

Summary
The cytoskeletal regulators that mediate the change in the neuronal cytoskeletal machinery from one that promotes oriented motility to one that facilitates differentiation at the appropriate locations in the developing neocortex remain unknown. We found that Nck associated protein 1 (Nap1), an adaptor protein thought to modulate actin nucleation, is selectively expressed in the developing cortical plate, where neurons terminate their migration and initiate laminar specific differentiation. Loss of function of Nap1 disrupts neuronal differentiation. Premature expression of Nap1 in migrating neurons retards migration and promotes post migratory differentiation. Nap1 gene mutation in mice leads to neural tube and neuronal differentiation defects. Disruption of Nap1 retards the ability to localize key actin cytoskeletal regulators such as WAVE1 to the protrusive edges where they are needed to elaborate process outgrowth. Thus, Nap1 plays an essential role in facilitating the neuronal cytoskeletal changes underlying the post-migratory differentiation of cortical neurons, a critical step in functional wiring of the cerebral cortex.
Keywords: Cortical development, neuronal differentiation, neuronal migration, Nap1
Introduction
Neurons generated in the proliferative ventricular zones of the developing cerebral cortex migrate in distinct radial and tangential routes to the top of the embryonic cortex where they terminate their migration and start to differentiate into distinct classes of cortical neurons. Dynamic regulation of neurons' cytoskeletal machinery in response to extracellular guidance or positional cues enables appropriate neuronal generation, migration and differentiation in the developing cerebral cortex. Molecular analyses of human and mouse cortical developmental disorders elegantly illustrate this. For example, mutations in microtubule associating protein, ASPM (abnormal spindle-like microcephaly associated protein: ASPM), lead to defective generation of cortical neurons, whereas mutations in actin-binding protein filamin a (FLNA) and microtubule associated proteins, doublecortin (Dcx) or Lis1 (non- catalytic subunit of platelet- activating factor acetylhydrolase isoform 1b), lead to disrupted initiation and maintenance of neuronal migration, respectively (reviewed in Marin and Rubenstein, 2003; Mochida and Walsh, 2004; Ayala et al., 2007). Although the significance of cytoskeletal dynamics during neuronal generation and maintenance of migration is well established, the cytoskeletal regulators and mechanisms that are needed to convert neurons engaged in oriented migration into neurons that are stably positioned and actively extending axons and dendrites in the appropriate laminar locations of the developing cerebral cortex remains unknown.
Here we found that Nck associated protein 1 (Nap1), an adaptor protein that is thought to modulate actin nucleation by forming a pentameric complex with WAVE, PIR121, Abi1/2 and HSPC300 (Baumgartner et al., 1995; Bladt et al., 2003; Stradal et al., 2004; Hummel et al., 2000, Bogdan and Klambt, 2003, Soto et al., 2002; Suzuki et al., 2000), is selectively expressed in the cortical plate region of the developing cortex, where neurons terminate their migration and begin their final laminar specific differentiation, characterized by the elaboration of distinct axonal and dendritic architecture. Functional analysis of Nap1 indicate that Nap1-mediated cytoskeletal rearrangements in the emerging cortical plate play an essential role in cortical neuronal differentiation underlying the formation of functional connectivity in cerebral cortex.
Results
Developmental expression of Nap1 in cerebral cortex
To study the cytoskeletal dynamics underlying how neurons terminate their migration and start their final differentiation in the developing cerebral cortex, we mapped the embryonic cortical expression profiles of twenty five murine orthologues of Drosophila or C. elegans cytoskeleton related genes that are known to regulate distinct stages of neuronal migration or differentiation. Among the proteins screened, Nck associated protein 1 (Nap1) was selectively expressed in the differentiating neurons of the embryonic cerebral cortex.
In situ hybridization analysis indicates that Nap1 is primarily expressed in the cortical plate (CP) region of the embryonic cortex (E14-18), where neurons terminate their migration and begin their final, layer specific phenotypic differentiation (Fig.1A-C.) Identical expression pattern of Nap1 is evident in cortical sections from Nap1 indicator mice (Nap1lacZ/+) in which β-gal expression is indicative of endogenous Nap1 expression pattern (Supplemental Fig.1). Co-immunolabelling with post- mitotic neuron specific Tuj-1 antibodies indicates that Nap1 is expressed specifically in cortical plate neurons, not by actively migrating neurons in the intermediate zone (Fig. 1D-F). Nap1 expression persists in postnatal cortical neurons as they differentiate and form mature synaptic connections (Supplemental Fig.1G-H). Co- immunolabelling with axonal and dendritic markers indicate that Nap1 is present in both axons and dendrites of differentiating cortical neurons. Prominent Nap1 expression is noticed in neuronal growth cones and in dendritic spine- like protrusions along neuritic shafts (Fig.1G-M). Immunoblots of whole cell extracts of cortices from different embryonic ages indicate a pattern of increased Nap1 expression corresponding to increased levels of cortical neuronal differentiation (Fig.1N). Together, these results indicate that during development Nap1 expression is induced in cortical neurons as they arrive in the cortical plate and initiate their post migratory differentiation, characterized by extension of processes and formation of functional synaptic connections.
Figure 1. Distribution of Nap1 in developing cerebral cortex.

(A- C) In situ hybridization mapping of Nap1 expression at E16 indicates prominent expression in the cortical plate region (arrowheads, A-C) throughout the entire rostro-caudal extent [rostral (A), middle (B), and caudal (C)] of the developing cerebral cortex. (D-F) In E16 cortex, co- labeling with neuron specific Tuj-1 antibodies indicate that Nap1 (red) is specifically expressed in the cortical plate (CP) neurons, and not in the intermediate zone (IZ) region containing the migrating neurons. (G, H) Co- labeling of differentiating cortical neurons with axonal (Tau-1) and dendritic (Map2) markers, indicate that Nap1 is present in both axons (G) and dendrites (H). Yellow indicates co-labeled sites. (I) Nap1 is prominently expressed in the tips of cortical neurites (arrowhead, I). Panels J-M are higher magnification images of Nap1 expression at the leading edges of differentiating cortical neurons. Cortical neurons in panels I-M were co-labeled with Tuj-1 antibodies. (N) Immunoblot analysis of Nap1 expression in the developing cortex indicates that increase in Nap1 expression parallels increased neuronal differentiation. VZ-ventricular zone, IZ-intermediate zone, CP-cortical plate. Scale bar: A-C, 400 μm, D-F, 250μm; G-H, 30 μm; I, 15 μm; J-M, 10 μm.
Defective neuronal differentiation following inhibition of Nap1
To evaluate the effect of loss of function of Nap1 in cortical neuronal differentiation, we utilized shRNA mediated knockdown of endogenous Nap1 in cortical neurons. We generated shRNA constructs targeted to different mouse Nap1 specific regions. As a negative control for the shRNA constructs, 3 nt mutations were made in each of the respective targeting sequences. The target sequence oligos and mutated target sequence oligos were subcloned into pCGLH vector, which contains chicken beta actin promoter driven EGFP and H1 promoter for shRNA transcription. Nap1 shRNA, but not the control shRNA, specifically reduced Nap1 levels (Supplemental Fig.2). Nap1 shRNA induced no changes in the expression levels of unrelated proteins such as tubulin (Supplemental Fig. 2) or ErbB4 (data not shown). Immunolabeling of control or Nap1 shRNA transfected neurons with Nap1 antibodies indicates similar reduction in Nap1 expression (data not shown). Furthermore, in embryonic cortical cells cotransfected with Nap1 or control shRNA (in pCRLH vector expressing RFP) and full length Nap1 –EGFP fusion plasmids, Nap1-EGFP expression was diminished only in Nap1shRNA expressing cells, but not in control shRNA expressing cells (data not shown). Together, these studies confirm that Nap1 shRNA constructs can specifically suppress endogenous Nap1 protein expression.
To determine the effect of Nap1 in post-mitotic differentiation of cortical neurons in vitro, dissociated E14 cortical neurons were transfected with either control or Nap1 shRNA. Three days later, neurons were immunolabelled with neuron specific Tuj1 antibodies to assess the extent of differentiation. The total length, the number of primary, secondary, and tertiary branches of both axons and dendrites, and the number of dendrites on these neurons was quantified. Axons and dendrites were identified based on their morphology (Gaudilliere et al., 2004). Suppressing Nap1 expression in post mitotic cortical neurons significantly reduced their ability to elaborate characteristic axons and dendrites as indicated by the marked reduction in the extent, branching, and numbers of dendrites and axons (Fig. 2). Similar retardation of neuronal differentiation was also noticed when Nap1 deficient neurons were seeded on cortical plate region of E16 embryonic cortical slices, a relevant substrate where cortical neuronal differentiation normally occurs in vivo (data not shown). These observations indicate that Nap1 deficiency significantly impaired the ability of neurons to differentiate in vitro.
Figure 2. Suppression of Nap1 expression disrupts embryonic cortical neuronal differentiation.
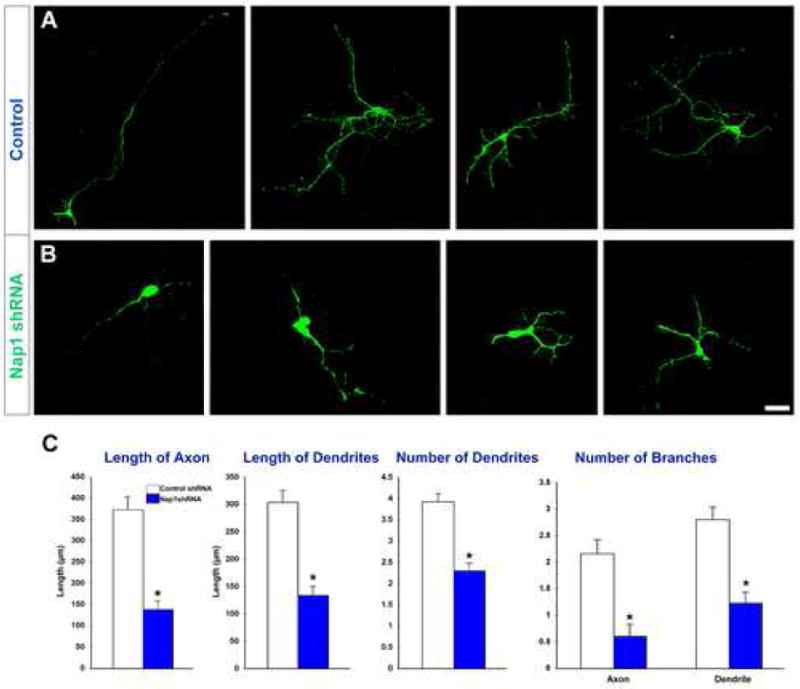
(A-B) E14.5 cortical neurons were transfected with control (A) or Nap1 shRNA (B). Compared to control neurons, Nap1 deficient neurons displayed reduced axonal and dendritic growth and branching. (C) Quantification of neuronal differentiation defects in Nap1 deficient neurons. Asterisk indicates significant when compared with controls at p<0.001 (Student's t test). Scale bar: 20μm.
To determine the effect of Nap1 in post-migratory differentiation of cortical neurons in vivo, E15 embryos were electroporated with Nap1 or control shRNA, allowed to survive till postnatal day 2 or 17, and the patterns of neuronal position, dendritic and axonal morphology of control and Nap1 shRNA expressing neurons were evaluated. Nap1 knockdown did not affect the positioning of neurons within the cortical plate. Quantitative analysis of neuronal position in the cortex indicates no difference between control and Nap1 shRNA expressing neurons (Fig. 3K). Furthermore, real time analysis of migration of Nap1 deficient and control neurons indicates that Nap1 knockdown did not affect neuronal migration. Control neurons migrated at an average rate of 21±2.8 μm/hr and Nap1 shRNA expressing cells migrated at a comparable rate of 19.2±2.4 μm/hr. However, Nap1 knock down significantly retarded all aspects of cortical neuronal differentiation in vivo (Fig. 3). Nap1 deficient neurons displayed significantly reduced axonal and dendritic process extension and branching. Furthermore, the terminal, post- migratory differentiation and maturation of cortical neurons in cerebral cortex is characterized by the elaboration of specialized dendritic protrusions essential for synaptic plasticity, i.e., dendritic spines. We therefore analyzed the effect of Nap1 on dendritic spine morphology in the above cortical neurons. Nap1 deficiency profoundly retarded the dendritic spine density in these cortical neurons (Supplemental Fig. 3). Together, these data demonstrate that Nap1 is critical for neuronal differentiation in the emerging cortical plate.
