

Abstract
In diethyl ether, N-Boc-2-lithio-2-arylpiperidines have been found to be configurationally stable at −80 °C whereas N-Boc-2-lithio-2-arylpyrrolidines are configurationally stable at −60 °C. Several tertiary benzylic carbanions derived from enantioenriched 2-aryl heterocycles have been successfully alkylated or acylated with little to no loss of enantiopurity. The scope of the reactions has been explored. The enantiomerization dynamics of N-Boc-2-lithio-2-phenylpyrrolidine and N-Boc-2-lithio-2-phenylpiperidine have been studied in the presence of different solvents and achiral ligands.
Introduction
Enantioenriched 2-aryl pyrrolidines and piperidines have recently become available by a lithiation/transmetalation/arylation sequence. Specifically, Campos demonstrated that enantioenriched N-Boc-2-aryl-pyrrolidines are available by enantioselective deprotonation using sec-butyllithium/(−)-sparteine followed by transmetalation with ZnCl2 and Pd-mediated arylation.1 Coldham and coworkers reported the dynamic thermodynamic resolution (DTR) of N-Boc-2-lithiopiperidine 1, achieving enantiomer ratios as high as 85:15 using stoichiometric amounts of monolithiated diaminoalkoxide ligands.2 Inspired by Coldham’s work, we showed that 1 is amenable to a catalytic dynamic resolution (CDR) using 10 mol% of diastereomeric ligands (S,S)-2 and (S,R)-2 (Figure 1A) at −45 °C in Et2O with 4 equivalents of TMEDA.3 Combination of the CDR with Campos’ arylation procedure effected enantioselective arylation of N-Boc-piperidine, affording compounds such as 3–7 in the er’s shown in Figure 1.4
Figure 1.

A) N-Boc-2-lithiopiperidine and chiral ligands used to resolve it under CDR conditions, B) Enantioenriched 2-Aryl piperidines.
If lithiation/alkylation occurred at the benzylic position without racemization of the intermediate carbanion, quaternary stereocenters could be obtained by electrophilic substitution. Benzylic organolithium compounds that exhibit significant configurational stability are relatively rare.5 A recent review cites several examples of quantitative studies on the inversion dynamics of tertiary benzylic organolithiums where free energy barriers ranging from 9 to 14 kcal/mol at −80 °C were measured.6 Such low barriers indicate a high degree of configurational instability of benzylic organolithiums (half-life for inversion ≤10 minutes), which provides a challenge to stereoselective synthesis.7 Approaches to lithiation/alkylation sequences at benzylic sites often employ chiral auxiliaries8 or chiral ligands such as (−)-sparteine9 or a bisoxazoline7e, 10 to impose configurational stability in the ground state or diastereomeric bias in the transition state, thus leading to diastereomerically or enantiomerically enriched products. Many years ago, the necessity of diastereomeric bias in the alkylation of a benzylic dipole-stabilized α-aminoorganolithium compound was demonstrated by Meyers, who showed that an enantioenriched tertiary tetrahydroisoquinoline formamidine (97:3 er), in which the lithium was also intramolecularly chelated, produced racemic product after a lithiation-substitution sequence in THF at −100 °C.11
In their synthesis of a 2,2-disubstituted piperidine NK1 antagonist, Xiao and coworkers reported that lithiation of rac-N-Boc-2-phenylpiperidine 3 at −78 °C using n-BuLi occurs exclusively at the benzylic position.12 We therefore decided to explore stereoselective lithiation-substitutions of N-Boc-2-arylpiperidines and pyrrolidines, in the absence of any chiral ligand or auxiliary. We find that enantioenriched 2,2-disubstituted pyrrolidines and piperidines are produced with little to no loss of enantiomeric purity. We then conducted a detailed study of the effects of ligands and solvents on the dynamics of enantiomerization of N-Boc-2-lithio-2-phenylpyrrolidine and N-Boc-2-lithio-2-phenylpiperidine, and determined the enthalpies and entropies of activation for carbanion inversion under several different conditions. While an earlier version of this manuscript was in preparation, we became aware of a related mechanistic study that included important details of deprotonation relative to rotamer interconversion of these same compounds.13
Results and Discussion
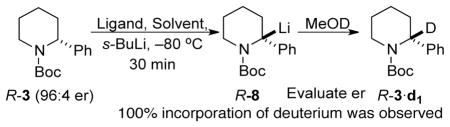
Previously, we and Coldham measured the dynamics of enantiomerization of N-Boc-2-lithiopiperidine 1 in Et2O and found that its configurational stability depended on the amount of TMEDA present.14 Given these results, we investigated the configurational stability of the benzylic lithiopiperidine 8 in the presence and absence of TMEDA.
After treatment of R-3 of 96:4 er (0.06 M in Et2O or THF) with 1 equiv s-BuLi at −80 °C in the presence or absence of TMEDA, 8 was generated and immediately trapped with MeOD. The extent of lithiation was monitored using GC-MS by comparing the integral ratio of the base beak for the singly deuterated product, 3·d1 to that of the starting material 3. After complete deuteration (as noted by the disappearance of m/z 206 and appearance of m/z 207, see Figure 2 and the Supporting Information), the enantiomer ratios of 3 and 3·d1 were evaluated by CSP-SFC. In the presence of TMEDA (1 or 4 equiv) and with Et2O as the solvent, quenching with MeOD showed complete deuteration after 30 min at −80 °C and 3·d1 of 96:4 er (entries 1 & 2) was obtained. When the reaction mixture was quickly transferred to a bath at −55 °C and stirred for 1 h, quenching at −80 °C with MeOD gave 3·d1 of 85:15 er (entry 3). In the absence of a ligand, after 60 min of lithiation, 3·d1 was obtained in 91:9 er (entry 4). In THF, slight loss of enantiopurity was observed (entries 5&6), which was most pronounced in the absence of TMEDA (entry 7).
Figure 2.

