

Abstract
Epidemiological studies have demonstrated that the consumption of fruits and vegetables is associated with reduced risk for cardiovascular disease and stroke. Detailed investigations into the specific dietary components of these foods have revealed that many polyphenolic constituents exert anti-oxidant effects on key substrates involved in the pathogenesis and progression of ischemic injury. These data have perpetuated the belief that the protective effects of flavonoids result from direct anti-oxidant actions at the levels of the cerebral vasculature and brain parenchyma. While many in vitro studies using purified extracts support this contention, first-pass metabolism alters the bioavailability of flavonoids such that the achievable concentrations after oral consumption are not consistent with this mechanism. Importantly, oral consumption of flavonoids may promote neural protection by facilitating the expression of gene products responsible for detoxifying the ischemic microenvironment through both anti-oxidative and anti-inflammatory actions. In particular, the transcriptional factor nuclear factor erythroid 2-related factor 2 has emerged as a critical regulator of flavonoid-mediated protection through the induction of various cytoprotective genes. The pleiotropic effects associated with potent transcriptional regulation likely represent the primary mechanisms of neural protection, as the flavonoid concentrations reaching ischemic tissues in vivo are sufficient to alter intracellular signal transduction but likely preclude the one-to-one stoichiometry necessary to confer protection by direct anti-oxidation. These data reflect an exciting new direction in the study of complementary and alternative medicine that may lead to the development of novel therapies for ischemic/hemorrhagic stroke, traumatic brain injury, and other neurological disorders.
Keywords: Ischemia, Nuclear factor (erythroid-derived 2)-like 2, Polyphenol, Stroke, Therapy, Complementary and alternative medicine
Introduction
The beneficial effects of fruits and vegetables are well recognized and have been widely attributed to poly-phenol constituents. Much research has been devoted to uncovering the mechanisms by which flavonoids confer beneficial effects on human health, particularly within the context of cancer and vascular disease.
Over the past two decades, epidemiological studies have linked the consumption of flavonoid-containing dietary sources to reduced risk of vascular/ischemic events. These data have been extensively reviewed and will not be detailed here. To briefly summarize these data, many studies showed that flavonoids promote vascular function, reduce hypertension,1 and lower the risk for cardiovascular disease and stroke.2,3 Although encouraging, these data have not aided in establishing causality or defining mechanism of action.
Understanding the mechanisms responsible for these health benefits is essential in developing novel approaches not only to mitigate the risk for stroke, but also to identify targets for therapies that can be applied after the insult. While studies using culture systems have established that flavonoids exert direct anti-oxidant actions, these effects have not been demonstrated in vivo after oral consumption. Importantly, it has become evident that in addition to exerting direct anti-oxidant effects, flavonoids induce nuclear translocation of Nrf2 (nuclear factor erythroid 2-related factor 2), a redox-sensitive transcription factor that facilitates the induction of Phase II detoxifying enzymes and other cytoprotective proteins through binding to anti-oxidant response elements (AREs).4 While this effect was first demonstrated in vitro, numerous studies have now verifiedthis mechanism as a target for stroke therapy through the use of experimental rodent stroke models.5
These data are provocative for several reasons. Identification of this alternative protective mechanism in vivo may allow for the determination of which flavonoids have the greatest potential as therapies to combat stroke-induced injury. Additionally, establishing a link between the protective effects of flavonoids and alterations in gene expression allows for further investigations of downstream targets that can be exploited in the development of novel therapeutics. Other possibilities include the synthesis of flavonoid analogs with increased bioavailability, or distinct synthetic agents that mimic the actions of flavonoids, which can be consumed either prophylactically by a high-risk population or possibly after stroke onset. This review summarizes the data obtained in preclinical injury models characterized by ischemic/hemorrhagic stroke and other insults resulting in similar neuropathology. Data from these studies offer a glimpse into promising avenues for future studies aimed at reducing neural injury through both prophylactic and delayed treatment regimens.
Can direct anti-oxidation account for the beneficial effects of dietary flavonoids?
