

Abstract
Kainate receptors containing the GluK1 subunit (GluK1Rs; previously known as GluR5 kainate receptors) are concentrated in certain brain regions, where they play a prominent role in the regulation of neuronal excitability, by modulating GABAergic and/or glutamatergic synaptic transmission. In the basolateral nucleus of the amygdala (BLA), which plays a central role in anxiety as well as in seizure generation, GluK1Rs modulate GABAergic inhibition via postsynaptic and presynaptic mechanisms. However, the role of these receptors in the regulation of glutamate release, and the net effect of their activation on the excitability of the BLA network are not well understood. Here, we show that in amygdala slices from 35 to 50 day-old rats, the GluK1 agonist ATPA (300 nM) increased the frequency of spontaneous excitatory postsynaptic currents (sEPSCs) and miniature EPSCs (mEPSCs) recorded from BLA principal neurons, and decreased the rate of failures of evoked EPSCs. The GluK1 antagonist UBP302 (25 or 30 μM) decreased the frequency of mEPSCs, reduced evoked field potentials, and increased the “paired-pulse ratio” of the field potential amplitudes. Taken together, these results suggest that GluK1Rs in the rat BLA are present on presynaptic terminals of principal neurons, where they mediate facilitation of glutamate release. In vivo bilateral microinjections of ATPA (250 pmol) into the rat BLA increased anxiety-like behavior in the open field test, while 2 nmol ATPA induced seizures. Similar intra-BLA injections of UBP302 (20 nmol) had anxiolytic effects in the open field and the acoustic startle response tests, without affecting pre-pulse inhibition. These results suggest that although GluK1Rs in the rat BLA facilitate both GABA and glutamate release, the facilitation of glutamate release prevails, and these receptors can have an anxiogenic and seizurogenic net function. Presynaptic facilitation of glutamate release may, in part, underlie the hyperexcitability-promoting effects of GluK1R activation in the rat BLA.
Keywords: GluK1 kainate receptors, basolateral amygdala, presynaptic glutamate release, anxiety, seizures
1. Knowledge of the functions of the GluK1-containing kainate receptors (GluK1Rs, formerly known as GluR5 kainate receptors; Collingridge et al., 2009; Jane et al., 2009) has expanded substantially in recent years, with the development of selective agonists and antagonists (Jane et al., 2009) and the use of GluK1-deficient mice (Mulle et al., 2000). GluK1Rs appear to play a prominent role in the regulation of neuronal excitability in different brain regions, affecting glutamatergic (Li and Rogawski, 1998; Vignes et al., 1998; Clarke and Collingridge, 2002; Gryder and Rogawski, 2003; Partovi and Frerking, 2006; Campbell et al., 2007; Miyata and Imoto, 2009; Sun et al. 2009) and/or GABAergic (Cossart et al., 1998; Rodriguez-Moreno et al., 2000; Braga et al., 2003; Clarke and Collingridge, 2004; Wu et al., 2007a,b; Wondolowski and Frerking, 2009) synaptic transmission via postsynaptic and/or presynaptic mechanisms.
GluK1Rs are not widely distributed in the brain; they are heavily expressed in certain brain regions (Bettler et al., 1990). The amygdala, a relatively small amygdaloid (Greek for almond-shaped) structure in the temporal lobe that plays a key role in emotional behavior and associated disorders - such as anxiety disorders (Davis et al., 1994; Davidson et al., 1999; Etkin and Wager, 2007; Garrett and Chang, 2008; Stein and Stein, 2008) - as well as in seizure generation and certain types of epilepsy (Pitkanen et al., 1998; Aroniadou-Anderjaska et al., 2008), has a markedly high expression of GluK1Rs (Bettler et al., 1990; Li et al., 2001; Braga et al., 2003). Of all the amygdalar nuclei, the basolateral nucleus (BLA) has the highest propensity to generate seizures (White and Price, 1993a,b; Handforth and Ackerman, 1995; Mohapel et al., 1996; Aroniadou-Anderjaska et al., 2008), and, along with the medial nucleus, display the highest expression of the GluK1 subunit (Li et al., 2001; Braga et al., 2003).
In the BLA, GluK1Rs are present on somatodendritic sites of GABAergic interneurons, where they can be activated by repetitive synaptic stimulation (Braga et al., 2003); activation of these receptors by exogenous application of an agonist increases the frequency and amplitude of spontaneous inhibitory postsynaptic currents (sIPSCs), in rats (Braga et al., 2003) and mice (Wu et al., 2007a). In the rat BLA, these receptors are also present on GABAergic presynaptic terminals; weak activation of these presynaptic heteroreceptors, including activation by ambient concentrations of extracellular glutamate in the basal state, facilitates GABA release, whereas strong activation by high agonist concentrations suppresses GABA release (Braga et al., 2003). Whether or not there are also GluK1 autoreceptors in the BLA, modulating glutamate release presynaptically, is not known. However, principal neurons in the rat BLA do carry somatodendritic GluK1Rs, which can be activated synaptically (Li and Rogawski, 1998; Gryder and Rogawski, 2003; Läck et al., 2008). Based on this information, we have suggested that when glutamate concentrations are low, as in the basal or “resting” state, GluK1R-function may be to suppress excitability in the BLA by facilitating GABA release; when glutamate concentrations are high, as during epileptic activity or high anxiety states, activation of these receptors may exacerbate hyperexcitability by presynaptic suppression of GABA release, and by contributing to depolarization of principal neurons (Aroniadou-Anderjaska et al., 2007; Fritsch et al., 2009).
The first evidence that activation of these glutamatergic receptors may suppress excitatory activity – due to excitation of GABAergic interneurons – was obtained in the hippocampus, where in an in vitro preparation of intact hippocampi from neonatal rats, application of the GluK1R agonist (RS)-2-amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl) propanoic acid (ATPA) suppressed seizure propagation to the contralateral hippocampus (Khalilov et al., 2002). Consistent with a suppressing role of GluK1Rs on neuronal excitability, the hippocampi of GluK1-knockout mice were found to have a higher susceptibility to kainate-induced epileptiform activity (Fisahn et al., 2004). On the other hand, GluK1R antagonists were shown to block hippocampal epileptiform activity in vitro and limbic seizures in vivo induced by electrical stimulation, or by the muscarinic agonist pilocarpine (Smolders et al., 2002), suggesting a hyperexcitability-promoting role for GluK1Rs. Contradictory findings in regard to the net function of GluK1Rs have also been reported in the amygdala. The GluK1R agonist ATPA increased anxiety-like behavior (assessed in the light/dark box) when microinjected bilaterally into the rat BLA (Läck et al., 2008), and systemically injected antagonists of GluK1Rs have anxiolytic effects when tested in the punished responding (Kotlinska and Liljequist, 1998; Alt et al., 2006, 2007) and the elevated plus maze (Kotlinska and Liljequist, 1998); these findings suggest that GluK1R activation increases amygdalar excitability. On the other hand, intraperitoneal injection of ATPA in mice had anxiolytic effects in the elevated plus maze, intra-BLA injection of a GluK1 antagonist increased anxiety, and GluK1-knockout mice display anxiety-like behavior (Wu et al. 2007a), results that suggest a suppressing role of GluK1Rs on amygdalar excitability.
In an attempt to shed more light into the mechanisms by which GluK1Rs participate in the regulation of neuronal excitability in the BLA, and understand the net effect that activation of these receptors has on the excitability of the BLA network, which may help explain some of the contradictory findings described above, we 1) examined if GluK1Rs in the rat BLA regulate glutamate release at the presynaptic level, and 2) studied the effects of activation or blockade of these receptors on evoked population responses in vitro and on anxiety-like behavior, in vivo. The results suggest that in the rat BLA, 1) there are GluK1Rs on glutamatergic terminals, where they mediate facilitation of glutamate release even in the basal state, and 2) although GluK1Rs can enhance both GABA and glutamate release, the hyperexcitability-promoting function prevails.
2. EXPERIMENTAL PROCEDURES
2.1. Electrophysiological experiments
Male, Sprague-Dawley rats (120 to 220 g) were anesthetized with isoflourane before decapitation. Coronal brain slices (400 μm thick; −2.64 to −3.36 from bregma) containing the amygdala were prepared in ice-cold artificial cerebrospinal fluid (ACSF). The cutting solution consisted of (in mM): 115 sucrose, 70 NMDG, 1 KCl, 2 CaCl2, 4 MgCl2, 1.25 NaH2PO4, 30 NaHCO3, 25 D-glucose. The slices were transferred to a holding chamber, at room temperature, in a bath solution containing: 125 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 21 mM NaHCO3, 2 mM CaCl2, 1 mM MgCl2, and 11 mM D-glucose. Recording solution was the same as the holding bath solution. For field potential recordings the bath/recording solution was same as above, except for the concentration of MgCl2 (1.5 mM). All solutions were saturated with 95% O2, 5% CO2 to achieve a pH near 7.4. For whole-cell recordings, the slice chamber (0.7 mL capacity) had continuously flowing ACSF (~5 mL/min) at temperature 32~33°C. The osmolarity of this external solution was adjusted to 325 mOsm with D-glucose. Field potential recordings were obtained in an interface-type chamber, maintained at 32~33°C, with a flow rate of the ACSF at 2 ml/min.
