

Summary
Recent studies indicate that binding of urokinase-type plasminogen activator (uPA) to its high affinity receptor (uPAR), orchestrates uPAR interactions with other cellular components that play a pivotal role in diverse (patho-)physiological processes including wound healing, angiogenesis, inflammation, and cancer metastasis. However, notwithstanding the wealth of biochemical data available describing the activities of uPAR, little is known as to the exact mode of uPAR-uPA interactions and the presumed conformational changes that accompanying uPA-uPAR engagement. Here we report the crystal structure of soluble urokinase plasminogen activator receptor (suPAR), which contains the three domains of the wild-type receptor but lacks the cell surface anchoring sequence, in complex with the amino terminal fragment of urokinase-type plasminogen activator (ATF), at the resolution of 2.8 Å. We also report the 1.9 Å crystal structure of the free ATF. Our results provide a structural basis, represented by conformational changes induced in uPAR, for several published biochemical observations describing the nature of uPAR-uPA interactions and provide insight into mechanisms that may be responsible for the cellular responses induced by uPA binding.
Keywords: urokinase receptor, X-ray crystallography, protein-protein interactions, kringle domain, plasminogen
Introduction
Urokinase plasminogen activator (uPA) is a 50 kDa serine protease that has been implicated in a number of physiological and disease processes including tumor growth and metastasis, angiogenesis, and inflammation1,2. Many of these activities of uPA are mediated at the cell surface through a specific receptor (uPAR, CD87). They include cell signaling leading to differentiation and proliferation, activation of cell surface protease cascades, and remodeling of extracellular matrix and basement membrane, leading to cell motility3. Binding of uPA has been proposed to regulate the association of uPAR with a number of other cell surface ligands including the low-density lipoprotein receptor (LRP)4, integrins5, vitronectin6, and kininogen7, as well as other receptors such as platelet-derived growth factor receptor (PDGFR), epidermal growth factor receptor (EGFR), and G-protein coupled receptors (GPCRs)8,9. How uPA mediates these changes in uPAR function requires further study.
uPA is a multidomain protein comprised of three domains: an N-terminal growth factor domain (GFD, amino acids 1 through 46), a kringle domain (KD, amino acids 47 through 135), and a serine protease domain. The GFD and the kringle domain represent the amino-terminal fragment of uPA (ATF), that contains all molecular determinants required for binding to uPAR10,11, and the interactions of the ATF and uPA with uPAR are indistinguishable kinetically10,12.
uPAR is also a protein composed of three domains: domain DI (amino acids 1–77), domain DII (amino acids 93–177) and domain DIII (amino acids 193–272) that are homologous based on their arrangement of disulfide bonds, but not on their amino acid sequence13. The high affinity interaction of uPAR with uPA has been demonstrated to require contributions from all three domains although the spatial nature of these interactions is not fully appreciated. Domains DI and DII of uPAR are connected by a linker sequence (amino acids Gln78-Tyr92), which, upon uPA binding, becomes exposed to proteolytic attack by various proteases including uPA, plasmin and matrix metalloproteases14,15. The physiological effects of the linker cleavage include abolision of uPA binding with concomitant loss of uPAR affinity for vitronectin16 and, depending on the exact cleavage sites, generation of chemotactically active fragments of uPAR17.
uPAR is bound to cell surfaces via a glycosyl-phosphatidyl-inositol (GPI) anchor linked to its C-terminal Gly283. Soluble forms of uPAR (suPAR), generated by either hydrolytic activity of GPI-specific phospholipases18 or juxtamembrane proteolytic cleavage19, have been identified in biological fluids, both in vitro and in vivo. These soluble receptor species retain high affinity binding for urokinase that is indistinguishable from the parental molecule. uPAR is extensively glycosylated with N-linked carbohydrates (linked to Asn52, Asn162, Asn172, Asn200, and Asn233) comprising up to 50 % of the molecular weight of the receptor20. Earlier biochemical studies revealed that only the N-glycosylation at the Asn52 site is required to retain wild-type uPA-binding affinity, whereas removal of other carbohydrates by either site-directed mutagenesis21 or by enzymatic treatment22 has no appreciable effect.
In this report, we describe the 2.8 Å crystal structure of ATF bound to suPAR2345 (a soluble fragment of uPAR comprising amino acids 1–277 with four N-glycosylation sites eliminated by the Asn → Gln mutations) and the structure of the unbound ATF at the resolution of 1.9 Å. Additionally, we report the 3.3 Å structure of the binary complex between ATF and the fully glycosylated soluble fragment of uPAR (suPARWT). Structural comparison of the two binary complexes confirms all conclusions based solely on analysis of the higher resolution ATF/suPAR2345.
During the preparation of this manuscript the structure of the ATF/suPAR/ATN615 ternary complex has been reported23, in which a Fab fragment of the monoclonal antibody ATN625 targeting the domain DIII of suPAR was co-crystallized with the ATF/suPAR. The incorporation of the antibody reportedly improved the resolution of X-ray data, however, it also raised the possibility of structural perturbations induced by the antibody-receptor interactions. Structures presented in our report not only translate the observations of Huai and collaborators to the physiologically more relevant binary complex, suPAR/ATF, but also provide additional information that were not derived from the ternary complex, such as conformational changes in the suPAR/ATF complex that localize to regions previously implicated in chemotaxis24 and integrin interactions5. Moreover, the structure of both unbound and the receptor-engaged ATF allows us to render conclusions linking the inter-domain contacts within ATF to physiological processes described in the literature.
Results
Protein Constructs and Heterologous Overexpression
To facilitate crystallography, we used a soluble form of uPAR (suPARWT, amino acids 1–277), containing the three domains of uPAR (DIDIIDIII), but lacking a GPI-anchor signal sequence. To further reduce receptor heterogeneity resulting from differences in N-linked oligosaccharide structures, four of the five N-glycosylation sites of the suPARWT were mutated (N162Q, N172Q, N200Q, and N233Q), and the resulting mutant was denoted suPAR2345 (reported earlier in ref.12). Both suPAR constructs were expressed in Drosophila Schneider’s S2 cells and purified to homogeneity by combination of immunoaffinity chromatography and size-exclusion chromatography (data not shown). Both receptor constructs contain all of the determinants required for binding to uPA/ATF and neither alteration perturbs uPA (ATF) binding affinity25–28. Thus, ATF and suPAR2345 are suitable surrogates for their full length parent proteins and contain all of the interacting residues required to form the high affinity uPA/uPAR complex.
Structure of the unbound ATF
The structure of unbound ATF was solved by molecular replacement and the refined model of the ATF includes residues 11 through 132. The 10 residues at the N-terminus and 11 residues C-terminus are not modeled due to the absence of the corresponding electron density. Comparison of our structure of the free ATF solved by X-ray crystallography and the NMR structure of ATF published previously29 reveals several differences. First of all, although the basic structural features and global fold are similar, the models do not display close structural alignment, with root-mean-square deviations (r.m.s.d.) of 2.7 Å (37 Cα pairs) and 3.4 Å (86 Cα pairs) for GFD and KD, respectively. This low structural similarity might be attributed to prevalence of coiled regions in the ATF molecule as well as lower quality of the final NMR model. For comparison, structural alignment between the four individual molecules in an asymmetric unit of the unbound ATF crystals results in r.m.s. deviations of maximum 2.0 Å (GFD, 37 Cα pairs) and 0.38 Å (KD, 86 Cα pairs). The former value suggests a fair degree of flexibility within the GFD (mainly confined to the Ω-loop), which is not unexpected, given the virtual absence of well-defined secondary structures within this domain.