Figure 3. Knockdown of Nap1 disrupts embryonic cortical neuronal differentiation in vivo.
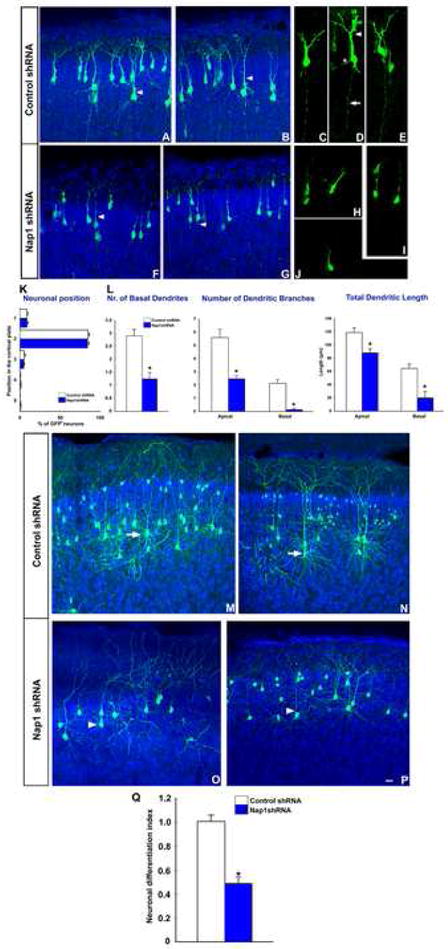
Cerebral cortices of E15.5 embryos were electroporated with control or Nap1 shRNA and differentiating neurons in the cortical plate were analyzed at post natal day 2 (A-L) or 17 (M-Q). (A-E) Cortical neurons expressing control shRNA displayed characteristic axons and dendrites (arrowheads, A, B) at their early stages of development. In higher magnification images of these cells (C-E), apical (arrowhead, D), basal dendrites (asterisk, D), and axon (arrow, D) are evident. (F-J) In contrast, Nap1 shRNA expressing neurons displayed significantly reduced axonal and dendritic growth and branching (arrowheads, F, G; H-J). (K) Analysis of neuronal position indicates no difference between control and Nap1 shRNA expressing neurons. (L) Quantification of dendritic numbers, length, and branches in control and Nap1 deficient neurons. (M-P) At post natal day 17, extensive dendritic arborization is evident in control neurons (arrows, L, M). Nap1 deficient neurons (arrowheads, N, O), however, displayed reduced dendritic growth and branching. (Q) Analysis of the extent of neuronal differentiation indicates a substantial reduction in the complexity of neuronal process growth and arborization in Nap1 deficient neurons. Asterisk indicates significant when compared with controls at p<0.001 (Student's t test). Scale bar: A-B, F-G, L-O 30 μm; C-E, H-J, 15μm.
Ectopic expression of Nap1 promotes neuronal differentiation
If Nap1 expression normally facilitates neuronal differentiation, premature Nap1 induction in migrating neurons in the intermediate zone may lead to changes in migration and premature initiation of neuronal differentiation. To test this, we ectopically induced Nap1 in migrating neurons in the intermediate zone using NeuroD promoter, which is active in post- mitotic, migratory neurons (Dr. F. Polleux [UNC Neuroscience Center], unpublished observations; Huang et al., 2000). By prematurely expressing Nap1 in migrating neurons before they arrive in the cortical plate, we asked if the induction of Nap1 will promote premature initiation of post migratory, differentiation state. Embryonic day 14 or 15 mouse cortices were in utero electroporated with either NeuroD promoter-Nap1-IRES-EGFP or control NeuroD promoter- IRES-EGFP plasmids. Forty eight hours after electroporation, cerebral cortices were removed, vibrotome sliced, and immunolabelled with markers that are normally expressed by neurons that are under going post migratory layer specific differentiation in the cortical plate (e.g., Tbr-1[layer VI], Brn-1[layer II-IV]). Compared to controls, migration of Nap1 expressing neurons was significantly curtailed. Quantification of the extent of neuronal migration indicates that most of Nap1 expressing neurons are found in the lower intermediate zone, whereas control neurons migrate well into the upper IZ and CP (Fig. 4A, C, E, F). Importantly, control neurons in the intermediate zone display the characteristic morphology of migrating neurons, with leading and trailing processes (Fig.4I, J). In contrast, Nap1 expressing neurons tend to have multiple long, branched processes, characteristic of differentiating neurons (Fig.4K-P). Furthermore, majority of Nap1 expressing neurons, but not control neurons, in the intermediate zone expressed molecular markers which are characteristically expressed by differentiating, post-migratory neurons in the cortical plate. When Nap1 was induced during early embryonic stages, significantly higher number of Nap1 expressing neurons in the IZ expressed Tbr-1, a T-domain transcription factor normally expressed in early generated glutaminergic cortical neurons (Fig.4B, G; Hevner et al., 2001). Similar induction of Brn-1, a POU- domain transcription factor normally expressed in upper layer cortical neurons (McEvilly et al., 2002; Sugitani et al., 2002), was evident when Nap1 was electroporated during late embryonic stages (Fig. 4D, H). Together, these observations suggest that ectopic Nap1 expression retards neuronal migration and promotes neuronal differentiation in vivo.
Figure 4. Ectopic expression of Nap1 in migrating neurons in the intermediate zone promotes premature neuronal differentiation.
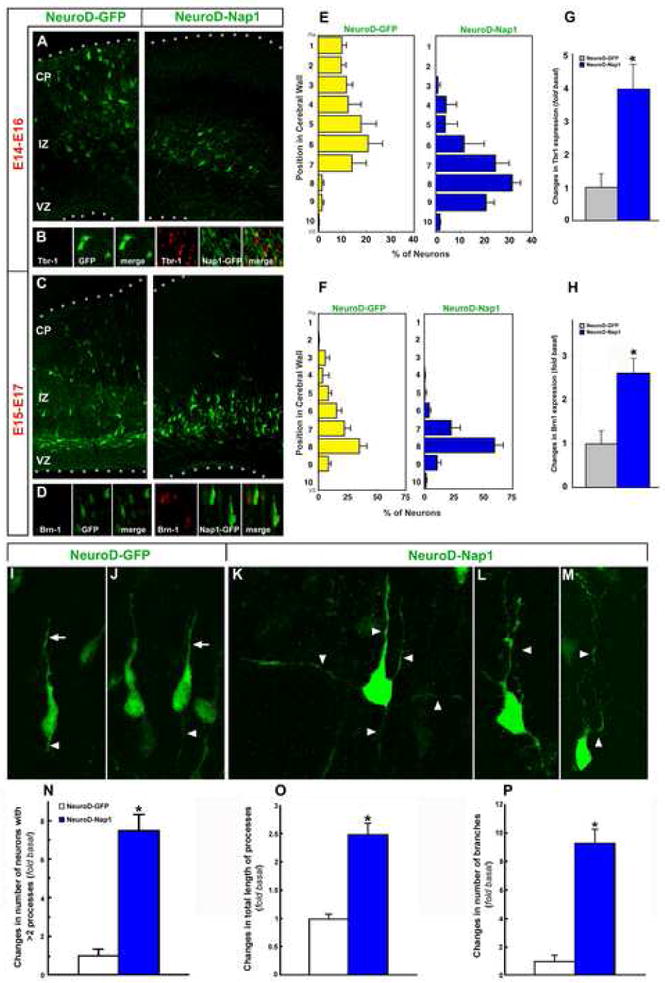
NeuroD promoter is active in post-mitotic, migratory neurons of the intermediate zone. E14 or E15 embryonic cortices were electroporated with NeuroD promoter-Nap1-IRES-EGFP or NeuroD promoter -IRES-EGFP and the position of GFP+ neurons and the expression of neuronal differentiation markers in GFP+ neurons in the IZ were analyzed 48 hours later. Nap1 expression significantly retards the migration of neurons generated at E14 (A, B) or E15 (E, F). Significantly higher number of Nap1 expressing neurons in the IZ also express markers (Tbr1, Brn1) that are normally expressed by differentiating neurons in the CP (B, D, G, H). Higher magnification images of GFP immunolabeled neurons in the intermediate zone indicates that control neurons display the characteristic morphology of migrating neurons, with leading (arrow, I-J) and trailing processes (arrowhead, I-J), whereas Nap1 overexpressing neurons in the IZ tend to have multiple, branched processes (arrowheads, K-L), characteristic of differentiating neurons. (N-P) Quantification of neurons with multiple processes, total process length, and branch numbers suggest that premature expression of Nap1 in migrating neurons promotes premature differentiation of neurons. Data shown are mean ± SEM (n=6); asterisk, significant when compared with controls at p<0.01 (Student's t test). VZ-ventricular zone, IZ-intermediate zone, CP-cortical plate. Dotted lines in panels A and C indicate pial (top) and ventricular (bottom) surfaces.
To further explore Nap1's role in neuronal differentiation, the intermediate zone containing control GFP or Nap1 over expressing neurons were micro dissected from the electroporated cortical slices, dissociated, plated at low density on laminin, and neuronal differentiation was monitored at different time points. We hypothesized that if Nap1 facilitates neuronal differentiation, we should notice rapid emergence of morphological differentiation in Nap1 expressing, but not control neurons. Immediately after attachment, both control and Nap1 expressing neurons display a smooth cell soma and are morphologically undifferentiated. However, within a few hours in vitro, in contrast to control neurons, Nap1 expressing neurons rapidly display signs of morphological differentiation as indicated by extension of multiple processes (Fig.5). These in vitro observations further suggest that cell autonomous Nap1 expression promotes neuronal differentiation.
Figure 5. Nap1 over expression promotes neuronal differentiation.
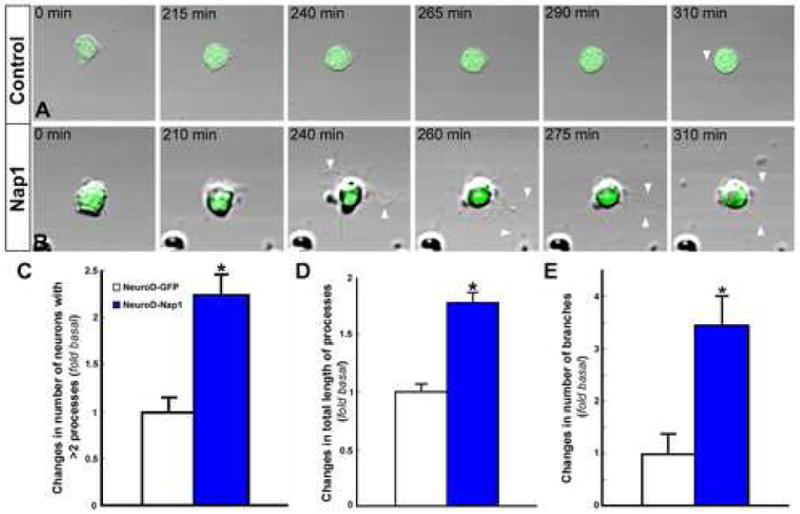
(A, B) Dissociated, control GFP or Nap1 expressing neurons from the intermediate zone of embryos electroporated with NeuroD promoter-Nap1-IRES-EGFP or NeuroD promoter -IRES-EGFP were plated on laminin and repeatedly monitored for several hours. Phase light images of GFP expressing neurons were collected. Immediately after adhesion, both control (A) and Nap1 expressing neurons (B) have smooth, round cell bodies. However, in contrast to control neurons, Nap1 expressing neurons rapidly extend multiple processes (arrowheads), suggesting that Nap1 expression promotes neuronal differentiation. (C-E) Quantification of neuronal differentiation after 7.5 hours in vitro indicates that Nap1 expression promotes process extension and branching. Data shown are mean ± SEM (n=6); asterisk, significant when compared with controls at p<0.01 (Student's t test).
