

Abstract
Sphingosine 1-phosphate (S1P) is a pleiotropic lipid mediator involved in numerous cellular and physiological functions. Notable among these are cell survival and migration as well as lymphocyte trafficking. S1P, which exerts its effects via five G protein coupled receptors (S1P1-5), is formed by the action of two sphingosine kinases (SphKs). While SphK1 is the more intensively studied isotype, SphK2 is unique in it nuclear localization and has been reported to oppose some of the actions ascribed to SphK1. While several scaffolds of SphK1 inhibitors have been described, there is a scarcity of selective SphK2 inhibitors that are necessary to evaluate the downstream effects of inhibition of this isotype. Herein we report a cationic amphiphilic small molecule that is a selective SphK2 inhibitor. In the course of characterizing this compound in wild type and SphK null mice we discovered that administration of the inhibitor to wild type mice resulted in a rapid increase in blood S1P, which is in contrast to our SphK1 inhibitor that drives circulating S1P levels down. Using a cohort of F2 hybrid mice, we confirmed, compared to wild type mice, that circulating S1P levels were higher in SphK2 null mice and lower in SphK1 null mice. Thus both SphK1 and SphK2 inhibitors recapitulate the blood S1P levels observed in the corresponding null mice. Moreover, circulating S1P levels mirror SphK2 inhibitor levels providing a convenient biomarker of target engagement.
Keywords: Sphingosine Kinase (SphK), Sphingosine 1-phosphate (S1P), SphK inhibitor
Sphingosine 1-phosphate (S1P) is a bioactive lipid involved in a host of cellular functions, including migration, differentiation, survival, angiogenesis and immune cell modulation. Extracellularly, S1P exerts its effects via five G-protein coupled receptors – S1P1-5 and intracellularly, acts to modulate transcription complexes [1]. Due in part to the remarkable clinical success of the S1P receptor agonist and immunomodulatory prodrug, fingolimod (FTY720), S1P signaling is currently the subject of many investigations.
Two sphingosine kinase isotypes (SphK1 [2] & SphK2 [3]) catalyze the phosphorylation of sphingosine and these enzymes are solely responsible for S1P synthesis [4]. Studies with Sphk1 null mice reveal that SphK1 is responsible for a substantial fraction of S1P production [5]. Not surprisingly, these kinases have come under increasing scrutiny as drug targets due to their role in the production of S1P, which is implicated in a variety of pathological conditions such as cancers, fibrosis, etc.
The relative importance of SphK1 versus SphK2 as potential drug targets remains a topic of debate. While SphK1 is reported to promote growth and survival [6–8], cell-based studies of SphK2 suggest that this enzyme is not protective, rather it opposes proliferation while enhancing apoptosis [9–11]. While several scaffolds of SphK1 inhibitors have been described [12–15], SphK2 inhibitors are less common. One compound, ABC294640, has a KI of 8 μM at SphK2 [16]. This adamantyl compound has been reported to be efficacious in several disease models including hepatic ischemia-reperfusion injury [17], osteoarthritis [18], Crohn’s disease [19], ulcerative colitis [20] and colon cancer [21] among others. However, ABC294640 was recently documented to bind to the estrogen receptor where it has tamoxifen-like properties [22].
In this study, we report the results of our characterization of another SphK2 inhibitor, SLR080811. SLR080811 is a guanidine-based SphK2 inhibitor with a T1/2 of 4–5 hours in mice. While this molecule lowers S1P levels in cultured cells, it drives a SphK1-dependent increase in S1P in mice and thus mimics the elevated S1P levels observed in SphK2 null mice.
MATERIALS AND METHODS
Materials
Sphk1−/− [5] and Sphk2−/− [4] mice were gifts from Dr. Richard Proia (NIH/NIDDK). C57BL/6j mice were from Jackson Laboratories (Bar Harbor, ME). The plasmid encoding diacylglycerol kinase alpha and was a gift from Dr. Kaoru Goto (Yamagata University School of Medicine, Yamagata, Japan). Adult mouse kidney fibroblast cultures were a gift from Dr. Amandeep Bajwa (University of Virginia). Deuterated (D7) S1P, S1P, sphingosine, C17 S1P and C17 sphingosine were purchased from Avanti Polar Lipids (Alabaster, AL, USA).
Synthesis of SLR080811
The details of the synthesis of SLR080811 ((S)-2-(3-(4-octylphenyl)-1,2,4-oxadiazol-5-yl)pyrrolidine-1-carboximidamide)) and related compounds will be reported elsewhere (Raje, Gao and Santos, in preparation).
Kinase assays
SphK activity was measured by a scintillation proximity assay as previously described [23]. Briefly, recombinant SphK1 or SphK2 were expressed in Sf9 insect cells, crude homogenates were prepared and incubated in 96 well FlashPlates (Perkin-Elmer) in a buffer containing D-erythro-sphingosine and γ-[33P]ATP. The [33P]S1P product, which adheres to the plate wall, was quantified by scintillation counting. To assay ceramide kinase or diacylglycerol kinases, the recombinant proteins were incubated with γ-[32P]ATP and substrate (C6 ceramide or 1-O-hexadecyl-2-acetyl-sn-glycerol, respectively) and the lipid product, after recovery by organic extraction, was resolved by thin layer chromatography, detected by autoradiography and quantified by liquid scintillation counting. These assays were performed with and without a fixed concentration of inhibitor and its effect on KM and Vmax determined.