More importantly, the crystal structure of free ATF enabled the relative orientation of the GFD and KD and the inter-domain contacts in the free ATF to be analyzed for the first time (Figure 1A). In contrast to the NMR data on ATF, the X-ray structure reveals rather extensive interactions between both domains, with all of the interacting residues clearly defined in the electron density. Notably, side-chains of Leu14 and Leu92 are engaged in close hydrophobic contacts, whereas the side-chain of Ile44 packs into the hydrophobic pocket formed by Lys61, His99 and Tyr101. In addition, the backbone carbonyl of Arg59 forms a short hydrogen bond with the amide group of Asp45 (2.88 Å), and the Asp45 β-carboxylate group is also involved in a network of hydrogen bonds with backbone amides of Ser47 (3.39 Å) and Lys48 (3.22 Å; see Figure 1B). These contacts are well conserved in the ATF bound to suPAR suggesting that as a whole the ATF molecule is less flexible than suggested previously. Relative orientation of the GFD and KD domains is quite similar for both the free and receptor bound forms of the ATF with r.m.s.d. below 2.0 Å for the 122 Cα pairs and this characteristic might have important biological implications (see the Discussion section).
Figure 1. Overall structure of unbound ATF.
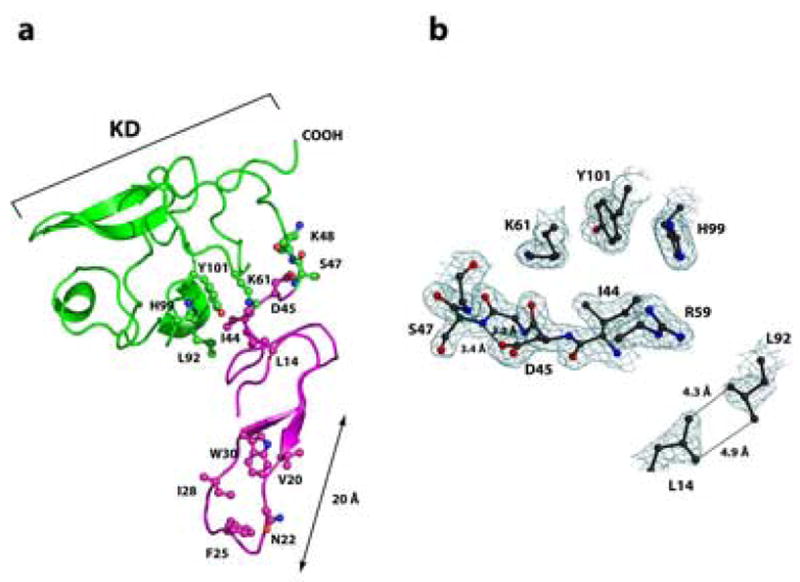
Panel A: The growth-factor-like domain (GFD, amino acids 10–46) is shown in magenta and the kringle domain (KD, amino acids 47–132), in green. The Ω-loop (amino acids 19–31) is responsible for the high-affinity interactions between uPA and uPAR. Amino acids Trp30, Ile28, Phe25, Asn22 and Val20, which form a hydrophobic patch within the Ω-loop that interacts primarily with the domain DI of uPAR, are shown in ball-and-stick representation. Ball-and-stick representation is also used to highlight amino acid residues forming the inter-domain interface between GFD and KD. The atoms are colored blue (nitrogen), red (oxygen), or according to the domain-color (carbons). Panel B: The representative electron density at the interface of GFD and KD. Interactions between amino acid residues implicated in inter-domain contacts between GFD and KD constrain the ATF in an invariant shape that is virtually identical for both unbound and receptor engaged ATF. Nitrogen atoms are blue, oxygen red, carbon atoms black. The 2Fo-Fc difference electron density map is contoured at the 1.2σ level. The picture was generated using Molscript56, Bobscript57 and rendered with PovRay.
suPAR Structure and Interdomain Contacts
The structure of the suPAR2345/ATF complex was solved by molecular replacement (for details see Materials and Methods). The overall bowl-like shape of the receptor in the suPAR/ATF complex, with the ‘fissure’ at the interface between domains DI and DIII and the central cavity approximately 23 Å deep, is very similar to the suPAR in the complex with an inhibitory peptide AE147 reported recently30. Domains DI and DII are primarily associated via interactions between amino acids of β-strands βIE and βIID (for nomenclature see refs.30,31) with the latter forming a ‘joint’ between these domains. Likewise, the β-strands βIIE and βIIID mediate contact between domains DII-DIII and contribute prominently to the interface of 1,360 Å2 connecting these two domains (Figure 2A). Finally, the ‘bowl’ is closed owing to hydrogen-bonding interactions between amino acid residues His47-Asn259 (NE2…OD1, 3.12 Å) and Arg53-Asp254 (NH2…OD2, 2.62 Å) of domains DI and DIII. Interestingly, no such interactions exist between DI and DIII in the suPAR/AE147 complex. ATF binding could thus invoke ’closure’ of the receptor by eliciting changes in relative orientation of the individual suPAR domains (see below).
Figure 2. Schematic representation of ATF binding to uPAR.
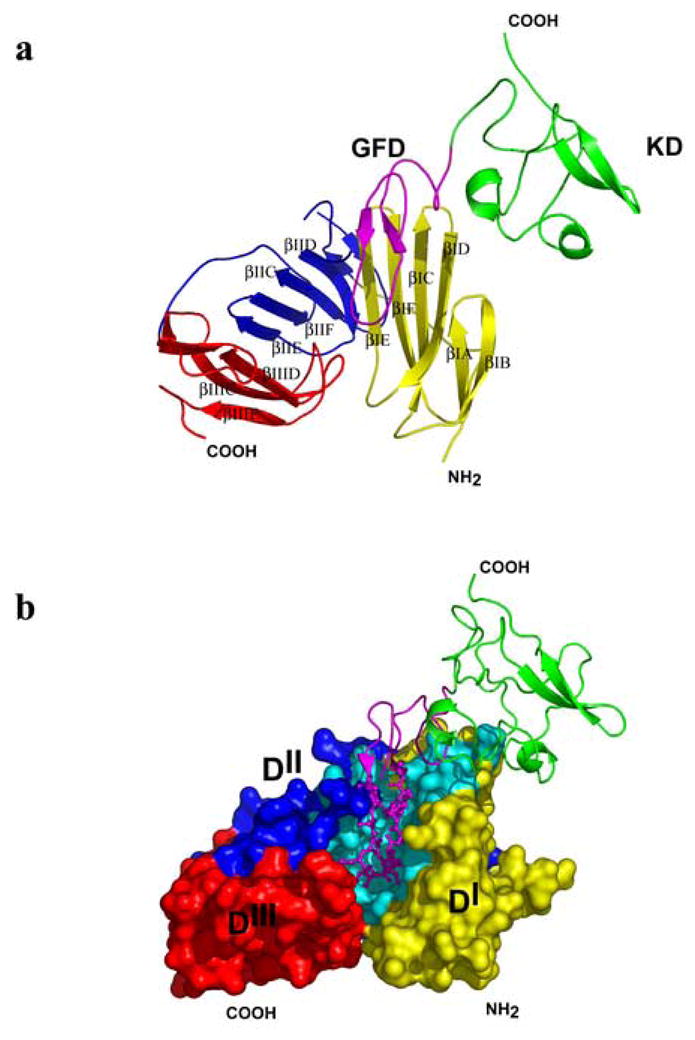
Domains DI (amino acids 1–93), DII (amino acids 94–191) and DIII (amino acids 192–277) of suPAR are shown in yellow, blue, and red, respectively; KD (47–132) is in green, GFD (10–46) in magenta. Panel A: A cartoon representation of the suPAR2345/ATF complex. The individual βstrands of suPAR2345 are labeled according to refs.30,31. Domain DI: βIA (residues 2–8), βIB (13–17), βIC (24–33), βID (38–45), βIE (53–59), and βIF (63–70); domain DII: βIIA (94–100), βIIB (112–115), βIIC (122–129), βIID (143–149), βIIE (156–161), and βIIF (163–171); domain DIII: βIIIA (195–199), βIIIB (211–214), βIIIC (222–229), βIIID (236–243), and βIIIE (259–267). Contacts between the domains are mediated via interactions βIE and βIID (domains DI and DII), βIIE and βIIID (domains DII and DIII). Panel B: The ATF (cartoon representation) binds to the central cavity of suPAR2345 (surface representation) and the Ω-loop (Cys19-Cys31, ball-and-sticks) is primarily responsible for the high affinity binding. Residues of suPAR2345 interacting with ATF are in cyan.
