

Abstract
Functionally selective G protein-coupled receptor (GPCR) ligands, which differentially modulate canonical and non-canonical signaling, are extremely useful for elucidating key signal transduction pathways essential for both the therapeutic actions and side-effects of drugs. However, few such ligands have been created and very little purposeful attention has been devoted to studying what we term: ‘structure-functional selectivity relationships’ (SFSR). We recently disclosed the first β-arrestin-biased dopamine D2 receptor (D2R) agonists UNC9975 (44) and UNC9994 (36), which have robust in vivo antipsychotic drug-like activities. Here we report the first comprehensive SFSR studies focused on exploring four regions of the aripiprazole scaffold, which resulted in the discovery of these β-arrestin-biased D2R agonists. These studies provide a successful proof-of-concept for how functionally selective ligands can be discovered.
INTRODUCTION
Classical notions of receptor pharmacology imply that when a ligand interacts with a receptor, only a unitary outcome is possible. Accordingly, a full or partial agonist can activate only a single signal transduction pathway while an antagonist can only block the actions of an agonist. The key theoretical construct underlying this model was the concept of intrinsic efficacy.1 According to this conceptualization, a full agonist has maximum intrinsic efficacy and maximally stimulates all cellular responses induced by ligand binding. A partial agonist possesses a lower degree of intrinsic efficacy and partially activates all cellular responses induced by an agonist. Antagonists, according to this schema, are neutral entities which possess no intrinsic activity but are able to block the receptor and preclude activation by full or partial agonists.1 An extension of this model has been that a single G protein-coupled receptor (GPCR) interacts with a single G protein subtype and that full and partial agonists activate only a single signal transduction pathway.
For many decades, however, it has been clear that these simplistic notions of GPCR function cannot account for the myriad of actions induced by agonist and antagonist binding. This was first convincingly demonstrated for the serotonin and dopamine families of receptors. For example, it has been demonstrated that: (1) agonists differentially activate distinct signal transduction pathways;2–9 (2) antagonists can possess negative intrinsic activity (e.g., function as inverse agonists) or be silent and thereby block the actions of an inverse agonist (e.g., neutral antagonists);10–14 and (3) antagonists can also induce receptor down regulation15 and receptor internalization in vitro16 and in vivo17, 18—properties typically associated with agonists.
Functional selectivity19, 20 refers to the process by which GPCR ligands differentially modulate canonical pathways involving heterotrimeric large G proteins and non-canonical G protein-independent pathways involving other signaling proteins including β-arrestins.6, 9, 21–24 GPCR ligands with distinct functional selectivity patterns will be extremely useful tools for elucidating the key signaling pathways essential for both the therapeutic actions and the side-effects of GPCR targeted drugs.20 Understanding which signaling pathways contribute to antipsychotic efficacy and side-effects, for instance, should enable the design of better antipsychotic drug candidates and may, ultimately, lead to safer and more effective therapies for patients. Despite the importance of functionally selective ligands, only a limited number have been reported.6, 19, 20, 24–27 In addition, to our knowledge, scant attention has been devoted to studying structure-functional selectivity relationships (SFSR). We recently reported the first β-arrestin-biased dopamine D2 receptor (D2R) agonists UNC9975 (44) and UNC9994 (36), which are simultaneously inactive toward Gi-regulated cyclic adenosine monophosphate (cAMP) production and partial agonists for D2R/β-arrestin-2 interactions.28 These β-arrestin-biased D2R agonists displayed robust antipsychotic drug-like activities in wild-type mice and the antipsychotic drug-like activities were significantly attenuated or completely abolished in β-arrestin-2 knockout mice, suggesting that β-arrestin recruitment and signaling can be a significant contributor to antipsychotic efficacy.28 Here we report our SFSR studies that resulted in the discovery of these β-arrestin-biased D2R agonists. We describe the design, synthesis, and in vitro and in vivo pharmacological evaluation of novel compounds that explore four regions of the scaffold represented by aripiprazole (1), an FDA-approved atypical antipsychotic drug.24, 29, 30 These first comprehensive SFSR studies provide a successful proof-of-concept for how functionally selective ligands of GPCRs can be discovered.
RESULTS AND DISCUSSION
We selected compound 1 as the starting point for the following reasons: (1) 1 is an FDA-approved drug with excellent pharmacokinetic (PK) properties including high oral bioavailability and central nervous system (CNS) penetration28, 31—PK properties likely to be retained by its close analogs;28 (2) although some structure-activity relationships (SAR) have been reported,32–34 the SFSR of the scaffold represented by compound 1 have not been studied; and (3) this core template is amenable to rapid multi-dimensional chemical modifications and optimization, which is ideal for exploring SFSR. To determine whether modifying various structural motifs of compound 1 could result in biased compounds that favor either cAMP or β-arrestin signaling, we intensively investigated the following four regions of compound 1: (1) the left-hand side (LHS) phenyl ring with various mono- or disubstitution; (2) the middle cyclic amino moiety; (3) the central linker; and (4) the right-hand side (RHS) bicyclic aromatic moiety (Figure 1).
Figure 1.

Four regions of compound 1 investigated for SFSR.
SFSR of the LHS Phenyl Moiety
To determine the effects of substituents in the LHS phenyl ring on D2R functional selectivity, we designed the compounds outlined in Figure 2. Because Oshiro and coworkers reported that 2-substituents at the LHS phenyl were preferred for D2R and enhanced in vivo activity,33 we explored a number of electron donating or withdrawing groups at the 2-position (compounds 2 – 10). In addition, 3- and 4-substitution and 2,3-disubsitution were also investigated (compounds 1, 11 – 13).
Figure 2.

Compounds designed to explore the LHS phenyl moiety.
A representative synthesis of these compounds is shown in Scheme 1. Compounds 4, 10, and 11 were produced via nucleophilic displacement of commercially available 7-(4-bromobutoxy)-3,4-dihydroquinolin-2(1H)-one with the corresponding 2-subsituted phenylpiperazines in moderate to good yields. The 2-isopropoxy-,35 2-trifluoromethyl-,36 and 3-ethoxyphenyl-37 piperazine intermediates were prepared according to literature procedures. Synthesis of compounds 1-3, 5-9, 12, and 13 were described previously.33, 34
Scheme 1.

Synthesis of compounds with various substituents on the LHS phenyl ring.a
a Reagents and conditions: (a) NaI/K2CO3, CH3CN, reflux, 6 h, 40–70%.
The synthesized compounds were evaluated in: (1) D2R radioligand binding assay to assess their binding affinity to the receptor; (2) D2R-mediated cAMP accumulation assay, which measures inhibition of isoproterenol-stimulated cAMP production via the Gi-coupled signaling pathway; and (3) D2R-mediated β-arrestin-2 recruitment Tango assay to determine their potency and efficacy in activating β-arrestin translocation. Quinpirole, a full agonist of D2R,38 was used as the positive control in both cAMP accumulation and β-arrestin-2 recruitment Tango assays. Details of these binding and functional assays were described in our recent paper.28
Similar to compound 1, which potently activated both D2R-mediated Gi-regulated cAMP accumulation and β-arrestin-2 recruitment, compounds with or without a 2-subsituent (compounds 2 – 10) had high D2R binding affinities (Ki < 10 nM) and high potencies for activating both Gi signaling and β-arrestin-2 recruitment (Table 1). These compounds did not display apparent bias for activating either Gi signaling or β-arrestin-2 recruitment, suggesting that neither the electronic nature (electron donating or withdrawing, compounds 2, 5 – 10) nor the steric bulkiness (compounds 2 – 4) of the 2-substituent significantly modulates functional selectivity profiles. Interestingly, compounds 5 and 6 activated both Gi and β-arrestin signaling with very high potencies (EC50 < 1 nM) and efficacies (Emax similar to the positive control quinpirole). On the other hand, compound 2 was a potent, low efficacy partial agonist (Emax = ~40%) at both signaling pathways. In addition, compounds 3, 4, and 10 were about 10-fold more potent at activating β-arrestin than Gi signaling. Effects of the substitution patterns were also evaluated. Moving the ethoxy group from the ortho-position (compound 3) to the meta- (compound 11) or para-position (compound 12) resulted in significant decreases in binding affinities and agonist potencies. However, these modifications did not lead to significant changes in functional selectivity patterns. Additionally, compound 13, which possesses a 2,3-dimethyl phenyl group, displayed similar efficacies (e.g., Emax values) for activating both Gi and β-arrestin pathways (similar to compound 1) although compound 13 was significantly more potent at activating β-arrestin than Gi signaling. Overall, the electronic nature and steric bulkiness of substituents on the LHS phenyl ring and various patterns of substitution did not significantly affect patterns of D2R functional selectivity for the tested compounds.
Table 1.
SFSR of the LHS phenyl moiety.a

| ||||||
|---|---|---|---|---|---|---|
| Cmpd | R | D2R Ki (nM) | β-arrestin | cAMP | ||
|
| ||||||
| EC50 (nM) | Emax (%) | EC50 (nM) | Emax (%) | |||
|
| ||||||
| 1 | 2,3-(Cl)2 | 3.9 | 4.0 | 62 | 1.0 | 51 |
| 2 | 2-OCH3 | 0.3 | 0.6 | 46 | 2.5 | 40 |
| 3 | 2-OEt | 2.8 | 0.8 | 66 | 7.9 | 46 |
| 4 | 2-OiPr | 3.1 | 2.0 | 63 | 25 | 43 |
| 5 | 2-H | 4.3 | 0.4 | 87 | 0.8 | 90 |
| 6 | 2-F | 5.5 | 0.8 | 89 | 0.3 | 93 |
| 7 | 2-Cl | 3.7 | 2.0 | 82 | 7.9 | 68 |
| 8 | 2-CN | 2.9 | 0.8 | 77 | 5.0 | 78 |
| 9 | 2-CH3 | 5.9 | 3.2 | 81 | 16 | 56 |
| 10 | 2-CF3 | 4.2 | 2.5 | 79 | 25 | 77 |
| 11 | 3-OEt | 21 | 10 | 40 | 158 | 76 |
| 12 | 4-OEt | 53 | 50 | 82 | 251 | 69 |
| 13 | 2,3-(CH3)2 | 8.1 | 5.0 | 74 | 200 | 65 |
Ki, EC50, and Emax values are the average of at least 2 duplicate experiments with standard deviations (SD) values that are 3-fold less than the average.
SFSR of the Middle Amino Moiety
We next investigated the middle amino moiety of compound 1. Compounds 18 and 19 (Scheme 2) were designed and synthesized to modulate the basicity of the inner nitrogen and the ring size of the cyclic amino motif, respectively. 1,4-Addition of the in situ generated Grignard reagent to activated pyridinium species gave the intermediate 14 in 76% yield, which was then converted to the desired 4-aryl piperidine 16 via hydrogenation and subsequent deprotection in good overall yield. The nucleophilic displacement of the commercially available 7-(4-bromobutoxy)-3,4-dihydroquinolin-2(1H)-one with the intermediate 16 afforded the targeted compound 18 in good yield. Synthesis of the intermediate 17 and the targeted compound 19 was reported in our previous paper.28
Scheme 2.