GC-MS and CSP-SFC traces for R-3 and R-3·d1
The following findings emerged from these experiments:
The tertiary α-amino benzylic organolithium 8 racemizes slowly in both Et2O and THF at −80 °C in the absence of TMEDA. TMEDA enhances the configurational stability of 8 in both solvents.
As expected, 8 is configurationally less stable at higher temperatures (Table 1, entry 2 vs 3).
Deuteration of 8 with MeOD is stereoretentive.
Table 1.
Evaluating the configurational stability of N-Boc-2-lithio-2-phenylpiperidine 8.
| |||
|---|---|---|---|
| Entry | Solvent | Ligand | er of 3·d1 (R:S) |
| 1 | Et2O | 1 equiv TMEDA | 96:4 |
| 2 | Et2O | 4 equiv TMEDA | 96:4 |
| 3a | Et2O | 4 equiv TMEDA | 85:15 |
| 4 | Et2O | Noneb | 91:9 |
| 5 | THF | 1 equiv TMEDA | 94:6 |
| 6 | THF | 4 equiv TMEDA | 95:5 |
| 7 | THF | Noneb | 92:8 |
transferred to a bath at −55 °C and stirred for 1 h,
lithiated for 1 h
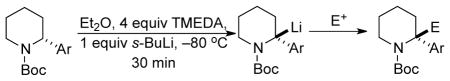
Several other electrophiles and aryl piperidines were investigated and the results are summarized in Table 2. In all cases, good yields were obtained and retention of configuration was observed. Methylation of 8 with Me2SO4 gave R-9 in 79% yield and 95:5 er (Table 2, entry 2). Removal of the Boc-group using gaseous HCl gave unprotected 9, with the same sign of specific rotation as that recently reported by Aggarwal for enantiopure deprotected R-9.5f Based on retentive deuteration and methylation, and the recently reported work of O’Brien and Coldham in which the steric course of several such substitutions were characterized,13 we have assigned all the substitutions reported herein as retentive. Quenching with Me3SiCl afforded S-10 in 88% yield and 96:4 er (entry 3). Quenching with ethyl chloroformate afforded the ester R-11 in 85% yield, with no loss of er (entry 4). A carbonyl electrophile was also evaluated. Quenching with (CD3)2CO15 and warming to room temperature before adding methanol gave rise to the oxazolidinone 12 in 90% yield and 95:5 er.
Table 2.
Lithiation-substitution of enantioenriched N-Boc-2-arylpiperidines (see also Figure 1B).
| ||||||
|---|---|---|---|---|---|---|
| Entry | Ar | er (R:S) | E+ | Product | % Yield | er of product |
| 1 | Ph | 96:4 | MeOD | R-3·d1 | 100a | 96:4 |
| 2 | Ph | 96:4 | Me2SO4 | R-9 | 79 | 95:5 |
| 3 | Ph | 96:4 | Me3SiCl | S-10 | 88 | 96:4 |
| 4 | Ph | 96:4 | EtOCOCl | R-11 | 85 | 96:4 |
| 5 | Ph | 96:4 | (CD3)2CO | R-12 | 90 | 95:5 |
| 6 | Ph | 96:4 | Allyl-Brb | S-13 | 66 | 92:8 |
| 7 | Ph | 96:4 | BnBrb | S-14 | 71 | 94:6 |
| 8 | 3,4-(MeO)2-C5H3 | 97:3 | MeOD | R-4·d1 | 100a | 97:3 |
| 9 | 3,4-(MeO)2-C6H3 | 97:3 | (CD3)2CO | R-15 | 93 | 97:3 |
| 10 | 4-(tBu)-C6H4 | 90:10 | MeOD | R-5·d1 | 100a | 90:10 |
| 11 | 4-(NC)-C6H4 | 90:10 | MeOD | R-6·d1 | 100a | 90:10 |
| 12 | 4-(NC)-C6H4 | 90:10 | Me2SO4 | R-16 | 71 | 90:10 |
| 13 | 1-Np | 97:3 | MeOD | R-7·d1 | 100a | 97:3 |
| 14 | 1-Np | 97:3 | Me2SO4 | R-17 | 74 | 93:7 |
% conversion by GC
via zinc and copper-mediated coupling.
After transmetalation of the benzylic organolithium R-8 to its organozinc counterpart, copper-mediated allylation and benzylation3 afforded S-13 (entry 6) and S-14 (entry 7), respectively, in good yields and er’s.
Lithiation-substitution of highly electron-rich R-4 of 97:3 er also proceeded easily, and the products R-4·d1 and R-15 were formed after quenching with MeOD and (CD3)2CO respectively (entries 8 and 9). The presence of a bulky electron rich substituent on the aryl moiety did not affect the configurational stability of the benzylic organolithium as R-5 of 90:10 er also gave R-5·d1 with no loss of er after deuteration with MeOD (entry 10). When electron-deficient R-6 of 91:9 er was employed, lithiation and deuteration or reaction with Me2SO4 provided R-6·d1 and R-16 respectively with no loss of enantiopurity (entries 11 and 12). Sterically crowded R-7 of 97:3 er gave R-7·d1 and R-17 after deuteration and methylation respectively (entries 13 and 14).
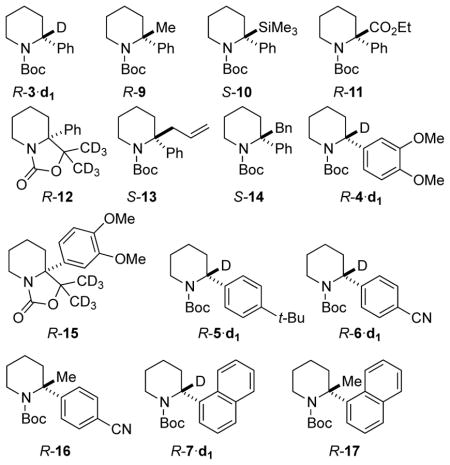
The configurational stability of the homologous N-Boc-2-arylpyrrolidines was also evaluated. Initially, R-18 of 96:4 er was synthesized using the Campos procedure, which employs a stoichiometric amount of (−)-sparteine.1a However, following a recent report by O’Brien and Campos on the use of catalytic amounts of the chiral ligand to effect a catalytic enantioselective arylation of N-Boc-pyrrolidine, subsequent (R)-N-Boc-2-arylpyrrolidines (R-18 to R-21, Figure 3) were synthesized using this method.1b, 16 The enantiomeric (S)-N-Boc-2-arylpyrrolidines (S-18 and S-22) were synthesized using Beak’s lithiation-cyclization method.17
Figure 3.