Sample preparation
Sample preparation protocols were from Shaner et al. [24] with minor modifications. Cell pellets (2–4 × 106 cells), whole blood (20 μl) or plasma (50 μl) was mixed with 2 ml of a methanol : chloroform solution (3:1) and transferred to a capped glass vial. Suspensions were supplemented with 10 μl of internal standard solution containing 10 pmoles each of C17 S1P or deuterated (D7) S1P, C17 sphingosine or deuterated (D7) sphingosine and an undecyl analogue of compound 1a [25,26]. The mixture was placed in a bath sonicator for 10 minutes and incubated at 48°C for 16 hours. The mixture was then cooled to ambient temperature and mixed with 200 μl of 1M KOH in methanol. The samples were again sonicated and incubated a further 2 hours at 37°C. Samples were then neutralized by the addition of 20 μl of glacial acetic acid and transferred to 2 ml microcentrifuge tubes. Samples were then centrifuged at 12,000 x g for 12 minutes at 4°C. The supernatant fluid was collected in a separate glass vial and evaporated under a stream of nitrogen gas. Immediately prior to LC-MS analysis, the dried material was dissolved in 0.3 ml of methanol and centrifuged at 12,000 x g for 12 minutes at 4°C. Fifty μl of the resulting supernatant fluid was analyzed.
LC/MS protocol
Analyses were performed by Liquid Chromatography-ESI Mass Spectrometry (LC-MS) using a triple quadrupole mass spectrometer (AB-Sciex 4000 Q-Trap) coupled to a Shimadzu LC-20AD LC system. A binary solvent gradient with a flow rate of 1 ml/min was used to separate sphingolipids and drugs by reverse phase chromatography using a Supelco Discovery C18 column (50 mm × 2.1 mm, 5 μm bead size). Mobile phase A consisted of water : methanol : formic acid (79:20:1) while mobile phase B was methanol : formic acid (99:1). The run started with 100% A for 0.5 minutes. Solvent B was then increased linearly to 100% B in 5.1 minutes and held at 100% for 4.3 minutes. The column was finally re-equilibrated to 100% A for 1 min. Natural sphingolipids were detected using multiple reaction monitoring (MRM) protocols previously described [24] as follows: C17S1P (366.4 → 250.4); S1P (380.4 → 264.4); dihydroS1P (382.4 → 266.4); deuterated (D7)C18S1P (387.4 → 271.3);, C17sphingosine (286.4 → 250.3); sphingosine (300.5 → 264.4); sphinganine (302.5 → 260.0), deuterated (D7) sphingosine (307.5 → 271.3). Fragmentation of compound SLR080811 was analyzed by direct infusion of a 1 μM solution in methanol:formic acid (99:1) and it was found that the transition (371.1 → 140.1) in positive mode provided the most intense signal at the following voltages, DP: 76; EP: 10; CE: 29; CXP: 10. All analytes were analyzed simultaneously using the aforementioned MRMs. Retention times for all analytes under our experimental conditions were between 5.1 and 5.6 min. Ceramide (16:0) was measured in positive mode by monitoring the m/z 264.4 product ion and using a Supelco Supelcosil LC-NH2 column (50mm × 2.1 mm, 3 μm bead size) as previously described [24]. ]. Quantification was carried out by measuring peak areas using commercially available software (Analyst 1.5.1).
Cell culture
U937 cells were grown in RPMI 1640 media supplemented with L-glutamate, 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37°C in an atmosphere containing 5% CO2 [12]. SKOV3 cells were grown in McCoy’s 5a media supplemented with 10% FBS and penicillin/streptomycin at 37°C in an atmosphere containing 5% CO2. Mouse kidney fibroblast were grown in DMEM low glucose media with 10% FBS. Twenty-four hours before adding inhibitors, the growth media was replaced with media containing 0.5% FBS.
Cell viability assay
U937 cells were plated in 96 well plates at a density of 60–80,000 cells per well. Cells were treated with the indicated concentration of compounds for 24 hours. Cell viability was assessed using TACS™ MTT assay according to the manufacturer’s protocol (R&D System, Minneapolis, MN). Briefly, 10 μl of MTT reagent was added to 100 μl of cell culture medium and the plate was incubated at 37°C for 4 hours, followed by incubation with 100 μl of Detergent Reagent at room temperature for 2 hours. Absorbance was measured at a wavelength of 570 nm.
Generation of F2 hybrid mice
Sphk1−/− and Sphk2−/− mice were mated and 15 (5 male, 10 female) of the resulting F1 heterozygotes were intercrossed to yield a cohort of F2 hybrid mice. Mice were genotyped at weaning (P21) and the various genotypes were found at the expected Mendelian frequencies except for the embryonic lethal double null genotype [4]. After 12–14 hours of fasting, blood was obtained from F2 mice from retro-orbital sinuses under light isofluorane anesthesia. Aliquots of whole blood and plasma were processed for LC-MS analysis of S1P levels as described above.
Pharmacokinetic analysis
Groups of 8–12 week old mice (strain: C57BL/6j) were injected (intraperitoneal route) with either SLR080811 (dose: 10 mg/kg) or an equal volume of vehicle (2% solution of hydroxypropyl-β-cyclodextrin (Cargill Cavitron 82004)). After injection, animals were bled at the specified time points (ASAP time points were 1–2 minutes after dosing). Whole blood was processed immediately for LC-MS analysis as described above. Animal protocols were approved prior to experimentation by the University of Virginia’s School of Medicine Animal Care and Use Committee.