Induction of Nap1 by BDNF
What induces Nap1 in the differentiating neurons of the cortical plate? Context dependent activity of extracellular cues in the developing CP, such as brain derived neurotrpohic factor (BDNF), are required to trigger cortical neuronal differentiation (McAllister et al., 1996; Ghosh et al., 1994; Reichardt et al., 2006) and thus may induce Nap1 in cortical neurons. To examine this, dissociated E16 cortical neurons were treated with 1, 5, 10, 15, and 25 ng/ml BDNF for 48 hours and the level of Nap1 expression in these cells was analyzed by immunoblotting. BDNF induced a dosage dependent increase in Nap1 levels (Fig. 6A). This increase in Nap1 was abolished when BDNF activity was blocked with TrkB receptor bodies (TrkB-IgG, Cabelli et al., 1995; Fig. 6A). To examine if Nap1 function is essential for BDNF's effect on neuronal differentiation, dissociated E16 cortical neurons were first transfected with either control or Nap1 shRNA and then maintained in BDNF (25ng/ml) supplemented or normal media. Two days later, neurons were immunolabelled with neuron- specific Tuj1 antibodies to assess the extent of differentiation. BDNF induced neuritic growth and differentiation in control neurons, but BDNF effect was absent in Nap1 deficient neurons (Fig. 6B, C). These data suggest that extracellular factors such as BDNF, which are known to play an essential role in the final post- migratory differentiation of neurons in cerebral cortex, can induce Nap1 expression in cortical neurons as they initiate their post- migratory terminal growth and differentiation in their appropriate laminar locations. Nap1 expression and function is critical to mediate the BDNF induced differentiation of cortical neurons.
Figure 6. BDNF induced Nap1 expression in embryonic cortical neurons.
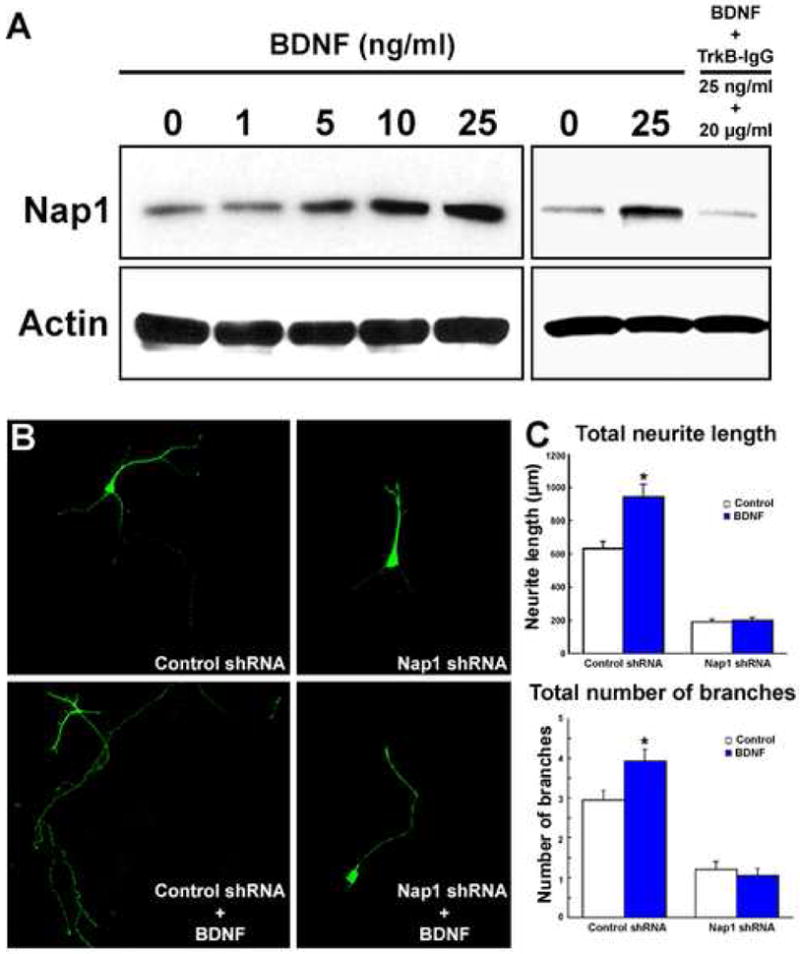
(A) E14 cortical neurons were treated with different concentrations of BDNF (0-25ng/ml) for 2 days. Immunoblot analysis of Nap1 expression in these neurons indicates a dosage dependent effect of BDNF on Nap1 protein level. The effect of BDNF was abolished when BDNF activity was neutralized with TrkB-IgG. Immunblotting for actin indicates equal loading. (B) BDNF promotes growth and differentiation of embryonic cortical neurons expressing control, but not Nap1 shRNA. (C) Analysis of neuronal differentiation indicates that Nap1 deficiency significantly retards BDNF induced neurite growth and branching. Data shown are mean ± SEM (n=3); asterisk, significant when compared with controls at p<0.01 (Student's t test).
Functional domains of Nap1
Having established the functional significance of Nap1 in cortical neuronal differentiation, we sought to determine the domains of Nap1 that are critical for its function. Initially, we generated serial deletion fragments of Nap1 fused to EGFP(pCMV-ΔNap1-EGFP), transfected Cos7 cells with Nap1 fragments, and analyzed the cellular localization of different Nap1 fragments. The full length Nap1 localized to the membrane edges (arrowheads, Supp.Fig. 4B). Deletion of one putative membrane association domain at C-terminus (1019 a.a. fragment) did not alter the localization to the membrane edges. However, deletion of all four membrane association domains in the C-terminal region lead to the association of the deleted Nap1 (910 a.a and 898 a.a fragments, Supp.Fig. 4D, E) with acetylated tubulin positive, stable microtubules. Shorter deletion fragments either associate with membrane edges (707 a.a. and 480 a.a. fragments, Supp.Fig. 4F, G) or were diffusely distributed through out the cell (315 a.a and 67 a.a fragments, Supp.Fig. 4H, J). Intriguingly, a 200 a.a fragment that contains one membrane association domain from the N-terminal region, localized to the microtubule organizing center ( Supp.Fig. 4I). This serial deletion analysis suggest that distinct domains of Nap1 may play a role in the specific targeting or association of Nap1 to distinct cellular compartments (i.e., membrane edges, microtubules, or microtubule organizing center) during neuronal development. Surprisingly, Nap1, in addition to its previously suggested role in actin dynamics, may also be capable of modulating microtubule cytoskeleton.
Defective neuronal differentiation in Nap1 mutant mice
Since Nap1's localization on membrane edges might be critical for its function to induce process outgrowth during neuronal differentiation in the cortical plate, we generated a Nap1 mutant mouse line from ES cells in which the function of the Nap1 gene has been disrupted by insertional mutagenesis with β- geo reporter (Leighton et al., 2001; BayGenomics), resulting in the deletion of Nap1 C-terminal region essential for the membrane localization of Nap1. The insertion site was mapped to the intronic region flanked by exons 24 and 25 (Fig. 7A). The resulting protein is thus a fusion containing the N-terminal 898 amino acids (a.a.) of Nap1 (ΔC Nap1), fused to the 1291 a.a. of the β-geo reporter (Fig. 7A, B). Northern analysis with a Nap1 probe demonstrates the presence of wild type Nap1 transcript in the wild type, but not in the homozygote embryos (Fig. 7D). Immunoblot analysis confirms the absence of wild type Nap1 protein and the presence of a mutant Nap1-ΔC-βgal fusion protein in homozygous mutants [Nap1lacZ/lacZ] (Fig. 7C)
Figure 7. Generation and characterization of Nap1 mutant mice.
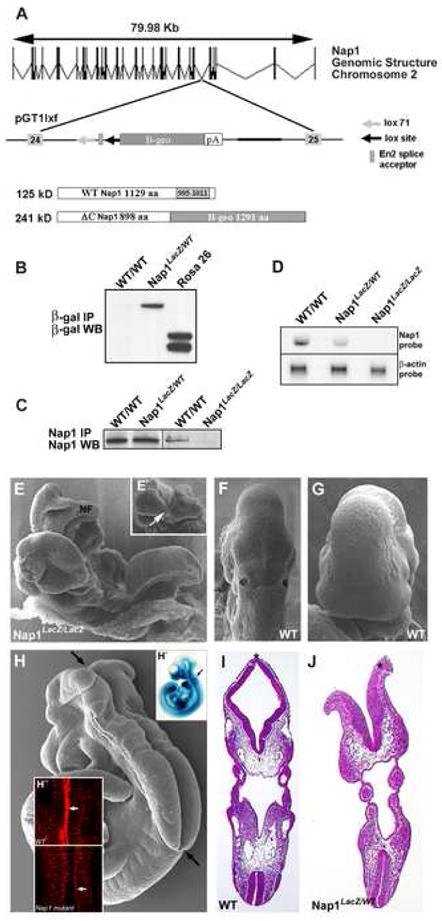
(A) Genomic structure of Nap1 locus on mouse chromosome 2, and insertion of gene trap vector pGT1lxf (BayGenomics) between exons 24 and 25. Wild-type Nap1 protein is approximately 125 kD, whereas mutant truncated-fusion protein is approximately 241 kD. (B) Brain extract from 3-week postnatal wild type (WT/WT) and heterozygous (Nap1LacZ/WT) mice was subjected to immunoprecipitation (IP) followed by Western blot (WB) analysis with an anti-β-galactosidase antibody. Truncated Nap1-β-gal fusion protein is expressed in Nap1LacZ/wt, but not in WT brains. Similar analysis using Rosa 26 brain extract is shown as a control. (C) Extract from E9.5 homozygous (Nap1LacZ/LacZ) and wild type mice were similarly subjected to immunoprecipitation followed by Western blotting with a C-terminal specific anti-Nap1 antibody to demonstrate the absence of wild-type Nap1 protein in homozygous mutant embryos. (D) Total RNA from wild type, heterozygous and homozygous embryos were analyzed by Northern blot using a Nap1 C-terminal probe, demonstrating the lack of wild type transcripts in mutants. β-actin is shown as a loading control. (E) Scanning electron micrograph (SEM) of the dorsal surface of a E9.5 Nap1LacZ/LacZ mutant embryo, where the neural folds (NF) remain unfused and wavy along the entire length of the anteroposterior axis. Anterior is to the left. (E') Anterior view of the cranial neural folds, which are completely open and curled over at the edges. White arrow indicates ventral midline. (F, G) SEM of the dorsal surface (F) and anterior view (G) of the neural tube in a wild type E9.5 embryo. The neural folds have completely closed, and the surface appears smooth. (H) SEM of the dorsal surface of a E10.5 embryo, which displays a neurulation defect. The luminal surface is completely exposed, and the edges of the neural folds have curled outward. (H') β-gal expression in Nap1LacZ/WT embryos indicates Nap1 expression in the developing neural tube (arrow). (H”) E9.5 wild type and mutant embryos were labeled with phalloidin and the midline region of anterior neural tube areas were imaged. Actin accumulation in the apical region of neuroepithelial cells is evident in wild type (arrows), but not Nap1 mutant (arrowhead) embryos. (I) H&E stained section of a wild type E9.5 embryo. Note the closed cranial neural tube (*). (J) Section from a E9.5 heterozygous animal, which demonstrates a neural tube defect. Note the neural folds which appear to curl outward, and remain unfused (*).
Analysis of litters derived from heterozygous crosses demonstrate that by embryonic day 8.5 (E8.5), Mendelian ratios of wild-type, heterozygous and homozygous embryos are detected; however, there was a drastic decrease in the number of homozygotes by E10.5, and no live Nap1lacZ/lacZ embryos were found at E11.5. Phenotypically normal Nap1lacZ/lacZ embryos could be recovered through E7.5, but embryos recovered from E8.5-E10.5 had varying degrees of morphological abnormalities ranging from severe neurulation defects to complete resorption. The most common phenotype observed in Nap1lacZ/lacZ E10.5 embryos is the strikingly open, undulating neural folds, which remain unfused along most of the rostral extent of the embryo, up to the mid spine (Fig. 7E). Wild type littermate controls at this stage display complete closure of the cranial neural tube and spinal cord (Fig. 7F, G). 12.4% of heterozygote mice show an open neural tube phenotype; however most heterozygous animals survive to adulthood (Fig. 7H). Histological analysis of these mutant heterozygous animals at E9.5 demonstrated dramatic abnormalities in the telencephalic neuroepithelium (Fig. 7I, J). Apical localization of actin filaments in neuroepithelial cells during neural tube closure is essential to complete this process (Ybot Gonzalez and Copp, 1999; Copp et al., 2003a, b; Rakeman and Anderson, 2006). Analysis of actin distribution with phalloidin labeling indicate that, in contrast to wt embryos, apical accumulation of actin filaments needed for neural tube closure is severely disrupted in Nap1 mutants (Fig. 7H″). To evaluate the neural tube defect further, we crossed Nap1 heterozygotes to ACTB-EGFP mice, expressing EGFP in all tissues under chicken β-actin promoter, to generate Nap1lacZ/+, ACTB-EGFP mice. These mice were intercrossed to generate Nap1lacZ/lacZ, ACTB-EGFP embryos. Live confocal imaging of apposing neural folds in the head region of these embryos indicate that the movement of neuroepithelial cells towards midline needed for neural tube closure is disrupted in Nap1 mutants (see Supplemental movies 1, 2, and 3).