For whole-cell recordings, neurons were visualized under infrared light, using Nomarski optics of an upright microscope (Zeiss Axioskop 2, Thronwood, NY) equipped with a CCD-100 camera (Dage-MTI, Michigan City, IN). The patch electrodes had resistances of 3.5–4.5 M Ω when filled with the internal solution: 60 mM CsCH3SO3, 60 mM KCH3SO3, 10 mM KCl, 10 mM EGTA, 10 mM HEPES, 5 mM Mg-ATP, 0.3 mM Na3GTP (pH 7.2), 290 mOsm. Tight-seal (over 1 GΩ) whole-cell recordings were obtained from the cell body of pyramidal-shaped neurons in the BLA region. Access resistance (5–24 MΩ) was regularly monitored during recordings, and cells were rejected if the resistance changed by more than 15% during the experiment. Ionic currents and action potentials were amplified and filtered (1 kHz) using the Axopatch 200B amplifier (Axon Instruments, Foster City, CA) with a four-pole, low-pass Bessel filter, were digitally sampled (up to 2 kHz) using the pClamp 10.2 software (Molecular Devices, Sunnyvale, CA), and further analyzed using the Mini Analysis program (Synaptosoft Inc., Fort Lee, NJ) and Origin (OriginLab Corporation, Northampton, MA).
Field potentials were evoked by stimulation of the external capsule at 0.05 Hz. Recording glass pipettes were filled with ACSF, and had a resistance of approximately 5 M. Stimulation was applied with a bipolar concentric stimulating electrode made of tungsten. Signals were digitized using the pClamp 10.2 software (Molecular Devices, Union City, CA), analyzed using Clampfit 10.2, and final presentation was prepared using Origin (OriginLab Corporation, Northampton, MA).
Drugs used were as follows: bicuculline methiodide, a GABAA receptor antagonist, and tetrodotoxin (TTX), a sodium channel blocker, both from Sigma-Aldrich (St. Louis, MO); D-AP5, an NMDA receptor antagonist, SCH50911, a GABAB receptor antagonist, (RS)-2-amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl) propanoic acid (ATPA), a selective GluK1R agonist (Clarke et al., 1997; Jane et al., 2009), and UBP302, a selective GluK1R antagonist, with similar affinity for homomeric and heteromeric GluK1 kainate receptors (More et al., 2004), were all obtained from Tocris (Ellisville, MO).
2.2. Behavioral experiments
Male, Sprague-Dawley rats (200 ~ 220 g) were individually housed in an environmentally controlled room (20–23°C, 12-h light/12-h dark cycle, lights on 06:00 pm), with food and water available ad libitum. All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the Uniformed Services University of the Health Sciences. The investigators adhered to the Guide for the Care and Use of Laboratory Animals, which requires humane treatment of all animals, with regard for alleviation of suffering (Institute of Laboratory Animal Research 1996).
The rats were surgically implanted with guide cannulae inserted bilaterally into the BLA (coordinates: 2.8 mm anteroposterior to the bregma, 4.6 mm lateral to the midline, and 8.0 mm in depth, based on Paxinos and Watson, 1997). Experiments were conducted in freely moving rats after 14 days of recovery, during the dark phase of the rats’ light/dark cycle. The rats were randomly assigned into groups that were administered the GluK1R agonist ATPA (250 nmol, 1 nmol, or 2 nmol) dissolved in artificial cerebrospinal fluid (ACSF; see field potential experiments in the electrophysiology section for composition), or the GluK1R antagonist UBP302 (20 nmol), dissolved in 50% ACSF / 50% DMSO solution, or vehicle (ACSF or a solution of 50% DMSO / 50% ACSF). The solutions were injected bilaterally into the BLA via a microinfusion pump (Harvard Apparatus, Holliston, MA) at a rate of 0.5 μl/min (total volume 1 μl for each solution), using a microinjection procedure as described by others (McCool and Chappell, 2007).
2.2.1 Open Field test
Anxiety-like behavior was assessed using an open field apparatus (40×40×30 cm clear Plexiglas arena), based on the procedure used by Grunberg and collaborators (Faraday et al., 2001). The animals were acclimated to the apparatus in one session. On test days, the rats were placed in the center of the open field, 2 min after drug or vehicle injection. Activity was measured and recorded for 20 min, using an Accuscan Electronics infrared photocell system (Accuscan Instruments Inc., Columbus, OH). Data were automatically collected and transmitted to a computer equipped with “Fusion” software (from Accuscan Electronics, Columbus, OH). Locomotion (distance traveled in cm), total movement time, and time spent in the center of the open field were analyzed. Anxiety behavior was measured as the ratio of the time spent in the center over the total movement time, expressed as a percentage of the total movement time. Subjects were exposed to the test two times, separated by about 1 week; each rat was tested only with a single dose of the drug and a single dose of the vehicle, and the order of testing was randomized.
2.2.2 Acoustic Startle Response test
Acoustic Startle Response (ASR) testing was conducted with the use of the Med Associates Acoustic Response Test System (Med Associates, Georgia, VT), which consists of weight-sensitive platforms inside individual sound-attenuating chambers. A ventilating fan built into the chamber provides background noise. Each rat was individually placed in a ventilated holding cage. The holding cages are small enough to restrict extensive locomotion, but large enough to allow the subject to turn around and make other small movements. Each cage was placed on a weight-sensitive platform. Subjects’ movements in response to stimuli were measured as a voltage change by a strain gauge inside each platform. Responses were recorded by an interfaced Pentium computer as the maximum response occurring during the no-stimulus periods, during the startle period, and during the pre-pulse period (see next paragraph), and were assigned a value based on an arbitrary scale used by the software of the Test System.
All animals were acclimated to the apparatus in two sessions. Then, animals received microinjections of UBP302 (or vehicle), and were placed in the testing apparatus, 5 min after the injection. A 3-min adaptation period was allowed, during which no startle stimuli were presented. Startle stimuli consisted of 120 or 110 dB Sound Pressure Level noise bursts of 20-ms duration. Each stimulus had a 2-ms rise and decay time, such that the onset and offset were abrupt, which is a primary requirement for startle. Trial types were as follows: 1) 110 dB stimulus, 2) 120 dB stimulus, or 3) 120 dB stimulus preceded by a 68 dB pure tone (“prepulse”). Each trial type was presented eight times. Trial types were presented in random to avoid order effects and habituation, and inter-trial intervals ranged randomly from 15 to 25 s. Each rat was tested after vehicle injection or UBP302, with an interval of 1 week between tests. The order of drugs was randomized between the subjects.
Only data from animals with histologically confirmed bilateral cannulae placements in the BLA were included in the analysis (38 of 45 animals).
2.3. Statistical analysis
Both behavioral and electrophysiological data were analyzed using the Paired Student’s t-tests. Results were considered statistically significant when p < 0.05. Data are presented as Mean ± Standard Error (SE) of the mean. For the in vitro experiments, sample size “n” refers to the numbers of slices. For the in vivo experiments, sample size “n” refers to the number of rats.
3. RESULTS
3.1. Effects of GluK1R activation on glutamate release
In brain slices containing the amygdala, whole-cell recordings were obtained from neurons in the BLA, which were identified as principal cells based on their size and pyramidal-like shape (Fig. 1A), as well as on their firing patterns in response to depolarizing current pulses in the current clamp mode (Fig. 1B) and the presence of a current activated by hyperpolarization (Ih current; Fig. 1C). It is known that over 85% of amygdala neurons are pyramidal cells displaying the Ih (Kroner et al. 2005; Park et al. 2007; Rainnie et al. 1993; Washburn and Moises 1992; Womble and Moises 1993). The presence of Ih was detected by applying four 1 sec-long hyperpolarizing steps from the holding potential of −70 mV, with 10 mV increments. Ih current profiles characteristic of principal pyramidal neurons are demonstrated in the left panel of Fig. 1C; for comparison, linear currents displayed by interneurons are demonstrated in the right panel of Fig. 1C.
Fig. 1.

