pathway (18). Moreover it has been shown that type I IFN amplifies the production of inflammatory cytokines IL-6, and IL-27 (19, 20). Thus the loss of function induced in our model can be through the action of type I IFNs or through the action of inflammatory cytokines amplified in presence of type I IFN or both. To determine this, gene expression levels of different cytokines in salivary glands of poly(I:C) treated IFNAR+/+ and IFNAR−/− were compared. Figure 2 shows that in presence of type I IFN signaling, significant upregulation was seen in the expression of Il-6 (34 fold, p=0.0159) and Il-27p28 (11 fold, p=0.0119) in IFNAR+/+ mice compared to IFNAR−/− mice. However, expression of Ebi3 or Il-27b, which is the common chain for IL-27 and IL-35, was not significantly different. A significant upregulation in the expression of non inflammatory or regulatory cytokine, Il-10 (16 fold, p=0.0079) was also seen. This difference in Il-10 expression between the two groups remains significant (3 fold, p=0.0159) even if the IFNAR+/+ mouse with the highest Il-10 expression is excluded from the analysis. The expression of Tnf-α was similar between the 2 groups, and Il-12a gene expression was significantly lower in the IFNAR+/+ mice. These data suggest that following poly(I:C) treatment, expression of Tnf-α and Il-12a is regulated differentially than that for Il-6, Il-27 and Il-10. There were no differences in the expression levels of cholinergic muscarinic receptor 3 (Chrm3). Mx1, an IFNα responsive gene was high only in IFNAR+/+ mice and served as a positive control (21).
Figure 2. In presence of type I IFN signaling, gene expression levels of Il-6, Il-10 and Il-27p28 are amplified within the salivary glands.

IFNAR+/+ and IFNAR−/− mice were injected with poly(I:C) and submandibular glands harvested after 3.5 hr. Expression levels of different genes were determined by real time PCR using Taqman gene expression assays. Gapdh was used as house keeping gene and results are expressed as relative change in gene expression. The ratio of mean gene expression level in IFNAR+/+ mice to that in IFNAR−/− mice is shown as fold (X) increase or decrease. Mx1 is an IFN responsive gene and was used as positive control. The relative gene expression levels of Il-6, Il-10 and Il-27p28 were 34, 16 and 11 folds higher in IFNAR+/+ mice. Similar results were obtained in an additional experiment performed in an independent cohort of mice.
IL-6 contributes towards glandular hypofunction
Since Il-6, Il-10 and Il-27 gene expression was elevated in IFNAR+/+ mice, their contribution to glandular hypofunction was studied in mice deficient in these cytokines. IL-6−/−, IL-10−/−, and IL-27B−/− mice were injected either with poly(I:C) or PBS and salivary gland function determined (figure 3). The loss of function in IL-27−/− (35%, p=0.0002) and IL-10−/− (38%, p<0.0001) mice was similar to that in the wild type mice (41%, p<0.0001). Thus, even in the absence of these cytokines, poly(I:C) had a significant effect on salivary gland function.
Figure 3. IL-6 contributes towards the loss of salivary gland function.

Mice deficient in IL-6, IL-10 and IL27B, were either injected with poly(I:C) or PBS and pilocarpine induced saliva measured. The mean saliva volume in all groups of poly(I:C) treated mice are significantly lower than the mean saliva volumes in respective PBS treated controls. Saliva collections in IL-6−/− mice were done at 2 time points (12wks and 30wks of age) and the loss in function was 26% and 21% respectively. The loss of function in IL-27−/− (35%) IL-10−/− (38%) mice was comparable to that observed in wild type mice (41%). Similar results were obtained in an additional experiment.
In the IL-6−/− mice at 12 weeks of age, the loss of function was around 25% (figure 3, lower panel). However, we noticed that overall saliva production in IL-6−/− mice was significantly lower than the age matched wild type mice. This data suggested that there might be defective or delayed development of salivary glands in absence of IL-6. Thus, the experiment was also conducted in older mice. The overall saliva produced by older IL-6−/− mice was significantly elevated. The reasons for this change are not known, but might involve some unknown compensatory mechanisms. Importantly, the loss of function in poly(I:C) treated group was still around 21%, reproducing results obtained at younger age. Regardless, at both time points despite intact type I IFN signaling, the loss of function in IL-6−/− mice did not reach the levels seen in wild type mice (~41%). Instead it was in between that observed for the wild type and the IFNAR−/− mice. These data suggest that IL-6 also contributes to some extent towards the induction of glandular hypofunction induced by activation of innate immunity.