Mode of suPAR2345/ATF interactions
The model of suPAR2345/ATF complex provides a mechanistic basis for a number of published biochemical observations in the literature. In agreement with these studies, GFD (in particular the β-hairpin – termed the Ω-loop and formed by the residues 19 through 31), plays a central role in the suPAR/ATF binding and is primarily responsible for the high affinity interactions between uPA and its receptor10,11 (Figure 2B). Upon binding of ATF to suPAR2345, approximately 2,340 Å2 of the combined molecular surface area is buried.
The patch of predominantly hydrophobic residues within the Ω-loop (Trp30, Ile28, Phe25, Asn22 and Val20) (Figure 1) faces a hydrophobic region of DI in suPAR2345 that is formed by the side chains of amino acid residues from the β-sheet strands βIC (Arg25, Thr27, Val29, Leu31), βID (Leu38, Leu40, Glu42), βIE (Arg53, Thr54, Leu55, Tyr57), and βIF (Lys62, Thr64, Leu66, Thr67, Glu68; Figures 2 and 3A). Particularly striking is the locking in of Trp30 (ATF) during complex formation by the receptor residues Val29, Leu31, Tyr57, Thr64, and Leu66. Whereas the side chain of Trp30 is highly mobile in free ATF, the indol moiety becomes well-ordered in the complex, contributing extensively to the ligand-receptor interface. The DI domain contributes 16 out of the total 27 residues interacting with the ATF ligand. The complementary surface area at this interface is 740 Å2, which comprises approximately 63% of the total complementary surface. This observation correlates closely with available biochemical data describing the prominent contribution of the DI domain and involvement of specific amino acids in uPA binding28,32 (Figure 3A).
Figure 3. Detailed view of amino acid residues implicated in ATF binding in the individual domains DI (A), and DII (B) of suPAR2345.

Isolated domains DI (1–77) and DII (94–177) are shown in cartoon representation and side chains of amino acids interacting with ATF (shown in magenta, amino acids 17–41) are shown as ball-and-sticks and are colored cyan (carbon), red (oxygen), and blue (nitrogen). Panel A: The DI-ATF interaction is stabilized mostly by the hydrophobic contacts involving 16 amino acid residues from domain the DI. Panel B: Amino acid residues within domain DII interacts primarily with Lys23 and Tyr24 of ATF (shown in magenta as balls-and-sticks). Additional interactions include contacts between the fragment Pro138-Asp140, which is a part of the uPAR loop implicated in integrin signaling, and the residues Cys19-Lys23 of the Ω-loop in ATF.
However, even though the domain DI plays a pivotal role in uPA/uPAR interactions28, simultaneous cooperation of all three uPAR domains in forming a composite binding site is needed to generate high affinity binding of ATF or uPA26. This observation is explained by our structure through identification of residues within the DIIDIII that are engaged in intermolecular contacts with ATF (Figure 3B) as well as interdomain interactions within the receptor. Buried deeply inside the suPAR cavity, Lys23 (of ATF) is hydrogen bonded to Thr127 (NZ…OG1, 2.9) and Asp140 (N…OD1, 2.55) and makes additional van der Waals contacts with His166. Furthermore, Tyr24 is tightly packed inside a hydrophobic pocket delineated by amino acid residues Val125, Leu150, Pro151, Leu168 and Asp256. Consistent with these observations, both Lys23 and primarily Tyr24 were shown to be important determinants of uPA-uPAR interactions33,34. At the same time, although the contribution of the above-mentioned uPAR residues to ATF binding is quite evident from the structural studies they eluded detection during mutagenesis analyses.
Structural Changes in uPAR upon ATF binding
Published biochemical data suggest substantial conformational changes in the uPAR upon uPA binding. This notion is fully supported by our findings, as comparison of the suPAR2345/ATF structure to the recently published structure of suPAR/AE147 complex30 shows that the ATF binding causes considerable structural alterations in suPAR.
Whereas structures of the individual isolated domains of suPAR2345 (in the suPAR2345/ATF complex) are very similar to the corresponding domains of the suPAR/AE147 complex and the domains DI, DII and DIII in both complexes could be superimposed with r.m.s. deviations of 1.6 Å, 1.9 Å, and 1.8 Å, respectively, significant changes are observed in the relative orientations of the domains within the two complexes. This observation can be well-illustrated by aligning both complexes based on the Cα-atoms of a single domain only. Thus, when the domains DI of the two complexes are superimposed, domains DIII align very poorly with a maximal positional difference between the corresponding amino acid residues exceeding 12 Å (for Gln234 Cα; see Figure 4A). The spatial divergence between the two complexes could be further documented by the r.m.s.d. of 4.1 Å for 258 equivalent Cα atoms of the whole receptor.
Figure 4. Binding of ATF induces substantial conformational changes in the suPAR.
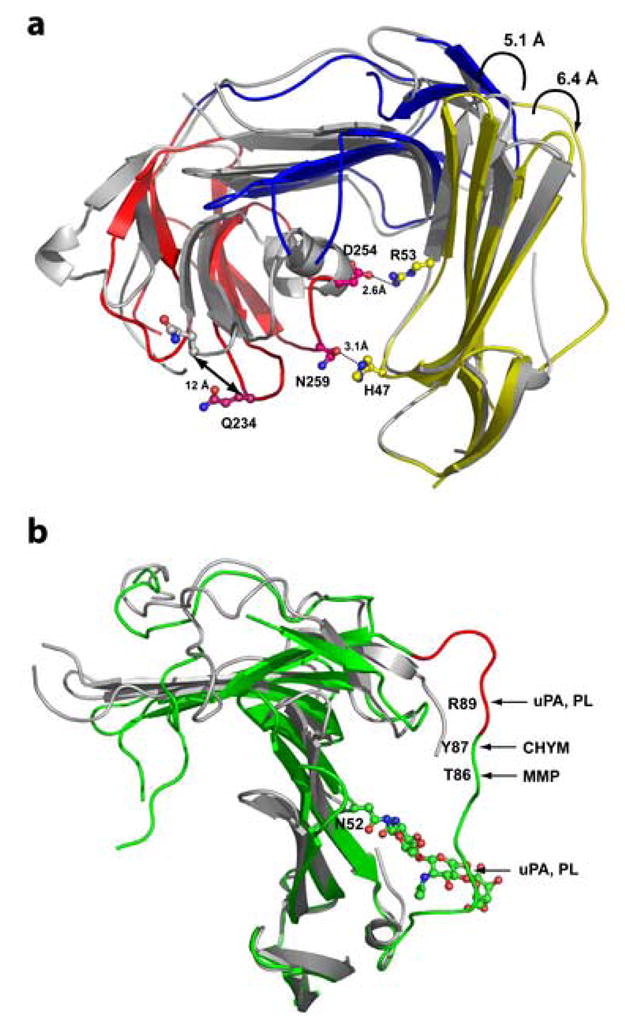
The domains DI (amino acids 1–77) of the suPAR2345/ATF and suPAR/AE147 (RCSB PDB code 1YWH) complexes were aligned using corresponding Cα atoms. Panel A: Ribbon representation of the receptors (ligands were omitted for clarity) with structurally aligned domains DI. Domains DI, DII and DIII of the suPAR2345/ATF complex are in shown in yellow, blue, and red, respectively, whereas receptor from the complex suPAR/AE147 is painted in gray. The binding of ATF results in the displacement of the domain the DIII (red) by more than 12 Å and shifts the domains DI and DIII close to each other , enabling a ‘closure’ of the suPAR. Interaction between DI and DIII is mediated by two pairs of residues, His47-Asn259 and Arg53-Asp254 (shown in ball-and-stick representation). Other structural changes include repositioning of hairpins βIC-βID, βIE-βIF as well as βIIC-βIID. Panel B: Displacement of the linker region (amino acid residues Gln78-Tyr92), connecting domains DI and DII of suPAR upon ATF binding (suPAR2345/ATF structure in green, 1YWH in grey). The epitope with chemotactic attributes (amino acids Ser88-Tyr92) is show in red and positions susceptible to hydrolysis by various proteases are indicated with arrows. The trisaccharide chain, attached to Asn52 and modeled in our structure, is also shown in ball-and-stick representation. Note that “typical” mammalian oligosaccharide structures are much larger and due to their flexibility could easily “shield” the linker region from a proteolytic attack. PL, uPA, MMP, and CHYM stand for plasmin, urokinase-type plasminogen activator, matrix metallopeptidases, and chymotrypsin, respectively. The domain DIII was omitted for clarity.