Activation of GluK1Rs by ATPA increases spontaneous EPSCs in principal BLA neurons. (A) Left: Rat brain slice showing the BLA (circle), where recordings were performed. Middle: The BLA nucleus at higher magnification; asterisk is marking the location of a patch-clamped neuron. Right: Patch-clamped pyramidal cell. Calibration bars: 1 mm, 500 μm, and 10 μm, from left to right, respectively. (B) Distinguishing BLA principal neurons from interneurons in current-clamp mode. Principal cells demonstrated variable patterns of accommodating spiking, while interneurons demonstrated fast, non-accomodating spiking. Responses were elicited by 500 ms-long current pulses, ranging from −300 pA to 300 pA with 100 pA steps. (C) Distinguishing BLA principal neurons from interneurons in voltage-clamp mode. Four 1s-long hyperpolarizing pulses starting from Vhold −70 mV to −80 mV and ending with −110 mV, elicited nonlinear current (Ih) in principal neurons and small linear current in interneurons. (D) Bath applied ATPA (300 nM) increased the frequency and amplitude of sEPSCs. Recordings are from a pyramidal-shaped neuron, in the presence of bicuculline (20 μM), D-AP5 (50 μM), and SCH50911 (20 μM). Example traces are shown in the left panel and group data from 7 cells in the bar graph to the right. *p < 0.05.
It has been shown previously that GluK1R-mediated EPSCs can be evoked in principal BLA neurons by stimulation of the external capsule (Gryder and Rogawski, 2003). This would predict that exogenous application of a GluK1R agonist will increase spontaneous excitatory postsynaptic currents (sEPSCs) by depolarizing principal neurons. Indeed, bath application of ATPA (300 nM), in the presence of bicuculline (20 μM), D-AP5 (50 μM), and SCH50911 (20 μM) increased the frequency of sEPSCs from 5.7 ± 0.4 Hz to 8.2 ± 0.9 Hz (n = 6, p = 0.012, Fig. 1D). ATPA also induced a non-desensitizing current of 8.1 ± 0.5 pA (n = 6), which was probably due to activation of somatodendritic GluK1Rs on the recorded neuron. The increase in the frequency of sEPSCs by ATPA can be attributed to activation of somatodendritic GluK1Rs on BLA principal neurons, which would depolarize the neurons producing an increase in action potential-dependent glutamate release. It is not known, however, if GluK1Rs are also present on presynaptic glutamatergic terminals, and if a presynaptic facilitation of glutamate release by ATPA also contributed to the increase of the sEPSCs.
To determine if GluK1Rs are present on glutamatergic terminals in the BLA, we used two approaches. First, we examined the effects of ATPA on the rate of failures of AMPA/kainate receptor-mediated synaptic transmission. In the presence of GABAA, GABAB and NMDA receptor antagonists, synaptic responses were evoked by minimal electric stimulation applied at 0.1 Hz, with a tungsten stimulating electrode placed approximately 50 μm away from the recorded cell, as described previously (Braga et al. 2003). Bath application of 300 nM ATPA decreased the number of failures of the evoked EPSCs (eEPSCs) from 57 ± 6 % in the control ACSF to 32 ± 6 % in the presence of ATPA (n = 7, p = 0.011, Fig. 2). The effect was at least partly reversible (it was not possible to maintain the cell until complete wash out of the ATPA).
Fig. 2.

Activation of GluK1Rs by ATPA decreases the rate of failures of AMPA/kainate receptor-mediated synaptic currents recorded from principal neurons in the BLA. (A) Superimposed traces are eEPSCs recorded before, during, and after bath application of 300 nM ATPA. The slice medium contains D-AP5 (100 μM), bicuculline (20 μM), and SCH50911 (10 μM); Vhold, −70 mV. (B) Time course of the effect of ATPA on the amplitude of the eEPSCs and the number of failures (same cell as in A). (C) Pooled data of the percentage of failures (Mean ± SE) before, during, and after ATPA (n = 7; *p < 0.05).
The most plausible interpretation of the reduction of the rate of failures of eEPSCs by ATPA is that the agonist acted on glutamatergic presynaptic terminals to facilitate glutamate release. It is also possible, however, that ATPA reduced the rate of failures by depolarizing principal cells via postsynaptic GluK1Rs; such depolarization of principal neurons by ATPA could produce either more glutamate release from the stimulated axon or the recruitment of an additional axon(s). To obtain direct evidence for the presence of GluK1Rs on glutamatergic terminals, we examined if ATPA affects the frequency of miniature EPSCs (mEPSCs). We recorded mEPSCs from BLA principal neurons in the presence of bicuculline (20 μM), D-AP5 (100 μM), SCH50911 (20 μM), and TTX (1 μM). Bath application of 300 nM ATPA increased the frequency of mEPSCs from 7.6 ± 1.7 Hz in control conditions to 14.9 ± 3.2 Hz in ATPA (n = 8, p = 0.0069, Fig. 3), with no significant effect on the amplitude or the kinetics of the mEPSCs. The increase in mEPSC frequency by ATPA was reversible upon addition to the slice medium of the GluK1R antagonist UBP302 (30 μM, Fig. 3; the reversal of the ATPA effect by UBP302 is only partial because, in some of the cells, recordings deteriorated before UBP302 took full effect). These results suggest that GluK1Rs are present on presynaptic glutamatergic terminals in the BLA and mediate facilitation of glutamate release.
Fig. 3.

The GluK1R agonist ATPA increases the frequency of mEPSCs. (A) Traces of mEPSCs recorded from a BLA pyramidal cell in control medium, during bath application of ATPA (300 nM), and after addition of UBP302 (30 μM). The slice medium contains bicuculline (20 μM), D-AP5 (100 μM), SCH50911 (20 μM), and TTX (1 μM); Vhold, −70 mV (B) Cumulative probability plot of inter-event intervals of mEPSCs in control medium, in the presence of ATPA, and in the presence of ATPA and UBP302 (data from the cell shown in A). (C) Group data showing the change in the frequency of mEPSCs by ATPA (n = 8, *p < 0.01).
3.2. Effects of GluK1R blockade on glutamate release
In the experiments described above, GluK1Rs on glutamatergic terminals were activated by exogenous application of an agonist. Next, we asked whether these receptors are activated by ambient concentrations of endogenous glutamate. To answer this question we examined the effects of GluK1R blockade by UBP302 on mEPSCs, recorded in the presence of bicuculline (20 μM), D-AP5 (100 μM), SCH50911 (20 μM), and TTX (1 μM). Application of 30 μM UBP302 to the slice medium reduced the frequency of mEPSCs from 7.8 ± 0.4 Hz to 5.7 ± 0.7 Hz (n = 7, p = 0.0073), with no significant effect on the amplitude; the reduction in mEPSC frequency by UBP302 was reversible (Fig. 4).
Fig. 4.

The GluK1R antagonist UBP302 decreases the frequency of mEPSCs. (A) Traces of mEPSCs recorded from a BLA pyramidal neuron before, during, and after bath application of 30 μM UBP302. The slice medium contains bicuculline (20 μM), D-AP5 (100 μM), SCH50911 (20 μM), and TTX (1 μM); Vhold, −70 mV. (B) Cumulative probability plot of inter-event intervals of mEPSCs in control medium, in the presence of UBP302, and after washing out UBP302 (data from the cell shown in A). (C) Group data showing the change in the frequency of mEPSCs by UBP302 (n = 7, *p < 0.01).
3.3. Effects of GluK1R blockade of field potentials
Taken together, the results described above suggest that GluK1Rs are present on glutamatergic presynaptic terminals in the BLA, and it appears that they are activated even by ambient concentrations of endogenous glutamate, facilitating glutamate release. We have previously shown that activation of GluK1Rs also enhances GABAergic activity, and that GluK1Rs on GABAergic presynaptic terminals are activated by ambient concentrations of extracellular glutamate, facilitating GABA release (Braga et al., 2003). The question then arises as to what is the net role of GluK1Rs in the regulation of the BLA excitability. The whole-cell recording experiments described above were performed in the presence of a GABAA receptor antagonist, in order for the role of GluK1Rs in the regulation of glutamatergic transmission to be revealed. But when all synaptic transmission in the BLA circuitry is left intact (no receptor antagonists present), does the activity of GluK1Rs enhance or suppress the excitability of the network?
It has been previously shown that when GluK1Rs are activated by perfusion of slices with ATPA (in the absence of GABA receptor antagonists), intracellularly-recorded principal cells in the BLA display epileptiform bursts (Li et al., 2001); we have also observed that bath application of ATPA induces epileptiform activity in BLA field potentials (V. Aroniadou-Anderjaska, unpublished results). In the present study, we wanted to determine whether the net effect of basal GluK1R activation by endogenous glutamate is suppression or enhancement of the BLA excitability, or if the GluK1R-mediated facilitation of both GABAergic and glutamatergic activity cancel each other with no significant net effect. For this purpose, we examined the effects of UBP302 on BLA population responses (in the absence of any other receptor antagonists), evoked by stimulation of the external capsule. We delivered paired-pulse stimulation in these experiments (inter-stimulus interval 60 ms or 120 ms) in order to obtain additional information on the effects of UBP302 on the probability of glutamate release and/or on evoked inhibition.