The early embryonic lethality prevents the use of this mouse model to study Nap1's role in cortical neuronal differentiation in vivo. However, to understand how Nap1 may influence neuronal differentiation, wild type and Nap1 mutant E9.5 telencephalic neuroepithelial cells, which eventually give rise to cortical neurons, were cultured for 4 days to allow for the generation of classIII β-tubulin (Tuj-1) positive neurons in vitro. Wild type neurons display the characteristic differentiated phenotype with elongated axons, dendrites, growth cones, and dendritic spines (Fig. 8A, B). In contrast, Nap1 mutant neurons are severely defective in their ability to differentiate and extend axons and dendrites (Fig. 8C-F). Instead, Nap1 mutant neurons extend short, ill-defined stumps of neurites. These data further support the hypothesis that Nap1 plays an essential role in the final phenotypic differentiation of cortical neurons.
Figure 8. Defective neuronal differentiation in Nap1 mutants.
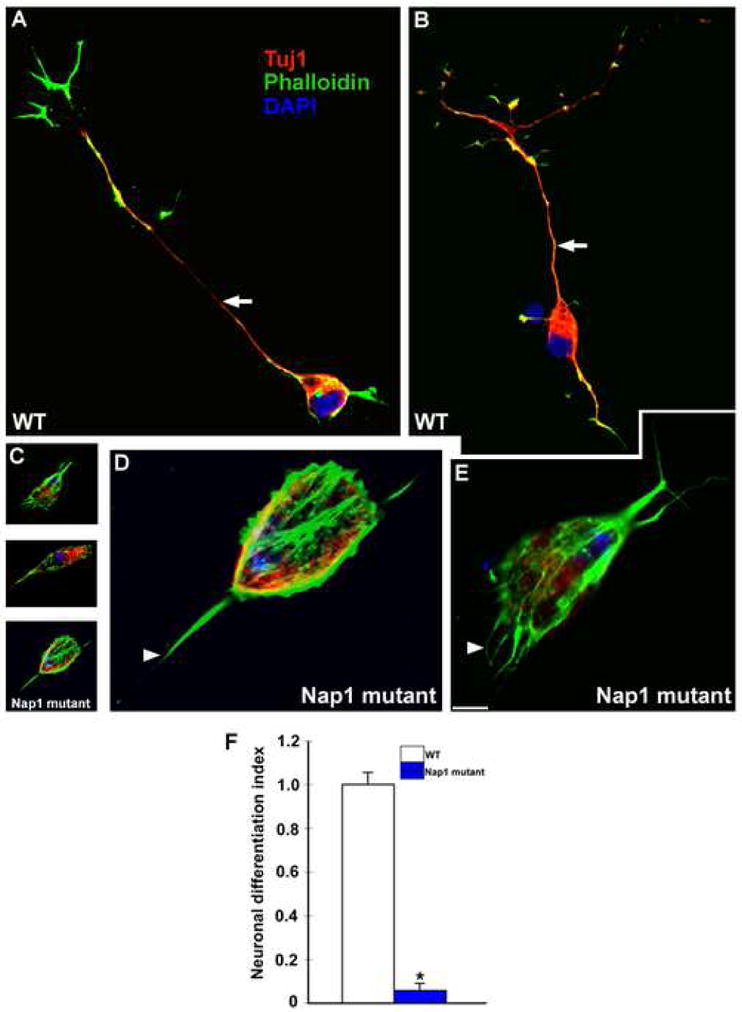
Telencephalic neuroepithelial cells from wild type and Nap1 mutant mice were maintained in vitro to generate neurons. Compared to wild type neurons (A, B, arrows), Nap1 mutant neurons (C) are severely defective in their ability to extend axons and dendrites. Higher magnification images of Nap1 mutant neurons (D, E) illustrate the ill- defined, short, stubby processes extended by these mutant neurons (arrowheads, D-E). (F) Assessment of neuronal differentiation (i.e., % of neurons with processes greater than 20μm length) indicates drastic deficits in Nap1 mutant neurons. Scale bar: A-C, 20 μm; D-E, 7μm.
Cellular mechanisms underlying Nap1 function
To evaluate the cellular mechanisms underlying Nap1's role in neuronal differentiation, we first analyzed the cytoskeletal organization of Nap1 mutant neuroepithelial cells. Since the early embryonic lethality of Nap1 mutants precludes the use of neurons, we used the telencephalic neuroepithelial cells that give rise to neurons in these studies. A significant disruption in actin cytoskeletal organization was noticed in Nap1 mutant cells. In contrast to wild type cells, actin filaments preferentially formed actin bundles around the cell cortex of the Nap1 mutant cells (Fig.9A, B). 94.6(±1.5) % of the mutant cells have actin filaments arrayed around the edges of the cells, compared to 2.96(±1.6) % of the wild type cells. Furthermore, the ratio of tyrosinated tubulin (i.e., newly polymerized tubulin) to acetylated tubulin (aged, stable form of tubulin) increases in Nap1 mutants and the acetylated tubulin fibers often form circular meshworks in Nap1 mutants (Fig.9C, D, G). Importantly, Nap1 mutant cells are mostly devoid of lamellipodia (Fig.9H-J). Live imaging of wild type and Nap1 mutant neuroepithelial cells indicate that wt cells display no defects in lamellipodial formation or activity, whereas Nap1 mutants are devoid of lamellipodia, and instead extend long, spiky processes which resemble flaccid filopodia (see Supplemental movies 4 and 5). In Nap1 mutant cells, immunoreactivity for cortactin, a marker of peripheral lamellipodia, is absent from the cell periphery, demonstrating that indeed these cells lack lamellipodia (Fig.9H, I). C-terminal deleted Nap1 (pCIG-ΔNap1-IRES-EGFP) overexpressing wild type cells displayed disrupted actin cytoskeletal phenotype similar to that of Nap1 mutant cells. shRNA mediated knockdown of Nap1 in wild type cells also disrupted actin and tubulin cytoskeletal organization and lamellipodial formation (Fig.9E, F, J). Conversely, expression of full length Nap1 (pCIG-Full Nap1-IRES-EGFP) rescued the Nap1 mutant phenotype (Fig.9J).
Figure 9. Actin, microtubule cytoskeletal organization and lamellipodial formation is disrupted in Nap1 mutants.

(A-B) Dissociated cells from the telencephalic neuroepithelium of E9.5 wild type and Nap1 mutant embryos were stained with phalloidin (green) and deoxyribonuclease (red). Phalloidin staining indicates F-actin, whereas deoxyribonuclease labels free actin monomers. Compared to wt cells (A), actin filaments (green) accumulate at the edges of Nap1 mutant cells (B). (C, D) Immunolabelling of wt and mutant cells with anti- acetylated tubulin antibodies, indicates disrupted acetylated tubulin organization in Nap1 mutants. Compared to the orderly array of stable microtubules in the wt cells (B, white arrowhead), acetylated microtubule strands appear to be disrupted and form concentric rings in Nap1 mutant cells (B, blue arrowhead). Similar disruptions in actin (E) and acetylated tubulin organization (F) is also evident in Nap1 shRNA expressing cells. Immunoblot analysis of acetylated tubulin and tyrosinated tubulin in wt and Nap1 mutant telencephalon indicates that Nap1 mutation reduced the level of stable, acetylated microtubules (blue arrowhead, G). (H-J) Analysis of lamellipodial formation in Nap1 disrupted cells. Primary neuroepithelial cells from wild type (F) and Nap1 mutant embryos (G) were labeled with phalloidin (green) and anti- cortactin antibodies (red). Wild type cells generate normal lamellipodia as seen by cortactin immunoreactivity and phalloidin staining (arrowheads ,H), whereas cells from Nap1 mutant cells generate abundant spiky protrusions (I), but not many lamellipodia. (J) Quantification of Nap1 effect. Analysis of cells with lamellipodia indicates a 90 % reduction in lamellipodial formation in Nap1 mutant cells. Expression of C-terminal deleted Nap1 or Nap1 shRNA in wild type neuroepithelial cells also leads to loss of lamellipodia . Expression of full length Nap1 rescues the mutant phenotype. Number of cells/ group> 3000. Data shown are mean ± SEM; asterisk, significant when compared with controls at p<0.001 (Student's t test). Scale bar: A, B, E, 25 μm; C, D, F, 3μm; H-I, 30μm. Also see movie files #4, and 5.
To further test the role of Nap1 in lamellipodial formation, we tested the ability of wild type, Nap1 mutant, or Nap1 shRNA expressing cells to form lamellipodia in response to PDGF. PDGF activates the Rac pathway and induces the formation of lamellipodia as well as ring ruffles (Krueger et al., 2003). Serum starved wild type and Nap1 disrupted cells were challenged with 10 ng/ml PDGF to assess their ability to form lamellipodia (Supplemental Fig. 5). 36% of wild type cells produced lamellipodia in response to PDGF treatment, but only 1.7% or 3.2% of the Nap1 mutant or Nap1 shRNA cells, respectively, formed lamellipodia under the same conditions (Supplemental Fig. 5). Nap1 disrupted cells are deficient in their ability to generate both dorsal and peripheral lamellipodial ruffles, two different types of lamellipodia noticed in cells undergoing active process extension (Suetsugu et al., 2003; Abercrombie et al., 1970). Deficits in lamellipodial activity were also noticed when Nap1 mutant cells were presented with PDGF coated beads (data not shown). Together, these data demonstrate that Nap1 protein is essential for the formation and activity of lamellipodia.
Lamellipodial activity and active, multiple membrane protrusions are essential steps in the initiation of neurite growth that occurs as neurons transform from migratory to post- migratory differentiation state in cerebral cortex. It is thought that during lamellipodial formation, Rac1 activation triggers active WAVE1 [(WASP (Wiskott–Aldrich syndrome protein)-family verprolin homologous protein1] complex to localize to membrane protrusions, causing actin nucleation in the protrusive edges of motile cells. The regulation of the sub cellular localization of WAVE1 plays an important role in the functional activity of WAVE1 (Eden et al., 2002, Stradal et al., 2004). Nap1, which forms a complex with WAVE1, is hypothesized to play a role in the functional status or subcellular targeting of WAVE1. Given the aberrant lamellipodial phenotype of the Nap1 disrupted cells, we tested the effect of Nap1 on WAVE1 localization. Biochemical analysis of WAVE1 expression in Nap1 mutants indicates that Nap1 mutant cells maintain the expression of WAVE1 (data not shown). We then immunolabelled wild type and Nap1 mutant cells with anti-WAVE1 antibodies and analyzed the pattern of WAVE 1 localization. In contrast to wild type cells, WAVE1 does not localize to the membrane ruffles at the leading, protrusive edges of the Nap1 mutant cells (Fig. 10A). Similar deficits in WAVE1 localization are also evident in Nap1 shRNA expressing cells (Fig.10B). This deficit was rescued by the re-expression of full length Nap1 (Fig.10B). To determine if Nap1 is essential to appropriately target WAVE1 to protrusive edges, we analyzed WAVE1 protein movement in wild type, Nap1 mutant, or Nap1 shRNA expressing cells. We generated WAVE1 fused with Kaede, a photoconvertible fluorescent protein which can be spectrally changed from green to red with UV light (Ando et al., 2002). Both wild type and Nap1 mutant cells were transfected with WAVE1-Kaeda. Some of the wild type cells were also co- transfected with WAVE1-Kaede and Nap1 shRNA. Localized green to red conversion of WAVE1 in transfected cells was induced with a 200msec. pulse of UV light. Time lapse analysis of the movement of photoconverted WAVE1 (red) indicates that in wild type cells WAVE1 gets targeted to and move towards protrusive membrane edges, whereas in Nap1 disrupted cells WAVE1 movement to membrane edges is severely retarded, thus confirming the essential role of Nap1 in the appropriate cellular localization of WAVE1 (Fig.10C-D; Supplemental movies 6, 7, and 8).
Figure 10. Defective localization of WAVE1 to protrusive membrane edges in Nap1 mutant cells.