RESULTS
Inhibitor design strategy
In the course of our previous studies of amidine-based SphK1 inhibitors [14,25] to identify compounds with longer half-lives, we reasoned that compounds containing an amidine or an amide might be rapidly hydrolyzed in vivo. Therefore, we synthesized compound SLR080811 (Table 1), which retained the pyrrolidine ring of our SphK1 inhibitor compound 1a [26] while replacing the amidine with the more stable guanidine isostere. Both compounds feature the carboximidamide ‘warhead’ [25]. Further, the amide in compound 1a was replaced by an oxadiazole group. SLR080811 was evaluated at recombinant SphKs, other lipid kinases, in whole cells and finally, in wild type and SphK null mice.
Table 1.
Chemical structure and inhibitory constants of compounds SLR080811 and 1a [26]. The chemical structure of compounds SLR080811 and 1a along with their inhibitory constants (KI) for recombinant SphK1 and SphK2 are shown. Inhibitory constants were obtained by kinetic analysis of S1P production using variable concentrations of sphingosine and a fixed concentration of ATP in presence and absence of compounds. These compounds exhibit a pattern of competitive inhibition therefore KI’s were calculated as KI = [I]/(KM′/KM− 1), where [I] is the concentration of inhibitor, and Km’ and Km are the Michaelis constants obtained in presence and absence of inhibitor. Measurements were carried out using [33P]-ATP as a tracer and a microplate-based scintillation proximity assay for the detection of [33P]-S1P as previously described [23].
| Compound | SphK1 KI (uM) | SphK2 KI (uM) |
|---|---|---|
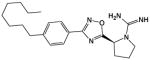 SLR080811 |
12 | 1.3 |
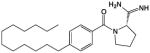 1a |
0.1 | 1.5 |
Evaluation of SLR080811 in vitro
SLR080811, prepared as the HCl salt, was tested first at recombinant SphK1 and SphK2 using a broken cell assay (see Methods). In these assays, SLR080811 was found to have inhibitory constants (KI) of 1.3 and 12 μM for SphK2 and SphK1, respectively (Table 1). Further, SLR080811 was found to be competitive with sphingosine, but not with ATP. Because SLR080811 is a sphingosine analog, we tested SLR080811 as an inhibitor of related lipid kinases including ceramide kinase and DGKα. At a concentration of 3 μM, no inhibition of either enzyme was observed (data not shown).
We characterized SLR080811 in detail because of its SphK2-selectivity that, although modest (10-fold), is unusual in our carboximidamide series. We chose human leukemia U937 cells for the evaluation of SphK2 inhibitors because they exhibit high SphK1 and SphK2 activities, can be cultured with ease, and more importantly, these cells have been used in the past by us and other authors [12] to test SphK1 inhibitors enabling comparisons of the effects of inhibitors.
We first treated U937 cultures with either vehicle or SLR080811 and measured the intracellular levels of S1P, sphinganine 1-phosphate (dhS1P), sphingosine (Sph), sphinganine (dhSph) and SLR080811. We observed that treatment of U937 cells with SLR080811, but not vehicle, resulted in decreased amounts of phosphorylated sphingolipids S1P and dhS1P (Figures 1a and 1c) and the concomitant increase of the corresponding non-phosphorylated precursors sphingosine and sphinganine (Figures 1b and 1d). The data in Figures 1a and 1c indicate that the IC50 values of SLR080811 are less than the its KI (1.3 μM) determined at recombinant SphK2. In addition to sphingolipids, we also measured the intracellular concentration of SLR080811. As shown in Figure 1e, SLR080811 accumulates inside U937 cells in a concentration dependent manner, which might explain its potent effect on the levels of intracellular sphingolipids shown in Figure 1a–1d.
Figure 1.

Levels of sphingolipids and compound SLR080811 in U937 treated with various concentrations of compound SLR080811 as indicated. After a 2 hour period of exposure, cells were harvested by centrifugation, lysed and the amounts of sphingolipids and SLR080811 in the lysates were measured by LC-MS as described in the Methods section. Amounts associated with cells are expressed as the number of pmoles per million cells. a: S1P; b: sphingosine; c: dihydroS1P; d: sphinganine: e: SLR080811. Data are presented as means ±SD of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 (one way ANOVA, and Bonferroni’s Multiple Comparison Test, compared to vehicle alone).
Treatment of human Jurkat T leukemia cells or human SKOV3 ovarian cancer cells with SLR080811 for 2 hours also resulted in decreased S1P (data not shown). In U937 cells, the effect of SLR080811 on intracellular sphingolipids was observed as early as 20 minutes after SLR080811 exposure (data not shown) and persisted for at least 72 hours as documented in Figures 2a and 2b. We also quantified one of the prominent ceramide species (C16:0) in these cells and found that this ceramide was significantly elevated, but only at the 48 and 72 hour time points (Figure 2c).
Figure 2.

Cultured U937 cells were treated with vehicle or SLR080811 (1 μM) for different times as indicated. Cells were harvested by centrifugation, lysed and the amounts of sphingolipids and SLR080811 in the lysates were measured by LC-MS as described in the Methods section. Amounts are expressed as the number of pmoles per million cells. a: S1P b: sphingosine; c: C16:0-ceramide. Data are presented as means ±SD of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 (Student t-test, compared with the control (no compound treatment)).
The most obvious explanation for the decline in U937 cell-associated S1P and dhS1P in response to SLR080811 is decreased synthesis, but it is conceivable that the decline was somehow the result of increased metabolism via, for example, S1P phosphatase or S1P lyase, or increased S1P export. To discriminate between these possibilities, we used FTY720, an SphK2-selective substrate [27,28]. We observed that treatment of U937 cells with SLR080811 impaired their ability to convert FTY720 into FTY720-phosphate. As shown in Figure 3, we observed much lower levels of intracellular FTY720-phosphate in SLR080811-treated cells that, as expected, correlated with correspondingly higher levels of FTY720. This suggests that the reduction of intracellular S1P levels in U937 cells produced by SLR080811 is due to SphK2 inhibition rather than the alternative mechanisms mentioned above.