Enantioenriched 2-Aryl pyrrolidines and diisopropylbispidine, 23.
Xiao, et al reported that lithiation/alkylation of rac-18 occurred at C2 at −78 °C using n-BuLi/TMEDA in THF.12 We found that lithiation of R-18, using s-BuLi/TMEDA in Et2O was somewhat slower than that of 3 under the conditions used for the piperidines. Although we were able to obtain 100% lithiation of 18 at −80 °C using s-BuLi/TMEDA, Coldham and O’Brien have shown that interconversion of rotamers at −80 °C is slow, and lithiation at C-5 is competitive at this temperature.13
We sought reaction conditions that would maximize the efficient generation of the C2-lithiated pyrrolidine 24 without compromising its configurational stability. After lithiation of a solution of R-18 of 96:4 er in Et2O using n-BuLi at −60 °C in the absence of a ligand, quenching of 24 with MeOD revealed complete lithiation after 3 h. CSP-SFC analysis also revealed stereoretentive deuteration and R-18·d1 was obtained in 96:4 er. Similar configurational stability was observed in the presence of stoichiometric or excess amounts of TMEDA. For example, when S-18 of 96:4 er, in Et2O containing 1 equivalent of TMEDA at −60 °C, was treated with n-BuLi for 3 hours and then quenched with MeOD, S-18·d1 having 96:4 er was obtained. In the presence of hindered diisopropylbispidine 23,18 R-18·d1 of 95:5 er was obtained. O’Brien and Coldham have shown that at this temperature, rotation of the Boc group is fast enough for interconversion, and C-5 deprotonation is not competitive when n-BuLi is used as the base.13
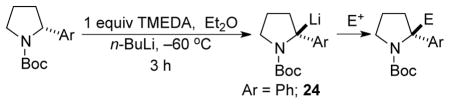
Knowing that configurationally stable R-24 can be efficiently generated after 3 hours at −60 °C in the presence or absence of TMEDA in Et2O, other electrophiles and aryl pyrrolidines were evaluated and the results are summarized in Table 3. Lithiation of R-18 of 96:4 er with n-BuLi/TMEDA followed by alkylation with Me2SO4 gave R-25 of 94:6 er in 86% yield (Table 3, entry 1). Lithiation with sec-BuLi/TMEDA and quenching with dimethyl formamide afforded crude aldehyde R-26 in 96:4 er. After column chromatography on silica, a slight improvement in the er was observed and enantiopure R-26 was obtained in 83% yield (entry 2), possibly due to self-disproportionation of enantiomers on the achiral stationary phase.19 Ethyl chloroformate gave ester R-27 in 70% yield and 95:5 er (entry 3). With (CD3)2CO, oxazolidinone R-28 was obtained in 85% yield and 96:4 er (entry 4). When R-24 was transmetalated to its organozinc counterpart followed by Pd-catalyzed coupling to 2-bromotoluene, R-29 was obtained in very low yield (entry 5). Entries 2–5 of Table 2 were initially conducted by deprotonationation using sec-BuLi/TMEDA at −80 °C, and probably produced a mixture of C2 and C5 lithiation/substitution products.13 The products of C2 substitution were characterized after chromatographic purification. Later, entries 2–5 were repeated using n-BuLi/TMEDA, which gives only C2 lithiation, but only analyzed for percent conversion by GC.
Table 3.
Lithiation-substitution of enantioenriched N-Boc-2-aryl pyrrolidines using indicated conditions, except as noted.
| ||||||
|---|---|---|---|---|---|---|
| Entry | Ar | er (R:S) | E+ | Product | % Yield | er |
| 1 | Ph | 96:4 | Me2SO4 | R-25 | 86a | 94:6 |
| 2 | Ph | 96:4 | DMF | R-26 | 83b (88c) | >99:1 |
| 3 | Ph | 96:4 | EtOCOCl | R-27 | 70b (79c) | 94:6 |
| 4 | Ph | 96:4 | (CD3)2CO | R-28 | 85b (92c) | 94:6 |
| 5 | Ph | 96:4 | 2-bromotoluene | R-29 | 8b,d | 92:8 |
| 6 | Ph | 50:50 | 2-bromotoluene | rac-29 | 12a,d,e | 50:50 |
| 7 | 2-tolyl | 90:10 | MeOD | R-19·d1 | 100c | 90:10 |
| 8 | 2-pyridyl | 90:10 | MeOD | R-20·d1 | 100c | 90:10 |
| 9 | 1-Np | 95:5 | MeOD | R-21·d1 | 100c | 95:5 |
| 10 | 1-Np | 95:5 | Me2SO4 | R-31 | 91a | 95:5 |
| 11 | 1-Np | 95:5 | PhBr | S-32 | <5c,d,e | 95:5 |
| 12 | 4-(NC)-C6H4 | 7:93 | MeOD | S-22·d1 | 100c | 93:7 |
isolated yield,
isolated yield of C2 and C5 substitution products after deprotonation by s-BuLi/TMEDA,
percent conversion by GC after deprotonation using n-BuLi/TMEDA,
via Pd-catalyzed coupling for 48 h at 40 °C,
lithiated in the absence of TMEDA.