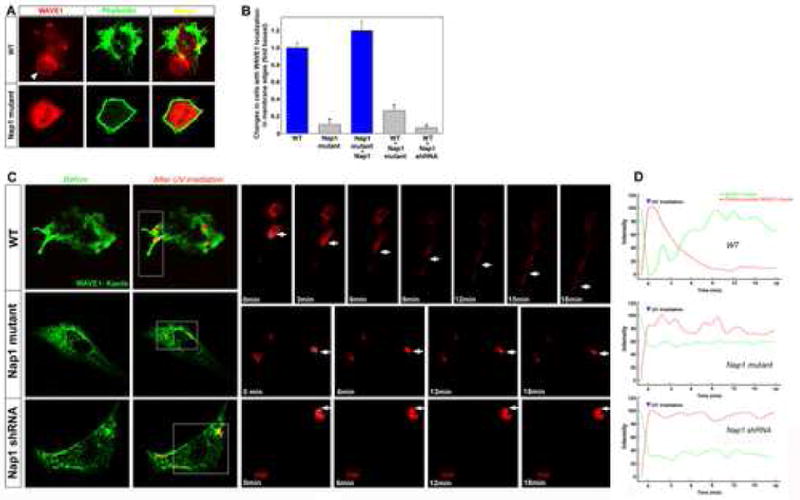
(A) Wild type and mutant telencephalic neuroepithelial cells were labeled with phalloidin (green) and anti-WAVE1 antibodies (red). In wt cells, WAVE1 predominantly localizes to lamellipodial membrane edges (arrowhead, A), whereas in Nap1 mutant cells, WAVE1 localization to lamellipodial protrusion is mostly absent. (B) Quantification of cells with WAVE1 localization on membrane edges indicates a significant deficit in Nap1 mutant or Nap1 deficient cells. Expression of C-terminal deleted Nap1 or Nap1 shRNA in wild type neuroepithelial cells leads to disrupted WAVE1 localization. This deficit can be rescued by expression of full length Nap1. (C, D) Tracking of WAVE1 localization in Nap1 disrupted cells. Wild type, Nap1 mutant, or Nap1 shRNA expressing cells were transfected with WAVE1- Kaede (green). After localized photoconversion with a UV laser , time-lapse images of photoconverted WAVE1- Kaeda (red) were obtained. In wild type cells WAVE1, actively moved towards the protrusive edges of the cells (arrows [wt panels], C; AVI movie file #6). In contrast WAVE1 movement is significantly retarded in Nap1 mutants (arrows [Nap1 mutant panels], C; AVI movie file #7) and in Nap1 knockdown cells (arrows [Nap1 shRNA panels], C; AVI movie file #8). Time after photoconversion is indicated in minutes. (D) Measurement of relative fluorescent intensities of WAVE1-green and WAVE1-red in defined areas within the photoconverted spots indicate that in wt cells both types of WAVE1 trafficked normally, whereas in Nap1 mutants or Nap1 shRNA expressing cells, movement of WAVE1 is highly restricted. Also see supplementary movie files #6, 7, and 8. Data shown are mean ±SEM; asterisk, significant when compared with controls at p<0.01 (Student's t test).
Discussion
The change in the neuronal cytoskeletal machinery from one that promotes oriented motility to one that facilitates elaboration of axons and dendrites is a critical step in the emergence of functional organization of neurons in cerebral cortex. What are the cytoskeletal regulators essential to affect this transition? Here we show that Nck associated protein 1 (Nap1), an adaptor protein that modulates cytoskeletal organization, is selectively expressed in the cortical plate region of the developing cortex, where neurons terminate their migration and initiate their final laminar specific differentiation. Nap1 is induced by BDNF, an essential mediator of cortical neuronal differentiation. Loss of Nap1 function inhibits post- migratory neuronal differentiation, whereas premature expression of Nap1 in migrating neurons retards their migration and promotes their differentiation. Furthermore, Nap1 mutation disrupts both actin and microtubule organization and the appropriate targeting of cytoskeletal regulators of process extension such as WAVE1. These findings imply that expression of Nap1 and the resultant changes in cytoskeletal dynamics is critical for the terminal, post-migratory differentiation of neurons in the developing cerebral cortex.
Cytoskeletal regulation during neuronal migration and differentiation in the cerebral cortex
Molecular analysis of human cortical developmental deficits suggests that dynamic regulation of neural cytoskeleton determines distinct aspects of the generation, migration, and differentiation of neurons in cerebral cortex. During neurogenesis, a microtubule associating protein (abnormal spindle-like microcephaly associated protein: ASPM) is expressed specifically in the VZ and is thought to modulate the spindle activity of the neuronal progenitor cells, resulting in the generation of appropriate numbers of post mitotic neurons (Bond et al., 2002). As newly generated neurons exit the VZ and embark on their journey towards the CP, appropriate expression of actin- binding protein, filamin A (FLNA) regulates the initiation of migration (reviewed in Marin and Rubenstein, 2003; Mochida and Walsh, 2004). Once neurons begin their migration, genes regulating microtubule cytoskeleton, including Lis1, doublecortin (Dcx), doublecortin like kinase (Dclk), Ndel1, MAP1b, MAP2, Tau, and mPAR6α, play an essential role in the maintenance of oriented neuronal motility (Solecki et al., 2004; Deuel at el., 2006; Koizumi et al., 2006; Shu et al., 2006; Tsai and Gleeson, 2005; Hatten, 2002). MAP1b, Tau, Filamin1, Nde1 and Dcx in migrating neurons are putative substrates for cyclin dependent kinase 5 (Cdk5), which together with its activating subunits, p35 and p39, functions to modulate normal neuronal migration in cerebral cortex (Tsai and Gleeson, 2005).
Though these observations clearly demonstrate that developmental stage specific expression and function of multiple cytoskeletal regulators critically influence the generation and migration of neurons, the cytoskeletal changes or regulators essential to convert neurons that are engaged in oriented motility into neurons that are capable of extending axons and dendrites in the developing cortex are unclear. Our analyses show that Nap1 is a cytoskeletal regulator essential for this step in the developing cerebral cortex. Suppression of Nap1 expression or mutations in Nap1 significantly retards neuronal differentiation (Fig.2, 3, 8). Nap1 knockdown does not affect the migration or the placement of neurons. Cohorts of neurons that arrive in the cortical plate at the same time begin their morphological differentiation at the same time (Bayer and Altman, 1991; Miller, 1981). Thus the differences in differentiation noticed between control and Nap1 deficient neurons are unlikely due to delayed migration and resultant late initiation of differentiation by Nap1 deficient neurons. Furthermore, ectopic, premature induction of Nap1 in migrating cortical neurons retards their migration and promotes premature neuronal differentiation (Fig.4, 5). Together, these studies suggest that Nap1 plays an essential role in the timely differentiation of neurons once they get to the cortical plate.
How does Nap1 modulate cortical neuronal differentiation? As a neuron undergoes terminal differentiation in distinct layers of the emerging cortical plate, it essentially transforms from a motile cell with a leading and trailing process, into one that is non-motile, but with multiple processes and branches. This requires generation of multiple membrane protrusive structures and coordinated changes in actin and microtubule cytoskeleton in response to activity dependent or extracellular neuronal differentiation cues expressed in the developing cortical plate. The induction of Nap1 in cortical plate, which is capable of organizing both actin and microtubule cytoskeleton (Fig.9), its ability to influence the cellular targeting of major cytoskeletal regulators of process outgrowth such as WAVE1 (Fig.10), and its essential role in inducing membrane protrusions (Fig.9, Supplemental Fig. 4, 5, 6), may thus lead to post migratory differentiation of neurons in the cerebral cortex. Consistent with this hypothesis, Nap1 is induced by BDNF, a potent cortical neuronal differentiation signal.
Nap1's function during neural tube development
Nap1 mutation in Nap1lacZ/llacZ mice clearly disrupts neural tube formation (Fig.7; Rakeman and Anderson, 2006). Although small amounts of normally spliced Nap1 can apparently be generated in Nap1 gene trap- insertion mutants (Rakeman and Anderson, 2006; Leighton et al., 2001) and Nap1 C-terminal deleted protein may have gain of function effects, similar neural tube defect was also noticed following loss of function missense mutation in the evolutionarily conserved L17P residue at the Nap1 N-terminal (Rakeman and Anderson, 2006). Normally, primary neural tube closure is initiated at the hindbrain/cervical boundary and proceeds in both rostral and caudal directions. Brain closure also depends on secondary closure events initiated at the midbrain/forebrain boundary and at the rostral tip of the forebrain (Copp et al., 2003). In Nap1 mutants, neural tube is open along most of the rostro-caudal extent (Fig.7), indicating a failure of normal neural tube closure events.
Completion of the neural tube closure depends on lamellipodial protrusions from the apical cells of the apposing neural folds. Interdigitation of these lamellipodial protrusions from across the midline facilitates cell- cell recognition and adhesion, leading to the fusion of the neural folds and the formation of neural tube (Copp et al., 2003; Geelen et al., 1979). The defective lamellipodial activity in Nap1 mutant neuroepithelial cells (Figs.9, Supp. Fig. 5, Supp. Movies 4-5) may have disrupted this process essential for neural tube closure in Nap1 mutants.
Cellular and molecular mechanisms underlying Nap1 function
Nap1's ability to promote neuronal differentiation may depend on its ability to appropriately target or control the functional status of associated components of cytoskeletal machinery essential for neuronal process elaboration and maintenance. Nap1 is a member of the WAVE complex. Nap1, which interacts directly with Sra1/PIR121 (which binds to GTP-bound Rac1) and Abi1 (which binds the SH3 domain of Nck) , forms a tetrameric complex containing Sra1/PIR121, Abi1/2, and HSPC300 to regulate WAVE1 activity (Kitamura et al., 1996; Kitamura et al., 1997; Kobayashi et al., 1998; Hummel et al., 2000; Soto et al., 2002; Yamamoto et al., 2001; Eden et al., 2002). WAVE1, in contrast to the related WASP proteins, which are autoinhibitory and are activated by binding GTP-bound Cdc42 to participate in the formation of filopodia, is constitutively active and acts downstream of Rac1 to initiate lamellipodia formation (Biyasheva et al, 2004; Blagg and Insall, 2004; Corey and Ridley, 2003; Machesky et al., 1999; Miki et al., 1998; Nakagawa et al., 2001; Innocenti et al., 2004; Kunda et al., 2003; Rogers et al., 2003; Rohatgi et al., 2000; Steffan et al., 2004). Nap1 containing WAVE complex regulates the functional status and cellular targeting of WAVE1. Upon activation of Rac and resultant changes in WAVE complex, WAVE1 binds and activates Arp2/3, leading to actin polymerization and branched actin filament formation at protrusive membrane edges and subsequent lamellipodial extension (Blanchoin et al., 2000; Eden et al., 2002; Gautreau et al., 2004; Bogdan and Klambt, 2003; Stradal et al., 2004; Innocenti et al., 2004; Kunda et al., 2003; Millard et al., 2004; Rogers et al., 2003; Steffan et al., 2004; Svetkina and Borrisey, 1999).
The localization of WAVE1 at the edges of extending processes is essential to drive the localized activation of Arp2/3 complex and actin polymerization at the protrusive edges (Hahne et al., 2001; Nakagawa et al., 2001). Of the three highly homologous members of WAVE proteins (WAVE1-3), only WAVE1's expression is limited to the developing brain (Dahl et al., 2003; Sossey-Alaoui et al., 2003). Loss of WAVE1 function disrupts cerebral cortical development and functions such as learning and memory (Dahl et al., 2003; Soderling et al., 2003, 2007). Nap1 appears to be essential not only for the targeting of WAVE1 to the membrane, but also for the stability of WAVE1 (Rakeman and Anderson, 2006; Steffen et al., 2004). Reduction in WAVE protein levels were noticed in Nap1 mutants or Nap1 shRNA expressing melanoma cells (Steffen et al., 2004; Rakeman and Anderson, 2006). However, the lack of WAVE targeting to the protrusive edges, not the reduced WAVE levels, appears to underlie the lamellipodial defects in Nap1 deficient cells (Steffen et al., 2004). Nap1 deficiency also disrupts the membrane localization of other WAVE complex components Sra1 and Abi1 (Steffan et al., 2004). In addition to being regulated by Nap1-Abi1/2-PIR121-HSPC300 complex (Echarri et al., 2004; Eden et al., 2002; Gautreau et al., 2004; Grove et al., 2004; Innocenti et al., 2004; Rakeman and Anderson, 2006; Steffen et al., 2004), WAVE1 can also be phosphorylated by Cdk5. Cdk5 can thus down modulate WAVE1's ability to activate Arp2/3 dependent actin polymerization (Kim et al., 2006). Interestingly, the decrease in dendritic spines following phosphorylation of CDK5 sites in WAVE1 is similar to that noticed in Nap1 deficient neurons (Supp.Fig.3). Furthermore, Cdk5 and its regulatory subunit, p35, can form a complex with PIR121, Nap1, and WAVE1 (Kim et al., 2006). Inactivation of Nap1 disrupts not only WAVE1 function (Fig.10), but may also inappropriately activate formins (Insall and Jones, 2006, Rakeman and Anderson, 2006). Thus, Nap1 by acting as a nodal point member of multiple complexes regulating the functional status of key cytoskeletal regulators such as WAVE1, may coordinate the cytoskeletal rearrangements needed to transform neurons from a migratory to post migratory differentiation state.