Figure 3.

Levels of FTY720-P and FTY720 in U397 cells treated with FTY720 and SLR080811. Cultured U937 cells were exposed to 1 μM of FTY720 and two concentrations of SLR080811 as indicated in the figure. After 2 hours of exposure, cells were harvested by centrifugation, lysed and the amounts of FTY720 and phospho-FTY720 were measured by LC-MS as described in the Methods section. a: accumulation of FTY720-P; b: accumulation of FTY720. Amounts are expressed as the number of pmoles per million cells. Drug and FTY720 concentrations on the x axis refer to the concentration of these molecules in the culture medium. Data are presente as means ±SD of three independent experiments. *p < 0.05, **p <0.01, ***p< 0.001 (one-way ANOVA, and Bonferroni’s multiple comparison test, compared with the control (no compound treatment)).
To evaluate further the selectivity of SLR080811 in vitro, we used SphK1 null and SphK2 null mouse kidney fibroblasts. Because the wild type fibroblasts derived from adult mouse kidney have both SphK1 and SphK2 activity (not shown), null cells are a useful model for testing compound selectivity. We found that SLR080811 reduces the levels of intracellular S1P in both wild type and SphK1 null cells but not in SphK2 null cells (Figure 4a). This suggests SphK2, but not SphK1, is a target for SLR080811. Contrary to our expectations however, the effect of SLR080811 on sphingosine levels was not selective for SphK2. As shown in Figure 4b, both SphK1 null and SphK2 null fibroblasts exhibited increased concentrations of sphingosine on treatment with SLR080811 suggesting that other mechanisms may be at play in the regulation of these sphingolipids.
Figure 4.

Selectivity of SLR080811 inhibition: WT, Sphk1−/− and Sphk2−/− adult mouse kidney fibroblast were exposed to 1 μM SLR080811 for 2 hours. Cells were harvested by centrifugation, lysed and sphingolipids were measured by LC-MS as described in the Methods section. a: S1P; b: sphingosine. Data are presented as means ±SD of three independent experiments. *p < 0.05, ***p < 0.001 (Student t-test, compared with the control (no inhibitor)).
Finally, we tested whether the effects of SLR080811 on U937 cells included cell toxicity. In general, we found that SLR080811 has no obvious cytotoxic effects on U397 cells. For example, cultures grew normally in medium containing up to 3 μM SLR080811 and there were no signs of cell growth inhibition (data not shown). Further, we investigated the effect of SLR080811 on U397 cells using a standard assay that correlates cell viability with their redox potential (MTT assay, see Methods section). We found that SLR080811 had a slight cytotoxic effect that is apparent even at the lowest concentration tested but was not concentration dependent (Figure 5).
Figure 5.

Viability of U937 cells treated with SLR080811 (a) or both SLR080811 and compound 1a (b). U937 cells were exposed to various concentrations of compounds for 24 hours as indicated. The viability of the cells was measured by MTT assay as described in the Methods section. Viability is directly proportional to the amount of formazan dye produced by live cells as measured by absorbance at 570 nm. Data are presented as means ±SD of three independent experiments. *p < 0.05, **p < 0.01 (one-way ANOVA, and Bonferroni’s multiple comparison post test, compared with the control (no inhibitor)).
Evaluation of SLR080811 in vivo: Isotype selectivity and pharmacokinetics
As an extension of our experiments in vitro, we sought to evaluate the sphingosine kinase isotype selectivity of SLR080811 in vivo. To this end, we injected groups of SphK1 null, SphK2 null, and wild type mice with a single intraperitoneal dose of SLR080811 and analyzed the blood levels of S1P. We observed (Figure 6a) that SphK1 null mice exhibit reduced levels of blood S1P after injection whereas in SphK2 null animals the blood levels of S1P did not change. This result once again suggests that SLR080811 is a selective inhibitor of SphK2. Levels of S1P in SphK1-null mice reached a nadir between 2 and 4 hours after injection and slowly returned to pre-treatment levels at approximately 24 hours. In addition to S1P, we also measured the blood levels of SLR080811. The kinetics of SLR080811 approximate that of S1P in the sense that we observed an early SLR080811 peak at 1 h, rather than 2 h (Figure 6b) as in the case of S1P, and then a slow disappearance of the compound over the subsequent 24 h.
Figure 6.

S1P and SLR080811 levels in the blood of mice injected with SLR080811. Wild type or Sphk1 or Sphk2 null mice were administered SLR080811 (dose: 10 mg/kg, intraperitoneal route). Blood samples were drawn at times 0, 1, 2, 4, 8 and 24 hours post injection. Levels of S1P (a) and SLR080811 in WT mice (b) in blood samples were measured by LC-MS. Data are presented as means ±SD of 3–5 mice per group. *p < 0.05, **p < 0.01 (repeated measures two way ANOVA, and Bonferroni’s multiple comparison test compared to ASAP time point after injection of the compound (time 0)).
The effect of a single dose of SLR080811 in wild type mice was surprising, in that S1P levels increased, rather than decreased (Figure 6a). To investigate this seemingly paradoxical result, we generated a cohort of age-matched F2 hybrid Sphk1 Sphk2 mice (see Methods) and measured their blood and plasma S1P levels. These mice constitute a genetically homogeneous population whose levels of blood S1P that should be less influenced by genetic variation than the parent strains. In this population we observed, as shown in Figure 7, the same pattern of blood S1P levels: high circulating S1P in SphK2 null animals, lower S1P levels in wild type animals and low levels of S1P in SphK1 null animals. Thus it appears that SLR080811, by inhibiting SphK2, mimics the effect of lack of functional Sphk2 alleles. However, we note that a different SphK2 inhibitor, ABC294640, was reported to lower circulating S1P, albeit after five weeks of daily dosing into xenograft-bearing SCID mice [29].