Poly(I:C) treatment suppresses agonist induced Ca2+ mobilization in acinar cells
In the stimulus-secretion coupling pathway for saliva production, intracellular Ca2+ levels critically regulate fluid transport within the acinar cells (22). Thus Ca2+ mobilization in acinar cells obtained from mice treated with poly(I:C) was investigated. Since, pilocarpine induced saliva was our indicator for salivary gland hypofunction, for in vitro Ca2+ mobilization studies, pilocarpine was used as an agonist. We conducted a total of 5 trials utilizing 8–22 acini per condition for a total of 70 untreated and 64 treated acini. In every trial, responses to pilocarpine stimulation were comparatively smaller in the poly(I:C)-treated group. The cumulative data in figure 4 shows that the pilocarpine-induced increase in intracellular calcium is significantly reduced in acinar cells exposed in vivo to poly(I:C) treatment in comparison with cells obtained from PBS treated mice. Since calcium levels are positively correlated with saliva production, these data demonstrate that poly(I:C) treatment produces measurable hypofunction at the level of acinar tissue.
Figure 4. Ca2+ mobilization is suppressed in acini from poly(I:C) treated mice.

C57BL/6 mice were either treated with poly(I:C) or PBS and submandibular glands were removed for acinar cell preparation. Cells were loaded with fura-2 AM and one set of cells were loaded with Cell Tracker Red. Following 30 min incubation, cells were washed, placed in recording chamber and calcium changes recorded following exposure to 5 μM of pilocarpine-hydrochloride. The data shown is mean ratio (340/380nm) ± SEM from 5 independent experiments with 70 acini from PBS treated mice and 64 acini from poly(I:C) treated mice. The panel on left shows the whole trace. The differences in baseline between poly(I:C) and PBS treated mice were statistically not significant. The right panel shows baseline subtracted mean ratio ± SEM seen at maximum stimulation. The mean Ca2+ 340/380nm ratio in acini from poly(I:C) treated mice is significantly lower (p=0.0003) than that observed in acini from PBS treated mice.
Discussion
In this study we have investigated the role of type I IFNs in poly(I:C) mediated induction of xerostomia. Poly(I:C) activates innate immunity through the engagement of TLR3, retinoic acid-inducible gene I (RIGI) and melanoma differentiation-associated antigen 5 (MDA5), and causes the production of type I IFN and inflammatory cytokines. Moreover, type I IFN further amplifies the production of other inflammatory cytokines such as IL-6 and IL-27 (19, 20). Thus, the loss of function induced by poly(I:C) can be a consequence of type I IFN action, or inflammatory cytokine action or both. We addressed this question by using mice deficient in type I IFN signaling and mice deficient in different inflammatory cytokines. Our data shows that the loss of function induced in IFNAR−/− mice was one fourth of that induced in the wild type mice, whereas in the IL-6−/−mice it was one half. These data indicate that both type I IFNs and IL-6 play a crucial role in interfering with salivary gland function.
Our findings provide some insights into possible mechanisms of salivary gland hypofunction induced by activation of innate immunity in both viral infections and autoimmune disorders like Sjögren’s syndrome. Recently, Kasman et al used murine cytomegalovirus (mCMV) infection model to mimic the effects of viral hepatitis in inducing salivary gland dysfunction (23). Following mCMV infection, salivary gland hypofunction was induced in BALB/c mice within 2 days. In this model, the glandular hypofunction only correlated with elevated liver enzymes. Since functional loss also occurred in mCMV infected BALB/c-c-Rel knockout mice, which are defective in proinflammatory Th1 cytokine production, it was suggested that disturbances in liver and unknown serum factors released by liver are important in mCMV induced salivary gland dysfunction. The role of type I IFNs was not investigated in this model and might also be critical for two reasons. First, mCMV infection rapidly induces type I IFN production (24, 25) and second, in c-Rel knockout mice IFNβ production is not affected following activation of innate immunity by different TLR ligands (26). Thus, it is possible that mCMV infection causes type I IFN production, which contributes towards hepatic injury and release of serum factors. In our model system poly(I:C) causes inflammatory cytokine responses both systemically as well as locally within the salivary glands.
Salivary gland hypofunction is a major characteristic of Sjögren’s syndrome. Although our acute model of salivary gland hypofunction does not represent the classic autoimmune phenotype of Sjögren’s syndrome, the finding that type I IFN can mediate salivary gland hypofunction is highly significant for understanding the pathogenesis of Sjögren’s syndrome. Analysis of salivary glands from Sjögren’s syndrome patients have shown increased expression levels of type I IFN responsive genes; increased accumulation of plasmacytoid dendritic cells; and a genetic predisposition to higher type I IFN responsiveness (8). Additional source of type I IFN can be through the activation of TLRs with immune-complexes containing ribonucleoproteins and chromatin. The major function ascribed to type I IFNs in Sjögren’s syndrome is in the initiation and amplification of the adaptive autoimmune responses. Our data shows for the first time that type I IFNs can directly influence salivary gland function. Thus, it will be important to scrutinize whether in Sjögren’s syndrome patients, elevated type I IFN and IL-6 levels in salivary glands correlate with functional loss.