Synthesis of compounds for exploring the middle amino moiety.a
a Reagents and conditions: (a) ClCOOCH2Ph, CuI, THF, −10 °C; (b) i-PrMgCl, THF, −20 °C, 76%; (c) H2, RhCl(PPh3)3, toluene, 70 °C, 4 d, 95%; (d) 6 N HCl, reflux, 93%; (e) NaI/K2CO3, CH3CN, reflux, 6 h, 60% for 18, 62% for 19.
These compounds were evaluated in the D2R radioligand binding, cAMP accumulation, and β-arrestin-2 recruitment assays to assess their binding affinities and patterns of D2R functional selectivity. Replacing the piperazine group (compound 1) with the more basic piperidine group (compound 18) did not result in significant changes in the efficacy of either Gi or β-arrestin signaling although compound 18 was significantly more potent at activating β-arrestin than Gi signaling (Table 2). On the other hand, replacing the piperazine group (compound 1) with the homopiperazine group (compound 19), which leads to substantial conformation changes, resulted in significant bias for β-arrestin over Gi signaling. Compound 19 was a potent partial agonist at activating β-arrestin-2 recruitment (EC50 = 2.0 nM, Emax = 41%) and was simultaneously inactive at Gi signaling. The effects of the homopiperazine moiety on biasing for β-arrestin signaling were also observed with other analogs (e.g., compounds 44 and 45 in Table 5). Both compounds 18 and 19 retained high binding affinity to D2R.
Table 2.
SFSR of the middle amino moiety.a

| ||||||
|---|---|---|---|---|---|---|
| Cmpd | Middle Amino Moiety | D2R Ki (nM) | β-arrestin | cAMP | ||
| EC50 (nM) | Emax (%) | EC50 (nM) | Emax (%) | |||
| 1 |

|
3.9 | 4.0 | 62 | 1.0 | 51 |
| 18 |
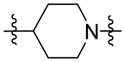
|
11 | 2.0 | 70 | 363 | 80 |
| 19 |

|
3.6 | 2.0 | 41 | N/A | < 20 |
Ki, EC50, and Emax values are the average of at least 2 duplicate experiments with standard deviations (SD) values that are 3-fold less than the average.
Table 5.
SFSR of combination compounds.a
| Cmpd | Structure | D2R Ki (nM) | β-arrestin | cAMP | ||
|---|---|---|---|---|---|---|
| EC50 (nM) | Emax (%) | EC50 (nM) | Emax (%) | |||
| 35 |

|
42 | 79 | 78 | N/A | < 20 |
| 36 |

|
75 | 50 | 97 | N/A | < 20 |
| 37 |

|
30 | 126 | 88 | N/A | < 20 |
| 38 |
|
20 | 63 | 71 | N/A | < 20 |
| 39 |
|
18 | 25 | 36 | N/A | < 20 |
| 40 |
|
11 | 20 | 80 | N/A | 32 |
| 41 |

|
104 | 200 | 78 | N/A | < 20 |
| 42 |
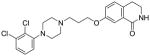
|
18 | 20 | 84 | N/A | < 20 |
| 43 |
|
5.7 | 6.3 | 41 | N/A | < 20 |
| 44 |
|
1.2 | 1.6 | 47 | N/A | < 20 |
| 45 |
|
3.4 | 2.5 | 49 | N/A | < 20 |
Ki, EC50, and Emax values are the average of at least 2 duplicate experiments with standard deviations (SD) values that are 3-fold less than the average.
SFSR of the Central Linker
To examine effects of the central linker on patterns of D2R functional selectivity, we designed the compounds outlined in Scheme 3, which contain either shorter or conformationally-constrained linkers. Compounds 22 – 24 were synthesized by a two-step sequence similar to that previously developed for compound 21.39 Thus, the commercially available 7-hydroxy-3,4-dihydroquinolin-2(1H)-one was refluxed with various dibromides or dichlorides to give the bromo or chloro intermediates, which were then reacted with commercially available 1-(2,3-dichlorophenyl)piperazine hydrochloride (20) to afford the target compounds 22 – 24. Synthesis of compounds 25 and 26 started from the piperazine 20 reacting with 1-bromo-3-(bromomethyl)benzene and 1-bromo-4-(bromomethyl)benzene to give the intermediates 27 and 28, which were then treated with 7-hydroxy-3,4-dihydroquinolin-2(1H)-one to afford the target compounds 25 and 26, respectively.
Scheme 3.

Synthesis of compounds for exploring the central linker.a
a Reagents and conditions: (a) K2CO3, EtOH, reflux, overnight, 35–80%; (b) NaI/K2CO3, CH3CN, reflux, 6 h, 40–70%; (c) Et3N, CH3CN, reflux, 4 h, 70–75%; (d) CuCl, Cs2CO3, TMHD, NMP, 120 °C, 7.5 h, 40–50%.
Results of these compounds in D2R binding, cAMP accumulation, and β-arrestin-2 recruitment assays are summarized in Table 3. Compound 21, which contains a shorter linker (3-carbon versus 4-carbon in compound 1), had slightly higher efficacy at activating β-arrestin-2 recruitment than Gi signaling although compound 21 had a lower binding affinity and agonist potency than compound 1. Interestingly, the cyclohexyl group containing compound 22, the acetylene containing compound 23, the meta-benzyl group containing compound 25, and the para-benzyl group containing compound 26 were all biased ligands for β-arrestin: partial agonists at β-arrestin-2 recruitment and simultaneously inactive at Gi signaling. However, all 4 compounds suffered from significant lower binding affinities and agonist potencies. On the other hand, the cyclopropyl group containing compound 24 did not display significant bias for activating β-arrestin-2 recruitment over Gi signaling. Taken together, these results suggest that the central linker plays an important role in modulating functional selectivity and potency of these compounds. This finding is consistent with previously reported results: the linker flexibility significantly affects ligand binding affinities and intrinsic efficacies.32–34, 40–42
Table 3.
SFSR of the central linker.a

| ||||||
|---|---|---|---|---|---|---|
| Cmpd | Middle Linker | D2R Ki (nM) | β-arrestin | cAMP | ||
| EC50(nM) | Emax (%) | EC50 (nM) | Emax (%) | |||
| 1 |
|
3.9 | 4.0 | 62 | 1.0 | 51 |
| 21 |
|
21 | 79 | 82 | 251 | 58 |
| 22 |
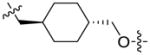
|
145 | 1,580 | 45 | N/A | < 20 |
| 23 |

|
1,004 | 199 | 45 | N/A | < 20 |
| 24 |

|
235 | 1,260 | 30 | N/A | < 20 |
| 25 |

|
113 | 316 | 57 | N/A | < 20 |
| 26 |

|
108 | 501 | 48 | N/A | < 20 |
Ki, EC50, and Emax values are the average of at least 2 duplicate experiments with standard deviations (SD) values that are 3-fold less than the average.
SFSR of the RHS Bicyclic Aromatic Moiety
We next explored the RHS bicyclic aromatic moiety of compound 1. We selected 6 different bicyclic aromatic groups (outlined in Scheme 4) to evaluate their effects on patterns of D2R functional selectivity. The previously described two-step nucleophilic displacement sequence for synthesizing compounds 29 and 3028 was applied to the preparation of compounds 31 – 34 (Scheme 4). The corresponding commercially available phenols were reacted with butyl-1,4-dibromide to give the bromo intermediates, which were then reacted with the piperazine 20 to yield the target compounds 31 – 34.
Scheme 4.

Synthesis of compounds with various RHS bicyclic aromatic groups.a
a Reagents and conditions: (a) K2CO3, EtOH, reflux, 6 h, 30–80%; (b) NaI/K2CO3, CH3CN, reflux, 6 h, 40–70%.
Results of these compounds in D2R binding, cAMP accumulation, and β-arrestin-2 recruitment assays are summarized in Table 4. In contrast to compound 1, compound 29, which contains a quinoline-2-one moiety, exhibited higher a efficacy at for activating D2R mediated β-arrestin translocation than D2R-mediated Gi activity. Similar effects were observed with compound 30 which contains a dihydro-1,8-naphthyridin-2-one moiety. Importantly, replacing the dihydroquinolin-2-one (1) with the dihydroisoquinolinone (compound 31), indazole (compound 32), benzimidazolone (compound 33), or benzothiazole (compound 34) resulted in apparent bias for D2R-mediated β-arrestin over Gi signaling. These 4 compounds were partial agonists at D2R-mediated β-arrestin-2 recruitment with moderate to high potencies and simultaneously inactive at D2R-mediated Gi signaling. In general, the RHS bicyclic aromatic moiety plays a critical role in modulating functional selectivity of these analogs. Thus, all 6 investigated RHS aryl groups exhibited some degree of bias for β-arrestin over Gi signaling.
Table 4.
SFSR of the RHS bicyclic aromatic moiety.a
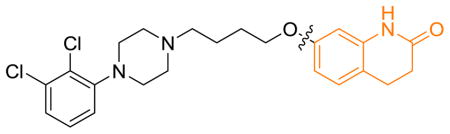
| ||||||
|---|---|---|---|---|---|---|
| Cmpd | RHS Aryl Group | D2R Ki (nM) | β-arrestin | cAMP | ||
| EC50 (nM) | Emax (%) | EC50 (nM) | Emax (%) | |||
| 1 |

|
3.9 | 4.0 | 62 | 1.0 | 51 |
| 29 |

|
7.3 | 6.3 | 79 | 158 | 29 |
| 30 |

|
3.4 | 3.2 | 73 | N/A | 41 |
| 31 |

|
18 | 4.0 | 46 | N/A | < 20 |
| 32 |
|
66 | 100 | 61 | N/A | < 20 |
| 33 |

|
15 | 16 | 65 | N/A | < 20 |
| 34 |

|
17 | 63 | 63 | N/A | < 20 |
Ki, EC50, and Emax values are the average of at least 2 duplicate experiments with standard deviations (SD) values that are 3-fold less than the average.
Synthesis and in vitro Pharmacological Evaluation of Combination Compounds
After exploring the above 4 regions of compound 1 and studying their SFSR, we next designed and synthesized a number of combination compounds (outlined in Scheme 5), which combined the structural motifs that contribute to the bias for β-arrestin recruitment into single molecules. We selected all 3 middle amino groups, 3- and 4-carbon linkers, and all 6 RHS aromatic groups for this study. The synthetic routes for these combination compounds were shown in Scheme 5. These compounds (35 – 45) were prepared following the same synthetic approach developed for compounds 29 – 34 using the corresponding amino groups, central linkers, and RHS aromatic groups.
Scheme 5.