Fig. 7.

S1P levels in 8 weeks old WT, Sphk1−/− and Sphk2−/− littermates. Blood were drawn from wild type, Sphk1 and SphK2 null mice and the S1P levels were measured by LC-MS as described in the Methods section. Data shown are independent measurements of whole blood (A) or plasma (B) from 4 WT, 9 Sphk1−/− and 6 Sphk2−/− mice. The null mice were either heterozygous or wild type at the other SphK locus. *p < 0.05; ***p < 0.001 (one-way ANOVA, and Bonferroni’s multiple comparison test, compared with WT).
Finally, in the course of the investigations described in this paper we noticed that the recovery of the C17-S1P internal standard was greater than 100% when samples from SphK2 −/− mice were analyzed. This prompted us to investigate the possible presence of endogenous C17-S1P or an interfering analyte in those samples. We found that Sphk2 −/− samples contain an analyte that is indistinguishable from authentic C17-S1P by LC/MS, i.e. their chromatographic retention times and transitions are indistinguishable. Because of these striking similarities we refer to this analyte as C17-S1P with the caveat that a definitive identification will require confirmation by additional analysis. Moreover we found that wild type and Sphk1 −/− mice samples also contain C17-S1P, albeit to a lesser extent. From the practical point of view, and regardless of the identity of this analyte, it was obvious to us that C17-S1P should not be used as an internal standard for our samples rather we used deuterated (D7) S1P. A representative experiment illustrating this technical issue can be seen in Figure 8.
Fig. 8.



Levels of S1P and C17-S1P in whole blood of wild type, SphK1 null and SphK2 null mice. Blood samples (20 μl) from a wild type (panel A), a Sphk1 −/− (panel B) and a Sphk2 −/− (panel C) mouse (all F2 hybrids) were analyzed by LC/MS for the presence of S1P and C17-S1P. Recovery was evaluated using deuterated (D7) S1P as an Internal Standard. The figure shows the chromatograms produced by the Analyst software for these three analytes, which were obtained using the following transitions: S1P (380.4→264.4), C17-S1P (366.4→250.4), and D7-S1P (387.4→271.4). The intensity of the signals corresponds to the number of counts per second (cps) recorded by the mass spectrometer’s ion detector. Because the Analyst software uses the height of the tallest peak as a full scale, the y-axis scale (cps) is different for each chromatogram. The numbers above the peaks correspond to the retention times in minutes. Retention time for authentic C17-S1P was 5.23 ± 0.01 min, n=3 (not shown). Amounts of analytes were calculated using the areas under the peaks and a standard curve. Areas in cps x min for the S1P, C17-S1P and D7-S1P peaks shown in the figure were as follows: wild type (7.93E+06, 1.37E+05, 2.36E+06), Sphk1 −/− (3.67E+06, 3.43E+04, 2.20E+06), Sphk2 −/− (4.23E+07, 2.95E+06, 2.07E+06). In these mice, which are representative of our F2 dihybrid cohort, C17-S1P is less than 2% of S1P in wild type mice and less than 1% in SphK1 null mice but is 7% in SphK2 null mice. In addition to revealing that the mole fraction of C17-S1P is higher in SphK2 null mouse blood, these chromatograms reveal also that the absolute concentration of S1P is 4–5-fold higher in SphK2 null mice than in wild type mice.
DISCUSSION
In this paper we disclose, SLR080811, a novel SphK2-selective inhibitor. SLR080811 was synthesized by modifying the chemical structure of compound 1a, a SphK1-selective inhibitor that we have reported previously [26]. The amidine and the amide group in compound 1a were replaced by guanidine and oxadiazole groups respectively in SLR080811. Although our original intention was to obtain a longer-lived version of compound 1a by switching to the more stable guanidine and oxadiazole groups, we unexpectedly found that these changes also brought about a reversal of selectivity for SphK isotypes. In the context of this report, “selectivity” refers not to absolute selectivity for SphK2, i.e. no effect on SphK1, but rather to relative selectivity. As shown in Table I, SLR080811 is about one order of magnitude more potent inhibitor of SphK2 as compared to SphK1 based on the KI values obtained in vitro with recombinant enzymes. In addition to the selectivity for SphK2, we found that SLR080811 does indeed have a longer half-life than compound 1a according to our original expectations. As shown in Figure 6 we estimate the half-life of SLR080811 to be 4–5 hours whereas the half-life for compound 1a is less than one hour [26]. We are currently testing SLR080811 analogs to identify compounds with enhanced potency, half-life and/or isotype selectivity.
Our contention that SLR080811 is a SphK2-selective inhibitor is based on observations carried out in different biological models both in vivo and in vitro. In agreement with our results with recombinant enzymes, the effects of SLR080811 in cultured cell lines such as U937 correspond to inhibition of S1P synthesis, namely reduction of S1P and dhS1P levels and concomitant elevation of their aminoalcohol precursors sphingosine and sphinganine (Figure 1). That these effects are mediated at least in part by SphK2 blockade is demonstrated by our experiments with FTY720. This drug is a selective SphK2 substrate and, as shown in Figure 3, its phosphorylation is greatly diminished in presence of SLR080811.