Bath application of UBP302 (25 μM) decreased the amplitude of the field response to the first stimulus pulse from 0.62 ± 0.06 mV to 0.50 ± 0.04 mV (n = 5, p = 0.024; Fig. 5), along with a reduction in the slope of the waveform (see example in Fig. 5B). In percentages, UBP302 reduced the field response to the first pulse to 81 ± 3.9 % of the control amplitude (p = 0.008; Fig. 5C). However, the response to the second pulse was not reduced significantly (from 0.73 ± 0.06 mV in control medium to 0.67 ± 0.07 mV in UBP302, n = 5, p = 0.092; Fig. 5A and C). As a result, the ratio of the amplitude of the response to Pulse 2 over the amplitude of the response to Pulse 1 (“paired-pulse ratio”) increased from 1.19 ± 0.12 in the control solution to 1.36 ± 0.13 in the presence of UBP302 (n = 5, p = 0.02, Fig. 5). Thus, blockade of GluK1Rs in the BLA produces a small but significant suppression of evoked responses and increases the paired-pulse ratio of their amplitudes.
Fig. 5.

Effects of UBP302 on field potentials evoked in the BLA by paired-pulse stimulation of the external capsule. (A) A representative example of BLA field synaptic responses to paired-pulse stimulation, before, during, and after wash out of bath-applied UBP302 (25 μM). Each trace is an average of 15 sweeps. Notice the reduction of the response to the first pulse by UBP302, without proportional reduction in the response to the second pulse. (B) An expanded view of field responses to the first stimulus pulse (different slice from the one shown in A), in control conditions and in the presence of UBP302 (red trace). Each of the two traces is the average of 15 sweeps. (C). Group data from 5 slices, showing the amplitude of the field responses to the first and the second stimulus pulses as percentages of the control response to the fist pulse, before, during, and after bath application of UBP302. Only the amplitude of the field response to the first stimulus pulse was significantly reduced by UBP302 (*p < 0.01). (D) Ratio of the amplitude of the synaptic response to the second pulse over the amplitude of the response to the first pulse, in control medium and in the presence of UBP302 (*p < 0.01).
3.4. Behavioral effects of GluK1R activation
Rats were administered 2 nmol, 1 nmol, or 250 pmol ATPA bilaterally into the BLA. The dose of 2 nmol induced “wet-dog shakes” in 8 out of the 10 rats, within less than 5 min after the micro-injection. The wet-dog shakes progressed to higher stages of behavioral seizures in 5 of these rats; the 2 rats that did not exhibit wet-dog shakes had the earliest onset of seizures. The average latency to the unilateral or bilateral forelimb clonus without rearing (stage 3 of the Racine scale; Racine, 1972; Racine et al., 1977) was 25 ± 5 min (n = 7). The observed behavioral seizures reached up to stage 5 of the Racine scale. The 1 nmol ATPA induced wet-dog shakes in all of the injected animals (6 out of 6), within 5 min after the injection, but there was no further progression of seizure activity. Rats injected with 250 pmol ATPA (n = 8) did not produce any behavior indicative of seizure activity for 2 hours of observation. These data are summarized in Table 1. When the highest dose of ATPA (2 nmol) was injected at 10 min after injection of UBP302 (20 nmol), the animals (n = 3) did not display any evidence of seizure activity during two hours of observation; thus, UBP302 was able to block the seizurogenic effects of ATPA.
Table 1.
Activation of GluK1Rs by bilateral injections of ATPA into the BLA can induce seizures.
| ATPA dose | Wet dog shakes | Seizures | Latency to Seizures (min) |
|---|---|---|---|
| 250 pmol | 0 (8) | 0 (8) | |
| 1 nmol | 6 (6) | 0 (6) | |
| 2 nmol | 8 (10) | 7 (10) | 25 ± 5 |
Shown are the number of animals out of the total sample size (in parenthesis) that presented “wet-dog shakes” or greater than stage 3 seizures, and the latency to the first unilateral/bilateral forelimb clonus. At the dose of 250 pmol, ATPA did not produce any indication of seizure activity, at 1 nmol, wet-dog shakes were induced in all animals, while at 2 nmol, ATPA induced stage 4 and 5 seizures in 70% of the animals.
The ATPA dose of 250 pmol was used to assess the anxiogenic properties of ATPA in the open field test. In this test, the more anxious the animal is, the less time it spends in the center of the field (for a review see Prut and Belzung, 2003). ATPA significantly reduced the amount of time the rats spent in the center of the open field (8.59 ± 1.83 % of the total movement time) compared to the time spent in the center after vehicle injection (15.86 ± 2.73 % of the total movement time; p = 0.03, n = 8, Fig. 6). The distance the rats traveled in the open field after ATPA injection (2092 ± 312 cm) was not significantly different from the distance they traveled after vehicle injection (1740 ± 189 cm).
Fig. 6.
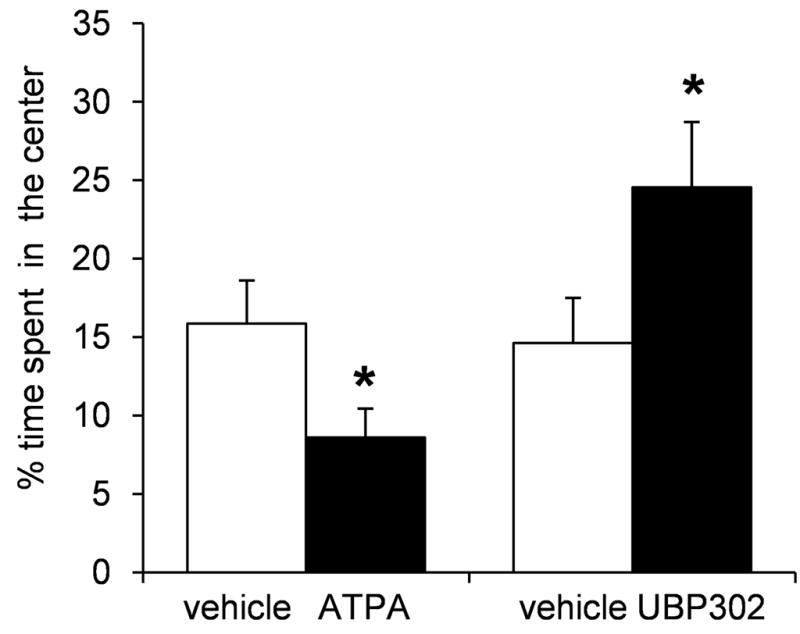
Activation of GluK1Rs by bilateral intra-BLA injections of ATPA increases anxiety, while blockade of these receptors by UBP302 reduces anxiety, in the open field test. When rats were injected with ATPA (250 pmol), they spent significantly less time in the center of the open field compared to the time they spent in the center when injected with the vehicle. Conversely, when rats were injected with UBP302 (20 nmol), they spent significantly more time in the center of the open field compared to the time they spent in the center when injected with the vehicle. Bars show the Mean ± SE of the percentage of time spent in the center, from a group of 8 rats tested with ATPA or vehicle, and another group of 7 rats tested with UBP302 or vehicle. *p < 0.05
3.5. Behavioral effects of GluK1R blockade
The concentration of UBP302 that must be used to block GluK1Rs in brain slices, in vitro, is at least 20× higher than the concentration needed for ATPA to activate GluK1 receptors. Therefore, in vivo, we used a concentration of UBP302 that was 20× higher than the 1 nmol of ATPA. When animals were injected with 20 nmol UBP302 bilaterally into the BLA, they spent significantly more time in the center of the open field (24.54 ± 4.16 % of the total movement time) compared to the time they spent in the center when injected with the vehicle (14.61 ± 2.87 % of the total movement time; p = 0.02, n = 7; Fig 6). The distance the rats traveled in the open field after UBP302 injection (1641 ± 134 cm) was not significantly different from the distance they traveled after vehicle injection (1742 ± 187 cm).
Another behavioral test that can assess the level of anxiety is the startle response to a strong acoustic stimulus (acoustic startle response; ASR). The neural circuitry mediating the startle reflex is relatively simple, but it is significantly modulated by activity in other brain regions and particularly the amygdala (for a review see Li et al., 2009); the greater the excitability of the amygdala (as during anxiety and/or fear), the larger the startle response (Hitchcock and Davis, 1987; Li et al., 1999). In rats injected in the BLA with 20 nmol of UBP302 or the vehicle, we measured the amplitude of the startle, and averaged the amplitude of 8 trials for each type of acoustic stimulus: 110 dB noise burst alone, 120 dB noise burst alone, and 120 dB preceded by a 68 dB prepulse. We hypothesized that if UBP302 has anxiolytic properties as the results from the open field test suggested, it should reduce the amplitude of the ASR. Delivery of a prepulse stimulus (a weaker acoustic stimulus preceding the strong stimulus) suppresses the startle response to the strong stimulus, and this effect has been termed “prepulse inhibition” (PPI; for reviews see Hoffman and Ison, 1980; Li et al., 2009). The amygdala plays a significant role in the modulation of PPI (for a review see Swerdlow et al., 2001). Therefore, the ASR test that included delivery of a prepulse aimed at determining if blockade of GluK1Rs in the BLA by UBP302 can affect the efficacy of prepulse inhibition.