Synthesis of combination compounds.a
a Reagents and conditions: (a) K2CO3, EtOH, reflux, 6 h, 30–80%; (b) NaI/K2CO3, CH3CN, reflux, 6 h, 40–70%.
We next evaluated compounds 35 – 45 in the D2R radioligand binding, cAMP accumulation, and β-arrestin-2 recruitment assays (results are summarized in Table 5). As expected, these combination compounds were all significantly biased for D2R-mediated β-arrestin over Gi signaling. With the exception of compound 40, all compounds were agonists at D2R mediated β-arrestin-2 recruitment with moderate to high potencies and simultaneously inactive at D2R-mediated Gi signaling. Most notably, compounds 36 and 37 were the most biased for β-arrestin with Emax values similar to the positive control quinpirole. Notably, compounds 44 and 45 had extremely high potencies (EC50 < 5 nM) but relatively low efficacies (Emax = 40 – 50%) at D2R-mediated β-arrestin translocation. These unique patterns of D2R-mediated functional selectivity profiles will be extremely useful for elucidating which signaling pathways contribute to antipsychotic efficacies and side-effects.
From these SFSR studies, we observed the following general trends: (1) the electronic nature (e.g., electron donating or withdrawing), steric bulkiness, and substitution pattern of substituents on the LHS phenyl ring did not, apparently, have significant effects on patterns of D2R functional selectivity; (2) the homopiperazine group as a middle amino moiety reduced efficacy for activating both β-arrestin and Gi pathways; (3) several conformationally-constrained central linkers could lead to significant bias for D2R-mediated β-arrestin-2 over Gi activities. However, these central linkers resulted in significant losses of potency; and (4) a number of RHS aromatic groups such as benzothiazole, dihydroisoquinolinone, indazole, and benzimidazolone led to significant bias for D2R-mediated β-arrestin-2 over Gi activities. Importantly, we observed that subtle structural changes could result in substantial changes in functional selectivity.
We subsequently confirmed that compounds 19, 36, and 44 were β-arrestin-biased agonists in several orthologous β-arrestin-2 translocation and signaling assays.28
In addition, we determined selectivity of compounds 19, 35, 36, and 44 against a number of dopamine and serotonin receptors. Compound 19 displayed high affinities to D2 and D3 receptors and low affinities to D1, D4, and D5 receptors while compound 35 displayed high affinities to D2, D3, and D4 receptors and low affinities to D1 and D5 receptors (Table S1). At serotonin receptors, although compounds 19 and 35 displayed moderate to high binding affinities (Ki’s: 0.6 – 138 nM) for 5-HT2A, 5-HT2B, 5-HT2C, and 5-HT1A (Table S1), compound 19 was significantly less potent in functional assays (Ca2+ mobilization fluorometric imaging plate reader (FLIPR) or cAMP biosensor).28 Similarly, compounds 36 and 44 displayed much lower functional potencies compared to their binding affinities to 5-HT2A, 5-HT2B, 5-HT2C, and 5-HT1A receptors.28 Selectivity of compounds 36 and 44 against dopamine D1 – D5 receptors was reported previously.
In vivo Behavioral Studies in Mice
We previously reported that compounds 36 and 44 displayed robust antipsychotic drug-like activities and did not induce catalepsy in wild-type mice.28 In β-arrestin-2 knockout mice, however, the antipsychotic drug-like activities of these compounds were significantly attenuated or completely abolished.28 To extend and confirm these findings, we evaluated the effect of two additional β-arrestin-biased D2R agonists (compound 19 and 35) on phencyclidine (PCP)-induced hyperlocomotion43 in β-arrestin-2 knockout mice and the wild-type littermate controls.44 Compound 19 (2 mg/kg, intraperitoneal (i.p.) administration) markedly inhibited PCP-induced hyperlocomotion in wild-type mice. Importantly, this significant antipsychotic-like activity of compound 19 was completely abolished in β-arrestin-2 knockout mice (Figure 3). Similarly, compound 35 (2 mg/kg, i.p.) significantly reduced PCP-induced hyperlocomotion in wild-type mice and this antipsychotic-like activity was significantly attenuated in β-arrestin-2 knockout mice (Figure 3). Taken together, these results suggest that β-arrestin recruitment and signaling can be a significant contributor to antipsychotic efficacy.
Figure 3. Compounds 19 and 35 exhibit potent antipsychotic-like activities in mouse hyperlocomotion studies which are completely abolished or significantly attenuated in β-arrestin-2 knockout mice.