In addition to overall realignment of entire domains of suPAR, more subtle conformational changes could be traced in the suPAR2345/ATF complex as compared to the suPAR complex with inhibitory peptide. Within the domain DI the hairpin formed by strands βIC and βID in suPAR2345/ATF is pushed outward from the central cavity by 6.4 Å (Glu34Cα), while the opposite shift is seen for the β-strands βIE and βIF (up to 5.1 Å for Leu61Cα). A highly flexible loop connecting β-strands βIIC and βIID in the domain DII of suPAR (Trp129 through Arg142) and harboring the Gly133-Glu-Glu-Gly136 motif implicated in integrin signaling5, which protrudes from the receptor in the suPAR/inhibitor complex, is now bent toward the central cavity of the receptor. This is due to interactions between Pro138, Lys139, and Asp140 of suPAR2345 and the amino acid stretch Cys19 through Lys23 of the Ω-loop of ATF (Figure 4B).
Another noticeable difference between the two structures is a readjustment of the highly mobile amino acid segment (Gln78-Tyr92) connecting domains DI and DII of uPAR. It has been proposed that this DIDII linker is displaced and exposed upon uPA (ATF) binding24. In our structure the Gln78-Tyr92 loop was ‘immobilized’ in its presumed ‘open’ conformation, i.e. in the conformation where the linker is distanced from the receptor body by more than 14 Å (Val85CA…Arg53CA; Figure 5). This spatial rearrangement makes the linker more susceptible to the hydrolysis by various proteases (including plasmin, uPA or matrix metalloproteases14,15) and as well it converts the chemotactic epitope Ser88-Tyr92 from its cryptic form to the form easily accessible to interactions with an fMLP receptors35,36. Although the ‘open’ conformation of the Gln78-Tyr92 linker observed in our structure results from the crystallographic contacts between different molecules of receptor, it likely represents a preferred conformation of the linker in the complex between suPAR and ATF (uPA). Furthermore, this conclusion is fully supported by the available biochemical data14,15,24,35,36. Therefore, under physiological conditions, the ATF (uPA) engagement with the receptor favors the ‘open’ conformation of he linker even though the previously reported ATF/suPAR/ATN615 ternary complex structure suggests that fair degree of flexibility of the linker is retained23. Taken together, depending on the nature of ligand/inhibitor occupying the central cavity, suPAR is capable of extensive structural transformations ensuring the most effective interactions with the corresponding ligand and these changes could further elicit altered affinity/specificity towards interaction partners with binding sites mapped to the outer portion of the receptor5,37.
Figure 5. Repositioning of the “integrin-interacting” loop (Trp129-Arg142) in suPAR2345 upon ATF binding.

Interactions between amino acid residues Cys19-Lys23 of the ATF and Pro138-Asp140 of suPAR2345 leads to bending of the loop towards the central cavity of the receptor. Panel A: The complexes, suPAR2345/ATF (domains DI, DII and DIII colored yellow, blue and red, respectively) and suPAR/AE147 (shown in grey), were aligned based on corresponding Cα atoms of the domain DI only. GFD is shown in combination of ball-and-sticks and semi-transparent surface. The βIIC-βIID hairpin is in cartoon representation and its residues interacting with GFD as ball-and-sticks. Panel B: Detailed view of residues engaged in the interactions between the strands βIIC and βIID of suPAR2345 and the Ω-loop of ATF. The Ω-loop is shown in surface representation and the interacting residues contributed by the domain DII are painted as balls-and-sticks. Note the major movement of the βIIC-βIID hairpin caused by interactions with the Ω-loop. In both structures amino acids 132 through 136 of suPAR are missing.
Discussion
In this report we present structural explanations for several important results of biological/biochemical studies reported in literature. In addition to the X-ray structure of the suPAR2345/ATF complex described in detail in the Results section, we solved the crystal structure of suPARWT/ATF complex, i.e. complex of the fully N-glycosylated receptor and ATF, at 3.3 Å resolution (see Table I). Comparison of the two ligand-receptor complexes confirms that mutations introduced in the suPAR2345 do not change the overall structural characteristics of the receptor as 1) binding modes between ATF and suPAR2345 or suPARWT, respectively, are identical and 2) both receptors have virtually identical topologies (data not shown). Due to the higher resolution only the complex of suPAR2345/ATF was used in the subsequent structural analysis.
Table 1.
. Data collection and refinement statistics
| ATF | suPAR2345/ATF | suPARWT/ATF | |
|---|---|---|---|
| Data collection | |||
| Space group | P1 | P21 | P3121 |
| Cell dimensions | |||
| a, b, c (Å) | 47.6, 64.3, 64.6 | 62.9, 281.9, 62.8 | 131.6, 131.6, 104.5 |
| α, β, γ (°) | 107.6, 92.1, 95.7 | 90.0, 105.4, 90.0 | 90.0, 90.0, 120 |
| Resolution (Å) | 40.0 - 1.9 (1.97 – 1.90)* | 50.0-2.8 (2.90 – 2.80) | 30.0-3.3 (3.42 – 3.30) |
| Rsym | 0.059 (0.356) | 0.047 (0.235) | 0.082 (0.553) |
| I/σI | 19.6 | 17.6 | 17.5 |
| Completeness (%) | 96.4 (87.0) | 91.2 (51.3) | 98.9 (90.7) |
| Redundancy | 3.8 (2.8) | 2.8 (1.8) | 5.0 (3.1) |
| Refinement | |||
| Resolution (Å) | 15.0 - 1.9 | 15.0 - 2.8 | 15.0 - 3.3 |
| No. reflections | 51,775 | 47,048 | 14,910 |
| R / Rfree | 0.182 (0.275)/ 0.208 (0.301) | 0.223 (0.352)/ 0.265 (0.479) | 0.249 (0.345) / 0.289 (0.411) |
| No. atoms | 4,278 | 11,900 | 2,986 |
| Protein | 3,892 | 11,838 | 2,986 |
| Ligand/ion | 25 | 10 | 0 |
| Water | 361 | 52 | 0 |
| B-factors (Å2) | |||
| Protein | 29.4 | 67.1 | |
| Ligand/ion | 57.8 | 68.8 | |
| Water | 33.7 | 76.3 | |
| R.m.s deviations | |||
| Bond lengths (Å) | 0.019 | 0.013 | |
| Bond angles (°) | 1.644 | 1.980 |
The highest resolution shells for ATF (1.97 – 1.90), suPAR2345/ATF (2.90 – 2.80) and suPARWT/ATF (3.42 – 3.30) are shown in parentheses.
Available biophysical data for uPA suggests high relative flexibility of its individual domains38,39. Although the possibility of some interaction between GFD and KD might have been suspected, specific contacts between these two domains were not elucidated. Similar conclusion were drawn from the NMR structural studies of ATF in solution, during which both domains were modeled as isolated entities due to lack of inter-domain NOEs29. Contrary to the NMR data, our structures revealed quite extensive interactions between GFD and KD, which constrain the ATF molecule in a virtually invariant shape. These contacts are likely of physiological relevance and are not attributable to the crystal packing, as they are well-conserved in all structures described in this report and also in the ternary ATF/suPAR/ATN615 complex published recently23.