Consistent with our observations with U937 cells, SLR080811 had no effect on S1P levels in SphK2 null fibroblasts while it reduced S1P levels in wild type and SphK1 null fibroblasts as shown in Figure 4a. Taken together these results corroborate the SphK inhibition by SLR080811 and its SphK2 selectivity. Our observations on sphingosine levels in these cells however were not completely consistent with this picture. Whereas we were able to detect the anticipated accumulation of sphingosine in both wild type and SphK1 null fibroblasts, we unexpectedly observed that sphingosine levels were higher in SphK2 null fibroblasts as well (Figure 4b). There is no simple explanation for this observation other than to suggest that some other mechanism(s) not presently known may be involved in the regulation of the metabolism of sphingolipids. One possibility is that SLR080811 might have some inhibitory effect on ceramide synthase.
Our in vivo observations support the notion that SLR080811 is a SphK2 selective inhibitor in the sense that it lowers the levels of blood S1P in of SphK1 null mice but does not have an effect on blood S1P when administered to mice lacking SphK2 (Figure 6). It is noteworthy that the basal levels of blood S1P in SphK2 null animals are much higher than those in SphK1 null or wild type mice. This was reported previously by Zemann et al. [30] with their SphK2 null mice and then by Olivera et al. [31] and Sensken et al. [32]. It may be hypothesized therefore that SphK1 is upregulated when SphK2 activity is reduced. However, increased SphK1 activity by itself seems to be insufficient to raise S1P levels as was demonstrated in transgenic mice with forced global expression of SphK1 [33]. These animals have high tissue SphK1 activity but have levels of plasma S1P that are not different from wild type animals. It appears that SphK2 suppression may have more complex consequences than just increasing SphK1 activity. A recent report showing that SphK2 may function as a transcriptional modulator [1] supports this contention, although this does not seem to be the case for the SphK2 null mice whose SphK1 mRNA levels were not different from wild type mice [30]. According to all these observations, it appears that there are factors at play in the regulation of S1P metabolism that we do not fully understand.
Our previous work with compound 1a, our SphK1-selective inhibitor, included the observation that this compound reduces the levels of blood S1P in wild type mice [26]. This effect appears to be the logical consequence of SphK inhibition in the context of a simple model of S1P metabolism whereby reducing the synthesis of S1P results in a drop in S1P levels. We expected SLR080811 to exhibit a qualitatively similar effect because both compounds are SphK inhibitors and because they exhibit other similarities. For example, both compounds accumulate to high levels in U937 cells possibly because they are recognized by the uptake system(s) that take up long chain bases such as sphingosine and sphinganine from the extracellular environment [34,35]. In addition, neither compound exerts major effects on cell viability at concentrations that result in extensive SphK blockade. However, we observed - contrary to our expectations - that SLR080811 increases the levels of blood S1P in wild type mice, i.e. the opposite effect of compound 1a. Obviously, this observation cannot be accommodated in a simple model of S1P metabolism. It seems that the most likely hypothesis that can be advanced to explain this effect is to postulate that SphK2 inactivation, whether genetic or pharmacological, leads to increased S1P levels. In this context, the effect of SLR080811 on wild type animals can be related to the increased levels of S1P in SphK2 null mice. In any event, if the effects of compounds 1a and SLR080811 prove to be a general property of SphK inhibitors and persist with chronic dosing then it should be possible to use SphK inhibitors to adjust the levels of S1P both positively and negatively. This may be useful not only in the investigation of S1P metabolism but in a clinical setting as well.
Acknowledgments
The authors thank Dr Richard Proia (NIH/NIDDK, Bethesda, MD, U.S.A.) for the gift of Sphk1 −/− and Sphk2 −/− mice, Dr Kaoru Goto (Yamagata University School of Medicine, Yamagata, Japan) and Dr Matthew Topham (University of Utah, Salt Lake City, UT, U.S.A.) for their gifts of DAG kinase plasmids, and Dr Amandeep Bajwa (University of Virginia, Charlottesville, VA, U.S.A.) for her gift of mouse kidney fibroblasts. The authors acknowledge the technical help of Ms Devon McCurdy.
FUNDING
The research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under the auspices of award number R01GM067958. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations used in the text
- SphK
sphingosine kinase
- S1P
sphingosine 1-phosphate
- LC/MS
liquid chromatography/mass spectrometry
Footnotes
AUTHOR CONTRIBUTIONS
Mithun Raje, Ming Gao and Webster Santos designed, synthesized and purified SLR080811. Yugesh Kharel and Kevin Lynch designed the experiments to characterize SLR080811. Yugesh Kharel performed the experiments except that Amanda Gellett did the DAG kinase assays and statistical analyses and Jose Tomsig did the LC/MS sphingolipid analyses. Yugesh Kharel and Kevin Lynch wrote the manuscript, all of the authors participated in its editing.