Locomotor activities shown as 5-min binned intervals for wild-type (WT) or β-arrestin-2 knockout (β-ARR2 KO) littermate mice given vehicle or 2.0 mg/kg 19 or 35 (i.p.) followed 30 min later with 6 mg/kg phencyclidine (PCP, i.p.). n = 8–13 WT and β-ARR2 KO pairs/group.
CONCLUSION
In summary, we designed and synthesized a series of novel compounds for exploring four regions of the scaffold represented by compound 1. Comprehensive evaluation of these compounds in D2R radioligand binding, cAMP accumulation, and β-arrestin-2 recruitment assays revealed a number of important SFSR findings. Combining the best structural motifs identified from these studies into single molecules resulted in the discovery of extremely β-arrestin-biased D2R agonists 35 – 37 and high potency and low efficacy β-arrestin-biased D2R agonists 19 and 44. Findings from our in vivo studies of these β-arrestin-biased D2R agonists in wild-type and β-arrestin-2 knockout mice suggest that β-arrestin recruitment and signaling can be a significant contributor to antipsychotic efficacy. Our combined medicinal chemistry and pharmacological profiling approach provides the biomedical community a successful proof-of-concept for how functionally selective ligands can be discovered.
EXPERIMENTAL SECTION
Chemistry General Procedures
Unless stated to the contrary, where applicable, the following conditions apply: all commercial grade reagents were used without further purification. MeCN and CH2Cl2 were distilled from CaH2 under a N2 atmosphere before use; THF was distilled from Na/benzophenone under N2. All other dry solvents were of anhydrous quality purchased from Sigma-Aldrich. Brine (NaCl), NaHCO3, and NH4Cl refer to saturated aqueous (sat aq.) solutions. Column chromatography was performed on silica gel G (200–300 mesh) with reagent grade solvents. Melting points were uncorrected. NMR spectra were recorded on a Varian spectrometer (400 MHz or 300 MHz for 1H NMR and 100 MHz or 75MHz for 13C NMR, respectively) at ambient temperature. All 1H and 13C chemical shifts are reported in ppm (δ) relative to CDCl3 (7.26 and 77.16, respectively) or CD3OD (3.30 and 49.00, respectively).45 HPLC data for all compounds were acquired using an Agilent 6110 series system with a UV detector set to 220 nm. Samples were injected (<10 μL) onto an Agilent Eclipse Plus 4.6 × 50 mm, 1.8 μm, C18 column at room temperature (rt) at a flow rate of 1.0 mL/min. A linear gradient from 10% to 100% (vol/vol) B over 5.0 min followed by 2.0 min at 100% B with a mobile phase of (A) H2O + 0.1% acetic acid and (B) MeOH + 0.1% acetic acid was used. Mass spectra (MS) data were acquired in positive ion mode using an Agilent 6110 series single quadrupole mass spectrometer with an electrospray ionization (ESI) source. High-resolution (positive ion) mass spectrum (HRMS) for compound 35 was acquired using a Shimadzu LCMS-IT-Tof time-of-flight mass spectrometer. HRMS (positive ion) for compounds 37, 38, 41 and 42 were recorded on Agilent 6210 ESI-LCT-TOF mass spectrometer with dual source for reference and sample. HPLC was used to establish the purity of targeted compounds. All compounds that were evaluated in biological assays had >95% purity using the HPLC methods described above.
Compounds 1-3, 5-9, 12 and 13 were synthesized as previously reported.31,33
7-(4-(4-(2-Isopropoxyphenyl)piperazin-1-yl)butoxy)-3,4-dihydroquinolin-2(1H)-one (4)
A mixture of intermediate 7-(4-bromobutoxy)-3,4-dihydroquinolin-2(1H)-one (119 mg, 0.4 mmol) and NaI (119.6 mg, 0.8 mmol) in CH3CN was heated to reflux for 30 min and then cooled to rt. Compound 1-(2-isopropoxyphenyl)piperazine (97.0 mg, 0.44 mmol) and anhydrous K2CO3 (121.6 mg, 0.88 mmol) were added to the mixture. The resulting mixture was heated to reflux and stirred for 6 h. Precipitated crystals were filtered off and the filtrate was evaporated under reduced pressure. The residue was extracted with EtOAc. The combined EtOAc layers were washed with brine, dried over anhydrous Na2SO4, concentrated in vacuo and purified by flash chromatography on silica gel column (elution with DCM/MeOH = 30:1) to give compound 4 as light yellow solid (75.3 mg, 43 %). 1H NMR (400 MHz, CDCl3): δ 8.59 (br s, 1H), 8.24 (d, J = 8.0 Hz, 1H), 7.41 (app t, J = 7.6 Hz, 1H), 7.06 (d, J = 8.4 Hz, 1H), 7.02 – 6.96 [m, 2H, containing a d at δ 7.00 (J = 8.4 Hz)], 6.49 (s, 1H), 6.46 (d, J = 8.4 Hz, 1H), 4.99 – 4.86 (m, 2H), 4.80 (sept, J = 6.0 Hz, 1H), 4.63 – 4.49 (m, 2H), 4.01 – 3.92 (m, 2H), 3.76 – 3.61 (m, 4H), 3.31 – 3.20 (m, 2H), 2.85 (app t, J = 7.4 Hz, 2H), 2.56 (app t, J = 7.4 Hz, 2H), 2.20 – 2.08 (m, 2H), 1.91 – 1.81 (m, 2H), 1.56 (d, J = 6.0 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ 171.7, 158.2, 151.1, 138.5, 132.3, 128.8, 128.2, 124.7, 121.4, 116.2, 114.7, 108.9, 102.6, 72.7, 67.2, 57.3, 49.8, 48.0, 31.2, 26.4, 24.7, 21.9, 20.9; HPLC 99%, RT 4.39 min; MS (ESI) m/z 438.3 [M + H]+.
7-(4-(4-(2-(Trifluoromethyl)phenyl)piperazin-1-yl)butoxy)-3,4-dihydroquinolin-2(1H)-one (10)
Compound 10 (188 mg) was prepared as white solid by the same procedure as preparing 4, yield 62%. 1H NMR (400MHz, CDCl3): δ 8.17 (br s, 1H), 7.62 (d, J = 8.0 Hz, 1H), 7.52 (app t, J = 7.8Hz, 1H), 7.40 (d, J = 8.0Hz, 1H), 7.23 (app t, J = 7.6Hz, 1H), 7.04 (d, J = 8.4 Hz, 1H), 6.52 (dd, J = 2.4, 8.4Hz, 1H), 6.34 (d, J = 2.4Hz, 1H), 4.01 – 3.92 (m, 2H), 3.18 – 2.93 (m, 4H), 2.92 – 2.44 [m, 10H, containing an app t at δ 2.89 (J = 7.6Hz) and an app t at δ 2.61 (J = 7.6Hz)], 1.93 – 1.71 (m, 4H); 13C NMR (101 MHz, CDCl3): δ 171.8, 158.7, 138.3, 133.0, 129.3, 128.8, 127.5 (q, JCF = 29.0 Hz), 127.3 (q, JCF = 5.4 Hz), 125.2, 124.3, 124.2 (q, JCF = 274.4 Hz), 115.9, 108.8, 102.3, 67.9, 58.2, 53.5, 53.0, 31.2, 27.3, 24.8, 23.1; HPLC 99%, RT 4.36 min; MS (ESI) m/z 448.3 [M + H]+.
7-(4-(4-(3-Ethoxyphenyl)piperazin-1-yl)butoxy)-3,4-dihydroquinolin-2(1H)-one (11)
Compound 11 (48 mg) was prepared as white solid by the same procedure as preparing compound 4, yield 57%. 1H NMR (400MHz, CDCl3): δ 7.76 (s, 1H), 7.15 (t, J = 8.1 Hz, 1H), 7.04 (d, J = 8.1 Hz, 1H), 6.56 – 6.49 (m, 2H), 6.47 (s, 1H), 6.40 (d, J = 8.1 Hz, 1H), 6.29 (s, 1H), 4.06 – 3.91 (m, 4H), 3.25 – 3.14 (m, 4H), 2.90 (t, J = 7.4 Hz, 2H), 2.67 – 2.56 (m, 6H), 2.46 (t, J = 7.2 Hz, 2H), 1.88 – 1.77 (m, 2H), 1.76 – 1.66 (m, 2H), 1.40 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 171.6, 160.1, 158.8, 152.8, 138.2, 129.9, 128.9, 115.9, 108.9, 108.8, 105.1, 103.2, 102.3, 68.1, 63.5, 58.4, 53.4, 49.19, 31.3, 27.4, 24.8, 23.6, 15.1; HPLC 99%, RT 4.26 min; MS (ESI) m/z 424.3 [M + H]+.
(4-(2,3-Dichlorophenyl)pyridin-1(4H)-yl)(phenyl)methanone (14)
To a −30 °C solution of 1-bromo-2,3-dichlorobenzene (10 g, 44 mmol) in THF (120 mL) was added i-PrMgCl (2.0 M in THF, 35 mL, 70 mmol) at a rate such that the temperature < −20 °C. Meanwhile, to a −10 °C solution of CuI (420 mg, 2.2 mmol) in THF (120 mL) was added pyridine (7.1 mL, 88 mmol) and then benzyl carbonochloridate (9.7 mL, 68 mmol) such that the temperature < 0 °C. To this heterogeneous mixture was added the initially formed Grignard at a rate such that the temperature < 0 °C. The resulting solution was stirred at 0 °C for 30 min and then allowed to warm to rt. The reaction was then quenched with 10% aq NH4Cl. EtOAc was added and the blue aqueous layer was removed. The organic layer was washed with 10% aq NH4Cl, 1 N HCl, and a 20% aq NaCl solution. The organic layer was then concentrated and the residue was dissolved and crystallized from MeOH. The slurry was filtered and the filtercake washed with MeOH to give benzyl 4-(2,3-dichlorophenyl)pyridine-1(4H)-carboxylate (14) (12 g, 76%) as an off-white solid. 1H NMR (300 MHz, CDCl3) δ 7.42 – 7.21 (m, 8H), 7.04 (d, J = 9.2 Hz, 1H), 6.92 (d, J = 6.4 Hz, 1H), 5.25 (s, 2H), 5.02 (d, J = 6.4 Hz, 1H), 4.93 (d, J = 9.2 Hz, 1H), 4.73 – 4.71 (m, 1H). HPLC: 99%, RT 4.069 min. MS (ESI) m/z 360.0 [M + H]+.
(4-(2,3-Dichlorophenyl)piperidin-1-yl)(phenyl)methanone (15)
To a solution of intermediate 15 (7.0 g, 19.5 mmol) in toluene (150 mL) was added RhCl(PPh3)3 (2.1 g, 2.0 mmol) as a slurry in toluene (50 mL). The reaction was subjected to an atmosphere of H2 at 40 psi and heated to 70 °C. After 6 h, the reaction was filtered through silica gel and washed with 1:9 EtOAc/toluene. The filtrate was dissolved in toluene, concentrated in vacuo, and purified by flash chromatography on silica gel column (elution with PE/EtOAc = 50:1) to give benzyl 4-(2,3-dichlorophenyl)piperidine-1-yl)(phenyl)methanone (15) (6.8 g, 96%) as an oil. 1H NMR (300 MHz, CDCl3) δ 7.41 – 7.28 (m, 5H), 7.26 – 7.25 (m, 1H), 7.18 (t, J = 7.8 Hz, 1H), 7.13 – 7.10 (m, 1H), 5.16 (s, 2H), 4.35 (d, J = 12.0 Hz, 2H), 3.24 – 3.19 (m, 1H), 2.93 (t, J = 12.6 Hz, 2H), 1.86 (d, J = 13.0 Hz, 2H), 1.59 (t, J = 10.5 Hz, 2H). HPLC: 99%, RT 3.881 min. MS (ESI) m/z 364.0 [M + H]+.
4-(2,3-Dichlorophenyl)piperidine hydrochloride (16)