Polyphenols have been identified as likely candidates in promoting the protective effects attributed to a healthy diet. Of the polyphenols, the most abundant in the human diet are the flavonoids. This general class of molecules includes the flavonols, flavanols, flavanones, flavones, anthocyanidins, isoflavones, and dihydroflavonols, as well as chalcones,6 which have been categorized based on the distinct chemical moieties linked to the basic flavonoid structure. In general, flavonoids are present in high concentrations in citrus fruits, apples, berries, grapes, red wine, olive oil, tea, chocolate, and cocoa. However, each of these dietary sources contains a unique flavonoid signature that is distinct in concentration and composition. Despite evidence that flavonoid-containing dietary sources are associated with a reduced risk of cardiovascular disease and stroke, identifying the mechanisms responsible has proven difficult mainly due to inherent caveats concerning the interpretation of epidemiological data, the considerable number of flavonoids identified to date, the variance of flavonoid distribution across dietary sources, and the manner in which these molecules are metabolized.1,2 For example, differences in the selection of flavonoid-containing foods make comparisons across studies difficult. Additionally, oral consumption subjects these molecules to first-pass metabolism, and consequently, gastrointestinal, bacterial, and hepatic enzymes catalyze structural alterations prior to systemic distribution. Several processes including sulfation, methylation, and glucuronidation occur within the small intestine and liver, forming sulfate and glucuronide conjugates from the original flavonoids.7 It is also noteworthy that the absorption profile of the procyanidins (polymeric flavanols) differs based upon size and composition, as monomeric and dimeric catechin polyphenols are readily absorbed8 while procyanidin oligomers must be hydrolyzed to monomers or dimers prior to absorption.9 Detailed investigations into the bioavailability of polyphenols10-12 highlight these complexities, and studies with rats preclinical animal models have further confirmed the importance of structure and formulation in achieving adequate absorption and bioavailability to neural tissues after stroke.13,14
For these reasons and others, questions remain as to the proportion of native flavonoids that reach the brain and whether the first-pass metabolites exert actions that also mediate cell viability. The prevailing notion that flavonoids confer protection through direct anti-oxidation arose from numerous in vitro biochemical experiments.15-17 Although these actions have also been demonstrated in cell culture systems, it is noteworthy that the stoichiometry for anti-oxidant quenching of pro-oxidative enzyme activity is one to one. Therefore, while direct anti-oxidation appears to be a mechanism of in vitro protection, it is unlikely that the bioavailability of orally consumed flavonoids is great enough to afford direct anti-oxidant rescue of neural cells from the degree of oxidation present.18 Furthermore, these actions would not target the delayed inflammatory component of infarct expansion that is characteristic of cerebral ischemia.
Despite these shortcomings, adopting more stringent criteria for the evaluation of compounds should provide insights into potential efficacy for therapeutic application in humans and mechanisms of action in vivo. For example, Schroeter et al.19 have developed criteria that must be met when assessing vascular effects in epidemiological studies, and through this method, data demonstrated that the flavanol (−)-epicatechin (EC) may promote vascular function through increased nitric oxide production. The necessary criteria proposed in this study included demonstrated absorption in humans, bioavailability in target tissues, measurement of circulating levels of the compound and its metabolites, pharmacokinetic, and pharmacodynamic assessments, reversal of effects using experimental strategies such as inhibitors of proposed mechanisms, and reversal of effects by withdrawing or withholding the compound.19
As vascular permeability is linked to several pathological cascades after ischemia, targeting vascular function through preventative or delayed therapeutic flavanol consumption is promising. Likewise, the ability of polyphenolic compounds to exert pleiotropic effects on cell viability through other substrates and mechanisms must be investigated. Importantly, Nrf2-mediated gene expression may represent one such mechanism.
The multiplicity of Nrf2 protection within the context of stroke
The neural response to stroke involves various pathological processes including the production of free oxygen and nitrogen radicals, excitotoxic injury,20 activation of matrix metalloproteinases (MMPs),21,22 entry of peripheral immune cells into the brain parenchyma,23-25 and upregulation of pro-inflammatory prostaglandins, cytokines, and chemokines,26 all of which contribute to expansion of the core infarct.27 Similar mechanisms contribute to intracerebral hemorrhage, traumatic brain injury, and other debilitating neuropathies characterized by cerebral ischemia. The clinical failures of various single-target therapeutic agents reflect the complexity of the neural sequelae and highlight the need for novel therapies that target multiple mechanisms.