To investigate mechanisms involved in type I IFN and IL-6 mediated salivary gland hypofunction several experiments were conducted. Our previous studies have demonstrated that submandibular glands from poly(I:C) treated mice do not show any gross histological abnormalities or aquaporin 5 redistribution (7). However, since type I IFNs can induce apoptosis, sections of submandibular glands were stained for apoptotic cells by Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) method. We did not observe any increase in apoptotic cell numbers in the submandibular glands of mice injected with poly(I:C) (data not shown). Exposure of salivary gland acinar cells to proinflammatory cytokines such as TNF-α and IFN-γ has been shown to affect integrity of tight junctions and thereby implied in disturbances in water transport (27, 28). We thus investigated tight junction integrity in submandibular gland acini from poly(I:C) and PBS treated mice by immunohistochemistry by staining for tight junction protein ZO-1. No disruption in tight junction integrity was observed in submandibular glands from poly(I:C) treated mice. Collectively these findings suggest that, type I IFN mediated salivary gland hypofunction must involve the non-apoptotic mechanisms (29).
Saliva production is regulated by parasympathetic and sympathetic stimulation (22). Following engagement of cholinergic muscarinic receptor 3 by acetylcholine, a sustained increase in intracellular Ca2+ dictates several downstream events that involve different ion channel proteins, which lead to increased fluid transport in acini and saliva production. We thus determined whether poly(I:C) treatment disturbs Ca2+ mobilization in acini. Our data shows that Ca2+ mobilization is significantly reduced in acini obtained from poly(I:C) treated mice. This can explain the decrease in pilocarpine mediated saliva production in poly(I:C) treated mice. The reasons for reduced Ca2+ mobilization in our model system are currently not known and might involve but are not limited to inositol 1, 4,5 triphosphate (IP3) levels, effects on IP3 receptors and ryanodine receptors (29, 30), and will be investigated in future studies. Recently it has been reported that poly(I:C) decreases histamine mediated Ca2+ mobilization in human gingival fibroblasts through the activation of protein kinase C and p38 (31). Considering phosphorylation of PKC is involved in agonist induced salivation (32), it is possible that poly(I:C) effects on this pathway are also involved in salivary gland hypofunction.
In summary our data suggests that systemic as well as local increase of type I IFNs and IL-6 within salivary glands in different chronic inflammatory conditions such as viral infections and autoimmune disorders like Sjögren’s syndrome have the potential to impact salivary gland function. This can happen without the involvement of tissue destruction. Recently, it was demonstrated that salivary gland hypofunction occurs in rats with experimental periodontitis, without any changes in acinar structure (33). Thus, elevated cytokine levels might be an important biomarker for investigating the causes of xerostomia as well as a therapeutic target.
Acknowledgments
This study was supported by grants from the National Institutes of Health, USA, R21DE019883 (USD), R01AI079621 (USD), R01DK069769 (HB) and an Innovative Research Grant from the Sjögren’s Syndrome Foundation USA (USD).
Footnotes
Conflict of Interest Statement
None of the authors have any conflict of interest.