To 6N HCl (30 mL) was added a solution of compound 15 (5.2 g, 14 mmol) in THF (10 mL). The mixture was heated to reflux for 3 h and then concentrated in vacuo. The residue was washed with Et2O to give 4-(2,3-dichlorophenyl)piperidine hydrochloride (16) (3.5 g, 92%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 7.48 – 7.42 (m, 1H), 7.36 – 7.31 (m, 2H), 3.51 – 3.41 (m, 3H), 3.33 – 3.15 (m, 3H), 2.11 – 2.07 (m, 2H), 1.99 – 1.85 (m, 2H). HPLC: 99%, RT 2.090 min. MS (ESI) m/z 230.0 [M + H]+.
7-(4-(4-(2,3-Dichlorophenyl)piperidin-1-yl)butoxy)-3,4-dihydroquinolin-2(1H)-one (18)
Compound 18 (106.8 mg) was prepared as white solid by the same procedure as preparing compound 4 from intermediate 7-(4-bromobutoxy)-3,4-dihydroquinolin-2(1H)-one (119 mg, 0.4 mmol) and compound 16 (117.3 mg, 0.44 mmol), yield 60%. 1H NMR(300 MHz, CDCl3): δ 7.85 (s, 1H), 7.33 (d, J = 8.1 Hz, 1H), 7.23 – 7.17 (m, 2H), 7.05 (d, J = 8.1 Hz, 1H), 6.54 (dd, J = 2.1 Hz, 8.1 Hz, 1H), 6.31 (d, J = 2.1 Hz, 1H), 3.96 (t, J = 6.0 Hz, 2H), 3.15-3.05 (m, 3H), 2.90 (t, J =7.3 Hz, 2H), 2.62 (t, J = 7.3 Hz, 2H), 2.50 (t, J = 7.2 Hz, 2H), 2.30-2.10 (m, 2H), 1.91 – 1.72 (m, 8H). HPLC: 99%, RT 2.504 min. MS (ESI) m/z 447.2 [M + H]+.
Intermdiate 17 and compounds 19, 21 were prepared according to previous procedures.27, 37,39
Trans-7-((4-((4-(2,3-dichlorophenyl)piperazin-1-yl)methyl)cyclohexyl)methoxy)-3,4-dihydroquinolin-2(1H)-one (22)
A mixture of 7-hydroxy-3,4-dihydroquinolin-2(1H)-one (105 mg, 0.65 mmol), 1,4-bis(bromomethyl)cyclohexane (523 mg, 1.9 mmol) and anhydrous K2CO3 (89 mg, 0.65 mmol) was dissolved in EtOH and the solution was heated to reflux for 6 hours. The solution was diluted with water and extracted with EtOAc. The combined organic layers were washed with saturated aq NaHCO3, brine, dried over anhydrous Na2SO4, concentrated in vacuo and purified by flash chromatography on silica gel column to give 7-((4-(bromomethyl)cyclohexyl)methoxy)-3,4-dihydroquinolin-2(1H)-one (70 mg, 30%, 0.2 mmole) as a white solid, which was re-dissolved in CH3CN. To this mixture was added NaI (60 mg, 0.4 mmol) and the reaction mixture was heated to reflux for 30 min and then cooled to rt. The commercial available compound 20 (81 mg, 0.3 mmol) and anhydrous K2CO3 (110 mg, 0.8 mmol) were added to the mixture. The resulting mixture was heated to reflux and stirred for 6 h. Precipitated crystals were filtered off and the filtrate was evaporated under reduced pressure. The residue was extracted with EtOAc. The combined EtOAc layers was washed with brine, dried over anhydrous Na2SO4, concentrated in vacuo and purified by flash chromatography on silica gel column (elution with DCM/MeOH = 50:1) to give trans-7-((4-((4-(2,3-dichlorophenyl)piperazin-1-yl)methyl)cyclohexyl)methoxy)-3,4-dihydroquinolin-2(1H)-one (22) as white solid (63 mg, yield 63%). 1H NMR (300 MHz, CDCl3): δ 8.06 (s, 1H), 7.15 – 7.13 (m, 2H), 7.04 (d, J = 8.4 Hz, 1H), 6.97 – 6.94 (m, 1H), 6.52 (dd, J = 2.4 Hz, 8.4 Hz, 1H), 6.32 (d, J = 2.1 Hz, 1H), 3.74 – 3.71 (m, 2H), 3.07 – 3.05 (m, 4H), 2.92 – 2.87 (m, 2H), 2.64 – 2.59 (m, 6H), 2.24 (d, J = 6.9 Hz, 2H), 1.83 (d, J = 6.9 Hz, 2H), 1.53 – 1.69 (m, 4H), 1.10 – 0.94 (m, 4H). HPLC: 99%, RT 2.652 min. MS (ESI) m/z 502.2 [M + H]+.
7-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)but-2-ynyloxy)-3,4-dihydroquinolin-2(1H)-one (23)
Compound 23 (280 mg) was prepared as yellow solid by the same procedure as prepaing 22, yield 79%. 1H NMR(400 MHz, CDCl3): δ 7.62 (s, 1H), 7.20 – 7.11 (m, 2H), 7.07 (d, J = 8.3 Hz, 1H), 6.96 (dd, J = 7.1, 2.5 Hz, 1H), 6.62 (dd, J = 8.3, 2.5 Hz, 1H), 6.39 (d, J = 2.4 Hz, 1H), 4.72 (t, J = 1.7 Hz, 2H), 3.41 (t, J = 1.7 Hz, 2H), 3.08 (bs, 4H), 2.93 – 2.85 (m, 2H), 2.78 – 2.68 (m, 2H), 2.63 – 2.57 (m, 2H). HPLC: 99%, RT 2.403 min. MS (ESI) m/z 444.1 [M + H]+.
Trans-7-((2-((4-(2,3-dichlorophenyl)piperazin-1-yl)methyl)cyclopropyl)methoxy)-3,4-dihydroquinolin-2(1H)-one (24)
Compound 24 (82 mg) was prepared as white solid by the same procedure as prepaing 22, yield 48%. 1H NMR (400 MHz, CDCl3): δ 7.69 (s, 1H), 7.19 – 7.09 (m, 2H), 7.03 (d, J = 8.2 Hz, 1H), 6.98 – 6.91 (m, 1H), 6.57 (d, J = 8.4 Hz, 1H), 6.35 (s, 1H), 3.89 (s, 2H), 3.04 (s, 4H), 2.88 (t, J = 7.3 Hz, 2H), 2.80 – 2.56 (m, 6H), 2.46 (s, 2H), 0.65 (s, 2H), 0.48 (s, 2H). 13C NMR (101 MHz, CDCl3): δ 171.6, 159.2, 151.5, 138.1, 134.2, 128.7, 127.6, 127.6, 124.6, 118.7, 115.8, 109.1, 102.5, 72.3, 62.8, 53.7, 51.4, 31.3, 24.8, 18.0, 9.5, 2.1. HPLC: 99%, RT 2.541 min. MS (ESI) m/z 460.1 [M + H]+.
1-(3-Bromobenzyl)-4-(2,3-dichlorophenyl)piperazine (27)
A mixture of 1-(2,3-dichlorophenyl)piperazine hydrochloride (20) (294 mg, 1.1 mmol), 1-bromo-3-(bromomethyl)benzene (250 mg, 1 mmol) and anhydrous triethylamine (253 mg, 2.5 mmol) was dissolved in CH3CN and the solution was heated to reflux for 4 h. The solution was diluted with water and extracted with EtOAc. The combined organic layers were washed with saturated aq NaHCO3, brine, dried over anhydrous Na2SO4, concentrated in vacuo and purified by flash chromatography on silica gel column (elution with PE/EtOAc = 8:1) to give 1-(3-bromobenzyl)-4-(2,3-dichlorophenyl)piperazine (27) (220 mg, 74%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 7.54 (s, 1H), 7.40 (d, J = 7.8 Hz, 1H), 7.28 (d, J = 7.8 Hz, 1H), 7.21 (d, J = 7.8 Hz, 1H), 7.17 – 7.10 (m, 2H), 6.97 – 6.94 (m, 1H), 3.55 (s, 2H), 3.07 (br, 4H), 2.64 (br, 4H). HPLC: 99%, RT 2.542 min. MS (ESI) m/z 399.1 [M + H]+.
7-(3-((4-(2,3-Dichlorophenyl)piperazin-1-yl)methyl)phenoxy)-3,4-dihydroquinolin-2(1H)-one (25)
To a solution of 7-hydroxy-3,4-dihydroquinolin-2(1H)-one (196 mg, 1.2 mmol in NMP was added Cs2CO3 (391 mg, 1.2 mmol). The slurry was degassed by evacuating and filling the reaction flask with N2 three times. Compound 27 (240 mg, 0.6 mmol) and TMHD (11 mg, 0.06 mmol) were added followed by the addition of CuCl (60 mg, 0.6mmol). The reaction mixture was degassed by evacuating and filling the reaction flask with N2 three times, and then warmed to 120 °C under N2 for 7.5 h. The reaction mixture was cooled to rt and diluted with Et2O. The slurry was filtered and the filtercake was washed with Et2O. Combined filtrates were washed with 2 N HCl, 0.6 N HCl, 2 M NaOH, and 10% aq NaCl. The resulting organic layer was dried, concentrated, and purified by flash chromatography on a silica gel column (elution with PE/EtOAc = 1:1) to give 7-(3-((4-(2,3-dichlorophenyl)piperazin-1-yl)methyl)phenoxy)-3,4-dihydro quinolin-2(1H)-one (Compound 25) (off-white solid, 110 mg, 38%). 1H NMR(300 MHz, CDCl3): δ 7.62 (s, 1H), 7.31 (d, J = 7.2 Hz, 2H), 7.15-7.05 (m, 4H), 6.96-6.89 (m, 2H), 6.62 (dd, J = 2.1 Hz, 8.1Hz, 1H), 6.41 (d, J = 2.1Hz, 1H), 3.57 (s, 2H), 3.06 (m, 4H), 2.94 (t, J = 7.5Hz, 2H), 2.66-2.61 (m, 5H). HPLC: 99%, RT 2.637 min. MS (ESI) m/z 482.1 [M + H]+. Mp: 182–183 °C.
1-(4-Bromobenzyl)-4-(2,3-dichlorophenyl)piperazine (28)
Compound 28 (220 mg) was prepared as white solid from 20 (294 mg, 1.1 mmol), 1-bromo-4-(bromomethyl) benzene (250 mg, 1.0 mmol) and anhydrous triethylamine (253 mg, 2.5 mmol) by the same procedure as preparing 27, yield 74%. 1H NMR (300 MHz, CDCl3) δ 7.46 (d, J = 8.1 Hz, 2H), 7.27 – 7.23 (m, 2H), 7.15 – 7.13 (m, 2H), 6.96 – 6.93 (m, 2H), 3.54 (s, 2H), 3.06 (br, 4H), 2.63 (m, 4H). HPLC: 99%, RT 2.541 min. MS (ESI) m/z 398.9 [M + H]+.
7-(4-((4-(2,3-Dichlorophenyl)piperazin-1-yl)methyl)phenoxy)-3,4-dihydroquinolin-2(1H)-one (26)
Compound 26 (120 mg) was prepared as off-white solid by the same procedure as preparing 25, yield 35%. 1H NMR (300 MHz, CDCl3): δ 7.67 (s, 1H), 7.32 (d, J = 8.7 Hz, 2H), 7.16 – 7.09 (m, 3H), 6.98 – 6.94 (m, 3H), 6.64 – 6.61 (m, 1H), 6.41 (d, J = 3.0Hz, 1H), 3.56 (s, 2H), 3.07 (m, 4H), 2.94 (t, J = 6.6 Hz, 2H), 2.66 – 2.61 (m, 6H). HPLC: 99%, RT 2.626 min. MS (ESI) m/z 482.1 [M + H]+. Mp: 185–186 °C.
Compounds 29 and 30 were synthesized as previously described.27,32
7-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butoxy)-3,4-dihydroisoquinolin-1(2H)-one (31)
Compound 31 (215 mg) was prepared as white solid by the same procedure as preparing 22, yield 65%. 1H NMR (300 MHz, CDCl3): δ 7.58 (d, J = 2.7 Hz, 1H), 7.14 – 7.11 (m, 3H), 7.01 – 6.94 (m, 2H), 6.18 (brs, 1H), 4.05 (t, J = 6.3 Hz, 2H), 3.56 – 3.51 (m, 2H), 3.07 (m, 4H), 2.90 (t, J = 8.1 Hz, 2H), 2.66 (m, 4H), 2.49 – 2.46 (m, 2H), 1.86 – 1.68 (m, 4H). HPLC: 99%, RT 2.374 min. MS (ESI) m/z 448.3 [M + H]+.
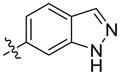
6-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butoxy)-1H-indazole (32)
Compound 32 (off-white solid, 35 mg) was prepared by the same procedure as preparing 22, yield 45%. 1H NMR (300 MHz, CDCl3): δ 7.97 (s, 1H), 7.61 (d, J = 8.7 Hz 1H), 7.17 – 7.15 (m, 2H), 6.96(dd, J = 3.0 Hz, 6.6 Hz, 1H), 6.86 – 6.81 (m, 2H), 4.05 (t, J = 5.4 Hz, 2H), 3.16 (s, 4H), 2.79 (s, 4H), 2.64 – 2.60 (m, 2H), 1.89 – 1.83 (m, 4H). HPLC: 99%, RT 2.491 min. MS (ESI) m/z 419.2 [M + H]+. mp: 101–103 °C.
5-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butoxy)-1H-benzo[d]imidazol-2(3H)-one (33)