Human uPAR contains five potential N-glycosylation sites, only first four of which appear to utilized22 and only glycosylation of Asn52 has a moderate impact on the affinity of uPA/uPAR interactions21. An analysis of presented structure suggests that Asn52 → Gln mutation would lead to the disruption of hydrogen bonding between Asn52 and Val70 (OD1…N, 2.86 Å) 21. Such change could cause local rearrangement of the receptor within the region involved in interactions with uPA (uPAR residues Arg53, Thr54 or Glu68). It is also possible that the presence of different oligosaccharide chains linked to Asn52 might regulate uPAR cleavage and, subsequently, its interaction with other partners such as integrins, vitronectin or G-protein-coupled receptors40. Such a possibility is conceivable due to the close proximity (about 10 Å in our complex) of the N-glycosylation attachment site and the DI-DII linker (see Figure 4B).
High affinity interaction between uPA and uPAR is mediated primarily via contacts of the Ω-loop with the receptor. Interactions between the ATF amino acids outside the Ω-loop and the suPAR likely play only a auxiliary (transient) role since they are not well-conserved between the four ATF/suPAR2345 complexes within the asymmetric unit and differ from the interactions observed in our ATF/suPARWT structure as well as in the ATF/suPAR/ATN615 ternary structure reported recently23. An absence of strong and conserved interactions between the kringle domain of uPA and the receptor molecule further underscores the importance of the inter-domain (GFD-KD) contacts constraining the invariant overall ATF shape, which might be of primary significance for efficient interactions with other physiological partners of the uPAR/uPA complex, including integrins5 and vitronectin6. For example, the conservation of the relative orientations of ATF domains leads to a series of weaker and not conserved interactions between the KD and DI of the receptor, which were previously suggested to stabilize the uPA-uPAR complex12. Additionally, the side-chain of Lys46 from the GFD is positioned in close proximity to the receptor, possibly explaining the impaired cleavage of uPA bound to uPAR that leads to a reduced rate and efficiency of uPA internalization/degradation41. The ATF (and probably KD) can also become engaged in a low affinity interaction with vitronectin that involves an interaction site distinct from the somatomedin B domain implicated in a high affinity uPAR-binding42. Consequently, under an assumption of favorable spatial orientation of the ATF domains, the KD-vitronectin interaction would contribute (in addition to structural changes in suPAR) to augmentation of the binding affinity in the uPA/uPAR/vitronectin ternary complex43. Also, the inter-domain forces in ATF may assure the proper orientation of KD (and the protease domain of uPA) for the interactions with an I-domain of the integrin αMβ2 and possibly with other integrins (see below).
Both uPA and uPAR are reported to interact with cell adhesion receptors of the integrin superfamily, including the subfamilies β1, β3, and β5, as well as the β2 integrin subfamily expressed in cells of hematopoietic lineage44–47. Functional uPA/uPAR/integrin association modulates various aspects of cell physiology such as motility, mitogenic activity or adhesive phenotype. At the molecular level, uPAR interacts with at least two distinct regions of the integrin molecule – the lectin domain48 and a site at the interface of α- and β-chains that is close to, but distinct from, the “classical” MIDAS integrin binding site47,49. Binding of uPA to uPAR typically strengthens uPAR-integrin interactions and could elicit the integrin-mediated signaling, but the mechanisms underlying these phenomenona are not clear. It is plausible that both conformational changes in the receptor and/or concurrent binding of suitably oriented KD to an integrin might be involved in these processes. In the latter case, the ‘favorable’ orientation of kringle (and protease) domains of uPA, enforced by the GFD-KD interdomain interactions, might be necessary to ensure specific kringle-integrin contacts. Additionally, a recent study suggests that the highly flexible loop between strands βIIC and βIID of the domain DII (amino acids Trp129-Arg142) plays an important role in binding of uPAR to the integrin αvβ35. Moreover, the Gly133-Glu-Glu-Gly136 motif activates αvβ3-dependent signaling pathways and stimulates cell migration5. As seen from our structure, binding of the ATF molecule induces a major conformational change of this loop (Figure 5). Although the structural comparison by itself is insufficient to explain an increased affinity of the interaction between suPAR and integrins or the integrin-mediated signaling upon uPA binding, it is likely that the loop repositioning, observed in our structure, contributes to these effects.
Overexpression of uPAR/uPA correlates with poor prognosis in a number of human tumor types. Thus, the development of agents that inhibit uPA/uPAR interactions has become an attractive therapeutic target2. In addition to uPA-derived linear and cyclic peptides, which should resemble uPA in their binding mode to uPAR, Plough and coworkers reported a series of peptidic antagonists derived from a phage display random library50. Based on the presence of the FXXYLW motif, a variation the uPAR binding motif in GFD, they proposed similar binding modes between the inhibitory peptides and uPA. Although direct comparison of uPAR2345/ATF and uPAR/AE147 complexes confirms a spatial overlap between the GFD and AE147 in the binding cavity of uPAR, pronounced differences exist in the positioning of the individual side chains and in their interactions with the receptor (Figure 6). These differences could be responsible for observed species-selectivity of the inhibitory peptides towards human uPAR. Consequently, the detailed analysis and comparison of binding modes of inhibitory peptides and ATF might be exploited in the rational design of novel inhibitors.
Figure 6. Binding of ATF in the central cavity of suPAR differs from the inhibitory peptide AE147.

The complexes suPAR2345/ATF and suPAR/AE147 were superimposed using the equivalent Cα atoms of the receptors. The AE147 peptide (green) and ATF (fragment Cys13-Cys31, magenta) are in cartoon representation. In the ball-and-stick representation are shown the residues marked in bold in the FXXYLW (AE147) or KYFXXIHW (ATF) motifs. The residues of receptor forming the binding pocket are displayed in surface representation. The labels of residues in AE147 are printed in blue and slanted, those of the ATF residues are shown in red, and the suPAR residues are printed in black. Due to presence of resembling sequence motifs (FXXXLW), similar binding modes were proposed for the ATF and AE14750. From this figure, it is clear that despite of a spatial overlap between the two ligands, except of Phe25 (ATF) and Phe5 (AE147), locations of the individual side chains differ markedly.
Materials and Methods
Protein Constructs
Recombinant soluble urokinase-type plasminogen activator receptor (suPARWT; amino acids 1–277) was expressed in Drosophila S2 cells and purified by a combination of immunoaffinity and size-exclusion chromatography. Cloning of the suPAR2345 construct (the mutant of suPARWT with N162Q, N172Q, N200Q, and N233Q mutations) was described elsewhere12, and the recombinant protein was expressed and purified as described for suPARWT. Amino-terminal fragment of full-length urokinase (ATF; amino acids 1–143) was heterologously over-expressed in Drosophila S2 cells12. The purification protocol included the ion-exchange chromatography using SP-Sepharose resin (GE Healthcare) and the size-exclusion chromatography using a Sephacryl 16/60 HR100 column (GE Healthcare). All of the purified protein constructs mentioned above were virtually pure as determined by Commassie stained SDS-PAGE (data not shown).
suPAR2345/ATF and suPARWT/ATF complexes were prepared by equilibration of the mixture of the receptor and ATF at a 1:2 molar ratio overnight at 4°C followed by size-exclusion chromatography using Superdex 10/30 HR200 (GE Healthcare). All size-exclusion steps were carried out in 20 mM Tris-HCl, 150 mM NaCl, pH 8.0, and the final protein concentrations determined by measuring absorbance at 280 nm.