References
- 1.Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, Luo C, Marmorstein R, Kordula T, Milstien S, Spiegel S. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science. 2009;325:1254–1257. doi: 10.1126/science.1176709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kohama T, Olivera A, Edsall L, Nagiec MM, Dickson R, Spiegel S. Molecular cloning and functional characterization of mammalian sphingosine kinase. J Biol Chem. 1998;273:23772–23728. doi: 10.1074/jbc.273.37.23722. [DOI] [PubMed] [Google Scholar]
- 3.Liu H, Sugiura M, Nava VE, Edsall LC, Kono K, Poulton S, Milstien S, Kohama T, Spiegel S. Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J Biol Chem. 2000;275:19513–19520. doi: 10.1074/jbc.M002759200. [DOI] [PubMed] [Google Scholar]
- 4.Mizugishi K, Yamashita T, Olivera A, Miller GF, Spiegel S, Proia RL. Essential role for sphingosine kinases in neural and vascular development. Mol Cell Biol. 2005;25:11113–11121. doi: 10.1128/MCB.25.24.11113-11121.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allende ML, Sasaki T, Kawai H, Olivera A, Mi Y, van Echten-Dekert G, Hajdu R, Rosenbach M, Keohane CA, Mandala S, Spiegel S, Proia RL. Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J Biol Chem. 2004;279:52478–52492. doi: 10.1074/jbc.M406512200. [DOI] [PubMed] [Google Scholar]
- 6.Limaye V, Li X, Hahn C, Xia P, Berndt MC, Vadas MA, Gamble JR. Sphingosine kinase-1 enhances endothelial cell survival through a PECAM-1-dependent activation of PI-3K/Akt and regulation of Bcl-2 family members. Blood. 2005;105:3169–3177. doi: 10.1182/blood-2004-02-0452. [DOI] [PubMed] [Google Scholar]
- 7.Olivera A, Kohama T, Edsall LC, Nava V, Cuvillier O, Poulton S, Spiegel S. Sphingosine kinase expression increases intracullular sphingosine-1-phosphate and promotes cell growth and survival. J Cell Biol. 1999;147:545–558. doi: 10.1083/jcb.147.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edsall LC, Cuvillier O, Twitty S, Spiegel S, Milstien S. Sphingosine kinase expression regulates apoptosis and caspase activation in PC12 cells. J Neurochem. 2001;76:1573–1584. doi: 10.1046/j.1471-4159.2001.00164.x. [DOI] [PubMed] [Google Scholar]
- 9.Liu H, Toman RE, Goparaju SK, Maceyka M, Nava BE, Sankala H, Payne SG, Bektas M, Ishi I, Chun J, Milstien S, Spiegel S. Sphingosine kinase type 2 is a putative BH-3 only protein that induces apoptosie. J Biol Chem. 2003;278:40330–40336. doi: 10.1074/jbc.M304455200. [DOI] [PubMed] [Google Scholar]
- 10.Igarashi N, Okada T, Hayashi S, Fujita T, Jahangeer S, Nakamura SI. Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. J Biol Chem. 2003;278:46832–46839. doi: 10.1074/jbc.M306577200. [DOI] [PubMed] [Google Scholar]
- 11.Maceyka M, Sankala H, Hait NC, Le Stunff H, Liu H, Toman RE, Collier C, Zhang M, Satin LS, Merrill AH, Jr, Milstien S, Spiegel S. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J Biol Chem. 2005;280:37118–37129. doi: 10.1074/jbc.M502207200. [DOI] [PubMed] [Google Scholar]
- 12.Paugh SW, Paugh BS, Rahmani M, Kapitonov D, Almenara JA, Kordula T, Milstien S, Adams JK, Zipkin RE, Grant S, Spiegel S. A selective sphingosine kinase 1 inhibitor integrates multiple molecular therapeutic targets in human leukemia. Blood. 2008;112:1382–1391. doi: 10.1182/blood-2008-02-138958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiang Y, Asmussen G, Booker M, Hirth B, Kane JL, Liao J, Noson KD, Yee C. Discovery of novel sphingosine kinase 1 inhibitors. Bioorg Med Chem Lett. 2009;19:6119–6121. doi: 10.1016/j.bmcl.2009.09.022. [DOI] [PubMed] [Google Scholar]
- 14.Kennedy AJ, Mathews TP, Kharel Y, Field SD, Moyer JL, East JE, Houck JD, Lynch KR, Macdonald TL. Development of amidine-based sphingosine kinase 1 nanomolar inhibitors and reduction of sphingosine 1-phosphate in human leukemia cells. J Med Chem. 2011;54:3524–3548. doi: 10.1021/jm2001053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schnute ME, McReynolds MD, Kasten T, Yates M, Jerome G, Rains JW, Hall T, Chrencik J, Kraus M, Cronin CN, Saabye M, Highkin MK, Broadus R, Ogawa S, Cukyne K, Zawadzke LE, Peterkin V, Iyanar K, Scholten JA, Wendling J, Fujiwara H, Nemirovskiy O, Wittwer AJ, Nagiec MM. Modulation of cellular levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem J. 2012 doi: 10.1042/BJ20111929. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 16.French KJ, Zhuang Y, Maines LW, Gao P, Wang W, Beljanski V, Upson JJ, Green CL, Keller SN, Smith CD. Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J Pharmacol Exp Therap. 2010;333:129–139. doi: 10.1124/jpet.109.163444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi Y, Rehman H, Ramshesh VK, Schwartz J, Liu Q, Krishnasamy Y, Zhang X, Lemasters JJ, Smith CD, Zhong Z. Sphingosine kinase-2 inhibition improves mitochondrial function and survival after hepatic ischemia-perfusion. J Hepatol. 2012;56:137–145. doi: 10.1016/j.jhep.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fitzpatrick LR, Green C, Maines LW, Smith CD. Experimental osteoarthritis in rats is attenuated by ABC294640, a selective inhibitor of sphingosine kinase-2. Pharmacology. 2011;87:135–143. doi: 10.1159/000323911. [DOI] [PubMed] [Google Scholar]