Post- migratory, differentiating cortical neurons undergo extensive neurite growth and guidance to generate the appropriate axon- dendritic architecture and connectivity. In general, microtubule polymerization is thought to drive neurite growth and elongation, whereas actin polymerization is critical for the ability of neurite growth cones to respond to guidance cues in the environment. Coordination of both actin and microtubule dynamics is essential for the differentiating cortical neurons to form and maintain appropriate patterns of connections in the embryonic cortex (Dent and Kalil, 2001; Marsh et al., 1984; Strasser et al., 2004; Rochlin et al., 1999). Nap1's ability to organize actin cytoskeleton and regulate microtubule stability places it in a unique position to influence both microtubule and actin dynamics during this process. Though the exact nature of actin- microtubule cross talk and coordination during cortical neuronal differentiation is yet to be fully elucidated, induction of Nap1 may influence neuronal cellular domains such as actin arcs or axon branch points, where actin and microtubules were found to modulate each other's organization and function during neuronal extension (Dent and Kalil, 2001; Schaefer et al., 2002). Elucidating how Nap1 differentially associates with and modulates the organization of actin and microtubule compartments in differentiating neurons will be essential to further delineate Nap1's significance during corticogenesis.
The growth and differentiation of cortical neurons rely on activity dependent neurotrophic factor (e.g., BDNF) signaling (McAllister et al., 1996; Ghosh et al., 1994; Reichardt et al., 2006). As such, the induction of Nap1, an essential cytoskeletal component of neuronal differentiation machinery, by BDNF in differentiating cortical neurons may involve correlated neuronal activity. Selective expression of Nap1 in cortical plate neurons and the resultant formation of multimeric complexes (e.g., Nap1-WAVE1, Nap1-Cdk5) capable of distinct cytoskeletal regulation may usher in the cytoskeletal rearrangements that are essential to change the neuronal cytoskeletal machinery from one that promotes oriented motility to one that facilitates elaboration of axons and dendrites, and interconnectivity between appropriate synaptic partners.
Materials and Methods
Generation of Nap1 mutant mice
Two independent ES cell lines, XE133 and XE68, containing identical insertions in the Nap1 locus were obtained (BayGenomics). These insertions utilized the pGT1lxf gene trap vector, which contains an En2 splice acceptor flanked by a 5′ lox71 and 3′ loxP site, and a gene fusion, β-geo, between β-galactosidase (β-gal) and the neomycin resistance gene (Mitchell et al., 2001). RT-PCR of the ES cells was used to confirm the presence of the insertion. Briefly, 5 ug total RNA isolated from ES cells using the Trizol method was reverse transcribed with MMLV reverse transcriptase using oligo 78 (5′-TAATGGGATAGGTTACGT-3′). PCR was then performed using cDNA derived from XE133 or XE68 ES cells as a template with the following primers: oligo 79 (common) 5′-AGTATCGGCCTCAGGAAGATCG-3′ and XE133FP1 5′-CTGTATACAAACTGGTACTTGG-3′ or XE68FP1 5′-AGACAGCCATGGAGAACCAACC-3′. To confirm that the XE133 and XE68 ES cells contained a single insertion site, Southern blotting was performed using genomic DNA isolated from ES cells digested with EcoRV and probed with a β-galactosidase specific [α-32p] dCTP labeled DNA probe. ES cells harboring gene trap insertions were used to generate chimeric mice by blastocyst injection, and germline transmission was achieved by mating male chimeric mice to wild-type C57Bl6 females. Additionally, F2 heterozygous males were outcrossed to wild type CD1 females; however, genetic background did not affect embryonic viability or phenotype.
Northern blot analysis and Nap1 mutant genotyping
Total RNA was isolated from E9 embryos using Trizol (Invitrogen) and analyzed by Northern blotting using αP32 dCTP DNA probes for Nap1 and β-actin. Genomic insertion site of the pGT1lxf gene trap vector was determined by sequence analysis of nested PCR products. PCR genotyping analysis of WT allele generates a 710 b.p. product using the following primers: Nap1-FPa (common) 5′-CTGTCTGCCTGCCTCTGCTTTTTG-3′ and Nap1-RP2a 5′-CCCACCCCCCAGGGTTTGAGTAGAG-3′, mutant allele generates 325 b.p. product using Nap1-FPa and pGT1lxf-RPa 5′-CCACCCTGGGGTTCGTGTCCTAC-3′.
Histology
For X-gal staining, embryos were fixed in 2% paraformaldehyde (PFA) and 0.1% gluteraldehyde in 0.1M Phosphate buffer, then stained with a β-Gal Staining Set (Roche). For Scanning Electron Microscopy, embryos were fixed as above, and processed according to standard procedures at UNC's electron microscope core facility. For H & E staining, embryos were fixed in 4% PFA, then dehydrated in ethanol, cleared in Histoclear (National Diagnostics), and embedded in paraffin. 7 um sections were mounted, deparaffinised, and stained with Harris hematoxylin and eosin-Y. Whole mount images were taken using a Leica MZFL III dissecting microscope and Nikon Coolpix 4500 digital camera.
Live imaging of neural tube development in Nap1 mutants
E8.5-9.5 embryos from Nap1lacZ/LacZ, ACTB-EGFP or Nap1wt/wt, ACTB-EGFP mice were immobilized with a mix of artificial cerebrospinal fluid and 1% low melting point agarose on a MatTek 35 mm glass bottom dish, immersed in OptiMEM/10% FBS media, and placed in a live incubation chamber attached to a Zeiss Pascal confocal microscope. The apposing neural folds of the head region of the embryos were repeatedly imaged every 3 minutes for up to 2 hours.
Immunoprecipitation and Western Blot Analysis
A polyclonal antibody to Nap1 was generated against the peptide sequence CHAVYKQSVTSSA (Covance). A monoclonal anti-β-galactosidase antibody (Promega) was used for immunoprecipitation of mutant fusion Nap1. For immunoprecipitation, anti-Nap1 antisera (1:500) or anti-β-gal antibody (1:1000) was added to extract prepared from wild type, heterozygous or homozygous telencephalon lysed in Buffer A (20 mM Tris, pH 7.5, 1 mM EDTA, 300 mM NaCl, 5% glycerol, 1% TritonX100, 0.2% Deoxycholate, 1 mM PMSF, 1 mM DTT). Antibody-Antigen complexes were collected with Protein A Sepharose (Zymed), followed by 3 washes with Buffer A. Nap1 was detected by Western blot with anti-Nap1 antisera (1:500) or anti-β-gal antibody (1:1000), followed by appropriate HRP-coupled secondary antibodies (Pierce). Protein concentration of lysates was determined using Nano-orange protein quantification kit (Molecular Probes) and was then normalized among all samples in each experiment prior to immunoprecipitation.
Immunohistochemistry
Primary neuroepithelial cells from E9.5 telencephalon was isolated and maintained in DMEM with 10% FBS and penicillin/streptomycin as described earlier (Schmid et al., 2003). The following antibodies were used: anti-cortactin (mouse, 4F11, Upstate), anti-WAVE1 (rabbit, gift from Dr. S. Kwak, Wyeth Laboratories), anti-Nestin (Iowa Hybridoma Bank), anti- acetylated tubulin (mouse, Sigma), anti-Tuj1 (mouse, Covance), anti-Tau1 (mouse, Sigma), anti-Map2 (mouse, Sigma) , anti β-galactosidase (rabbit, MP biomedicals), anti-Nap1 (rabbit, gift from Dr. Stradal, German Research Centre for Biotechnology), anti-Brn1 (gunny pig, gift from Dr. Ryan, McGill University), anti-Tbr1 (rabbit, Chemicon), anti-GFP (Chicken, rabbit, Abcam). Appropriate Alexa-dye labeled secondary antibodies (Molecular Probes) were used to detect primary antibody binding. For phalloidin staining, either an Alexa 488-phalloidin (Molecular Probes) or a TRITC-phalloidin (Sigma) was used.
In situ hybridization
Nap1 probe spans sequence #2281-3170 of the mouse Nap1 cDNA sequence. Nap1 cDNA was cut with EcoRV/EcoRI sites and cloned into pBluescript SK(−). After linearization with EcoRV, antisense transcripts were produced using T7 RNA polymerase. In situ hybridization was performed as previously described (Anton et al., 2004) at the UNC Neuroscience Center's In situ hybridization core facility.
PDGF assays
For PDGF treatment, telencephalic neuroepithelial cells were cultured in DMEM (Gibco) without serum for 12 hrs, treated with 10ng/ml PDGF-BB for 15 min, then fixed in 4% PFA and processed as above for immunofluorescence with phalloidin or anti-cortactin antibodies. In some experiments, PDGF (10 μg/ml) coated heparin beads were placed adjacent to cells and they were repeatedly imaged at 1-5 minute intervals (Suetsugu et al., 2003) with Zeiss confocal microsope. The rate of membrane extension towards the beads as well as the number of cells responding to PDGF with lamellipodial formation was measured.
Generation and characterization of Nap1 specific shRNA
The Nap1 unique target sequences, GCTCACCATCCTCAACGAC, GTTGCACACTGCACTTTCG, GTTCCTGAGTGAGAGCCTT, CCAGATTGCTGCAGCTTTG, and GGAATTCCTGGCGCTTGCA are located at 48-66 bp, 2031-2049 bp, 2547-2565 bp, 3111-3129 bp and 3168-3186 bp, respectively of Nap1 cDNA. As a negative control for each of the shRNA construct, 3 nt mutations were made in the respective targeting sequence (e.g., GCTTACCATTCTCAATGAC [control for 48-66bp target sequence]). The target sequence oligos and mutated target sequence oligos were subcloned into pCGLH vector (gift from Dr. Sestan, Yale University), which contains chicken β-actin promoter driven EGFP and H1 promoter for shRNA transcription. To test the efficiency of shRNA mediated Nap1 knockdown, targeting constructs were transfected into CAD cells or E16 primary cortical cells using Effectene (Qiagen) as per manufacturer's instructions. High transfection efficiency (>80% of total cells on the dish) was confirmed by EGFP expression. Four days later, cell extracts were immunoblotted with anti-Nap1, tubulin, or ErbB4 antibodies. Some of the transfected cortical neurons were fixed in 4% paraformaldehyde and immunolabelled with anti-Nap1 antibodies to further confirm that the Nap1 protein expression is diminished specifically in Nap1 shRNA expressing cells. Cortical neurons were also co-transfected with Nap1 shRNA (in pCRLH vector expressing RFP) and full length Nap1 –EGFP fusion plasmids. Four days later, ShRNA expressing neurons (red) were evaluated for reduced GFP expression, compared to control shRNA expressing neurons.
Generation and characterization of Nap1 fragments
To characterize the domains of Nap1 that are critical for its function, serial deletion fragments of Nap1 were subcloned into pCMV-EGFP plasmid (Clonetech) to generate Nap1 fragments fused to EGFP (pCMV-ΔNap1-EGFP). Cos7 cells or E9.5 neuroepithelial cells were transfected with Nap1 fragments to analyze the cellular localization of these EGFP tagged constructs. Nap1 deletion fragments were also subcloned into pCIG-IRES-EGFP plasmids and used to transfect wild type or mutant cells, alone or in combination with Nap1shRNA, to analyze the effect of Nap1 deletion fragments.