As such, Nrf2 has emerged as an attractive target for the treatment of stroke in that this transcription factor can exert downstream effects on both acute injury mechanisms, such as cytotoxicity resulting from free oxygen and nitrogen radicals, as well as delayed inflammatory signaling. Indeed, Nrf2 induces the expression of genes that provide cellular detoxification through the glutathione (glutathione (GSH), glutathione S-transferase (GST), glutamate cysteine ligase catalytic, and modulatory subunits (Gclc, Gclm)) and thioredoxin (thioredoxin reductase (TrxR, peroxiredoxin 1 (Prx1)) systems, facilitation of NAD(P)H production (required electron donor for these enzymes), catabolism of pro-oxidant heme (heme oxygenase 1 (HO1)), free iron, and copper/zinc sequestration (ferritin light chain 1, metallothionein 1 (MT1)), protection from superoxide radicals (superoxide dismutase 1 (SOD1)), and prevention of aberrant intracellular protein accumulation (heat shock protein 40) and proteasomal subunits (19S, 20S).28-31 Therefore, the induction of Phase II detoxifying enzymes, as well as other protective proteins that contain ARE-driven promoters, may provide an avenue to target multiple cellular processes required for protection from stroke-induced oxidative stress and similar insults (Fig. 1).
Figure 1.

Schematic representation depicting some of the various cytoprotective proteins that are upregulated by Nrf2. Flavonoid-mediated protection from ischemic/hemorrhagic stroke, traumatic brain injury, and/or other neuropathies may result in large part from Nrf2 regulation of these pathways.
In addition to these mechanisms, Nrf2 appears to be an important regulator of the peripheral inflammatory responses and thus may also contribute to the neuroinflammatory component of ischemic brain injury. Studies using Nrf2 deficient mice have demonstrated that this transcription factor regulates immune cell responsiveness, prostanoids, cytokine, and chemokine expression in response to various insults. For example, mice deficient in Nrf2 showed upregulated gene expression of pro-inflammatory cytokines, interferon-inducible transcripts, and nuclear factor kappa B (NFκB)-binding activity in peritoneal macrophages after administration of bacterial endotoxin or lipopoly-saccharide,32 while ovalbumin sensitization and challenge increased the severity of airway inflammation and increased cytokine production by splenocytes and Th2 lymphocytes in the same knockout strain.33 In a separate study of lipopolysaccharide-induced acute ocular inflammation, Nrf2−/− mice showed increased mRNA transcripts for intercellular adhesion molecule-1 and several pro-inflammatory mediators that occurred concomitantly with increased leukocyte adherence to the retinal vascular endothelium.34 As multiple mechanisms mediate the neural response to injury, future studies investigating the various cellular and molecular contributors to immune cell signaling and vascular permeability of the various cell layers of the blood-brain barrier (BBB) will likely uncover novel targets for therapeutic intervention.5
Despite the current lack of data defining specific mechanism, experimental in vivo models have provided some insights regarding the downstream effects of Nrf2-mediated alterations in gene transcription that likely mediate the neuroprotective effects of flavonoid administration. The text that follows provides a summary of the major findings thus far concerning the protective effects of flavonoids and their association with Nrf2-associated signaling. Taken together, these data suggest pleiotropic mechanisms by which natural polyphenolic compounds protect neural cells from the deleterious effects of stroke, thus extending the therapeutic potential of polyphenols beyond direct anti-oxidation.
Flavonoids, oxidation, and cell death
The field of stroke research has recognized the potential for polyphenols, particularly flavonoids, in treating ischemic/reperfusion-associated neurodegeneration. Among the animal models employed, middle cerebral artery occlusion (MCAO) is perhaps the most commonly utilized to mimic both transient and permanent embolic stroke. Although there are many differences across studies and laboratories, this general approach has aided in the identification of several downstream mechanisms by which flavonoids may confer protection (Table 1).
Table 1. Flavonoid/stilbene administration in rodent stroke models: comparison of treatment regimens, outcomes, and suggested mechanisms.