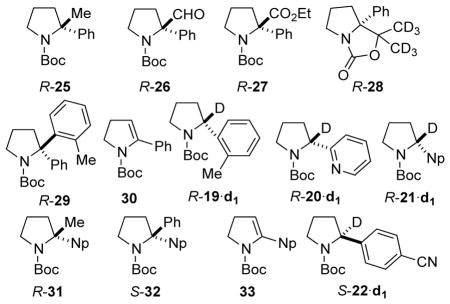
We reasoned that the low yield in entry 5 could be due to steric crowding or the presence of TMEDA, known to be problematic in the Pd-catalyzed arylation of racemic N-Boc-2-lithiopyrrolidine, obtained from deprotonation of N-Boc pyrrolidine using sec-BuLi/TMEDA.1b To test this possibility, we generated rac-24 by deprotonation using n-BuLi and subjected it to the same arylation conditions, affording rac-29 in just 12% yield (entry 6). GC-MS analysis of the crude product mixture showed complete consumption of R-18 but a significant byproduct was found, dehydropyrrolidine 30, probably produced by β-hydride elimination of the organopalladium intermediate.
Other aryl pyrrolidines with varying stereoelectronic properties were also evaluated after deprotonation using n-BuLi/TMEDA in Et2O. Lithiation-deuteration of electron-rich R-19 and hetereoaromatic R-20 proceeded with high levels of configurational stability (entries 7 and 8 respectively). The naphthyl substituent was found to be compatible as R-21 of 95:5 er gave R-21·d1 and R-31 after deuteration and methylation respectively (entries 9 and 10). When R-21 was lithiated in the absence of TMEDA, transmetalation to the organozinc and palladium-catalyzed coupling to bromobenzene afforded only trace amounts of S-32 (entry 11). Enamide 33 was also detected, consistent with undesirable β-hydride elimination during the arylation. We therefore concluded that the low yield in the arylation at C-2 of the 2-aryl pyrrolidines was largely due to steric hindrance. Complete and efficient lithiation of S-22 of 93:7 er followed by quenching with MeOD gave S-22·d1 (entry 12).
Enantiomerization dynamics of α-amino tertiary benzylic organolithiums 24 and 8
The dynamics of enantiomerization of benzylic organolithiums through a solvent-separated ion pair has been reported by Peoples and Grutzner using a benzylic lithiohydrocarbon,20 and by Ahlbrecht et al. using α-thio and α-seleno compounds.21 Cram and coworkers reported the enantiomerization of a chelated α-silylbenzylic organolithium22 via the conducted tour mechanism.23 In all of these examples, negative entropies of activation were observed, consistent with additional solvation in the transition state. In collaboration with the Coldham group, we have studied the dynamics of carbanion inversion of α-amino organolithium compounds. The activation parameters for enantiomerization of N-substituted-2-lithiopyrrolidines24 and for dynamic thermodynamic resolution (DTR) and racemization of N-Boc-2-lithiopiperidine3, 14 have been measured. It became clear to us that the role of an additive such as TMEDA in the racemization of α-amino organolithium compounds can change dramatically, depending on conditions. For example, whereas TMEDA catalyzes the racemization of N-Boc-2-lithiopyrrolidine,24c the effect of TMEDA on the racemization of N-Boc-2-lithiopiperidine is dependent on the stoichiometry. TMEDA catalyzes the racemization of N-Boc-2-lithiopiperidine up to 1 equiv,14 but two equivalents of TMEDA retards racemization.3 In another striking difference, when generated by tin-lithium exchange, enantioenriched N-Boc-2-lithiopiperidine racemizes in a matter of minutes at −80 °C in the presence of 1 equivalent of diisopropylbispidine (DIB) in Et2O whereas N-Boc-2-lithiopyrrolidine exhibits configurational stability under identical conditions.24c We therefore undertook an investigation of the activation parameters for enantiomerization of pyrrolidine 24 and piperidine 8 in the presence of varying solvents and ligands.
Dynamics of inversion of 24
Detailed kinetic measurements on the effects of solvents and ligands on the rate of enantiomerization of R-24 were performed using previously reported methods.14, 24c, d Thus stock solutions of R-18 of 96:4 er (0.06 M in Et2O, THF, or 2-methyltetrahydrofuran (2-MeTHF)) were prepared and placed in six oven-dried, septum capped test tubes each equipped with a stir bar. Prior to embarking on the time-dependent studies, we performed exploratory runs using different solvents and ligands at −43 °C. The results are summarized in Scheme 1. These data clearly indicate configurational stability in Et2O, even at −43 °C, and significantly faster racemization at the same temperature in either THF or 2-MeTHF. As a result of these experiments, we realized that the dynamics of inversion in Et2O would have to be studied at temperatures higher than −43 °C.
Scheme 1.

Enantiomer ratios for the racemization of 24 under different reactions conditions at −43 °C.
In order to determine the rate constants for enantiomerization at specified temperatures, the organolithium R-24 was generated using n-BuLi at −60 °C for 3 h followed by transferring to a bath set at the desired temperature to monitor inversion. At various time intervals, duplicate tubes were transferred to a bath at −80 °C and quenched with MeOD. GC-MS analysis of the crude mixture of each tube was carried out to ensure 100% deuterium incorporation, prior to evaluation of the enantiomer ratio by CSP-SFC. The GC traces showed evidence of some decomposition at higher temperatures but we were still able to obtain good kinetic data. Due to the slow racemization in Et2O at −43 °C, the dynamics were subsequently studied at temperatures ranging from −20 °C to 18 °C. After generating 24 using n-BuLi at −60 °C for 1 h, we followed the evolution of er as a function of time in the absence of any ligand in THF at −57 °C, −43 °C, and −31 °C. With 2-MeTHF, the kinetics were studied at −31 °C. The zero-order plots for the enantiomerization are in the SI. Eyring analysis of the rate constants at their respective temperatures provided the activation parameters listed in Table 4 (see SI for details on the analysis of the kinetic data). Examination of the enthalpic and entropic contributions to the free energies for the enantiomerization revealed significant differences. In the absence of any ligand in Et2O, carbanion inversion is mostly enthalpy controlled (Table 4, entry 1). Addition of TMEDA leads to little or no change in the enthalpy of activation but the entropic barrier changes by a factor of two (entry 2). With the hindered and weakly coordinating 23, the racemization is enthalpy controlled (entry 3). Comparing Et2O and THF, the enthalpy of activation in THF drops from 18.0 to 8.6 kcal/mol and the entropy of activation changes from −8.6 to −37.5 cal/mol·K (entries 1 vs. 4). The activation parameters for enantiomerization of 24 in the absence of any ligand in 2-MeTHF were not determined but we found that at −31 °C, the rate of enantiomerization of 24 is nine times faster in THF (t1/2 = 11.7 min) than in 2-MeTHF (t1/2 = 1.8 h).