Compound 33 (54 mg) was prepared as white solid by the same procedure as preparing 22, yield 42%. 1H NMR (300 MHz, CDCl3): δ 9.53 (s, 1H), 9.24 (s, 1H), 7.17 – 7.13 (m, 2H), 6.97 – 6.91 (m, 2H), 6.66 – 6.60 (m, 2H), 3.95 (t, J = 5.7 Hz, 2H), 3.20 – 3.01 (m, 4H), 2.80 – 2.61 (m, 4H), 2.52 (t, J = 7.5 Hz, 2H), 1.82 – 1.73(m, 4H). HPLC: 99%, RT 2.325 min. MS (ESI) m/z 435.1 [M + H]+.
6-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butoxy)benzo[d]thiazole (34)
Compound 34 (light yellow soild, 98 mg) was prepared by the same procedure as preparing 22, yield 60%. 1H NMR (300 MHz, CDCl3): δ 8.97 (s, 1H), 7.79 (d, J = 8.7 Hz, 1H), 7.61 (s, 1H), 7.16 – 7.08 (m, 3H), 6.97 – 6.94 (m, 1H), 4.11 (t, J = 5.7 Hz, 2H), 3.09 (br, 4H), 2.68 (brs, 4H), 2.55 – 2.50 (m, 2H), 1.92 – 1.76 (m, 4H). HPLC: 99%, RT 2.651 min. MS (ESI) m/z 436.3 [M+H]+. mp: 93–94.5 °C.
5-(4-(4-(2,3-Dichlorophenyl)piperidin-1-yl)butoxy)benzo[d]thiazole (35)
Compound 35 (144 mg) was prepared as light yellow solid by the same procedure as preparing 22, yield 66%. 1H NMR (400 MHz, CDCl3): δ 8.97 (s, 1H), 7.79 (d, J = 8.8 Hz, 1H), 7.61 (d, J = 2.4 Hz, 1H), 7.31 (dd, J = 7.6, 1.8 Hz, 1H), 7.23 – 7.12 (m, 2H), 7.09 (dd, J = 8.8, 2.5 Hz, 1H), 4.10 (t, J = 6.3 Hz, 2H), 3.16 – 3.01 (m, 3H), 2.55 – 2.45 (m, 2H), 2.14 (t, J = 10.9 Hz, 2H), 1.96 – 1.68 (m, 8H). 13C NMR (101 MHz, CDCl3) δ 158.6, 155.0, 154.8, 133.2, 131.9, 128.2, 127.5, 125.6, 125.5, 122.1, 116.6, 106.6, 68.3, 58.7, 54.4, 39.9, 32.1, 27.4, 23.7. HPLC: 99%, RT 2.689 min. MS (ESI) m/z 435.3 [M + H]+. HRMS m/z [M + H]+ calcd for C22H25Cl2N2OS 435.1065, found 435.1039. mp:79–81 °C.
Synthesis of compound 36 was described previously.27
5-(3-(4-(2,3-Dichlorophenyl)piperazin-1-yl)propoxy)benzo[d]thiazole (37)
Compound 37 (light yellow solid, 209 mg) was prepared by the same procedure as preparing 22, yield 62%. 1H NMR (400 MHz, CDCl3): δ 8.97 (s, 1H), 7.80 (d, J = 8.8 Hz, 1H), 7.62 (d, J = 2.4 Hz, 1H), 7.15 (dd, J = 7.0, 4.7 Hz, 2H), 7.10 (dd, J = 8.8, 2.5 Hz, 1H), 6.97 (dd, J = 6.6, 2.8 Hz, 1H), 4.15 (t, J = 6.3 Hz, 2H), 3.11 (bs, 4H), 2.81 – 2.61 (m, 6H), 2.15 – 2.05 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 158.5, 155.1, 154.8, 151.3, 134.2, 127.7, 127.6, 125.7, 124.8, 122.2, 118.8, 116.5, 106.7, 66.7, 55.3, 53.4, 51.3, 26.7. HPLC: 99%, RT 2.607 min. MS (ESI) m/z 422.0 [M + H]+. HRMS m/z [M + H]+ calcd for C20H22Cl2N3OS 422.0861, found 422.0885. mp: 127–129 °C.
5-(3-(4-(2,3-Dichlorophenyl)-1,4-diazepan-1-yl)propoxy)benzo[d]thiazole (38)
Compound 38 (light brown solid, 98 mg) was prepared by the same procedure as preparing 22, yield 60%. 1H NMR (400 MHz, CD3OD) δ 9.91 (s, 1H), 8.10 (t, J = 8.7 Hz, 1H), 7.65 (s, 1H), 7.34 (t, J = 7.7 Hz, 1H), 7.29 – 7.15 (m, 3H), 4.31 (t, J = 5.2 Hz, 2H), 3.87 – 3.71 (m, 2H), 3.67 – 3.43 (m, 6H), 3.42 – 3.29 (m, 2H), 2.48 – 2.24 (m, 4H). 13C NMR (101 MHz, CD3OD) δ 162.1, 160.9, 152.9, 134.9, 129.1, 128.6, 126.4, 125.8, 125.2, 122.1, 119.5, 119.2, 103.6, 67.2, 56.5, 56.3, 54.6, 53.4, 50.9, 25.8, 25.5. HPLC: 99%, RT 2.601 min. MS (ESI) m/z 436.3 [M + H]+. HRMS m/z [M + H]+ calcd for C21H24Cl2N3OS 436.1017, found 436.1038. mp: 93–94 °C.
5-(4-(4-(2,3-Dichlorophenyl)-1,4-diazepan-1-yl)butoxy)benzo[d]thiazole (39)
Compound 39 (light brown solid, 98 mg) was prepared by the same procedure as preparing 22, yield 60%. 1H NMR (300 MHz, CDCl3) δ 8.97(s, 1H), 7.80 (d, J = 8.7 Hz, 1H), 7.60 (d, J = 3.0 Hz, 1H), 7.11 – 7.07 (m, 3H), 7.01 – 6.98 (m, 1H), 4.10 (t, J = 5.7 Hz, 2H), 3.76 – 3.68 (m, 2H), 3.33 – 3.27 (m, 4H), 2.96 – 2.95 (m, 4H), 2.73 – 2.71 (m, 2H), 2.06 – 2.04 (m, 2H), 1.92 – 1.80 (m, 2H). HPLC: 99%, RT 2.702 min. MS(ESI) m/z 450.1[M + H]+. mp: 82–84°C.
5-(4-(4-(2,3-Dichlorophenyl)piperidin-1-yl)butoxy)-1H-benzo[d]imidazol-2(3H)-one (40)
Compound 40 (61 mg) was prepared as off-white solid by the same procedure as preparing 22, yield 40%. 1H NMR (300 MHz, CDCl3): δ 9.59 (brs, 1H), 9.29 (brs, 1H), 7.33 – 7.30 (m, 2H), 7.20 – 7.13 (m, 1H), 6.92 (d, J = 8.7 Hz, 1H), 6.65 – 6.59 (m, 2H), 3.94 – 3.91 (m, 2H), 3.15 – 3.04 (m, 3H), 2.51 – 2.49 (m, 2H), 2.15(t, J = 11.1Hz, 2H), 1.90 – 1.74 (m, 8H). HPLC: 99%, RT 2.332 min. MS (ESI) m/z 434.0 [M + H]+. Mp: 180–182 °C.
6-(3-(4-(2,3-Dichlorophenyl)piperazin-1-yl)propoxy)-1H-indazole (41)
Compound 41 (90 mg) was prepared by the same procedure as preparing 22, yield 71%. 1H NMR (400 MHz, CDCl3): δ 10.38 (bs, 1H), 7.97 (s, 1H), 7.60 (d, J = 8.7 Hz, 1H), 7.20 – 7.08 (m, 2H), 6.95 (dd, J = 7.1, 2.5 Hz, 1H), 6.90 – 6.78 (m, 2H), 4.08 (t, J = 6.3 Hz, 2H), 3.09 (bs, 4H), 2.77 – 2.61 (m, 6H), 2.12 – 2.00 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 159.2, 151.3, 141.5, 135.0, 134.2, 127.6, 127.5, 124.7, 121.7, 118.7, 118.1, 113.5, 91.7, 66.6, 55.3, 53.5, 51.4, 26.8. HPLC: 99%, RT 2.462 min. MS (ESI) m/z 405.2 [M + H]+. HRMS m/z [M + H]+ calcd for C22H26Cl2N3OS 405.1249, found 405.1273. mp: 159–161 °C.
7-(3-(4-(2,3-Dichlorophenyl)piperazin-1-yl)propoxy)-3,4-dihydroisoquinolin-1(2H)-one (42)
Compound 42 (88 mg) was prepared as white solid by the same procedure as preparing 22, yield 52%. 1H NMR (400 MHz, CDCl3): δ 7.60 (d, J = 2.7 Hz, 1H), 7.18 – 7.09 (m, 3H), 7.01 (dd, J = 8.3, 2.8 Hz, 1H), 6.96 (dd, J = 6.2, 3.4 Hz, 1H), 6.15 (bs, 1H), 4.10 (t, J = 6.4 Hz, 2H), 3.54 (td, J = 6.6, 2.8 Hz, 2H), 3.08 (bs, 4H), 2.93 (t, J = 6.6 Hz, 2H), 2.73 – 2.57 (m, 6H), 2.06 – 1.97 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 166.4, 158.3, 151.5, 134.1, 131.1, 129.9, 128.5, 127.7, 127.6, 124.7, 120.4, 118.8, 112.0, 66.7, 55.2, 53.5, 51.4, 40.7, 27.7, 26.9. HPLC: 99%, RT 2.612 min. MS (ESI) m/z 434.1 [M + H]+. HRMS m/z [M + H]+ calcd for C22H26Cl2N3O2 434.1402, found 434.1425. mp: 162–164 °C.
7-(3-(4-(2,3-Dichlorophenyl)-1,4-diazepan-1-yl)propoxy)-3,4-dihydroisoquinolin-1(2H)-one (43)
Compound 43 (125 mg) was prepared as white solid by the same procedure as preparing 22, yield 71%. 1H NMR (300 MHz, CDCl3) δ 7.60 (d, J = 2.1 Hz, 1H), 7.14 – 7.08 (m, 3H), 7.02 – 7.00 (m, 2H), 6.02 (br., 1H), 4.10 (t, J = 6.3 Hz, 2H), 3.56 – 3.53 (m, 2H), 3.31-3.27 (m, 4H), 2.96 – 2.88 (m, 6H), 2.77 – 2.72 (m, 2H), 2.01(m, 4H). HPLC: 99%, RT 2.578 min. MS (ESI) m/z 448.1 [M + H]+. mp: 116–117 °C. Synthesis of compound 44 was described previously.27
7-(4-(4-(2,3-Dichlorophenyl)-1,4-diazepan-1-yl)butoxy)quinolin-2(1H)-one (45)
Compound 45 (50 mg) was prepared as white solid by the same procedure as preparing 22, yield 46%. 1H NMR (300 MHz, CDCl3) δ 11.54 (br., 1H), 7.71 (d, J = 6.9 Hz, 1H) 7.44 (d, J = 8.1 Hz, 1H), 7.09 – 7.07 (m, 2H), 7.01 – 6.97 (m, 1H), 6.82 – 6.78 (m, 2H), 6.53 (d, J = 9.3 Hz, 1H), 4.10 (t, J = 6.6 Hz, 2H), 3.30 (d, J = 5.1 Hz, 4H), 2.87 – 2.81 (m, 4H), 2.62 (t, J = 7.5 Hz, 2H), 2.01 – 1.99 (m, 2H), 1.92 – 1.82 (m, 2H), 1.76 – 1.68 (m, 2H). HPLC: 99%, RT 2.461 min. MS(ESI) m/z 460.2 [M + H]+. mp: 55–57°C.
Experimental procedures for in vitro biochemical assays
General Procedures
Experimental procedures for the radioligand binding assays for the GPCRs listed in Table S1 (including D1, D3, D4, D5, 5-HT2A, 5-HT2B, 5-HT2C, and 5-HT1A) are available online through the Psychoactive Drug Screening Program (PDSP) website: http://pdsp.med.unc.edu/. The PDSP Assay Protocol book is freely available at http://pdsp.med.unc.edu/UNC-CH%20Protocol%20Book.pdf. The dopamine D2 radioligand binding, cAMP biosensor, and β-arrestin recruitment Tango assays are detailed below.
CHO-D2 Membrane Preparation and Radioligand Binding Assay
CHO-D2 membrane preparation. Cells stably expressing human D2L receptors (CHO-D2L) were plated in 15-cm dishes (in DMEM containing 10% FBS) and grown to 90% confluence. Then, cells were washed with PBS, pH 7.4, and harvested by scraping into PBS, pH 7.4. Harvested cells were centrifuged at 1,000 × g for 10 min and then hypotonically lysed by resuspension into ice-cold 50 mM Hepes, 1% BSA, pH 7.4. Membranes were isolated by centrifugation at 21,000 × g for 20 min. The supernatant was removed and the membrane pellets were stored at −80 °C until used for radioligand binding assays.
Radioligand binding assay
Membranes prepared as above were resuspended to 1 μg protein/μL (measured by Bradford assay using BSA as standard), and 50 μL was added to each well of a polypropylene 96-well plate containing (per well) 50 μL of buffer (20 mM Hepes, 10 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 100 mM N-methyl-D-gluconate, pH 7.4), 50 μL of 1.5 nM [3H]N-methylspiperone (final concentration 0.3 nM), and reference or D2 test ligands at various concentrations ranging from 50 pM to 50 μM (final concentrations ranging from 10 pM to 10 μM, triplicate determinations for each concentration of D2 test ligand). After a 1.5-h incubation in the dark at room temperature, the reactions were harvested onto 0.3% PEI-soaked Filtermax GF/A filters (Wallac) and washed three times with ice-cold 50 mM Tris, pH 7.4, using a Perkin-Elmer Filtermate 96-well harvester. The filters were subsequently dried and placed on a hot plate (100 °C), and Melitilex-A (Wallac) scintillant was applied. The filters were then removed from the hot plate and allowed to cool. The filters were counted on a Wallac TriLux microbeta counter (3 min/well). Residual [3H]N-methylspiperone binding to filtered membranes was plotted as a function of log [reference] or log [D2 test ligand] and the data were regressed using the one-site competition model built into Prism 4.0 (GraphPad software).