The amplitude of the ASR to the 110 dB noise burst was reduced from 2.31 ± 0.38 (arbitrary units; see methods) after vehicle injection to 1.34 ± 0.28 after UBP302 injection (p = 0.007, n = 7, Fig. 7). The amplitude of the ASR to the 120 dB noise burst was reduced from 5.60 ± 1.25 after vehicle injection to 3.36 ± 1.17 after UBP302 injection (p = 0.029, n = 7, Fig. 7). The 68 dB prepulse reduced the amplitude of the ASR to the 120 dB noise burst (p = 0.004, n = 7); UBP302 had no significant effect on this reduction (Fig. 7).
Fig. 7.

Blockade of GluK1Rs by bilateral intra-BLA injections of UBP302 reduces the amplitude of the acoustic startle response (ASR). The startle stimulus was 110 dB or 120 dB. In some trials, the 120 dB stimulus was preceded by a 68 dB prepulse (100 ms interval). The group data are Mean ± SE of the average ASR amplitude (arbitrary units) from 7 rats. UBP302 significantly reduced the amplitude of the ASR to both the 120 and 110 dB stimuli, but had no significant effect when the 120 dB noise burst stimuli were preceded by a prepulse. *p < 0.05, in comparison to the vehicle-injected rats; #p < 0.05 in comparison to the 120 dB stimulus that was not preceded by a prepulse.
4. DISCUSSION
It is well known that the amygdala plays a central role in emotional behavior, and, therefore, in disorders related to emotion such as the anxiety disorders (Davis et al., 1994; Davidson et al., 1999; Etkin and Wager, 2007; Garrett and Chang, 2008; Stein and Stein, 2008); excitability of the BLA, in particular, is closely associated with the expression of anxiety (Sanders and Shekhar, 1995; Menard and Treit, 1999; Läck et al., 2007). The BLA also has a high propensity to generate and propagate seizure activity (White and Price, 1993a,b; Handforth and Ackerman, 1995; Mohapel et al., 1996; Pitkanen et al., 1998; Aroniadou-Anderjaska et al., 2008). Therefore, understanding the mechanisms regulating BLA excitability can have significant bearing on the treatment of anxiety and seizure disorders. Here, we showed that one of the mechanisms by which GluK1Rs participate in the regulation of neuronal excitability in the BLA is the facilitation of presynaptic glutamate release. We also provided the first demonstration that local blockade of GluK1Rs in the BLA reduces anxiety. Further, we showed that local activation of GluK1Rs in the BLA, by ATPA, increases anxiety and can induce seizures.
4.1 GluK1Rs in the rat BLA mediate presynaptic facilitation of glutamate release
In addition to modulating GABAergic synaptic transmission – in the hippocampus (Clarke and Collingridge 2004; Cossart et al. 1998; Rodriguez-Moreno et al. 2000; Wondolowski and Frerking 2009) and the amygdala (Braga et al. 2003; Wu et al. 2007a,b) – GluK1Rs have also been reported to presynaptically regulate glutamate release. Activation of GluK1Rs suppresses glutamate release in CA3 to CA1 pyramidal cell synapses (Clarke and Collingridge, 2002; Partovi and Frerking, 2006; Vignes et al., 1998) and in cortical layer VI pyramidal cell synapses onto thalamic relay neurons (Miyata and Imoto, 2009). In contrast, in the prefrontal cortex (Campbell et al., 2007), entorhinal cortex (Chamberlain et al., 2011), and in the Schaffer collateral synapses onto somatostatin interneurons in the CA1 hippocampal area (Sun et al., 2009), GluK1R activation facilitates glutamate release. The evidence presented in the present study suggesting that GluK1Rs also mediate facilitation of glutamate release in the BLA, was: 1) ATPA decreased the rate of failures of eEPSCs, 2) mEPSCs were increased by ATPA, and 3) mEPSCs were decreased by UBP302. The increase in the paired-pulse ratio of the evoked field potentials by UBP302 is also consistent with a reduction in the probability of glutamate release by the antagonist; by blocking the GluK1R-mediated basal facilitation of glutamate release, UBP302 reduced the release of glutamate in response to the first pulse, which enhanced glutamate release in response to the second stimulus pulse (see Debanne et al., 1996; Thomson, 2000). Since we – purposely – did not block GABAergic inhibition in these experiments, a decrease of evoked inhibition by UBP302 (see Braga et al., 2003) could also contribute to increasing the paired-pulse ratio. However, this possibility is less likely because if blockade of the GluK1R-mediated facilitation of GABA release was the primary action of UBP302, then the population response to the first pulse would also increase. This was not the case; the amplitude of the field potential evoked by the first pulse was decreased, suggesting that the prevailing consequence of GluK1R blockade by UBP302 was a reduction in glutamate release and, probably, a reduced depolarization of principal BLA neurons as well, due to blockade of somatodendritic GluK1Rs.
4.2. Facilitation of both glutamatergic and GABAergic synaptic transmission via GluK1Rs and the net effect of GluK1R activation
In the rat BLA, GluK1Rs are present on both somatodendritic and presynaptic sites of GABAergic interneurons, and their activation increases GABA release (Braga et al., 2003). The role of the somatodendritic GluK1Rs in tonic depolarization of the interneurons is not known, but the presynaptic GluK1Rs facilitate GABA release even in the basal state, activated by ambient concentrations of extracellular glutamate. Strong activation however of these presynaptic GluK1 heteroreceptors by high concentrations of agonists inhibit GABA release (Braga et al. 2003). Principal neurons in the rat BLA also carry GluK1Rs on somatodendritic sites, and these receptors can be activated synaptically (Li and Rogawski, 1998; Gryder and Rogawski 2003), but it is not known if they are activated by basal concentrations of glutamate, and, therefore, if they contribute to tonic excitation of the BLA network. In the present study, we found that GluK1 autoreceptors facilitate glutamate release, even in the basal state. Thus, both GABAergic and glutamatergic activity can increase in the rat BLA via activation of GluK1Rs. We have previously suggested that when GluK1Rs are weakly activated in the basal state they enhance inhibition – by facilitating GABA release presynaptically and, possibly, postsynaptically as well via interneuronal depolarization –, while when they are activated by high glutamate concentrations as during seizure activity or in a high anxiety state they promote hyperexcitability, by inhibiting presynaptic GABA release and depolarizing BLA principal cells (Braga et al., 2003; Aroniadou-Anderjaska et al., 2007; Fritsch et al., 2009). The present results suggest that a third mechanism by which GluK1Rs can promote hyperexcitability is presynaptic facilitation of glutamate release. However, if the in vitro slices in normal ACSF can be considered to be similar to “the basal, resting state”, the present results also suggest that even in the basal state, the GluK1R-mediated facilitation of glutamate release overrides the facilitation of GABA release, since blocking GluK1Rs did not increase BLA excitability, as it can be concluded from the reduction in the amplitude of the field potential, which was probably due to presynaptic reduction of glutamate release and/or due to less postsynaptic postsynaptic depolarization of principal cells.
In vivo blockade of GluK1Rs by UBP302 also suppressed amygdalar excitability as evidenced by the reduction in anxiety-like behavior in the open field and the ASR tests, while activation of GluK1Rs by bilateral intra-BLA injections of ATPA, increased anxiety in the open field test, and, at higher concentrations of the agonist, induced seizures. At high concentrations, ATPA can also activate AMPA receptors. However, since the seizurogenic action of the highest concentration of ATPA used in the present study was blocked by pre-administration of UBP302, both the anxiogenic and seizurogenic effects of ATPA were probably due to activation of GluK1Rs.