Table 4.
Thermodynamic parameters for inversion of 24
| Entry | Description | ΔH‡ (kcal/mol) | ΔS‡ (cal/mol·K) |
|---|---|---|---|
| 1 | No ligand in Et2O | 18.0 ± 1.7 | −8.6 ± 0.3 |
| 2 | 1 equiv TMEDA in Et2O | 16.0 ± 1.3 | −18.7 ± 1.6 |
| 3 | 1 equiv 23 in Et2O | 21.3 ± 0.2 | −1.0 ± 0.4 |
| 4 | No ligand in THF | 8.6 ± 1.6 | −37. 5 ± 2.5 |
A plot of ΔG‡ vs. temperature for the different systems studied is illustrated in Figure 4. Although the observed rate constants increase with an increase in temperature, the negative entropies of activation reveal that the free energy barriers increase with temperature. Interestingly, O’Brien and Coldham have recently reported deprotonation of N-Boc-2-phenylpyrrolidine using n-BuLi in THF at −50 °C for 5 minutes, followed by electrophilic quench.13 The half-life for enantiomerization at this temperature is 50 minutes.
Figure 4.

Relationship between free energy of activation and temperature for inversion of 24. The solid lines indicate free energy barriers in the temperature region where the kinetics were measured; dashed lines are extrapolations.
Dynamics of inversion of 8
Using the method described above for the dynamics of inversion of 24, the barriers to enantiomerization of piperidine 8 in the absence of any ligands in Et2O or THF, and in the presence of TMEDA in Et2O, were measured. With Et2O as the solvent, the dynamics of inversion of 8 were studied at temperatures ranging from −40 °C to −20 °C with and without TMEDA. In THF, colder temperatures (−60 °C to −40 °C) were required due to faster racemization. The activation parameters are listed in Table 5 (details are in the SI) and a plot of ΔG‡ vs. temperature for the three systems studied is shown in Fig. 5. The significantly higher barriers in Et2O reveal significantly higher configurational stability than in THF. Our results in THF, from Tables 1 and 5, and Figure 5 are at somewhat at odds with the results of O’Brien and Coldham.13 Their conditions for deprotonation and alkylation of 3 in THF are −50 °C for 30 minutes, and they report 94:6 to 99:1 er’s after alkylation. In our hands, we begin to see racemization of 8 even at −80 °C (Table 1, entry 7). One of our kinetic runs was conducted at −50 °C in THF; after 30 minutes, we observe 66:34 er. From the parameters of entry 3 of Table 5, we calculate a half-life for enantiomerization of 22 minutes. Slight differences in temperature produce small differences in free energy barriers, but these can result in significant differences in half-life due to the exponential relationship between ΔG‡ and t1/2.
Table 5.
Thermodynamic parameters for inversion of 8
| Entry | Description | ΔH‡ (kcal/mol) | ΔS‡ (cal/mol·K) |
|---|---|---|---|
| 1 | No ligand in Et2O | 17.5 ± 0.8 | −2.0 ± 0.06 |
| 2 | 1 equiv TMEDA in Et2O | 16.0 ± 1.3 | −9.6 ± 0.5 |
| 3 | No ligand in THF | 10.1 ± 0.9 | −29.1 ± 4.2 |
Figure 5.
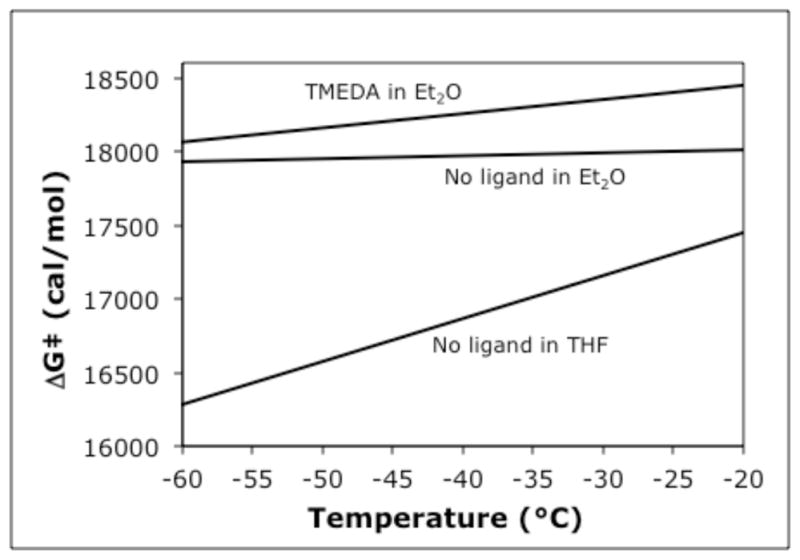
Relationship between free energy of activation and temperature for inversion of 8.
In Et2O at similar temperatures, we find that the rate of inversion of piperidine 8 is significantly faster than that of pyrrolidine 24. Curiously, the observed trend is similar to that reported for the racemization of the secondary, non benzylic organolithiums N-Boc-2-lithiopiperidine14 and N-Boc-2-lithiopyrrolidine24c in Et2O.