D2-Mediated cAMP Assay
HEK293T cells coexpressing the cAMP biosensor GloSensor-22F (Promega) and hD2L receptors were seeded (20,000 cells/20 μL/well) into white, clear-bottom, tissue culture plates in HBSS, 20 mM Hepes, pH 7.4. After 30 min of recovery, cells were treated with 10 μL of 3× test or reference drug prepared in HBSS, 20 mM Hepes, pH 7.4. After 30 min, cells were treated with 10 μL of 1,200 nM (4×) isoproterenol in 8% (4×) GloSensor reagent. Luminescence per well per second was read on a Wallac TriLux microbeta plate counter. Data were normalized to the isoproterenol response (100%) and the maximal quinpirole-induced inhibition thereof (0%) and regressed using the sigmoidal dose-response function built into GraphPad Prism 4.0. Notably, HEK293T cells expressing the GloSensor-22F alone (no hD2) were assayed in parallel and displayed no inhibition of isoproterenol-stimulated cAMP, either by quinpirole or by the test compounds, suggesting that the effect observed in hD2L-expressing cells was due to compound acting via the recombinant receptor.
D2 β-Arrestin Recruitment Tango Assay
Recruitment of β-arrestin to agonist-stimulated D2L receptors was performed using a previously described “Tango”-type assay.46 Briefly, HTLA cells stably expressing β-arrestin-TEV protease and a tetracycline transactivator-driven luciferase were plated in 15-cm dishes in DMEM containing 10% FBS and transfected (via calcium phosphate) with 16 μg of a D2V2-TCS-tTA construct.46 The next day, cells were plated in white, clear-bottom, 384-well plates (Greiner; 15,000 cells/well, 50 μL/well) in DMEM containing 1% dialyzed FBS. The following day, the cells were challenged with 10 μL/well of reference agonist (6 μM) or D2 test ligand (6 μM) prepared in HBSS, 20 mM Hepes, pH 7.4, and 6% DMSO (final ligand concentrations are 1 μM, final DMSO concentration is 1%). After 18 h, the medium was removed and replaced with 1× BriteGlo reagent (Promega), and luminescence per well was read using a TriLux plate reader (1 s/well). Data were normalized to vehicle (0%) and quinpirole (100%) controls and regressed using the sigmoidal dose-response function built into GraphPad Prism 4.0.0
Experimental procedures for in vivo Studies in Mice
General Procedures
All experiments were approved by the Institutional Animal Care and Use Committees at the University of North Carolina, Chapel Hill and Duke University. Wild-type and β-arrestin-2 knockout mice were housed under standard conditions – 14 h light/dark cycle (lights on 0600 hr) with food and water provided ad libitum. Adult, age-matched male and female wild-type and β-arrestin-2 knockout drug-naive mice were used for all behavioral testing.
Locomotor Activity of compounds 19 and 35
Wild-type and β-arrestin-2 knockout mice were treated with vehicle or 2 mg/kg compounds 19 or 35 (i.p.) and were immediately placed into an open field. Thirty minutes later the animals were administered 6 mg/kg PCP (i.p.) and were immediately returned to the open field for 90 minutes. Horizontal activity, measured as distance traveled, was recorded over 5-min segments for the duration of testing. RMANOVA for the first 30 min of testing (baseline) revealed a significant within subject effects of time [F(5,300) = 38.344, p<0.001]; the time by genotype, time by treatment, or the time by treatment by genotype interactions were not significant (Fig. 2).
A second analysis was run to analyze the locomotor activity of the mice following PCP treatment over the entire 90 min following injection. A RMANOVA noted a significant within subject effects of time [F(17,1020) = 64.520, p<0.001] and significant time by genotype [F(17,1020) = 1.592, p<0.012] and time by treatment interactions [F(34,1020) = 2.316, p<0.001]. Since these analyses indicated significant genotype and treatment effects, the RMANOVA was run within each treatment condition as a function of genotype. For the PCP-treated mice, no genotype differences were discerned across time (Fig. 2). For mice treated with 19 there was a significant within subjects effect of time [F(17,340) = 26.012, p<0.001] and time by genotype interaction [F(17,340) = 1.901, p<0.017]. The Bonferroni test reported that 19 suppressed PCP-induced locomotion in the WT relative to the β-arrestin 2 knockout mice at 50, 55, 60, 65, 75, and 105 min (ps<0.042). For mice treated with 35 there was a significant within subject effect of time [F(17,272) = 14.194, p<0.001] and time by genotype interaction [F(17,272) = 1.729, p<0.038]. The Bonferroni test found that 35 suppressed PCP-induced locomotion in the WT relative to the β-arrestin 2 knockout mice at 90 min (p<0.050). Hence, these data show that at 2 mg/kg 19 is more efficacious than 35 in reducing PCP-stimulated locomotion in wild-type mice; neither compound affected PCP-induced activity in the β-arrestin-2 knockout mice.
Supplementary Material
Acknowledgments
We thank the NIH (U19MH082441S1 and U19MH082441) for financial support and Dr. Weihe Zhang for critical reading of synthetic procedures and characterization data.
ABBREVIATIONS USED
- GPCR
G protein-coupled receptor
- SFSR
structure-functional selectivity relationships
- D2R
dopamine D2 receptor
- TM
transmembrane domain
- cAMP
cyclic adenosine monophosphate
- PK
pharmacokinetic
- CNS
central nervous system
- SAR
structure-activity relationship
- LHS
left-hand side
- RHS
right-hand side
- i.p
intraperitoneal
Footnotes
Supporting Information. Radioligand binding affinities of compounds 19 and 35 at select dopamine and serotonin receptors. 1H and 13C NMR spectra of compound 35 and 37. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Furchgott RF. The use of haloalkylamines in the differentiation of receptors and in the determination of dissociation constants of agonist-receptor complexes. In: Harper N, Simmonds A, editors. Advances in Drug Research. Vol. 3. Academic Press; New York: 1966. pp. 21–55. [Google Scholar]
- 2.Roth BL, Chuang DM. Multiple mechanisms of serotonergic signal transduction. Life Sci. 1987;41:1051–1064. doi: 10.1016/0024-3205(87)90621-7. [DOI] [PubMed] [Google Scholar]
- 3.Berg KA, Maayani S, Goldfarb J, Scaramellini C, Leff P, Clarke WP. Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: evidence for agonist-directed trafficking of receptor stimulus. Mol Pharmacol. 1998;54:94–104. [PubMed] [Google Scholar]
- 4.Parrish J, Braden M, Gundy E, Nichols DE. Differential phospholipase C activation by phenylalkylamine serotonin 5-HT2A receptor agonists. J Neurochem. 2005;95:1575–1584. doi: 10.1111/j.1471-4159.2005.03477.x. [DOI] [PubMed] [Google Scholar]
- 5.Ghosh D, Snyder SE, Watts VJ, Mailman RB, Nichols DE. 9-Dihydroxy-2,3,7,11b-tetrahydro-1H-naph[1,2,3-de]isoquinoline: a potent full dopamine D1 agonist containing a rigid-beta-phenyldopamine pharmacophore. J Med Chem. 1996;39:549–555. doi: 10.1021/jm950707+. [DOI] [PubMed] [Google Scholar]
- 6.Kilts JD, Connery HS, Arrington EG, Lewis MM, Lawler CP, Oxford GS, O’Malley KL, Todd RD, Blake BL, Nichols DE, Mailman RB. Functional selectivity of dopamine receptor agonists. II. Actions of dihydrexidine in D2L receptor-transfected MN9D cells and pituitary lactotrophs. J Pharmacol Exp Ther. 2002;301:1179–1189. doi: 10.1124/jpet.301.3.1179. [DOI] [PubMed] [Google Scholar]
- 7.Mottola DM, Kilts JD, Lewis MM, Connery HS, Walker QD, Jones SR, Booth RG, Hyslop DK, Piercey M, Wightman RM, Lawler CP, Nichols DE, Mailman RB. Functional selectivity of dopamine receptor agonists. I. Selective activation of postsynaptic dopamine D2 receptors linked to adenylate cyclase. J Pharmacol Exp Ther. 2002;301:1166–1178. doi: 10.1124/jpet.301.3.1166. [DOI] [PubMed] [Google Scholar]
- 8.Beaulieu JM, Tirotta E, Sotnikova TD, Masri B, Salahpour A, Gainetdinov RR, Borrelli E, Caron MG. Regulation of Akt signaling by D2 and D3 dopamine receptors in vivo. J Neurosci. 2007;27:881–885. doi: 10.1523/JNEUROSCI.5074-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 10.Chidiac P, Hebert T, Valiquette M, Dennis M, Bouvier M. Inverse agonist activity of Beta-adrenergic antagonists. Molecular Pharmacology. 1993;45:490–499. [PubMed] [Google Scholar]
- 11.Barker EL, Westphal RS, Schmidt D, Sanders-Bush E. Constitutively active 5-hydroxytryptamine2C receptors reveal novel inverse agonist activity of receptor ligands. J Biol Chem. 1994;269:11687–11690. [PubMed] [Google Scholar]
- 12.Rauser L, Savage JE, Meltzer HY, Roth BL. Inverse agonist actions of typical and atypical antipsychotic drugs at the human 5-hydroxytryptamine(2C) receptor. J Pharmacol Exp Ther. 2001;299:83–89. [PubMed] [Google Scholar]
- 13.Gilliland SL, Alper RH. Characterization of dopaminergic compounds at hD2short, hD4.2 and hD4. 7 receptors in agonist-stimulated [35S]GTPgammaS binding assays. Naunyn Schmiedebergs Arch Pharmacol. 2000;361:498–504. doi: 10.1007/s002100000224. [DOI] [PubMed] [Google Scholar]