References
- 1.HOPCRAFT MS, TAN C. Xerostomia: an update for clinicians. Aust Dent J. 2010;55:238–44. doi: 10.1111/j.1834-7819.2010.01229.x. [DOI] [PubMed] [Google Scholar]
- 2.PEDERSEN AM, REIBEL J, NAUNTOFTE B. Primary Sjogren's syndrome (pSS): subjective symptoms and salivary findings. J Oral Pathol Med. 1999;28:303–11. doi: 10.1111/j.1600-0714.1999.tb02045.x. [DOI] [PubMed] [Google Scholar]
- 3.DANIELS TE, COX D, SHIBOSKI CH, et al. Associations between salivary gland histopathologic diagnoses and phenotypic features of Sjogren's syndrome among 1,726 registry participants. Arthritis Rheum. 2011;63:2021–30. doi: 10.1002/art.30381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.BOOKMAN AA, SHEN H, COOK RJ, et al. Whole stimulated salivary flow: correlation with the pathology of inflammation and damage in minor salivary gland biopsy specimens from patients with primary Sjogren's syndrome but not patients with sicca. Arthritis Rheum. 2011;63:2014–20. doi: 10.1002/art.30295. [DOI] [PubMed] [Google Scholar]
- 5.HAYASHI T. Dysfunction of lacrimal and salivary glands in Sjogren's syndrome: nonimmunologic injury in preinflammatory phase and mouse model. J Biomed Biotechnol. 2011;2011:407031. doi: 10.1155/2011/407031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.GROSSMANN SM, TEIXEIRA R, OLIVEIRA GC, et al. Xerostomia, hyposalivation and sialadenitis in patients with chronic hepatitis C are not associated with the detection of HCV RNA in saliva or salivary glands. J Clin Pathol. 2010;63:1002–7. doi: 10.1136/jcp.2010.080036. [DOI] [PubMed] [Google Scholar]
- 7.DESHMUKH US, NANDULA SR, THIMMALAPURA PR, SCINDIA YM, BAGAVANT H. Activation of innate immune responses through Toll-like receptor 3 causes a rapid loss of salivary gland function. J Oral Pathol Med. 2009;38:42–7. doi: 10.1111/j.1600-0714.2008.00700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.MAVRAGANI CP, CROW MK. Activation of the type I interferon pathway in primary Sjogren's syndrome. J Autoimmun. 2010;35:225–31. doi: 10.1016/j.jaut.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 9.SOZZANI S, BOSISIO D, SCARSI M, TINCANI A. Type I interferons in systemic autoimmunity. Autoimmunity. 2010;43:196–203. doi: 10.3109/08916930903510872. [DOI] [PubMed] [Google Scholar]
- 10.GOTTENBERG JE, CAGNARD N, LUCCHESI C, et al. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjogren's syndrome. Proc Natl Acad Sci US A. 2006;103:2770–5. doi: 10.1073/pnas.0510837103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.NORDMARK G, ALM GV, RONNBLOM L. Mechanisms of disease: Primary Sjogren's syndrome and the type I interferon system. Nat Clin Pract Rheumatol. 2006;2:262–9. doi: 10.1038/ncprheum0173. [DOI] [PubMed] [Google Scholar]
- 12.ITTAH M, MICELI-RICHARD C, GOTTENBERG JE, et al. Viruses induce high expression of BAFF by salivary gland epithelial cells through TLR- and type-I IFN-dependent and -independent pathways. Eur J Immunol. 2008;38:1058–64. doi: 10.1002/eji.200738013. [DOI] [PubMed] [Google Scholar]
- 13.VOULGARELIS M, TZIOUFAS AG. Pathogenetic mechanisms in the initiation and perpetuation of Sjogren's syndrome. Nat Rev Rheumatol. 2010;6:529–37. doi: 10.1038/nrrheum.2010.118. [DOI] [PubMed] [Google Scholar]
- 14.MULLER U, STEINHOFF U, REIS LF, et al. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–21. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 15.KONDO Y, NAKAMOTO T, MUKAIBO T, KIDOKORO M, MASAKI C, HOSOKAWA R. Cevimeline-induced monophasic salivation from the mouse submandibular gland: decreased Na+ content in saliva results from specific and early activation of Na+/H+ exchange. J Pharmacol Exp Ther. 2011;337:267–74. doi: 10.1124/jpet.110.174946. [DOI] [PubMed] [Google Scholar]
- 16.CORBIN KL, HALL TE, HAILE R, NUNEMAKER CS. A novel fluorescence imaging approach for comparative measurements of pancreatic islet function in vitro. Islets. 2011;3:14–20. doi: 10.4161/isl.3.1.14133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.JAHANSHAHI P, WU R, CARTER JD, NUNEMAKER CS. Evidence of diminished glucose stimulation and endoplasmic reticulum function in nonoscillatory pancreatic islets. Endocrinology. 2009;150:607–15. doi: 10.1210/en.2008-0773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.VERCAMMEN E, STAAL J, BEYAERT R. Sensing of viral infection and activation of innate immunity by toll-like receptor 3. Clin Microbiol Rev. 2008;21:13–25. doi: 10.1128/CMR.00022-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.YASUDA K, RICHEZ C, MACIASZEK JW, et al. Murine dendritic cell type I IFN production induced by human IgG-RNA immune complexes is IFN regulatory factor (IRF)5 and IRF7 dependent and is required for IL-6 production. J Immunol. 2007;178:6876–85. doi: 10.4049/jimmunol.178.11.6876. [DOI] [PubMed] [Google Scholar]