Mechanistic Hypothesis
A possible mechanism for the enantiomerization of 8 or 24, which is consistent with the observed negative entropies is via the conducted tour, known to be operative in the inversion of N-substituted-2-lithio pyrrolidines and piperidines.24a, b Infrared studies have shown that the carbonyl oxygen chelates the lithium in both 8 and 24.13, 25 In the conducted tour, the lithium–oxygen distance is shortened and the lithium–carbon distance is lengthened as the transition state is approached. This necessitates a reorganization of the coordination sphere around the lithium, but the lithium remains coordinated to the carbonyl oxygen as it moves from one face of the carbanion to the other. This is consistent with the observation that a strongly coordinating ligand (TMEDA) and a more coordinating solvent (THF) result in more negative entropies for the enantiomerization of 24 or 8 than in Et2O alone. Since the coordinating ability of 2-MeTHF is intermediate between Et2O and THF,26 it is perhaps not surprising that the rate of enantiomerization of 24 is faster than with Et2O but slower than with THF.
Summary and conclusions
In summary, the configurational stability of several α-amino tertiary benzylic organolithiums has been demonstrated at low temperatures on two different heterocycles, in the absence of any chiral ligand or auxiliary. TMEDA enhances the configurational stability of both 8 and 24. A variety of 2,2-disubstituted piperidines and pyrrolidines have been synthesized bearing α-amino quaternary stereocenters. The thermodynamic parameters for racemization of 8 and 24 in the absence of any ligand in Et2O and THF are significantly different. In THF, the racemization is faster than in Et2O and mostly entropy controlled.
Experimental Section
For general experimental conditions, see the Supporting Information.
Lithiation of (R)-N-Boc-2-aryl piperidine followed by Direct Trapping with the Electrophile
General Procedure
To an oven-dried, septum-capped round bottom flask equipped with a stir bar, was added freshly distilled TMEDA (4.0 equiv) and Et2O under argon. The solution was cooled to −80 °C and a solution of s-BuLi in cyclohexane (1.0 equiv) was added. A precooled solution of the N-Boc-2-arylpiperidine (1.0 equiv) in Et2O was added to the flask containing the TMEDA/s-BuLi solution. After 30 min at this temperature, the reaction was quenched with the electrophile (~1.1 to 1.5 equiv). After 2–16 h, MeOH was added and the mixture was stirred for 5 min. After warming to room temperature, 2 M HCl was added. The layers were separated and the aqueous layer was extracted with Et2O. The combined organic layers were dried over MgSO4 and evaporated to obtain the crude product. The er was determined before and after purification by column chromatography. See Supporting Information for details.
Lithiation of (R)-N-Boc-2-arylpiperidine followed by Zinc and Copper-Mediated Allylation or Benzylation
General Procedure
To an oven-dried, septum-capped round bottom flask equipped with a stir bar, was added freshly distilled TMEDA (4.0 equiv) and Et2O under argon. The solution was cooled to −80 °C and a solution of s-BuLi in cyclohexane (1.0 equiv) was added. A precooled solution of the N-Boc-2-arylpiperidine (1.0 equiv) in Et2O was added to the flask containing the TMEDA/s-BuLi mixture. After 30 min, a solution of ZnCl2 (1.3 equiv, 1.0 M in Et2O was added slowly. After 30 min, a solution of CuCN·2LiCl [prepared from CuCN (1.2 equiv) and LiCl (2.5 equiv)] in THF was added. After 30 min, allyl bromide or benzyl bromide (1.1 equiv) was added. The mixture was allowed to stir for 10 h at this temperature prior to addition of MeOH and warming to room temperature. A solution of NH4Cl was added and the aqueous layer was extracted with Et2O. The combined organic layers were dried over Na2SO4 and evaporated to give the crude product. The er was determined before and after purification by column chromatography. See Supporting Information for details.
Lithiation of (R)-N-Boc-2-aryl pyrrolidine followed by Direct Trapping with the Electrophile
General Procedure
To an oven-dried, septum-capped round bottom flask equipped with a stir bar, was added freshly distilled TMEDA (1.0 equiv) and Et2O under argon. The mixture was cooled to −60 °C and a solution of n-BuLi in hexanes (1.0 equiv) was added. A precooled solution of the N-Boc-2-arylpyrrolidine (1.0 equiv) in Et2O was added to the flask containing the TMEDA/n-BuLi mixture. After 3 h at this temperature, the mixture was quenched with the electrophile (~1.1 to 1.5 equiv). After 2–16 h, MeOH was added and the mixture was stirred for 5 min. After warming to room temperature, 2 M HCl was added. The layers were separated and the aqueous layer was extracted with Et2O. The combined organic layers were dried over MgSO4 and evaporated to obtain the crude product. The er was determined before and after column chromatography.
Lithiation of (R)-N-Boc-2-aryl pyrrolidine followed by Zinc and Palladium-Catalyzed Arylation
Attempted Synthesis of 32
To an oven-dried, septum-capped round bottom flask equipped with a stir bar, was added R-21 of 95:5 er (75 mg, 0.25 mmol, 1.0 equiv) in Et2O (2 mL) under argon. The mixture was cooled to −60 °C and a solution of n-BuLi in hexanes (0.1 mL, 0.25 mmol, 2.5 M, 1.0 equiv) was added slowly. After 3 h at this temperature, a solution of ZnCl2 (0.15 mL, 1.0 M solution in Et2O, 0.6 equiv), was added slowly over a two minute period and the mixture was stirred for 30 minutes followed by warming to room temperature. After 30 minutes, Pd(OAc)2 (2.5 mg, 4 mol%), t-Bu3P·HBF4 (6 mg, 8 mol%) and phenyl bromide (0.033 mL, 0.28 mmol, 1.1 equiv) were added sequentially. After stirring for 48 h at 40 °C, the heterogenous mixture was returned to room temperature. NH4OH (2 mL, 10% aqueous solution) was added dropwise and the mixture was stirred for 30 minutes. The resulting slurry was filtered through Celite and rinsed with 5 mL Et2O. The filtrate was washed with 1 M HCl(aq) (10 mL), then with water (2 × 5 mL), dried over Na2SO4 and evaporated under reduced pressure to obtain the crude product. Analysis of the crude product by CG-MS showed complete conversion of 21 but less than 5% yield of 32 was present.