- 19.Maines LW, Fitzpatrick LR, Green CL, Zhuang Y, Smith CD. Efficacy of a novel sphingosine kinase inhibitor in experimental Crohn’s disease. Inflammopharmacology. 2010;18:73–85. doi: 10.1007/s10787-010-0032-x. [DOI] [PubMed] [Google Scholar]
- 20.Maines LW, Fitzpatrick LR, French KJ, Zhuang Y, Xia Z, Keller SN, Upson JJ, Smith CD. Suppression of ulcerative colitis in mice by orally available inhibitors of sphingosine kinase. Dig Dis Sci. 2008;53:997–1002. doi: 10.1007/s10620-007-0133-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chumanevich AA, Poudyal D, Cui X, Davis T, Wood PA, Smith CD, Hofseth LJ. Suppression of colitis-driven colon cancer in mice by a novel small molecule inhibitor of sphingosine kinase. Carcinogenesis. 2010;31:1787–1793. doi: 10.1093/carcin/bgq158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Antoon JW, White MD, Meacham WD, Slaughter EM, Muir SE, Elliott S, Rhodes LB, Ashe HB, Wiese TE, Smith CD, Burow ME, Beckman BS. Anti-estrogenic effects of the novel sphingosine kinase-2 inhibitor ABC294640. Endocrinology. 2010;151:5124–5135. doi: 10.1210/en.2010-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kharel Y, Mathews TP, Kennedy AJ, Macdonald TL, Lynch KR. A rapid assay for assessment of sphingosine kinase inhibitors and substrates. Anal Biochem. 2011;411:230–235. doi: 10.1016/j.ab.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaner RL, Allegood JC, Park H, Wang E, Kelly S, Haynes CA, Sullards MC, Merrill AH. Quantitative analysis of sphingolipids for lipidomics using triple quadrupole and quadrupole linear ion trap mass spectrometers. J Lipid Res. 2009;50:1692–1707. doi: 10.1194/jlr.D800051-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mathews TP, Kennedy AJ, Kharel Y, Kennedy PC, Nicoara O, Sunkara M, Morris AJ, Wamhoff BR, Lynch KR, Macdonald TL. Discovery, biological evaluation, and structure-activity relationship of amidine based sphingosine kinase inhibitors. J Med Chem. 2010;53:2766–2778. doi: 10.1021/jm901860h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kharel Y, Mathews TP, Gellett AM, Tomsig JL, Kennedy PC, Moyer ML, Macdonald TL, Lynch KR. Sphingosine kinase type 1 inhibition reveals rapid turnover of circulating sphingosine 1-phosphate. Biochem J. 2011;440:345–353. doi: 10.1042/BJ20110817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Billich A, Bornancin F, Dévay P, Mechtcheriakova D, Urtz N, Baumruker T. Phosphorylation of the immunomodulatory drug FTY720 by sphingosine kinases. J Biol Chem. 2003;278:47408–47415. doi: 10.1074/jbc.M307687200. [DOI] [PubMed] [Google Scholar]
- 28.Paugh SW, Payne SG, Barbour SE, Milstein S, Spiegel S. The immunosuppressant FTY720 is phosphorylated by sphingosine kinase type 2. FEBS Lett. 2003;554:189–193. doi: 10.1016/s0014-5793(03)01168-2. [DOI] [PubMed] [Google Scholar]
- 29.Beljanski V, Lewis CS, Smith CD. Antitumor activity of sphingosine kinase 2 inhibitor ABC294640 and sorafenib in heptocellular carcinoma xenografts. Can Biol & Therap. 2011;11:524–534. doi: 10.4161/cbt.11.5.14677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zemann B, Kinzel B, Müller M, Reuschel R, Mechtcheriakova D, Urtz N, Bornancin F, Baumruker T, Billich A. Sphingosine kinase type 2 is essential for lymphodepletion induced by the immunomodulatory drug FTY720. Blood. 2006;107:1454–1458. doi: 10.1182/blood-2005-07-2628. [DOI] [PubMed] [Google Scholar]
- 31.Olivera A, Mizugishi K, Tikhonova A, Ciaccia L, Odom S, Proia RL, Rivera J. The sphingosine kinase-sphingosine-1-phosphate axis is a determinant of mast cell function and anaphylaxis. Immunity. 2007;26:287–297. doi: 10.1016/j.immuni.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 32.Sensken SC, Bode C, Nagarajan M, Peest U, Pabst O, Gräler MH. Redistribution of sphingosine 1-phosphate by sphingosine kinase 2 contributes to lymphopenia. J Immunol. 2010;184:4133–4142. doi: 10.4049/jimmunol.0903358. [DOI] [PubMed] [Google Scholar]
- 33.Takuwa N, Ohkura SI, Takashima SE, Ohtani K, Okamoto Y, Tanaka T, Hirano K, Usui S, Wang F, Du W, Yoshioka K, Banno Y, Sasaki M, Ichi I, Okamura M, Sugimoto N, Mizugishi K, Nakanuma Y, Ishii I, Takamura M, Kaneko S, Kojo S, Satouchi K, Mitumori K, Chun J, Takuwa Y. S1P3-mediated cardiac fibrosis in sphingosine kinase 1 transgenic mice involves reactive oxygen species. Cardiovas Res. 2010;85:484–493. doi: 10.1093/cvr/cvp312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanada K, Nishijima M, Fujita T, Kobayashi S. Specificity of inhibitors of serine palmitoyltransferase (SPT), a key enzyme in sphingolipid biosynthesis, in intact cells. Biochem Pharmacol. 2000;59:211–216. doi: 10.1016/s0006-2952(00)00251-3. [DOI] [PubMed] [Google Scholar]
- 35.Chigorno V, Giannotta C, Ottico E, Sciannambio M, Mikulak J, Prinetti A, Sonnino S. Sphingolipid uptake by cultured cells. J Biol Chem. 2005;280:2668–2675. doi: 10.1074/jbc.M407749200. [DOI] [PubMed] [Google Scholar]