Crystallization
Crystals of ATF were grown at room temperature using the hanging drop vapor diffusion method from 3 μl droplets obtained by mixing equal volumes of protein (20 mg/ml) and the reservoir solutions (1.2 M sodium dihydrogen phosphate, 0.8 M potassium hydrogen phosphate, 200 mM lithium sulfate, 100 mM CHES, pH 10.5). For diffraction experiments, a single crystal was briefly soaked in the reservoir solution enriched with 15% (v/v) glycerol and flash-frozen in a stream of nitrogen cooled to 100 K.
For crystallization of the suPAR2345/ATF complex, the hanging droplets were formed from the equal volumes of protein (8 mg/ml) and the reservoir (22.5% w/v PEG3350, 200 mM ammonium sulfate, 100 mM Bis-Tris, pH 5.5) solutions at room temperature. Typically, pyramidal crystals grew within a week. Crystals were transferred to the reservoir solution containing 15% (v/v) glycerol and flash-frozen in liquid nitrogen.
suPARWT/ATF crystals were obtained at room temperature using the hanging drop vapor diffusion from drops containing equal volumes of protein complex (8 mg/ml) and the reservoir solutions (1.54 M ammonium sulfate, 100 mM HEPES, pH 7.4). Rod-like crystals that belong to the space group P3121 grew within two days. For the data collection, the crystals were flash-frozen in the reservoir solution supplemented with 25% (v/v) glycerol.
Data collection
Diffraction data for the native crystals of ATF and the suPAR2345/ATF complex were collected at the SER-CAT beamline at the Advanced Photon Source (Argonne, USA) at an X-ray wavelength of 1.135 Å. Data for the suPARWT/ATF complex were collected at the beamline X25 at the National Synchrotron Light Source, Brookhaven National Laboratory using an ADSC Quantum Q210 CCD detector and an X-ray wavelength of 1.254 Å. All experiments were performed at temperature 100 K. All data were processed and scaled using HKL200051 (HKL Research). Crystals of ATF belong to the space group P1 with unit cell dimensions of a = 47.6 Å, b = 64.3 Å, c = 64.6 Å, α = 107.62°, β = 92.11°, γ = 95.75°, and four molecules ATF are present in the unit cell. Crystals of the suPAR2345/ATF complex belong to the space group P21 (a = 62.9 Å, b = 281.9 Å, c = 62.8 Å, β = 105.4°) with four complex molecules found in the asymmetric unit. Finally, crystals of the suPARWT/ATF complex belong to the space group P3121 (a = 131.6 Å, c = 104.5 Å) with a single molecule of the complex present in the asymmetric unit. Detailed data collection statistics are shown in Table 1.
Structure Solving
The structure of ATF was solved by molecular replacement at the resolution of 2.5 Å using program Phaser52. Models of both GFD and KD domains based on the NMR structure of ATF29 were used as independent search probes. Initial refinement, accompanied by extensive model rebuilding and modeling the fragments of the ATF molecule, was carried out with the program CNS53. At later stages of the CNS refinement, carried out at 1.9 Å resolution, solvent molecules were incorporated in the model. The final refinement was conducted with the program Refmac 5.054, resulting in an R value of 0.182 (Rfree= 0.208). Due to a disorder, amino acids 1–10 and 133–143 are not modeled in the final structure. The Rachamadran plot shows 87.9% and 12.1% of the residues in favored and allowed regions, respectively, and the additional refinement statistics are shown in Table 1.
Structure of the suPAR2345/ATF complex was solved by molecular replacement with the program Phaser at the resolution 3.3 Å. Five independent models, subsequently incorporated in partial solutions, were those of individual domains of the ATF (GFD and KD from the crystal structure solved earlier) and suPAR (DIDIIDIII). Models of DI, DII, and DIII domains of suPAR, comprising residues 1–82, 91–173, and 190–240, respectively, were generated from the PDB entry 1YHW. All initial corrections and extensions to the model were assisted by refinement (CNS) consisting a simulated annealing, a steepest-gradient energy minimization, and the overall B-factor refinement, with a very gradual extension of the resolution. During further improvement of the structure, refinement was carried with the program Refmac 5.0. At the final cycles of refinement, partial solvent structure was modeled, which consisted of just the water molecules occupying sites indicated by strong Fo-Fc electron density peaks and a suitable chemical environment. The final model with four molecules of suPAR2345/ATF complexes in an asymmetric unit is characterized by an R value of 0.222 (Rfree= 0.265) with 75.5%, 21.5%, and 3.0% of the residues in favorable, allowed and generously allowed regions, respectively. Additionally, due to a lack of the interpretable electron density peaks, amino acids 1–10 and 133–143 of the ATF and 108–111, 134–138, and 248–251 of suPAR2345 are not included in the final model.
Initial attempts to solve the crystal structure of the suPARWT/ATF complex were based on the anomalous data for the Ta6Br12-derivatized trigonal crystals. Analysis of the initial electron density maps revealed several regions clearly identifiable with structural features of the complex. A combination of low resolution and relatively weak phases, however, prevented further building and extension of the model. The suPARWT/ATF structure was also solved by molecular replacement with the program Phaser, according to the protocol described earlier for the suPAR2345/ATF complex. Subsequently, very preliminary refinement of the model (CNS, Refmac 5), carried out at resolution 3.3 Å, resulted in an R value of 0.249 (Rfree= 0.289). Most importantly, however, these preliminary results already showed the topological identity of both suPAR2345/ATF and suPARWT/ATF, complexes. Due to significantly lower resolution of X-ray data (3.3 Å) available for suPARWT/ATF crystals as compared to the suPAR2345/ATF complex, and apparent identity of topologies of both complexes, further refinement of the trigonal crystal form was abandoned.
The quality of the final models was assessed using PROCHECK55, and the atomic coordinates together with the experimental diffraction amplitudes were deposited at the RCSB Protein Data Bank with accession numbers XXX (unbound ATF) and YYY (suPAR2345/ATF complex).
Acknowledgments
We would like to thank Dr. Joseph Tropea and Mr. Scott Cherry for help in preparation of the complex samples, Dr. Zbigniew Dauter for his help with collection of X-ray data for the suPARWT/ATF crystals, and Dr. Peter Zwart for help with initial molecular replacement searches. Diffraction data were collected at the South-East Regional Collaborative Access Team (SER-CAT) beamline 22-ID, located at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng38. This project was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (JL), by grants HL60169, HL076206, CA83121, HL76406 and a grant from the University of Pennsylvania Research Foundation (DBC).