Functional analysis of Nap1 in the developing cerebral cortex
To determine the effect of ectopic, premature induction of Nap1 in migrating neurons, NeuroD promoter-Nap1-IRES-EGFP or control NeuroD promoter-IRES-EGFP plasmids were in utero electroporated into E14-15 cerebral cortex (Gongidi et al., 2004). Forty eight hours after electroporation, cerebral cortices were removed, vibrotome sliced, and immunolabeled with antibodies to markers that are normally expressed by neurons that are under going post migratory differentiation (i.e., Tbr-1, Brn-1) to evaluate if premature induction of Nap1 in migrating neurons induce premature differentiation as evidenced by premature layer specific molecular marker expression. The morphology of control or Nap1 expressing neurons in the intermediate zone was evaluated by immunolabeling them with anti-GFP antibodies to obtain higher magnification images of these neurons. The extent of migration of the control and Nap1 expressing neurons within the cerebral wall was analyzed as described in Gongidi et al., 2004, and Schmid et al., 2004. In some experiments, intermediate zone containing GFP+ control and Nap1 expressing neurons were microdissected, dissociated, and plated at a density of 500K cells/ml on laminin coated MatTek 35 mm glass bottom dishes. GFP+ neurons were then repeatedly imaged using a Zeiss Pascal confocal microscope equipped with live incubation chamber. Some of these cultures were fixed in 4% paraformaldehyde after 7.5 hours in vitro and immunolabeled with neuron specific Tuj1 antibodies to evaluate the morphological differentiation of control and Nap1 expressing neurons.
To determine the effect of Nap1 during post migratory differentiation of cortical neurons in vitro, dissociated E14 cortical neurons were transfected with either control, or Nap1 shRNA plasmids. Three days after transfection, neurons were fixed and immunolabelled with Tuj1. The total length, the number of primary, secondary, and tertiary branches of both axons and dendrites, and the number of dendrites on these neurons were measured as described in Schmid et al., 2004. Axons and dendrites were identified based on their morphology (Gaudilliere et al., 2004).
To determine the effect of Nap1 in post-migratory differentiation of cortical neurons in vivo, E15 embryos were electroporated with Nap1 or control shRNA, allowed to survive till postnatal day 2 or 17 and the patterns of dendritic and axonal morphology (i.e., length, numbers, branching patterns, and orientation of apical processes) of control and Nap1 shRNA expressing neurons (GFP+) in cerebral cortex was evaluated as described earlier (Schmid et al., 2004; Anton et al., 2004). Neuronal differentiation index at P17 was measured as the percentage of area occupied by the GFP positive neurons per 25K μm2 total area. Neuronal position within the cortex was measured as described earlier (Gongidi et al., 2004). In some experiments, cortices were removed from the embryos forty eight hours after electroporation, coronally sectioned, mounted on nucleopore membrane filters, and GFP labeled control or Nap1shRNA+ neurons in the intermediate zone of the slices were repeatedly imaged using a Zeiss inverted microscope (attached to a confocal laser scanning system and a live cell incubation chamber) for 2-3 hours. The rate of migration of the monitored cells was measured using LSM5 Pascal program (Zeiss).
WAVE1 protein tracking
To characterize the movement of WAVE1 protein in normal and Nap1 disrupted cells, WAVE1 (gift from Dr. Terada, University of Texas Southwestern) and Kaede (MBL Co.) were subcloned into pCAGS plasmid to generate WAVE1 fused to Kaede. E9.5 neuroepithelial cells from wild type and Nap1 mutant cells were transfected with WAVE1-Kaede (green). Some of the wild type cells were also co-transfected with Nap1 shRNA and WAVE- Kaede. Twenty four hours later, localized spots in transfected cells were photoconverted with a 200m.sec pulse of UV laser (351-364nm) attached to a Leica SP2 Laser Scanning confocal microscope. The movement of converted WAVE1-Kaede (red) was evaluated by time lapse imaging of WAVE1 green/red fluorescence. The relative changes in WAVE1 fluorescence intensity in the photoconverted regions of the cells were measured using Zeiss LSM image browser and image J program.
Supplementary Material
Acknowledgments
This research was supported by NIH grant MH060929 to EA and by the confocal imaging core of an NINDS institutional center core grant. We thank F. Polleaux, A- S. Lamantia, M. Deshmukh, N. Sestan, W. Snider, J. Anderson, and P. Manness for helpful comments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abercrombie M, Heaysman JE, Pegrum SM. The locomotion of fibroblasts in culture. II. “Ruffling.”. Exp Cell Res. 1970;60:437–444. doi: 10.1016/0014-4827(70)90537-9. [DOI] [PubMed] [Google Scholar]
- Ando R, Hama H, Yamamoto-Hino M, Mizuno H, Miyawaki A. An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein. Proc Natl Acad Sci U S A. 2002;99:12651–6. doi: 10.1073/pnas.202320599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anton ES, Ghashghaei HT, Weber JL, McCann C, Fischer TM, Cheung ID, Gassmann M, Messing A, Klein R, Schwab MH, Lloyd KC, Lai C. Receptor tyrosine kinase ErbB4 modulates neuroblast migration and placement in the adult forebrain. Nat Neurosci. 2004;7:1319–28. doi: 10.1038/nn1345. [DOI] [PubMed] [Google Scholar]
- Ayala R, Shu T, Tsai LH. Trekking across the brain: the journey of neuronal migration. Cell. 2007;128:29–43. doi: 10.1016/j.cell.2006.12.021. [DOI] [PubMed] [Google Scholar]
- Baumgartner S, Martin D, Chiquet-Ehrismann R, Sutton J, Desai A, Huang I, Kato K, Hromas R. The HEM proteins: a novel family of tissue-specific transmembrane proteins expressed from invertebrates through mammals with an essential function in oogenesis. J Mol Biol. 1995;251:41–9. doi: 10.1006/jmbi.1995.0414. [DOI] [PubMed] [Google Scholar]
- Bayer SA, Altman J. Development of the cortical plate. In: Bayer SA, Altman J, editors. Neocortical Development. Raven Press; NY: 1991. [Google Scholar]
- Biyasheva A, Svitkina T, Kunda P, Baum B, Borisy G. Cascade pathway of filopodia formation downstream of SCAR. J Cell Sci. 2004;117:837–48. doi: 10.1242/jcs.00921. [DOI] [PubMed] [Google Scholar]
- Bladt F, Aippersbach E, Gelkop S, Strasser GA, Nash P, Tafuri A, Gertler FB, Pawson T. The murine Nck SH2/SH3 adaptors are important for the development of mesoderm-derived embryonic structures and for regulating the cellular actin network. Mol Cell Biol. 2003;23:4586–97. doi: 10.1128/MCB.23.13.4586-4597.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagg SL, Insall RH. Solving the WAVE function. Nat Cell Biol. 2004;6:279–81. doi: 10.1038/ncb0404-279. [DOI] [PubMed] [Google Scholar]
- Blanchoin L, Amann KJ, Higgs HN, Marchand JB, Kaiser DA, Pollard TD. Direct observation of dendritic actin filament networks nucleated by Arp2/3 complex and WASP/Scar kinases. Nature. 2000;404:1007–1011. doi: 10.1038/35010008. [DOI] [PubMed] [Google Scholar]
- Bogdan S, Klambt C. Kette regulates actin dynamics and genetically interacts with Wave and Wasp. Development. 2003;130:4427–37. doi: 10.1242/dev.00663. [DOI] [PubMed] [Google Scholar]
- Bond J, Roberts E, Mochida GH, Hampshire DJ, Scott S, Askham JM, Springell K, Mahadevan M, Crow YJ, Markham AF, Walsh CA, Woods CG. ASPM is a major determinant of cerebral cortical size. Nat Genet. 2002;32:316–20. doi: 10.1038/ng995. [DOI] [PubMed] [Google Scholar]
- Cabelli RJ, Shelton DL, Segal RA, Shatz CJ. Blockade of endogenous ligands of trkB inhibits formation of ocular dominance columns. Neuron. 1997;19:63–76. doi: 10.1016/s0896-6273(00)80348-7. [DOI] [PubMed] [Google Scholar]
- Copp AJ, Greene ND, Murdoch JN. Dishevelled: linking convergent extension with neural tube closure. Trends Neurosci. 2003a;26:453–5. doi: 10.1016/S0166-2236(03)00212-1. [DOI] [PubMed] [Google Scholar]
- Copp AJ, Greene ND, Murdoch JN. The genetic basis of mammalian neurulation. Nat Rev Genet. 2003b;4:784–93. doi: 10.1038/nrg1181. [DOI] [PubMed] [Google Scholar]
- Cory GO, Ridley AJ. Cell motility: braking WAVEs. Nature. 2002;418(6899):732–3. doi: 10.1038/418732a. [DOI] [PubMed] [Google Scholar]
- Dahl JP, Wang-Dunlop J, Gonzales C, Goad ME, Mark RJ, Kwak SP. Characterization of the WAVE1 knock-out mouse: implications for CNS development. J Neurosci. 2003;23:3343–52. doi: 10.1523/JNEUROSCI.23-08-03343.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent EW, Kalil K. Axon branching requires interactions between dynamic microtubules and actin filaments. J Neurosci. 2001;21:9757–9769. doi: 10.1523/JNEUROSCI.21-24-09757.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuel TA, Liu JS, Corbo JC, Yoo SY, Rorke-Adams LB, Walsh CA. Genetic interactions between doublecortin and doublecortin-like kinase in neuronal migration and axon outgrowth. Neuron. 2006;49:41–53. doi: 10.1016/j.neuron.2005.10.038. [DOI] [PubMed] [Google Scholar]
- Echarri A, Lai MJ, Robinson MR, Pendergast AM. Abl interactor 1 (Abi-1) wave-binding and SNARE domains regulate its nucleocytoplasmic shuttling, lamellipodium localization, and wave-1 levels. Mol Cell Biol. 2004;24:4979–93. doi: 10.1128/MCB.24.11.4979-4993.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden S, Rohatgi R, Podtelejnikov AV, Mann M, Kirschner MW. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature. 2002;418:790–3. doi: 10.1038/nature00859. [DOI] [PubMed] [Google Scholar]
- Gaudilliere B, Konishi Y, de la Iglesia N, Yao G, Bonni A. A CaMKII-NeuroD signaling pathway specifies dendritic morphogenesis. Neuron. 2004;41:229–41. doi: 10.1016/s0896-6273(03)00841-9. [DOI] [PubMed] [Google Scholar]
- Gautreau A, Ho HY, Li J, Steen H, Gygi SP, Kirschner MW. Purification and architecture of the ubiquitous Wave complex. Proc Natl Acad Sci U S A. 2004;101:4379–83. doi: 10.1073/pnas.0400628101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geelen JA, Langman J. Ultrastructural observations on closure of the neural tube in the mouse. Anat Embryol (Berl) 1979;156:73–88. doi: 10.1007/BF00315716. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–23. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- Gongidi V, Ring C, Moody M, Brekken R, Sage EH, Rakic P, Anton ES. SPARC-like 1 regulates the terminal phase of radial glia-guided migration in the cerebral cortex. Neuron. 2004;41:57–69. doi: 10.1016/s0896-6273(03)00818-3. [DOI] [PubMed] [Google Scholar]
- Grove M, Demyanenko G, Echarri A, Zipfel PA, Quiroz ME, Rodriguiz RM, Playford M, Martensen SA, Robinson MR, Wetsel WC, Maness PF, Pendergast AM. ABI2-deficient mice exhibit defective cell migration, aberrant dendritic spine morphogenesis, and deficits in learning and memory. Mol Cell Biol. 2004;24:10905–22. doi: 10.1128/MCB.24.24.10905-10922.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne P, Sechi A, Benesch S, Small JV. Scar/WAVE is localised at the tips of protruding lamellipodia in living cells. FEBS Lett. 2001;492:215–220. doi: 10.1016/s0014-5793(01)02239-6. [DOI] [PubMed] [Google Scholar]
- Hatten ME. New directions in neuronal migration. Science. 2002;297:1660–3. doi: 10.1126/science.1074572. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Shi L, Justice N, Hsueh Y, Sheng M, Smiga S, Bulfone A, Goffinet AM, Campagnoni AT, Rubenstein JL. Tbr1 regulates differentiation of the preplate and layer 6. Neuron. 2001;29:353–66. doi: 10.1016/s0896-6273(01)00211-2. [DOI] [PubMed] [Google Scholar]