| Class | Model | Species | Extract | Dose/Route | Infarct | Function | Mechanism | Ref. |
|---|---|---|---|---|---|---|---|---|
| Flavanols | tMCAO (90 minutes) |
C57 mouse (m): WT, Nrf2−/−, HO1−/− |
EC | Dose response (2.5, 5, 15, 30 mg/kg); 90 minutes pre; 3.5 or 6 hours post; p.o. |
+WT pre (5, 15, 30 mg/ kg) and post (30 mg/kg); UC Nrf2−/−; UC HO1−/− |
+WT NDS pre and post; UC Nrf2−/−; UC HO1−/− |
Nrf2 and HO1 required for protection |
35 |
| BCCAO (10 minutes) |
ddY mouse (m) | EC or CA |
100 mg/kg ~5 minutes pre; s.c. ori.v. |
UC necrosis of cortex or hippocampus |
+Retention of passive avoidance (EC and CA); UC locomotor activity |
NA | 36 | |
| tMCAO (80 minutes) |
C57 mouse (m) | EGCG | 50 mg/kg immediately post; i.p. |
+Total volume | N/A | Reduced MMP-9 activity | 37 | |
| tMCAO (30, 60, or 90 minutes) |
Gerbil (m) | EGCG | Dose response (25 or 50 mg/kg); 30 minutes pre and immediately post; i.p. |
+50 mg/kg total volume (60 minutes tMCAO) |
N/A | Reduced lipid peroxidation (MDA) and BBB degradation (water content) |
38 | |
| BCCAO (3 minutes) |
Gerbil (m) | EGCG | Dose response (10, 25, or 50 mg/kg) immediately post; i.p. |
+CA1 hippocampal neurons (25 and 50 mg/kg) |
N/A | N/A | 39 | |
| tMCAO (2 hours) |
SD rat (m) | EGCG | 50 mg/kg immediately post; i.p. |
+Cortex; UC total and striatum |
+Sticky tape 10 days post but UC 1, 5, 14 days; UC hindlimb weight bearing |
N/A | 40 | |
| BCCAO (5 minutes) |
Gerbil (f) | GTE | Dose response (0.5, 2%); 3 weeks pre; oral ad libitum |
+Total and cortex(0.5 and 2%); +striatum (2%) |
+Locomotor activity (blocked increase) |
Reduced H2O2, lipid peroxidation, and apoptotic cells (TUNEL) (0.5 and 2%) |
41 | |
| tMCAO (2 hours) |
SD rat (m) | TOFs | Dose response (100 or 200 mg/kg 4 days pre and post; p.o. |
+Total injury, cortex, and striatum neuronal injury (100 and 200 mg/kg) |
+NDS, elevated plus maze, open field |
Reduced excitotoxicity and oxidative stress (reduced glutamate and GSH; increased SOD); reduced lipid peroxidation |
42 | |
| Flavones | pMCAO | SD rat (m) | Bai | 30 mg/kg immediately post; i.v. |
+Total infarct | +NDS | Reduced p38 MAPK, 12/15-LOX, and cPLA2α (lipid peroxidation) |
43 |
| pMCAO or tMCAO (2 hours) |
SD rat (m) | Bai | 20mg/kg 30 minutes pre, 2 and 4 hours post; i.p. |
+Total, cortex for tMCAO; UC for pMCAO |
+NDS for pMCAO (3 and 24 hours) and tMCAO (24 hours) |
Reduced apoptosis for tMCAO: caspase 3 and TUNEL |
44 | |
| tMCAO (2 hours) |
CD1 mouse (m) | Bai | 300 mg/kg immediately pre; i.p. |
N/A | N/A | Reduced apoptosis (AIF expression and colocalization with 12/15-LOX) |
45 | |
| tMCAO (2 hours) |
Wistar rat (m) | Bai | Dose response (50, 100, or 200 mg/kg immediately post); i.v. |
+Total infarct (dose dependent) |
N/A | Reduced NFkB p65 subunit (all doses) |
46 | |
| tMCAO (90 minutes) |
C57 mouse (m): WT, LOX15−/− |
Bai | 300 mg/kg immediately pre; i.v. |
+total volume for Bai and LOX15−/− |
N/A | Reduced claudin 5 degradation and edema (retention of BBB) |
47 | |
| pMCAO | SD rat (m) | Wog | 20 mg/kg 30 minutes pre and 4 hours post; i.p. |
+Total area, cortex and striatum |
+NDS | N/A | 48 | |
| tMCAO | SD rat (f) | Lut | 5 or 20 mg/kg; 6 hours pre and daily for 13 days; i.p. |
+Total area and striatum (5 and 20 mg/kg) |
+NDS (20 mg/kg at 7 days and both doses at 14 days); +Beam balance (7 and 14 days) |
Decreased ROS, GSH, and catalase (20 mg/kg effective for all; 5 mg/kg only in some brain regions) |
49 | |
| Flavonols | Focal ischemia (photo- thromb) |
SD rat (m) | Quer | 25 μmol/kg 1 hour post then daily for 3 days; i.p. |
N/A | +Locomotor activity | Reduced BBB permeability, brain water content, neuronal MMP-9 protein expression |