Abbreviations footnote
- uPA
urokinase-type plasminogen activator
- GFD
the growth factor-like domain of uPA
- KD
the kringle domain of uPA
- ATF
the amino terminal fragment of urokinase-type plasminogen activator
- uPAR
urokinase-type plasminogen activator receptor
- suPARWT
soluble fragment of urokinase-type plasminogen activator receptor
- suPAR2345
mutant of suPARWT with four (of five) N-glycosylation sites mutated (N162Q, N172Q, N200Q, and N233Q)
- r.m.s.d
root-mean-square deviation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mondino A, Blasi F. uPA and uPAR in fibrinolysis, immunity and pathology. Trends Immunol. 2004;25:450–455. doi: 10.1016/j.it.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 2.Romer J, Nielsen BS, Ploug M. The urokinase receptor as a potential target in cancer therapy. Curr Pharm Des. 2004;10:2359–2376. doi: 10.2174/1381612043383962. [DOI] [PubMed] [Google Scholar]
- 3.Rabbani SA, Mazar AP. The role of the plasminogen activation system in angiogenesis and metastasis. Surg Oncol Clin N Am. 2001;10:393–415. [PubMed] [Google Scholar]
- 4.Czekay RP, Kuemmel TA, Orlando RA, Farquhar MG. Direct binding of occupied urokinase receptor (uPAR) to LDL receptor-related protein is required for endocytosis of uPAR and regulation of cell surface urokinase activity. Mol Biol Cell. 2001;12:1467–1479. doi: 10.1091/mbc.12.5.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Degryse B, Resnati M, Czekay RP, Loskutoff DJ, Blasi F. Domain 2 of the urokinase receptor contains an integrin-interacting epitope with intrinsic signaling activity: generation of a new integrin inhibitor. J Biol Chem. 2005;280:24792–24803. doi: 10.1074/jbc.M413954200. [DOI] [PubMed] [Google Scholar]
- 6.Wei Y, Waltz DA, Rao N, Drummond RJ, Rosenberg S, Chapman HA. Identification of the urokinase receptor as an adhesion receptor for vitronectin. J Biol Chem. 1994;269:32380–32388. [PubMed] [Google Scholar]
- 7.Colman RW, Pixley RA, Najamunnisa S, Yan W, Wang J, Mazar A, McCrae KR. Binding of high molecular weight kininogen to human endothelial cells is mediated via a site within domains 2 and 3 of the urokinase receptor. J Clin Invest. 1997;100:1481–1487. doi: 10.1172/JCI119669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kiyan J, Kiyan R, Haller H, Dumler I. Urokinase-induced signaling in human vascular smooth muscle cells is mediated by PDGFR-beta. EMBO J. 2005;24 :1787–1797. doi: 10.1038/sj.emboj.7600669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mazzieri R, Blasi F. The urokinase receptor and the regulation of cell proliferation. Thromb Haemost. 2005;93:641–646. doi: 10.1160/TH05-01-0021. [DOI] [PubMed] [Google Scholar]
- 10.Appella E, Robinson EA, Ullrich SJ, Stoppelli MP, Corti A, Cassani G, Blasi F. The receptor-binding sequence of urokinase. A biological function for the growth-factor module of proteases. J Biol Chem. 1987;262:4437–4440. [PubMed] [Google Scholar]
- 11.Quax PH, Grimbergen JM, Lansink M, Bakker AH, Blatter MC, Belin D, van Hinsbergh VW, Verheijen JH. Binding of human urokinase-type plasminogen activator to its receptor: residues involved in species specificity and binding. Arterioscler Thromb Vasc Biol. 1998;18:693–701. doi: 10.1161/01.atv.18.5.693. [DOI] [PubMed] [Google Scholar]
- 12.Bdeir K, Kuo A, Sachais BS, Rux AH, Bdeir Y, Mazar A, Higazi AA, Cines DB. The kringle stabilizes urokinase binding to the urokinase receptor. Blood. 2003;102:3600–3608. doi: 10.1182/blood-2003-03-0949. [DOI] [PubMed] [Google Scholar]
- 13.Casey JR, Petranka JG, Kottra J, Fleenor DE, Rosse WF. The structure of the urokinase-type plasminogen activator receptor gene. Blood. 1994;84:1151–1156. [PubMed] [Google Scholar]
- 14.Andolfo A, English WR, Resnati M, Murphy G, Blasi F, Sidenius N. Metalloproteases cleave the urokinase-type plasminogen activator receptor in the D1–D2 linker region and expose epitopes not present in the intact soluble receptor. Thromb Haemost. 2002;88:298–306. [PubMed] [Google Scholar]
- 15.Hoyer-Hansen G, Ploug M, Behrendt N, Ronne E, Dano K. Cell-surface acceleration of urokinase-catalyzed receptor cleavage. Eur J Biochem. 1997;243:21–26. doi: 10.1111/j.1432-1033.1997.0021a.x. [DOI] [PubMed] [Google Scholar]
- 16.Hoyer-Hansen G, Behrendt N, Ploug M, Dano K, Preissner KT. The intact urokinase receptor is required for efficient vitronectin binding: receptor cleavage prevents ligand interaction. FEBS Lett. 1997;420:79–85. doi: 10.1016/s0014-5793(97)01491-9. [DOI] [PubMed] [Google Scholar]
- 17.Gargiulo L, Longanesi-Cattani I, Bifulco K, Franco P, Raiola R, Campiglia P, Grieco P, Peluso G, Stoppelli MP, Carriero MV. Cross-talk between fMLP and vitronectin receptors triggered by urokinase receptor-derived SRSRY peptide. J Biol Chem. 2005;280:25225–25232. doi: 10.1074/jbc.M412605200. [DOI] [PubMed] [Google Scholar]
- 18.Wilhelm OG, Wilhelm S, Escott GM, Lutz V, Magdolen V, Schmitt M, Rifkin DB, Wilson EL, Graeff H, Brunner G. Cellular glycosylphosphatidylinositol-specific phospholipase D regulates urokinase receptor shedding and cell surface expression. J Cell Physiol. 1999;180:225–235. doi: 10.1002/(SICI)1097-4652(199908)180:2<225::AID-JCP10>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 19.Beaufort N, Leduc D, Rousselle JC, Magdolen V, Luther T, Namane A, Chignard M, Pidard D. Proteolytic regulation of the urokinase receptor/CD87 on monocytic cells by neutrophil elastase and cathepsin G. J Immunol. 2004;172:540–549. doi: 10.4049/jimmunol.172.1.540. [DOI] [PubMed] [Google Scholar]
- 20.Behrendt N, Ronne E, Ploug M, Petri T, Lober D, Nielsen LS, Schleuning WD, Blasi F, Appella E, Dano K. The human receptor for urokinase plasminogen activator. NH2-terminal amino acid sequence and glycosylation variants. J Biol Chem. 1990;265:6453–6460. [PubMed] [Google Scholar]
- 21.Moller LB, Pollanen J, Ronne E, Pedersen N, Blasi F. N-linked glycosylation of the ligand-binding domain of the human urokinase receptor contributes to the affinity for its ligand. J Biol Chem. 1993;268:11152–11159. [PubMed] [Google Scholar]
- 22.Ploug M, Rahbek-Nielsen H, Nielsen PF, Roepstorff P, Dano K. Glycosylation profile of a recombinant urokinase-type plasminogen activator receptor expressed in Chinese hamster ovary cells. J Biol Chem. 1998;273:13933–13943. doi: 10.1074/jbc.273.22.13933. [DOI] [PubMed] [Google Scholar]
- 23.Huai Q, Mazar AP, Kuo A, Parry GC, Shaw DE, Callahan J, Li Y, Yuan C, Bian C, Chen L, Furie B, Furie BC, Cines DB, Huang M. Structure of human urokinase plasminogen activator in complex with its receptor. Science. 2006;311:656–659. doi: 10.1126/science.1121143. [DOI] [PubMed] [Google Scholar]
- 24.Resnati M, Guttinger M, Valcamonica S, Sidenius N, Blasi F, Fazioli F. Proteolytic cleavage of the urokinase receptor substitutes for the agonist-induced chemotactic effect. EMBO J. 1996;15:1572–1582. [PMC free article] [PubMed] [Google Scholar]
- 25.Bdeir K, Kuo A, Mazar A, Sachais BS, Xiao W, Gawlak S, Harris S, Higazi AA, Cines DB. A region in domain II of the urokinase receptor required for urokinase binding. J Biol Chem. 2000;275:28532–28538. doi: 10.1074/jbc.M001595200. [DOI] [PubMed] [Google Scholar]
- 26.Behrendt N, Ronne E, Dano K. Domain interplay in the urokinase receptor. Requirement for the third domain in high affinity ligand binding and demonstration of ligand contact sites in distinct receptor domains. J Biol Chem. 1996;271:22885–22894. doi: 10.1074/jbc.271.37.22885. [DOI] [PubMed] [Google Scholar]