- 20.MOLLE C, GOLDMAN M, GORIELY S. Critical role of the IFN-stimulated gene factor 3 complex in TLR-mediated IL-27p28 gene expression revealing a two-step activation process. J Immunol. 2010;184:1784–92. doi: 10.4049/jimmunol.0902005. [DOI] [PubMed] [Google Scholar]
- 21.STAEHELI P, DANIELSON P, HALLER O, SUTCLIFFE JG. Transcriptional activation of the mouse Mx gene by type I interferon. Mol Cell Biol. 1986;6:4770–74. doi: 10.1128/mcb.6.12.4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MELVIN JE, YULE D, SHUTTLEWORTH T, BEGENISICH T. Regulation of fluid and electrolyte secretion in salivary gland acinar cells. Annu Rev Physiol. 2005;67:445–69. doi: 10.1146/annurev.physiol.67.041703.084745. [DOI] [PubMed] [Google Scholar]
- 23.KASMAN LM, LONDON LL, LONDON SD, PILGRIM MJ. A mouse model linking viral hepatitis and salivary gland dysfunction. Oral Dis. 2009;15:587–95. doi: 10.1111/j.1601-0825.2009.01600.x. [DOI] [PubMed] [Google Scholar]
- 24.DELALE T, PAQUIN A, ASSELIN-PATUREL C, et al. MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-alpha release and initiation of immune responses in vivo. J Immunol. 2005;175:6723–32. doi: 10.4049/jimmunol.175.10.6723. [DOI] [PubMed] [Google Scholar]
- 25.HOKENESS-ANTONELLI KL, CRANE MJ, DRAGOI AM, CHU WM, SALAZAR-MATHER TP. IFN-alphabeta-mediated inflammatory responses and antiviral defense in liver is TLR9-independent but MyD88-dependent during murine cytomegalovirus infection. J Immunol. 2007;179:6176–83. doi: 10.4049/jimmunol.179.9.6176. [DOI] [PubMed] [Google Scholar]
- 26.WANG X, HUSSAIN S, WANG EJ, WANG X, LI MO, GARCIA-SASTRE A, BEG AA. Lack of essential role of NF-kappa B p50, RelA, and cRel subunits in virus-induced type 1 IFN expression. J Immunol. 2007;178:6770–6. doi: 10.4049/jimmunol.178.11.6770. [DOI] [PubMed] [Google Scholar]
- 27.BAKER OJ, CAMDEN JM, REDMAN RS, et al. Proinflammatory cytokines tumor necrosis factor-alpha and interferon-gamma alter tight junction structure and function in the rat parotid gland Par-C10 cell line. Am J Physiol Cell Physiol. 2008;295:C1191–201. doi: 10.1152/ajpcell.00144.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.EWERT P, AGUILERA S, ALLIENDE C, et al. Disruption of tight junction structure in salivary glands from Sjogren's syndrome patients is linked to proinflammatory cytokine exposure. Arthritis Rheum. 2010;62:1280–9. doi: 10.1002/art.27362. [DOI] [PubMed] [Google Scholar]
- 29.DAWSON LJ, FOX PC, SMITH PM. Sjogrens syndrome--the non-apoptotic model of glandular hypofunction. Rheumatology. 2006;45:792–8. doi: 10.1093/rheumatology/kel067. [DOI] [PubMed] [Google Scholar]
- 30.CAULFIELD VL, BALMER C, DAWSON LJ, SMITH PM. A role for nitric oxide-mediated glandular hypofunction in a non-apoptotic model for Sjogren's syndrome. Rheumatology. 2009;48:727–33. doi: 10.1093/rheumatology/kep100. [DOI] [PubMed] [Google Scholar]
- 31.GUTIERREZ-VENEGAS G, RODRIGUEZ-PEREZ CE. Toll-like receptor 3 activation promotes desensitization of histamine response in human gingival fibroblasts: Poly (I:C) induces histamine receptor desensitization in human gingival fibroblasts. Cell Immunol. 2012;273:150–7. doi: 10.1016/j.cellimm.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 32.SOLTOFF SP, TOKER A. Carbachol, substance P, and phorbol ester promote the tyrosine phosphorylation of protein kinase C delta in salivary gland epithelial cells. J Biol Chem. 1995;270:13490–5. doi: 10.1074/jbc.270.22.13490. [DOI] [PubMed] [Google Scholar]
- 33.AMER M, ELVERDIN JC, FERNANDEZ-SOLARI J, MEDINA VA, CHIARENZA AP, VACAS MI. Reduced methacholine-induced submandibular salivary secretion in rats with experimental periodontitis. Arch Oral Biol. 2011;56:421–27. doi: 10.1016/j.archoralbio.2010.11.004. [DOI] [PubMed] [Google Scholar]