In agreement with our findings, Läck et al. (2008) showed that local application of ATPA in the rat BLA increases anxiety as assessed in the light/dark box, while Kaminski et al. (2004) reported that intravenous infusion of ATPA induces clonic seizures. Other studies have shown that systemically administered GluK1R antagonists have an anxiolytic effect on punished responding (Kotlinska and Liljequist, 1998; Alt et al., 2006, 2007) and the elevated plus maze (Kotlinska and Liljequist, 1998) behavioral tests, while an AMPA receptor antagonist did not have anxiolytic action (Alt et al., 2007). Our findings indicate that blockade of GluK1Rs in the BLA, alone, is sufficient for the anxiolytic effect. Taken together, these studies suggest that GluK1Rs in the rat BLA can promote hyperexcitability. Further support for this conclusion comes from the ability of GluK1R antagonists to suppress nerve agent-induced epileptiform activity in the BLA, in vitro (Apland et al., 2009) and nerve agent-induced seizures, in vivo (Figueiredo et al., 2009), as well as pilocarpine-induced limbic seizures (Smolders et al., 2002). It is also relevant and worth noting that the anxiolytic effects of ethanol appear to be mediated, at least in part, via blockade of GluK1Rs in the BLA (Läck et al., 2008), and the anticonvulsant properties of the well-known antiepileptic topiramate also involve blockade of GluK1Rs in the BLA (Braga et al., 2009; Gryder and Rogawski, 2003; Kaminski et al., 2004).
Other studies, however, contradict a hyperexcitability-promoting role of GluK1Rs in the BLA. Thus, injection of a GluK1R antagonist into the BLA of wild-type mice increased anxiety, and intraperitoneal injection of ATPA produced an anxiolytic effect in the elevated plus maze test; in addition, GluK1-knockout mice display anxiety-like behavior (Wu et al., 2007a). Consistent with these findings, stress/anxiety-induced hyperthermia in mice was not decreased by a GluK1R antagonist (Rorick-Kehn et al., 2005). The discrepant results between rats and mice may imply a species-difference in the function of GluK1Rs. Indeed, in the rat BLA, ATPA increases both sIPSCs (Braga et al., 3002) and sEPSCs (present study), while in the mice BLA, ATPA increases sIPSCs, but not sEPSCs (Wu et al., 2007a). Similarly, GluK1R-mediated currents can be evoked on BLA principal cells by synaptic stimulation (Gryder and Rogawski, 2003), or by puff-application of ATPA (Läck et al., 2008), while this is not the case in mice where such currents can be evoked in BLA interneurons, but they are of very low amplitude or absent in principal neurons (Wu et al., 2007a).
4.3. The effects of GluK1R blockade on the ASR and PPI
The amygdala plays an important role in the modulation of the ASR, and the amplitude of ASR correlates positively with the degree of amygdalar excitation (Hitchcock and Davis, 1987; Li et al., 1999). The amygdala also modulates PPI, which is produced if a weaker stimulus precedes the strong stimulus that evokes the ASR (Hoffman and Ison 1980; Swerdlow et al. 2001; Li et al. 2009). The results from the use of the 68 dB prepulse preceding the 120 dB noise burst suggest that UBP302 neither enhances nor interferes with PPI. We can only speculate on the mechanism by which UBP302 reduced the ASR (and, by inference, the excitation of the amygdala) when the strong stimulus arrived as a single pulse, but not significantly so – compared to the corresponding vehicle conditions – when the strong stimulus came after the prepulse. The mechanism may involve reduction in the probability of glutamate release by UBP302. Thus, in the presence of UBP302, the reduced glutamate release in the BLA in response to the prepulse, results in more glutamate release in response to the second, strong stimulus, which reduces the difference in the ASR amplitude from the vehicle conditions; this would be analogous to the observations in the field potential experiments, where UBP302 did not reduce significantly the response to Pulse 2, probably due to reduction of glutamate release in the response to Pulse1 (which results in more glutamate release by Pulse 2).
If the purpose of the PPI is to protect the processing of the prepulse stimulus from the disrupting effect of the startle stimulus (Graham, 1975), or if – again, teleologically speaking – the focus is on the startle response, and the purpose of the PPI is to prepare the organism for a more “controlled” ASR, then it appears that blockade of GluK1Rs neither facilitates nor interferes with these processes, but it does help control (reduce) the ASR when a pre-stimulus is not present.
4.5. Conclusions
The present study demonstrated the anxiogenic and seizurogenic role of GluK1Rs in the rat BLA, and identified one of the mechanisms that can underlie this function, namely the presynaptic facilitation of glutamate release The data presented lend further support to the emerging evidence that GluK1R antagonists offer promise for the treatment of anxiety and seizure disorders, particularly considering the excellent tolerability of these compounds (Sang et al. 1998, 2004).
GluK1 receptors facilitate presynaptic glutamate release in the basolateral amygdala
Activation of GluK1 receptors in the basolateral amygdala increases anxiety
Blockade of GluK1 receptors in the basolateral amygdala reduces anxiety
Acknowledgments
We thank Dr. Neil E. Grunberg, Erin Barry, and Daniel Stevens for assistance with the behavioral experiments. This study is part of the research supported by the CounterACT Program, National Institutes of Health, Office of the Director, through the National Institute of Neurological Disorders and Stroke (award # 5U01NS058162-06), and the Defense Threat Reduction Agency-Joint Science and Technology Office, Medical S&T Division (grants # CBM.NEURO.01.10.US.15 and CBM.NEURO.01.10.US.18).
Abbreviations
- BLA
basolateral nucleus of the amygdala
- GluK1Rs
kainate receptors containing the GluK1 subunit
- sEPSCs
spontaneous excitatory post-synaptic currents
- mEPSCs
miniature excitatory post-synaptic currents
- ATPA
(RS)-2-amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl) propanoic acid
- UBP(302)
(S)-1-(2-amino-2-carboxyethyl)-3-(2-carboxybenzyl) pyrimidine-2,4-dione
- ACSF
artificial cerebrospinal fluid
- ASR
acoustic startle response
- PPI
pre-pulse inhibition
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alt A, Weiss B, Ogden AM, Li X, Gleason SD, Calligaro DO, Bleakman D, Witkin JM. In vitro and in vivo studies in rats with LY293558 suggest AMPA/kainate receptor blockade as a novel potential mechanism for the therapeutic treatment of anxiety disorders. Psychopharmacology (Berl) 2006;185:240–247. doi: 10.1007/s00213-005-0292-0. [DOI] [PubMed] [Google Scholar]
- Alt A, Weiss B, Ornstein PL, Gleason SD, Bleakman D, Stratford RE, Jr, Witkin JM. Anxiolytic-like effects through a GLUK5 kainate receptor mechanism. Neuropharmacology. 2007;52:1482–1487. doi: 10.1016/j.neuropharm.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Apland JP, Aroniadou-Anderjaska V, Braga MF. Soman induces ictogenesis in the amygdala and interictal activity in the hippocampus that are blocked by a GluR5 kainate receptor antagonist in vitro. Neuroscience. 2009;159:380–389. doi: 10.1016/j.neuroscience.2008.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroniadou-Anderjaska V, Qashu F, Maria Braga MF. Mechanisms Regulating GABAergic Inhibitory Transmission in the Basolateral Amygdala: Implications for Epilepsy and Anxiety Disorders. Amino Acids. 2007;32:305–15. doi: 10.1007/s00726-006-0415-x. [DOI] [PubMed] [Google Scholar]
- Aroniadou-Anderjaska V, Fritsch B, Qashu F, Braga MF. Pathology and pathophysiology of the amygdala in epileptogenesis and epilepsy. Epilepsy Res. 2008;78:102–116. doi: 10.1016/j.eplepsyres.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettler B, Boulter J, Hermans-Borgmeyer I, O’Shea-Greenfield A, Deneris ES, Moll C, Borgmeyer U, Hollmann M, Heinemann S. Cloning of a novel glutamate receptor subunit, GluR5: expression in the nervous system during development. Neuron. 1990;5:583–595. doi: 10.1016/0896-6273(90)90213-y. [DOI] [PubMed] [Google Scholar]