Activation Parameters for Racemization of 24 with 1.0 equiv TMEDA in Et2O
Typical kinetic run
In oven-dried, septum-capped tubes equipped with a stir bar, R-18 (0.06 M in ether, 0.5 mL) and 0.06 M TMEDA (1.00 equiv) were treated with n-BuLi (1.0 equiv) at −60 °C for 3 h under nitrogen. The total volume of each tube was maintained at 1.0 mL. The tubes were quickly transferred to a second bath thermostatted at the desired temperature. At various time intervals, a tube was transferred to the bath at −80 °C and rapidly quenched with MeOD. Each tube was analyzed by GC-MS to ensure 100% deuterium incorporation (indicative of complete lithiation). The enantiomer ratio (er) of 18·d1 was determined by CSP-SFC monitoring at 210 nm under the following column conditions: Column: Pirkle Whelk-O-1, Flow Rate = 2.0 mL/min, Polarity Modifier = 2.0% EtOH. S-18·d1 elutes after ~4.2 min and R-18·d1 elutes after ~5.7 min. The rate constants were determined by non-linear fitting of the zero-order plots using reversible first-order kinetics. See Supporting Information for details.
Activation Parameters for Racemization of 8 without a Ligand in Et2O
Typical kinetic run
In oven-dried, septum-capped tubes equipped with a stir bar, R-3 (0.06 M in Et2O, 1.0 mL) was treated with n-BuLi (1.0 equiv) at −80 °C for 1 h under nitrogen. The tubes were quickly transferred to a second bath thermostatted at the desired temperature. At various time intervals over a four–hour period, a tube was transferred to the bath at −80 °C and rapidly quenched with MeOD. Each tube was analyzed by GC-MS to ensure 100% deuterium incorporation (indicative of complete lithiation). The enantiomer ratio (er) of 3·d1 was determined by CSP-SFC monitoring at 210 nm under the following column conditions: Column: Pirkle Whelk-O-1, Flow Rate = 0.5 mL/min, Polarity Modifier = 10.0% IPA. S-3·d1 elutes after ~17.2 min and R-3·d1 elutes after ~21 min. In some cases, the enantiomer ratio (er) of 3·d1 was determined by CSP-HPLC monitoring at 254 nm. The rate constants were determined by non-linear fitting of the zero-order plots using reversible first-order kinetics. See Supporting Information for details.
Supplementary Material
Acknowledgments
We thank the Arkansas Biosciences Institute and the National Science Foundation (CHE 1011788) for direct support of this work. Core facilities were funded by the Arkansas Biosciences Institute and the National Institutes of Health (P30 RR031154 and GM103450). JSW thanks the NSF-REU for a summer fellowship (CHE 0851505). The authors are grateful to Professors Peter O’Brien and Iain Coldham for sharing their related manuscript13 prior to publication.
Footnotes
Supporting Information Available. Full experimental details and spectroscopic and kinetic data. This information is available free of charge via the internet at http://pubs.acs.org
Notes and References
- 1.(a) Campos KR, Klapars A, Waldman JH, Dormer PG, Chen C-y. J Am Chem Soc. 2006;128(11):3538–3539. doi: 10.1021/ja0605265. [DOI] [PubMed] [Google Scholar]; (b) Barker G, McGrath JL, Klapars A, Stead D, Zhou G, Campos KR, O’Brien P. J Org Chem. 2011;76(15):5936–5953. doi: 10.1021/jo2011347. [DOI] [PubMed] [Google Scholar]
- 2.Coldham I, Raimbault S, Chovatia PT, Patel JJ, Leonori D, Sheikh NS, Whittaker DTE. Chem Commun. 2008:4174–4176. doi: 10.1039/b810988e. [DOI] [PubMed] [Google Scholar]
- 3.Beng TK, Gawley RE. J Am Chem Soc. 2010;132(35):12216–12217. doi: 10.1021/ja105772z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beng TK, Gawley RE. Org Lett. 2011;13:394–397. doi: 10.1021/ol102682r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Hoppe D, Carstens A, Krämer T. Angew Chem Int Ed Engl. 1990;29:1424–1425. [Google Scholar]; (b) Carstens A, Hoppe D. Tetrahedron. 1994;50:6097–6108. [Google Scholar]; (c) Derwing C, Hoppe D. Synthesis. 1996;1996(01):149–154. [Google Scholar]; (d) Derwing C, Frank H, Hoppe D. Eur J Org Chem. 1999;1999(12):3519–3524. [Google Scholar]; (e) Stymiest JL, Bagutski V, French RM, Aggarwal VK. Nature. 2008;456(7223):778–782. doi: 10.1038/nature07592. [DOI] [PubMed] [Google Scholar]; (f) Bagutski V, Elford TG, Aggarwal VK. Angew Chem Int Ed. 2011;50(5):1080–1083. doi: 10.1002/anie.201006037. [DOI] [PubMed] [Google Scholar]