- 27.Gardsvoll H, Werner F, Sondergaard L, Dano K, Ploug M. Characterization of low-glycosylated forms of soluble human urokinase receptor expressed in Drosophila Schneider 2 cells after deletion of glycosylation-sites. Protein Expr Purif. 2004;34:284–295. doi: 10.1016/j.pep.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 28.Higazi AA, Mazar A, Wang J, Quan N, Griffin R, Reilly R, Henkin J, Cines DB. Soluble human urokinase receptor is composed of two active units. J Biol Chem. 1997;272:5348–5353. doi: 10.1074/jbc.272.8.5348. [DOI] [PubMed] [Google Scholar]
- 29.Hansen AP, Petros AM, Meadows RP, Nettesheim DG, Mazar AP, Olejniczak ET, Xu RX, Pederson TM, Henkin J, Fesik SW. Solution structure of the amino-terminal fragment of urokinase-type plasminogen activator. Biochemistry. 1994;33:4847–4864. doi: 10.1021/bi00182a013. [DOI] [PubMed] [Google Scholar]
- 30.Llinas P, Le Du MH, Gardsvoll H, Dano K, Ploug M, Gilquin B, Stura EA, Menez A. Crystal structure of the human urokinase plasminogen activator receptor bound to an antagonist peptide. EMBO J. 2005;24:1655–1663. doi: 10.1038/sj.emboj.7600635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Low BW, Preston HS, Sato A, Rosen LS, Searl JE, Rudko AD, Richardson JS. Three dimensional structure of erabutoxin b neurotoxic protein: inhibitor of acetylcholine receptor. Proc Natl Acad Sci U S A. 1976;73:2991–2994. doi: 10.1073/pnas.73.9.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gardsvoll H, Dano K, Ploug M. Mapping part of the functional epitope for ligand binding on the receptor for urokinase-type plasminogen activator by site-directed mutagenesis. J Biol Chem. 1999;274:37995–38003. doi: 10.1074/jbc.274.53.37995. [DOI] [PubMed] [Google Scholar]
- 33.Magdolen V, Rettenberger P, Koppitz M, Goretzki L, Kessler H, Weidle UH, Konig B, Graeff H, Schmitt M, Wilhelm O. Systematic mutational analysis of the receptor-binding region of the human urokinase-type plasminogen activator. Eur J Biochem. 1996;237:743–751. doi: 10.1111/j.1432-1033.1996.0743p.x. [DOI] [PubMed] [Google Scholar]
- 34.Ploug M, Rahbek-Nielsen H, Ellis V, Roepstorff P, Dano K. Chemical modification of the urokinase-type plasminogen activator and its receptor using tetranitromethane. Evidence for the involvement of specific tyrosine residues in both molecules during receptor-ligand interaction. Biochemistry. 1995;34:12524–12534. doi: 10.1021/bi00039a006. [DOI] [PubMed] [Google Scholar]
- 35.de Paulis A, Montuori N, Prevete N, Fiorentino I, Rossi FW, Visconte V, Rossi G, Marone G, Ragno P. Urokinase induces basophil chemotaxis through a urokinase receptor epitope that is an endogenous ligand for formyl peptide receptor-like 1 and -like 2. J Immunol. 2004;173:5739–5748. doi: 10.4049/jimmunol.173.9.5739. [DOI] [PubMed] [Google Scholar]
- 36.Resnati M, Pallavicini I, Wang JM, Oppenheim J, Serhan CN, Romano M, Blasi F. The fibrinolytic receptor for urokinase activates the G protein-coupled chemotactic receptor FPRL1/LXA4R. Proc Natl Acad Sci U S A. 2002;99:1359–1364. doi: 10.1073/pnas.022652999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Y, Lawrence DA, Zhang L. Sequences within domain II of the urokinase receptor critical for differential ligand recognition. J Biol Chem. 2003;278:29925–29932. doi: 10.1074/jbc.M300751200. [DOI] [PubMed] [Google Scholar]
- 38.Novokhatny V, Medved L, Mazar A, Marcotte P, Henkin J, Ingham K. Domain structure and interactions of recombinant urokinase-type plasminogen activator. J Biol Chem. 1992;267:3878–3885. [PubMed] [Google Scholar]
- 39.Nowak UK, Li X, Teuten AJ, Smith RA, Dobson CM. NMR studies of the dynamics of the multidomain protein urokinase-type plasminogen activator. Biochemistry. 1993;32:298–309. doi: 10.1021/bi00052a038. [DOI] [PubMed] [Google Scholar]
- 40.Montuori N, Rossi G, Ragno P. Cleavage of urokinase receptor regulates its interaction with integrins in thyroid cells. FEBS Lett. 1999;460:32–36. doi: 10.1016/s0014-5793(99)01314-9. [DOI] [PubMed] [Google Scholar]
- 41.Poliakov A, Tkachuk V, Ovchinnikova T, Potapenko N, Bagryantsev S, Stepanova V. Plasmin-dependent elimination of the growth-factor-like domain in urokinase causes its rapid cellular uptake and degradation. Biochem J. 2001;355:639–645. doi: 10.1042/bj3550639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moser TL, Enghild JJ, Pizzo SV, Stack MS. Specific binding of urinary-type plasminogen activator (u-PA) to vitronectin and its role in mediating uPA-dependent adhesion of U937 cells. Biochem J. 1995;307:867–873. doi: 10.1042/bj3070867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sidenius N, Andolfo A, Fesce R, Blasi F. Urokinase regulates vitronectin binding by controlling urokinase receptor oligomerization. J Biol Chem. 2002;277:27982–27990. doi: 10.1074/jbc.M111736200. [DOI] [PubMed] [Google Scholar]
- 44.May AE, Kanse SM, Lund LR, Gisler RH, Imhof BA, Preissner KT. Urokinase receptor (CD87) regulates leukocyte recruitment via beta 2 integrins in vivo. J Exp Med. 1998;188:1029–1037. doi: 10.1084/jem.188.6.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reuning U, Magdolen V, Hapke S, Schmitt M. Molecular and functional interdependence of the urokinase-type plasminogen activator system with integrins. Biol Chem. 2003;384:1119–1131. doi: 10.1515/BC.2003.125. [DOI] [PubMed] [Google Scholar]
- 46.Simon DI, Rao NK, Xu H, Wei Y, Majdic O, Ronne E, Kobzik L, Chapman HA. Mac-1 (CD11b/CD18) and the urokinase receptor (CD87) form a functional unit on monocytic cells. Blood. 1996;88:3185–3194. [PubMed] [Google Scholar]
- 47.Wei Y, Czekay RP, Robillard L, Kugler MC, Zhang F, Kim KK, Xiong JP, Humphries MJ, Chapman HA. Regulation of alpha5beta1 integrin conformation and function by urokinase receptor binding. J Cell Biol. 2005;168:501–511. doi: 10.1083/jcb.200404112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xue W, Kindzelskii AL, Todd RF, III, Petty HR. Physical association of complement receptor type 3 and urokinase-type plasminogen activator receptor in neutrophil membranes. J Immunol. 1994;152:4630–4640. [PubMed] [Google Scholar]
- 49.Simon DI, Wei Y, Zhang L, Rao NK, Xu H, Chen Z, Liu Q, Rosenberg S, Chapman HA. Identification of a urokinase receptor-integrin interaction site. Promiscuous regulator of integrin function. J Biol Chem. 2000;275:10228–10234. doi: 10.1074/jbc.275.14.10228. [DOI] [PubMed] [Google Scholar]
- 50.Ploug M, Ostergaard S, Gardsvoll H, Kovalski K, Holst-Hansen C, Holm A, Ossowski L, Dano K. Peptide-derived antagonists of the urokinase receptor. affinity maturation by combinatorial chemistry, identification of functional epitopes, and inhibitory effect on cancer cell intravasation. Biochemistry. 2001;40:12157–12168. doi: 10.1021/bi010662g. [DOI] [PubMed] [Google Scholar]
- 51.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CW Jr, Sweet RM, editors. Methods in Enzymology. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 52.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 53.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 54.Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr D Biol Crystallogr. 1999;55:247–255. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- 55.Laskowski RA, McArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 56.Kraulis PE. MOLSCRIPT: A Program to Produce Both Detailed and Schematic Plots of Protein Structures. J App Crystal. 1991;24:946–950. [Google Scholar]
- 57.Esnouf RM. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr D Biol Crystallogr. 1999;55:938–940. doi: 10.1107/s0907444998017363. [DOI] [PubMed] [Google Scholar]