- Braga MF, Aroniadou-Anderjaska V, Xie J, Li H. Bidirectional modulation of GABA release by presynaptic glutamate receptor 5 kainate receptors in the basolateral amygdala. J Neurosci. 2003;23:442–452. doi: 10.1523/JNEUROSCI.23-02-00442.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga MF, Aroniadou-Anderjaska V, Li H, Rogawski MA. Topiramate reduces excitability in the basolateral amygdala by selectively inhibiting GluK1 (GluR5) kainate receptors on interneurons and positively modulating GABAA receptors on principal neurons. J Pharmacol Exp Ther. 2009;330:558–566. doi: 10.1124/jpet.109.153908. [DOI] [PubMed] [Google Scholar]
- Campbell SL, Mathew SS, Hablitz JJ. Pre- and postsynaptic effects of kainate on layer II/III pyramidal cells in rat neocortex. Neuropharmacology. 2007;53:37–47. doi: 10.1016/j.neuropharm.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain SE, Jane DE, Jones RS. Pre- and post-synaptic functions of kainate receptors at glutamate and GABA synapses in the rat entorhinal cortex. Hippocampus. 2012;22:555–576. doi: 10.1002/hipo.20921. [DOI] [PubMed] [Google Scholar]
- Clarke VR, Ballyk BA, Hoo KH, Mandelzys A, Pellizzari A, Bath CP, Thomas J, Sharpe EF, Davies CH, Ornstein PL, Schoepp DD, Kamboj RK, Collingridge GL, Lodge D, Bleakman D. A hippocampal GluR5 kainate receptor regulating inhibitory synaptic transmission. Nature. 1997;389:599–603. doi: 10.1038/39315. [DOI] [PubMed] [Google Scholar]
- Clarke VR, Collingridge GL. Characterisation of the effects of ATPA, a GLU(K5) receptor selective agonist, on excitatory synaptic transmission in area CA1 of rat hippocampal slices. Neuropharmacology. 2002;42:889–902. doi: 10.1016/s0028-3908(02)00039-4. [DOI] [PubMed] [Google Scholar]
- Clarke VR. Characterisation of the effects of ATPA, a GLU(K5) kainate receptor agonist, on GABAergic synaptic transmission in the CA1 region of rat hippocampal slices. Neuropharmacology. 2004;47:363–372. doi: 10.1016/j.neuropharm.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Olsen RW, Peters J, Spedding M. A nomenclature for ligand-gated ion channels. Neuropharmacology. 2009;56:2–5. doi: 10.1016/j.neuropharm.2008.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart R, Esclapez M, Hirsch JC, Bernard C, Ben Ari Y. GluR5 kainate receptor activation in interneurons increases tonic inhibition of pyramidal cells. Nat Neurosci. 1998;1:470–478. doi: 10.1038/2185. [DOI] [PubMed] [Google Scholar]
- Davidson RJ, Abercrombie H, Nitschke JB, Putnam K. Regional brain function, emotion and disorders of emotion. Curr Opin Neurobiol. 1999;9:228–234. doi: 10.1016/s0959-4388(99)80032-4. [DOI] [PubMed] [Google Scholar]
- Davis M, Rainnie D, Cassell M. Neurotransmission in the rat amygdala related to fear and anxiety. Trends Neurosci. 1994;17:208–214. doi: 10.1016/0166-2236(94)90106-6. [DOI] [PubMed] [Google Scholar]
- Debanne D, Guérineau NC, Gähwiler BH, Thompson SM. Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: quantal fluctuation affects subsequent release. J Physiol. 1996;491:163–176. doi: 10.1113/jphysiol.1996.sp021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etkin A, Wager TD. Functional neuroimaging of anxiety: a meta-analysis of emotional processing in PTSD, social anxiety disorder, and specific phobia. Am J Psychiatry. 2007;164:1476–1488. doi: 10.1176/appi.ajp.2007.07030504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraday MM, Elliott BM, Grunberg NE. Adult vs. adolescent rats differ in biobehavioral responses to chronic nicotine administration. Pharmacol Biochem Behav. 2001;70:475–89. doi: 10.1016/s0091-3057(01)00642-6. [DOI] [PubMed] [Google Scholar]
- Figueiredo TH, Qashu F, Apland JP, Aroniadou-Anderjaska V, Souza AP, Braga MF. The GluR5 kainate /AMPA receptor antagonist LY293558 reduces soman-induced seizures and neuropathology. J Pharmacol Exp Ther. 2011;336:303–312. doi: 10.1124/jpet.110.171835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisahn A, Contractor A, Traub RD, Buhl EH, Heinemann SF, McBain CJ. Distinct roles for the kainate receptor subunits GluR5 and GluR6 in kainate-induced hippocampal gamma oscillations. J Neurosci. 2004;24:9658–9668. doi: 10.1523/JNEUROSCI.2973-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch B, Qashu F, Figueiredo TH, Aroniadou-Anderjaska V, Rogawski MA, Braga MF. Pathological alterations in GABAergic interneurons and reduced tonic inhibition in the basolateral amygdala during epileptogenesis. Neuroscience. 2009;163:415–429. doi: 10.1016/j.neuroscience.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett A, Chang K. The role of the amygdala in bipolar disorder development. Dev Psychopathol. 2008;20:1285–1296. doi: 10.1017/S0954579408000618. [DOI] [PubMed] [Google Scholar]
- Graham FK. The more or less startling effects of weak prestimulation. Psychophysiology. 1975;12:238–248. doi: 10.1111/j.1469-8986.1975.tb01284.x. [DOI] [PubMed] [Google Scholar]
- Gryder DS, Rogawski MA. Selective antagonism of GluR5 kainate receptor-mediated synaptic currents by topiramate in rat basolateral amygdala neurons. J Neurosci. 2003;23:7069–7074. doi: 10.1523/JNEUROSCI.23-18-07069.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handforth A, Ackermann RF. Mapping of limbic seizure progressions utilizing the electrogenic status epilepticus model and the 14C-2-deoxyglucose method. Brain Res Brain Res Rev. 1995;20:1–23. doi: 10.1016/0165-0173(94)00003-8. [DOI] [PubMed] [Google Scholar]
- Hitchcock JM, Davis M. Fear-potentiated startle using an auditory conditioned stimulus: effect of lesions of the amygdala. Physiol Behav. 1987;39:403–408. doi: 10.1016/0031-9384(87)90242-3. [DOI] [PubMed] [Google Scholar]
- Hoffman HS, Ison JR. Reflex modification in the domain of startle: I. Some empirical findings and their implications for how the nervous system processes sensory input. Psychol Rev. 1980;87:175–189. [PubMed] [Google Scholar]
- Jane DE, Lodge D, Collingridge GL. Kainate receptors: pharmacology, function and therapeutic potential. Neuropharmacology. 2009;56:90–113. doi: 10.1016/j.neuropharm.2008.08.023. [DOI] [PubMed] [Google Scholar]
- Kaminski RM, Banerjee M, Rogawski MA. Topiramate selectively protects against seizures induced by ATPA, a GluR5 kainate receptor agonist. Neuropharmacology. 2004;46:1097–1104. doi: 10.1016/j.neuropharm.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Khalilov I, Hirsch J, Cossart R, Ben-Ari Y. Paradoxical anti-epileptic effects of a GluR5 agonist of kainate receptors. J Neurophysiol. 2002;88:523–527. doi: 10.1152/jn.2002.88.1.523. [DOI] [PubMed] [Google Scholar]
- Kotlinska J, Liljequist S. The putative AMPA receptor antagonist, LY326325, produces anxiolytic-like effects without altering locomotor activity in rats. Pharmacol Biochem Behav. 1998;60:119–124. doi: 10.1016/s0091-3057(97)00565-0. [DOI] [PubMed] [Google Scholar]
- Kroner S, Rosenkranz JA, Grace AA, Barrionuevo G. Dopamine modulates excitability of basolateral amygdala neurons in vitro. J Neurophysiol. 2005;93:1598–1610. doi: 10.1152/jn.00843.2004. [DOI] [PubMed] [Google Scholar]
- Läck AK, Diaz MR, Chappell A, Dubois DW, McCool BA. Chronic ethanol and withdrawal differentially modulate pre- and post-synaptic function at glutamatergic synapses in rat basolateral amygdala. J Neurophysiol. 2007;98:3185–3196. doi: 10.1152/jn.00189.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Läck AK, Ariwodola OJ, Chappell AM, Weiner JL, McCool BA. Ethanol inhibition of kainate receptor-mediated excitatory neurotransmission in the rat basolateral nucleus of the amygdala. Neuropharmacology. 2008;55:661–668. doi: 10.1016/j.neuropharm.2008.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Chen A, Xing G, Wei ML, Rogawski MA. Kainate receptor-mediated heterosynaptic facilitation in the amygdala. Nat Neurosci. 2001;4:612–620. doi: 10.1038/88432. [DOI] [PubMed] [Google Scholar]
- Li H, Rogawski MA. GluR5 kainate receptor mediated synaptic transmission in rat basolateral amygdala in vitro. Neuropharmacology. 1998;37:1279–1286. doi: 10.1016/s0028-3908(98)00109-9. [DOI] [PubMed] [Google Scholar]
- Li L, Fulton JD, Yeomans JS. Effects of bilateral electrical stimulation of the ventral pallidum on acoustic startle. Brain Res. 1999;836:164–172. doi: 10.1016/s0006-8993(99)01651-0. [DOI] [PubMed] [Google Scholar]