- 6.Gawley RE. Top Stereochem. 2010;26:93–133. doi: 10.1002/9783906390628.ch3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Hoppe D, Hense T. Angew Chem Int Ed. 1997;36(21):2283–2316. [Google Scholar]; (b) Hoppe D, Marr F, Brüggemann M. Top Organomet Chem. 2003;5:61–138. [Google Scholar]; (c) Beak P, Johnson TA, Kim DD, Lim SH. Top Organomet Chem. 2003;5:139–176. [Google Scholar]; (d) Hoppe D, Christoph G. Asymmetric deprotonation with alkyllithium-(−)-sparteine. In: Rappoport Z, Marek I, editors. The chemistry of organolithium compounds. John Wiley and Sons, Ltd; Oxford: 2004. pp. 1055–1164. [Google Scholar]; (e) Lange H, Huenerbein R, Wibbeling B, Frölich R, Grimme S, Hoppe D. Synthesis. 2008:2905–2918. [Google Scholar]
- 8.(a) Highsmith TK, Meyers AI. The Asymmetric Synthesis of Alkaloids: the α-Alkylation of Nitrogen Heterocycles via Formamidine-Mediated Chiral Carbanions. In: Pearson WH, editor. Advances in Heterocyclic Natural Product Synthesis. Vol. 1. JAI; Greenwich, CT: 1991. pp. 95–135. [Google Scholar]; (b) Meyers AI. Tetrahedron. 1992;48:2589–2612. [Google Scholar]; (c) Pippel DJ, Curtis MD, Du H, Beak P. J Org Chem. 1998;63(1):2–3. doi: 10.1021/jo972042k. [DOI] [PubMed] [Google Scholar]; (d) Bragg RA, Clayden J, Bladon M, Ichihara O. Tetrahedron Lett. 2001;42(20):3411–3414. [Google Scholar]
- 9.(a) Basu A, Beak P. J Am Chem Soc. 1996;118:1575–1576. [Google Scholar]; (b) Nakamura S, Nakagawa R, Watanabe Y, Toru T. Angew Chem Int Ed. 2000;39:353–355. doi: 10.1002/(sici)1521-3773(20000117)39:2<353::aid-anie353>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]; (c) Nakamura S, Nakagawa R, Watanabe Y, Toru T. J Am Chem Soc. 2000;122:11340–11347. [Google Scholar]; (d) Beak P, Anderson DR, Curtis MD, Laumer JM, Pippel DJ, Weisenburger GA. Acc Chem Res. 2000;33:715–727. doi: 10.1021/ar000077s. [DOI] [PubMed] [Google Scholar]; (e) Lee WK, Park YS, Beak P. Acc Chem Res. 2009;42(2):224–234. doi: 10.1021/ar8000662. [DOI] [PubMed] [Google Scholar]
- 10.Schlosser M, Limat D. J Am Chem Soc. 1995;117:12342–12343. [Google Scholar]
- 11.Meyers AI, Guiles J, Warmus JS, Gonzalez MA. Tetrahedron Lett. 1991;32:5505–5508. [Google Scholar]
- 12.Xiao D, Lavey BJ, Palani A, Wang C, Aslanian RG, Kozlowski JA, Shih NY, McPhail AT, Randolph GP, Lachowicz JE, Duffy RA. Tetrahedron Lett. 2005;46(44):7653–7656. [Google Scholar]
- 13.Sheikh NS, Leonori D, Barker G, Firth JD, Campos KR, Meijer AJHM, O’Brien P, Coldham I. J Am Chem Soc. 2012;134(11):5300–5308. doi: 10.1021/ja211398b. [DOI] [PubMed] [Google Scholar]
- 14.Coldham I, Leonori D, Beng TK, Gawley RE. Chem Commun. 2009:5239–5240. doi: 10.1039/b911024k. corrigendum with correct enthalpy and entropy values: 2010, 9267–9268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.When deuterated acetone was used, less reprotonation of the organolithium was observed. This could be due to an isotope effect or simply more thoroughly dried acetone in a small vial of the NMR-grade solvent.
- 16.McGrath MJ, O’Brien P. J Am Chem Soc. 2005;127(47):16378–16379. doi: 10.1021/ja056026d. [DOI] [PubMed] [Google Scholar]
- 17.Wu S, Lee S, Beak P. J Am Chem Soc. 1996;118:715–721. [Google Scholar]
- 18.Bilke JL, O’Brien P. J Org Chem. 2008;73(16):6452–6454. doi: 10.1021/jo8010655. [DOI] [PubMed] [Google Scholar]
- 19.(a) Soloshonok VA. Angew Chem Int Ed. 2006;45(5):766–769. doi: 10.1002/anie.200503373. [DOI] [PubMed] [Google Scholar]; (b) Soloshonok VA, Berbasov DO. J Fluorine Chem. 2006;127(4/5):597–603. [Google Scholar]; (c) Trapp O, Schurig V. Tetrahedron: Asymmetry. 2010;21(11–12):1334–1340. [Google Scholar]
- 20.Peoples PR, Grutzner JB. J Am Chem Soc. 1980;102:4709–4715. [Google Scholar]
- 21.Ahlbrecht H, Harbach J, Hoffmann RW, Ruhland T. Liebigs Ann. 1995:211–216. [Google Scholar]
- 22.Cram DJ, Gosser L. J Am Chem Soc. 1964;86:5457–5465. [Google Scholar]
- 23.(a) Fraenkel G, Cabral JA. J Am Chem Soc. 1993;115:1551–1557. [Google Scholar]; (b) Fraenkel G, Martin KV. J Am Chem Soc. 1995;117:10336–10344. [Google Scholar]
- 24.(a) Hæffner F, Brandt P, Gawley RE. Org Lett. 2002;4(12):2101–2104. doi: 10.1021/ol026041c. [DOI] [PubMed] [Google Scholar]; (b) Ashweek NJ, Brandt P, Coldham I, Dufour S, Gawley RE, Hæffner F, Klein R, Sanchez-Jimenez G. J Am Chem Soc. 2005;127:449–457. doi: 10.1021/ja048090l. [DOI] [PubMed] [Google Scholar]; (c) Yousaf TI, Williams RL, Coldham I, Gawley RE. Chem Commun. 2008:97–98. doi: 10.1039/b714302h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Beng TK, Yousaf TI, Coldham I, Gawley RE. J Am Chem Soc. 2009;131:6908–6909. doi: 10.1021/ja900755a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stead D, Carbone G, O’Brien P, Campos KR, Coldham I, Sanderson A. J Am Chem Soc. 2010;132:7260–7261. doi: 10.1021/ja102043e. [DOI] [PubMed] [Google Scholar]
- 26.Remenar JF, Lucht BL, Collum DB. J Am Chem Soc. 1997;119(24):5567–5572. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.