- 14.Burstein ES, Ma J, Wong S, Gao Y, Pham E, Knapp AE, Nash NR, Olsson R, Davis RE, Hacksell U, Weiner DM, Brann MR. Intrinsic efficacy of antipsychotics at human D2, D3, and D4 dopamine receptors: identification of the clozapine metabolite N-desmethylclozapine as a D2/D3 partial agonist. J Pharmacol Exp Ther. 2005;315:1278–1287. doi: 10.1124/jpet.105.092155. [DOI] [PubMed] [Google Scholar]
- 15.Peroutka SJ, Snyder SH. Long-term antidepressant treatment decreases spiroperidol-labeled serotonin receptor binding. Science. 1980;210:88–90. doi: 10.1126/science.6251550. [DOI] [PubMed] [Google Scholar]
- 16.Berry S, Shah M, Khan N, Roth B. Rapid agonist-induced internalization of the 5-hydroxytryptamine2A receptor occurs via the endosome pathway in vitro. Mol Pharmacol. 1996;50:306–313. [PubMed] [Google Scholar]
- 17.Willins DL, Alsayegh L, Berry SA, Backstrom JR, Sanders-Bush E, Friedman L, Khan N, Roth BL. Serotonergic antagonist effects on trafficking of serotonin 5-HT2A receptors in vitro and in vivo. Ann N Y Acad Sci. 1998;861:121–127. doi: 10.1111/j.1749-6632.1998.tb10182.x. [DOI] [PubMed] [Google Scholar]
- 18.Willins D, Berry S, Alsayegh L, Backstrom J, Sanders-Bush E, Roth B. Clozapine and other 5-hydroxytryptamine2A receptor antagonists alter the subcellular distribution of 5-hydroxytryptamine2A receptors in vitro and in vivo. Neurosci. 1999;91:599–606. doi: 10.1016/s0306-4522(98)00653-8. [DOI] [PubMed] [Google Scholar]
- 19.Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, Miller KJ, Spedding M, Mailman RB. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- 20.Mailman RB. GPCR functional selectivity has therapeutic impact. Trends Pharmacol Sci. 2007;28:390–396. doi: 10.1016/j.tips.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 22.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 23.Abbas A, Roth BL. Arresting serotonin. Proc Natl Acad Sci U S A. 2008;105:831–832. doi: 10.1073/pnas.0711335105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Urban JD, Vargas GA, von Zastrow M, Mailman RB. Aripiprazole has functionally selective actions at dopamine D2 receptor-mediated signaling pathways. Neuropsychopharmacology. 2007;32:67–77. doi: 10.1038/sj.npp.1301071. [DOI] [PubMed] [Google Scholar]
- 25.Violin JD, Lefkowitz RJ. [beta]-Arrestin-biased ligands at seven-transmembrane receptors. Trends in Pharmacological Sciences. 2007;28:416–422. doi: 10.1016/j.tips.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 26.Gesty-Palmer D, Flannery P, Yuan L, Corsino L, Spurney R, Lefkowitz RJ, Luttrell LM. A beta-arrestin-biased agonist of the parathyroid hormone receptor (PTH1R) promotes bone formation independent of G protein activation. Sci Transl Med. 2009;1:1ra1. doi: 10.1126/scitranslmed.3000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Butini S, Gemma S, Campiani G, Franceschini S, Trotta F, Borriello M, Ceres N, Ros S, Coccone SS, Bernetti M, De Angelis M, Brindisi M, Nacci V, Fiorini I, Novellino E, Cagnotto A, Mennini T, Sandager-Nielsen K, Andreasen JT, Scheel-Kruger J, Mikkelsen JD, Fattorusso C. Discovery of a new class of potential multifunctional atypical antipsychotic agents targeting dopamine D3 and serotonin 5-HT1A and 5-HT2A receptors: design, synthesis, and effects on behavior. J Med Chem. 2009;52:151–169. doi: 10.1021/jm800689g. [DOI] [PubMed] [Google Scholar]
- 28.Allen JA, Yost JM, Setola V, Chen X, Sassano MF, Chen M, Peterson S, Yadav PN, Huang XP, Feng B, Jensen NH, Che X, Bai X, Frye SV, Wetsel WC, Caron MG, Javitch JA, Roth BL, Jin J. Discovery of Beta-Arrestin-Biased Dopamine D2 Ligands for Probing Signal Transduction Pathways Essential for Antipsychotic Efficacy. Proc Natl Acad Sci U S A. 2011;108:18488–18493. doi: 10.1073/pnas.1104807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lawler CP, Prioleau C, Lewis MM, Mak C, Jiang D, Schetz JA, Gonzalez AM, Sibley DR, Mailman RB. Interactions of the novel antipsychotic aripiprazole (OPC-14597) with dopamine and serotonin receptor subtypes. Neuropsychopharmacology. 1999;20:612–627. doi: 10.1016/S0893-133X(98)00099-2. [DOI] [PubMed] [Google Scholar]
- 30.Mailman RB, Murthy V. Third generation antipsychotic drugs: partial agonism or receptor functional selectivity? Curr Pharm Des. 2010;16:488–501. doi: 10.2174/138161210790361461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mallikaarjun S, Salazar DE, Bramer SL. Pharmacokinetics, Tolerability, and Safety of Aripiprazole following Multiple Oral Dosing in Normal Healthy Volunteers. The Journal of Clinical Pharmacology. 2004;44:179–187. doi: 10.1177/0091270003261901. [DOI] [PubMed] [Google Scholar]
- 32.Johnson DS, Choi C, Fay LK, Favor DA, Repine JT, White AD, Akunne HC, Fitzgerald L, Nicholls K, Snyder BJ, Whetzel SZ, Zhang L, Serpa KA. Discovery of PF-00217830: Aryl piperazine napthyridinones as D2 partial agonists for schizophrenia and bipolar disorder. Bioorganic & Medicinal Chemistry Letters. 2011;21:2621–2625. doi: 10.1016/j.bmcl.2011.01.059. [DOI] [PubMed] [Google Scholar]
- 33.Oshiro Y, Sato S, Kurahashi N, Tanaka T, Kikuchi T, Tottori K, Uwahodo Y, Nishi T. Novel antipsychotic agents with dopamine autoreceptor agonist properties: synthesis and pharmacology of 7-[4-(4-phenyl-1-piperazinyl)butoxy]-3,4-dihydro-2(1H)-quinolinone derivatives. J Med Chem. 1998;41:658–667. doi: 10.1021/jm940608g. [DOI] [PubMed] [Google Scholar]
- 34.Vangveravong S, Zhang Z, Taylor M, Bearden M, Xu J, Cui J, Wang W, Luedtke RR, Mach RH. Synthesis and characterization of selective dopamine D(2) receptor ligands using aripiprazole as the lead compound. Bioorg Med Chem. 2011;19:3502–3511. doi: 10.1016/j.bmc.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin GE, Elgin RJ, Mathiasen JR, Davis CB, Kesslick JM, Baldy WJ, Shank RP, Distefano DL, Fedde CL, Scott MK. Activity of Aromatic Substituted Phenylpiperazines Lacking Affinity for Dopamine Binding-Sites in a Preclinical Test of Antipsychotic Efficacy. J Med Chem. 1989;32:1052–1056. doi: 10.1021/jm00125a020. [DOI] [PubMed] [Google Scholar]
- 36.Elworthy TR, Ford APDW, Bantle GW, Morgans DJ, Ozer RS, Palmer WS, Repke DB, Romero M, Sandoval L, Sjogren EB, Talamas FX, Vazquez A, Wu H, Arredondo NF, Blue DR, DeSousa A, Gross LM, Kava MS, Lesnick JD, Vimont RL, Williams TJ, Zhu QM, Pfister JR, Clarke DE. N-arylpiperazinyl-N′-propylamino derivatives of heteroaryl amides as functional uroselective alpha(1)-adrenoceptor antagonists. J Med Chem. 1997;40:2674–2687. doi: 10.1021/jm970166j. [DOI] [PubMed] [Google Scholar]
- 37.Fu XT, Lei, Yuan Mu, Shi Jingshan. Design, synthesis and vasorelaxant activity of R, S-1-(substituted phenyl)-4-[3-(naphtha-1-yl-oxy)-2-hydroxypropyl]-piperazine derivatives. Yaoxue Xuebao. 2007;42:735–740. [PubMed] [Google Scholar]
- 38.Tsuruta K, Frey EA, Grewe CW, Cote TE, Eskay RL, Kebabian JW. Evidence that LY-141865 specifically stimulates the D-2 dopamine receptor. Nature. 1981;292:463–465. doi: 10.1038/292463a0. [DOI] [PubMed] [Google Scholar]
- 39.Banno K, Fujioka T, Kikuchi T, Oshiro Y, Hiyama T, Nakagawa K. Studies on 2(1H)-quinolinone derivatives as neuroleptic agents I. Synthesis and biological activities of (4-phenyl-1-piperazinyl)-propoxy-2(1H)-quinolinone derivatives. Chem Pharm Bull. 1988;36:4377–4388. doi: 10.1248/cpb.36.4377. [DOI] [PubMed] [Google Scholar]
- 40.Kiss B, Horvath A, Nemethy Z, Schmidt E, Laszlovszky I, Bugovics G, Fazekas K, Hornok K, Orosz S, Gyertyan I, Agai-Csongor E, Domany G, Tihanyi K, Adham N, Szombathelyi Z. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333:328–340. doi: 10.1124/jpet.109.160432. [DOI] [PubMed] [Google Scholar]
- 41.van Wijngaarden I, Kruse CG, van der Heyden JA, Tulp MT. 2-Phenylpyrroles as conformationally restricted benzamide analogues. A new class of potential antipsychotics. 2. J Med Chem. 1988;31:1934–1940. doi: 10.1021/jm00118a011. [DOI] [PubMed] [Google Scholar]
- 42.van Wijngaarden I, Kruse CG, van Hes R, van der Heyden JA, Tulp MT. 2-Phenylpyrroles as conformationally restricted benzamide analogues. A new class of potential antipsychotics. 1. J Med Chem. 1987;30:2099–2104. doi: 10.1021/jm00394a028. [DOI] [PubMed] [Google Scholar]
- 43.Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- 44.Nordquist RE, Risterucci C, Moreau JL, von Kienlin M, Kunnecke B, Maco M, Freichel C, Riemer C, Spooren W. Effects of aripiprazole/OPC-14597 on motor activity, pharmacological models of psychosis, and brain activity in rats. Neuropharmacology. 2008;54:405–416. doi: 10.1016/j.neuropharm.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 45.Gottlieb HE, Kotlyar V, Nudelman A. NMR chemical shifts of common laboratory solvents as trace impurities. J Org Chem. 1997;62:7512–7515. doi: 10.1021/jo971176v. [DOI] [PubMed] [Google Scholar]
- 46.Barnea G, Strapps W, Herrada G, Berman Y, Ong J, Kloss B, Axel R, Lee KJ. The genetic design of signaling cascades to record receptor activation. P Natl Acad Sci USA. 2008;105:64–69. doi: 10.1073/pnas.0710487105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.