- Li L, Du Y, Li N, Wu X, Wu Y. Top-down modulation of prepulse inhibition of the startle reflex in humans and rats. Neurosci Biobehav Rev. 2009;33:1157–1167. doi: 10.1016/j.neubiorev.2009.02.001. [DOI] [PubMed] [Google Scholar]
- McCool BA, Chappell A. Strychnine and taurine modulation of amygdala-associated anxiety-like behavior is state-dependent. Behav Brain Res. 2007;178:70–81. doi: 10.1016/j.bbr.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menard J, Treit D. Effects of centrally administered anxiolytic compounds in animal models of anxiety. Neurosci Biobehav Rev. 1999;23:591–613. doi: 10.1016/s0149-7634(98)00056-6. [DOI] [PubMed] [Google Scholar]
- Miyata M, Imoto K. Contrary roles of kainate receptors in transmitter release at corticothalamic synapses onto thalamic relay and reticular neurons. J Physiol. 2009;587:999–1012. doi: 10.1113/jphysiol.2008.164996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapel P, Dufresne C, Kelly ME, McIntyre DC. Differential sensitivity of various temporal lobe structures in the rat to kindling and status epilepticus induction. Epilepsy Res. 1996;23:179–187. doi: 10.1016/0920-1211(95)00084-4. [DOI] [PubMed] [Google Scholar]
- More JC, Nistico R, Dolman NP, Clarke VR, Alt AJ, Ogden AM, Buelens FP, Troop HM, Kelland EE, Pilato F, Bleakman D, Bortolotto ZA, Collingridge GL, Jane DE. Characterisation of UBP296: a novel, potent and selective kainate receptor antagonist. Neuropharmacology. 2004;47:46–64. doi: 10.1016/j.neuropharm.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Mulle C, Sailer A, Swanson GT, Brana C, O’Gorman S, Bettler B, Heinemann SF. Subunit composition of kainate receptors in hippocampal interneurons. Neuron. 2000;28:475–484. doi: 10.1016/s0896-6273(00)00126-4. [DOI] [PubMed] [Google Scholar]
- Park K, Lee S, Kang SJ, Choi S, Shin KS. Hyperpolarization-activated currents control the excitability of principal neurons in the basolateral amygdala. Biochem Biophys Res Commun. 2007;361:718–24. doi: 10.1016/j.bbrc.2007.07.064. [DOI] [PubMed] [Google Scholar]
- Partovi D, Frerking M. Presynaptic inhibition by kainate receptors converges mechanistically with presynaptic inhibition by adenosine and GABAB receptors. Neuropharmacology. 2006;51:1030–1037. doi: 10.1016/j.neuropharm.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; London: 1997. [Google Scholar]
- Pitkanen A, Tuunanen J, Kalviainen R, Partanen K, Salmenpera T. Amygdala damage in experimental and human temporal lobe epilepsy. Epilepsy Res. 1998;32:233–253. doi: 10.1016/s0920-1211(98)00055-2. [DOI] [PubMed] [Google Scholar]
- Prut L, Belzung C. The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: a review. Eur J Pharmacol. 2003;463:3–33. doi: 10.1016/s0014-2999(03)01272-x. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation: II. Motor Seizure. Electroencephalography and Clinical Neurophysiology. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Racine RJ, Rose PA, Burnham WM. Afterdischarge thresholds and kindling rates in dorsal and ventral hippocampus and dentate gyrus. Can J Neurol Sci. 1977;4:273–278. doi: 10.1017/s0317167100025117. [DOI] [PubMed] [Google Scholar]
- Rainnie DG, Asprodini EK, Shinnick-Gallagher P. Intracellular recordings from morphologically identified neurons of the basolateral amygdala. J Neurophysiol. 1993;69:1350–1362. doi: 10.1152/jn.1993.69.4.1350. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Moreno A, Lopez-Garcia JC, Lerma J. Two populations of kainate receptors with separate signaling mechanisms in hippocampal interneurons. Proc Natl Acad Sci USA. 2000;97:1293–1298. doi: 10.1073/pnas.97.3.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorick-Kehn LM, Hart JC, McKinzie DL. Pharmacological characterization of stress-induced hyperthermia in DBA/2 mice using metabotropic and ionotropic glutamate receptor ligands. Psychopharmacology (Berl) 2005;183:226–240. doi: 10.1007/s00213-005-0169-2. [DOI] [PubMed] [Google Scholar]
- Sanders SK, Shekhar A. Regulation of anxiety by GABAA receptors in the rat amygdala. Pharmacol Biochem Behav. 1995;52:701–706. doi: 10.1016/0091-3057(95)00153-n. [DOI] [PubMed] [Google Scholar]
- Sang CN, Hostetter MP, Gracely RH, Chappell AS, Schoepp DD, Lee G, Whitcup S, Caruso R, Max MB. AMPA/kainateantagonist LY293558 reduces capsaicin-evoked hyperalgesiabut not pain in normal skin in humans. Anesthesiology. 1998;89:1060–1067. doi: 10.1097/00000542-199811000-00005. [DOI] [PubMed] [Google Scholar]
- Sang CN, Ramadan NM, Wallihan RG, Chappell AS, Freitag FG, Smith TR, Silberstein SD, Johnson KW, Phebus LA, Bleakman D, Ornstein PL, Arnold B, Tepper SJ, Vandenhende F. LY293558, a novel AMPA/GluR5 antagonist, is efficacious and well-tolerated in acute migraine. Cephalalgia. 2004;24:596–602. doi: 10.1111/j.1468-2982.2004.00723.x. [DOI] [PubMed] [Google Scholar]
- Smolders I, Bortolotto ZA, Clarke VR, Warre R, Khan GM, O’Neill MJ, Ornstein PL, Bleakman D, Ogden A, Weiss B, Stables JP, Ho KH, Ebinger G, Collingridge GL, Lodge D, Michotte Y. Antagonists of GLU(K5)-containing kainate receptors prevent pilocarpine-induced limbic seizures. Nat Neurosci. 2002;5:796–804. doi: 10.1038/nn880. [DOI] [PubMed] [Google Scholar]
- Stein MB, Stein DJ. Social anxiety disorder. Lancet. 2008;371:1115–1125. doi: 10.1016/S0140-6736(08)60488-2. [DOI] [PubMed] [Google Scholar]
- Sun HY, Bartley AF, Dobrunz LE. Calcium-permeable presynaptic kainate receptors involved in excitatory short-term facilitation onto somatostatin interneurons during natural stimulus patterns. J Neurophysiol. 2009;101:1043–1055. doi: 10.1152/jn.90286.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA, Braff DL. Neural circuit regulation of prepulse inhibition of startle in the rat, current knowledge and future challenges. Psychopharmacology. 2001;156:194–215. doi: 10.1007/s002130100799. [DOI] [PubMed] [Google Scholar]
- Thomson AM. Facilitation, augmentation and potentiation at central synapses. Trends Neurosci. 2000;23:305–312. doi: 10.1016/s0166-2236(00)01580-0. [DOI] [PubMed] [Google Scholar]
- Vignes M, Clarke VRJ, Parry MC, Bleakman D, Lodge D, Ornstein PL, Collingridge GL. The GluR5 subtype of kainate receptor regulates excitatory synaptic transmission in areas CA1 and CA3 of the rat hippocampus. Neuropharmacology. 1998;37:1269–1277. doi: 10.1016/s0028-3908(98)00148-8. [DOI] [PubMed] [Google Scholar]
- Washburn MS, Moises HC. Electrophysiological and morphological properties of rat basolateralamygdaloid neurons in vitro. J Neurosci. 1992;12:4066–4079. doi: 10.1523/JNEUROSCI.12-10-04066.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White LE, Price JL. The functional anatomy of limbic status epilepticus in the rat. I. Patterns of 14C-2-deoxyglucose uptake and Fos immunocytochemistry. J Neurosci. 1993a;13:4787–4809. doi: 10.1523/JNEUROSCI.13-11-04787.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White LE, Price JL. The functional anatomy of limbic status epilepticus in the rat. II. The effects of focal deactivation. J Neurosci. 1993b;13:4810–4830. doi: 10.1523/JNEUROSCI.13-11-04810.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womble MD, Moises HC. Hyperpolarization-activated currents in neurons of the rat basolateral amygdala. J Neurophysiol. 1993;70:2056–2065. doi: 10.1152/jn.1993.70.5.2056. [DOI] [PubMed] [Google Scholar]
- Wondolowski J, Frerking M. Subunit-dependent postsynaptic expression of kainate receptors on hippocampal interneurons in area CA1. J Neurosci. 2009;29:563–574. doi: 10.1523/JNEUROSCI.4788-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LJ, Ko SW, Toyoda H, Zhao MG, Xu H, Vadakkan KI, Ren M, Knifed E, Shum F, Quan J, Zhang XH, Zhuo M. Increased anxiety-like behavior and enhanced synaptic efficacy in the amygdala of GluR5 knockout mice. PLoS One. 2007a;2:e167. doi: 10.1371/journal.pone.0000167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LJ, Xu H, Ren M, Zhuo M. Genetic and pharmacological studies of GluR5 modulation of inhibitory synaptic transmission in the anterior cingulate cortex of adult mice. Dev Neurobiol. 2007b;67:146–157. doi: 10.1002/dneu.20331. [DOI] [PubMed] [Google Scholar]