- Huang HP, Liu M, El-Hodiri HM, Chu K, Jamrich M, Tsai MJ. Regulation of the pancreatic islet- specific gene BETA2 (neuroD) by Neurogenein 3. Mol Cell Biol. 2000;20:3292–3307. doi: 10.1128/mcb.20.9.3292-3307.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel T, Leifker K, Klambt C. The Drosophila HEM-2/NAP1 homolog KETTE controls axonal pathfinding and cytoskeletal organization. Genes Dev. 2000;14:863–73. [PMC free article] [PubMed] [Google Scholar]
- Innocenti M, Zucconi A, Disanza A, Frittoli E, Areces LB, Steffen A, Stradal TE, Di Fiore PP, Carlier MF, Scita G. Abi1 is essential for the formation and activation of a WAVE2 signalling complex. Nat Cell Biol. 2004;6:319–27. doi: 10.1038/ncb1105. [DOI] [PubMed] [Google Scholar]
- Insall RH, Jones GE. Moving matters: signals and mechanisms in directed cell migration. Nat Cell Biol. 2006;8:776–9. doi: 10.1038/ncb0806-776. [DOI] [PubMed] [Google Scholar]
- Kim Y, Sung JY, Ceglia I, Lee KW, Ahn JH, Halford JM, Kim AM, Kwak SP, Park JB, Ho Ryu S, Schenck A, Bardoni B, Scott JD, Nairn AC, Greengard P. Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature. 2006;442:814–7. doi: 10.1038/nature04976. [DOI] [PubMed] [Google Scholar]
- Kitamura T, Kitamura Y, Yonezawa K, Totty NF, Gout I, Hara K, Waterfield MD, Sakaue M, Ogawa W, Kasuga M. Molecular cloning of p125Nap1, a protein that associates with an SH3 domain of Nck. Biochem Biophys Res Commun. 1996;219:509–14. doi: 10.1006/bbrc.1996.0264. [DOI] [PubMed] [Google Scholar]
- Kitamura Y, Kitamura T, Sakaue H, Maeda T, Ueno H, Nishio S, Ohno S, Osada S, Sakaue M, Ogawa W, et al. Interaction of Nck-associated protein 1 with activated GTP-binding protein Rac. Biochem J. 1997;322:873–8. doi: 10.1042/bj3220873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Kuroda S, Fukata M, Nakamura T, Nagase T, Nomura N, Matsuura Y, Yoshida-Kubomura N, Iwamatsu A, Kaibuchi K. p140Sra-1 (specifically Rac1-associated protein) is a novel specific target for Rac1 small GTPase. J Biol Chem. 1998;273:291–5. doi: 10.1074/jbc.273.1.291. [DOI] [PubMed] [Google Scholar]
- Koizumi H, Tanaka T, Gleeson JG. Doublecortin-like kinase functions with doublecortin to mediate fiber tract decussation and neuronal migration. Neuron. 2006;49:55–66. doi: 10.1016/j.neuron.2005.10.040. [DOI] [PubMed] [Google Scholar]
- Krueger EW, Orth JD, Cao H, McNiven MA. A dynamin-cortactin-Arp2/3 complex mediates actin reorganization in growth factor-stimulated cells. Mol Biol Cell. 2003;14:1085–96. doi: 10.1091/mbc.E02-08-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunda P, Craig G, Dominguez V, Baum B. Abi, Sra1, and Kette control the stability and localization of SCAR/WAVE to regulate the formation of actin-based protrusions. Curr Biol. 2003;13:1867–75. doi: 10.1016/j.cub.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Leighton PA, Mitchell KJ, Goodrich LV, Lu X, Pinson K, Scherz P, Skarnes WC, Tessier-Lavigne M. Defining brain wiring patterns and mechanisms through gene trapping in mice. Nature. 2001;410:174–9. doi: 10.1038/35065539. [DOI] [PubMed] [Google Scholar]
- Machesky LM, Mullins RD, Higgs HN, Kaiser DA, Blanchoin L, May RC, Hall ME, Pollard TD. Scar, a WASp-related protein, activates nucleation of actin filaments by the Arp2/3 complex. Proc Natl Acad Sci U S A. 1999;96:3739–44. doi: 10.1073/pnas.96.7.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin O, Rubenstein JL. Cell migration in the forebrain. Annu Rev Neurosci. 2003;26:441–83. doi: 10.1146/annurev.neuro.26.041002.131058. [DOI] [PubMed] [Google Scholar]
- Marsh L, Letourneau PC. Growth of neurites without filopodial or lamellipodial activity in the presence of cytochalasin B. J Cell Biol. 1984;99:2041–2047. doi: 10.1083/jcb.99.6.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophin regulation of cortical dendritic growth requires activity. Neuron. 1996;17:1057–64. doi: 10.1016/s0896-6273(00)80239-1. [DOI] [PubMed] [Google Scholar]
- McEvilly RJ, de Diaz MO, Schonemann MD, Hooshmand F, Rosenfeld MG. Transcriptional regulation of cortical neuron migration by POU domain factors. Science. 2002;295:1528–32. doi: 10.1126/science.1067132. [DOI] [PubMed] [Google Scholar]
- Miki H, Suetsugu S, Takenawa T. WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. Embo J. 1998;17:6932–41. doi: 10.1093/emboj/17.23.6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard TH, Sharp SJ, Machesky LM. Signalling to actin assembly via the WASP (Wiskott-Aldrich syndrome protein)-family proteins and the Arp2/3 complex. Biochem J. 2004;380:1–17. doi: 10.1042/BJ20040176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. Maturation of rat visual cortex. I. A quantitative study of Golgi-impregnated pyramidal neurons. J Neurocytol. 1981;10:859–878. doi: 10.1007/BF01262658. [DOI] [PubMed] [Google Scholar]
- Mitchell KJ, Pinson KI, Kelly OG, Brennan J, Zupicich J, Scherz P, Leighton PA, Goodrich LV, Lu X, Avery BJ, Tate P, Dill K, Pangilinan E, Wakenight P, Tessier-Lavigne M, Skarnes WC. Functional analysis of secreted and transmembrane proteins critical to mouse development. Nat Genet. 2001;28:241–9. doi: 10.1038/90074. [DOI] [PubMed] [Google Scholar]
- Mochida GH, Walsh CA. Genetic basis of developmental malformations of the cerebral cortex. Arch Neurol. 2004;61:637–40. doi: 10.1001/archneur.61.5.637. [DOI] [PubMed] [Google Scholar]
- Nakagawa H, Miki H, Ito M, Ohashi K, Takenawa T, Miyamoto S. N-WASP, WAVE and Mena play different roles in the organization of actin cytoskeleton in lamellipodia. J Cell Sci. 2001;114:1555–65. doi: 10.1242/jcs.114.8.1555. [DOI] [PubMed] [Google Scholar]
- Rakeman AS, Anderson KV. Axis specification and morphogenesis in the mouse embryo require Nap1, a regulator of WAVE-mediated actin branching. Development. 2006;133:3075–83. doi: 10.1242/dev.02473. [DOI] [PubMed] [Google Scholar]
- Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361:1545–64. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochlin MW, Dailey ME, Bridgman PC. Polymerizing microtubules activate site-directed F-actin assembly in nerve growth cones. Mol Biol Cell. 1999;10:2309–2327. doi: 10.1091/mbc.10.7.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers SL, Wiedemann U, Stuurman N, Vale RD. Molecular requirements for actin-based lamella formation in Drosophila S2 cells. J Cell Biol. 2003;162:1079–88. doi: 10.1083/jcb.200303023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R, Ho HY, Kirschner MW. Mechanism of N-WASP activation by CDC42 and phosphatidylinositol 4, 5-bisphosphate. J Cell Biol. 2000;150:1299–310. doi: 10.1083/jcb.150.6.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer AW, Kabir N, Forscher P. Filopodia and actin arcs guide the assembly and transport of two populations of microtubules with unique dynamic parameters in neuronal growth cones. J Cell Biol. 2002;158:139–52. doi: 10.1083/jcb.200203038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid RS, McGrath B, Berechid BE, Boyles B, Marchionni M, Sestan N, Anton ES. Neuregulin 1-erbB2 signaling is required for the establishment of radial glia and their transformation into astrocytes in cerebral cortex. Proc Natl Acad Sci U S A. 2003;100:4251–6. doi: 10.1073/pnas.0630496100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid RS, Shelton S, Stanco A, Yokota Y, Kreidberg JA, Anton ES. alpha3beta1 integrin modulates neuronal migration and placement during early stages of cerebral cortical development. Development. 2004;131:6023–31. doi: 10.1242/dev.01532. [DOI] [PubMed] [Google Scholar]
- Shu T, Tseng HC, Sapir T, Stern P, Zhou Y, Sanada K, Fischer A, Coquelle FM, Reiner O, Tsai LH. Doublecortin-like kinase controls neurogenesis by regulating mitotic spindles and M phase progression. Neuron. 2006;49:25–39. doi: 10.1016/j.neuron.2005.10.039. [DOI] [PubMed] [Google Scholar]
- Soderling SH, Langeberg LK, Soderling JA, Davee SM, Simerly R, Raber J, Scott JD. Loss of WAVE-1 causes sensorimotor retardation and reduced learning and memory in mice. Proc Natl Acad Sci U S A. 2003;100:1723–8. doi: 10.1073/pnas.0438033100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderling SH, Guire ES, Kaech S, White J, Zhang F, Schutz K, Langeberg LK, Banker G, Raber J, Scott JD. A WAVE-1 and WRP signaling complex regulates spine density, synaptic plasticity, and memory. J Neurosci. 2007;27:355–65. doi: 10.1523/JNEUROSCI.3209-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solecki DJ, Model L, Gaetz J, Kapoor TM, Hatten ME. Par6alpha signaling controls glial-guided neuronal migration. Nat Neurosci. 2004;7:1195–203. doi: 10.1038/nn1332. [DOI] [PubMed] [Google Scholar]
- Sossey-Alaoui K, Head K, Nowak N, Cowell JK. Genomic organization and expression profile of the human and mouse WAVE gene family. Mamm Genome. 2003;14:314–22. doi: 10.1007/s00335-002-2247-7. [DOI] [PubMed] [Google Scholar]
- Soto MC, Qadota H, Kasuya K, Inoue M, Tsuboi D, Mello CC, Kaibuchi K. The GEX-2 and GEX-3 proteins are required for tissue morphogenesis and cell migrations in C. elegans. Genes Dev. 2002;16:620–32. doi: 10.1101/gad.955702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen A, Rottner K, Ehinger J, Innocenti M, Scita G, Wehland J, Stradal TE. Sra-1 and Nap1 link Rac to actin assembly driving lamellipodia formation. Embo J. 2004;23:749–59. doi: 10.1038/sj.emboj.7600084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stradal TE, Rottner K, Disanza A, Confalonieri S, Innocenti M, Scita G. Regulation of actin dynamics by WASP and WAVE family proteins. Trends Cell Biol. 2004;14:303–11. doi: 10.1016/j.tcb.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Strasser GA, Rahim NA, VanderWaal KE, Gertler FB, Lanier LM. Arp2/3 is a negative regulator of growth cone translocation. Neuron. 2004;43:81–94. doi: 10.1016/j.neuron.2004.05.015. [DOI] [PubMed] [Google Scholar]
- Suetsugu S, Yamazaki D, Kurisu S, Takenawa T. Differential roles of WAVE1 and WAVE2 in dorsal and peripheral ruffle formation for fibroblast cell migration. Dev Cell. 2003;5:595–609. doi: 10.1016/s1534-5807(03)00297-1. [DOI] [PubMed] [Google Scholar]
- Sugitani Y, Nakai S, Minowa O, Nishi M, Jishage K, Kawano H, Mori K, Ogawa M, Noda T. Brn-1 and Brn-2 share crucial roles in the production and positioning of mouse neocortical neurons. Genes Dev. 2002;16:1760–5. doi: 10.1101/gad.978002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Nishiyama K, Yamamoto A, Inazawa J, Iwaki T, Yamada T, Kanazawa I, Sakaki Y. Molecular cloning of a novel apoptosis-related gene, human Nap1 (NCKAP1), and its possible relation to Alzheimer disease. Genomics. 2000;63:246–54. doi: 10.1006/geno.1999.6053. [DOI] [PubMed] [Google Scholar]
- Svitkina TM, Borisy GG. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J Cell Biol. 1999;145:1009–1026. doi: 10.1083/jcb.145.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai LH, Gleeson JG. Nucleokinesis in neuronal migration. Neuron. 2005;46:383–8. doi: 10.1016/j.neuron.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Suzuki T, Sakaki Y. Isolation of hNap1BP which interacts with human Nap1 (NCKAP1) whose expression is down-regulated in Alzheimer's disease. Gene. 2001;271:159–69. doi: 10.1016/s0378-1119(01)00521-2. [DOI] [PubMed] [Google Scholar]
- Ybot-Gonzalez P, Copp AJ. Bending of the neural plate during mouse spinal neurulation is independent of actin microfilaments. Dev Dyn. 1999;215:273–83. doi: 10.1002/(SICI)1097-0177(199907)215:3<273::AID-AJA9>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.