50 |
| tMCAO (1 hour) |
Wistar rat (m) | Kae | Dose response (50, 100, or 200 μM 30 minutes pre and immediately post); i.v. |
+Total area, cortex and striatum (100 μM only) |
N/A | Reduced MMP activity, nitrosylation, apoptosis (100 μM only) |
51 | |
| Flavanones | BCCAO (5 minutes) |
Laca mouse (m) |
Nar | Dose response (50, 100 mg/kg) 7 days pre and post; i.p. |
N/A | +NDS; +locomotor activity | Altered levels of various ROS/ redox enzymes and substrates |
52 |
| Stilbenes | pMCAO or tMCAO (2 hours) |
Balb/c mouse (m) |
Resv | Time course 50 mg/kg (5 minutes pre; 24 or 72 hours post); p.o. |
+Total volume pre- pMCAO and pre-tMCAO; UC post |
+NDS pre-pMCAO and pre-tMCAO; UC post |
Reduced MMP-2 and increased VEGF (pre only) |
53 |
| tMCAO (2 hours) |
Balb/c mouse (m) |
Resv | 50 mg/kg 7 days pre; p.o. | +Total area and total % | N/A | Reduced MMP-9 | 54 | |
| tMCAO (90 minutes) |
C57 mouse (m): WT, HO1−/− |
Resv | Dose response (5, 10, 20 mg/kg); Acute or chronic (2 hours or 7 days pre); p.o. |
+Cortex, hemisphere and striatum for WT (20 mg/ kg); UC for HO1−/− |
N/A | HO1 required for protection | 55 | |
| tMCAO (30 minutes) |
C57 mouse (m and f) |
Resv | Dose response (1, 2.5 or 5 mg/kg); Time course (3 or 6 hours post); i.v. |
+(m): total volume and cortex (5 mg/kg 3 hours post only); UC striatum and 6 hours treatments; (f): total volume (1 mg/kg only) |
N/A | Reduced ROS (superoxide) and inflammation (TNFalpha, IL1beta, Iba1) (5 mg/kg 3 hours post); showed (m) only |
56 | |
| tMCAO (2 hours) |
Wistar rat (m) | Resv | 20 mg/kg 3 weeks pre; i.p. |
+Infarct volume | +Rotorod, locomotor activity |
Reduced lipid peroxidation and oxidative stress (MDA and GSH) |
57 | |
| Isoflavones | tMCAO (90 minutes) |
Wistar rat (m) | TIF (high- soy diet) |
5 weeks pre and post; oral ad libitum |
+Cortex, striatum and total volume; +total % but UC cortex and striatum % |
+NDS | N/A | 58 |
| tMCAO (90 minutes) |
SD rat (f) (ovx) | TIF (high- soy diet) |
3 weeks pre and post; oral ad libitum |
+Total volume | N/A | Reduced apoptosis (caspase 3, TUNEL, spectrin cleavage, Bcl2, nuclear AIF); increased Bcl-xL |
59 | |
| tMCAO (90 minutes) |
SD rat (f) (ovx) | TIF (high- soy diet) |
5 weeks pre and post; oral ad libitum |
+Cortex, striatum and total % |
N/A | N/A | 60 |
−/−, knockout; +, improved outcome; AIF, apoptosis inducing factor; Bai, baicalein; BBB, blood–brain barrier; C57, C57BL/6; CA, (+)-catechin; EC, (−)-epicatechin; EGCG, (−)-epigallocatechin gallate; f, female; GTE, green tea extract; HO1, hemeoxygenase 1; i.p., intraperitoneal administration; i.v., intravenous administration; Kae, kaempferol; LOX, lipoxygenase; Lut, luteolin; m, male; MMP, matrix metalloproteinase; N/A, not applicable; Nar, naringin; NDS, neurological deficit scores; ovx, ovariectomized; p.o., oral administration; photo-thromb, cortical microvessel photothrombosis; Quer, quercetin; Resv, resveratrol; ROS, reactive oxygen species; s.c., subcutaneous administration; SD, Sprague Dawley; TIF, total isoflavones; TOF, total oligomeric flavonoids; UC, unchanged outcome; VEGF, vascular endothelial growth factor; Wog, wogonin.
The role of oxidative stress in exacerbating stroke injury is well documented, and several flavonoids have demonstrated neuroprotection associated with reduced reactive oxygen species (ROS) and/or alterations in redox-associated enzymes. For example, chronic pretreatment (3 weeks) with 20 mg/kg resveratrol-linked neuroprotection and functional recovery with reduced oxidation and lipid peroxidation, as measured by malondialdehyde (MDA) and GSH assays, in the rat transient MCAO (tMCAO) (2 hours MCA occlusion) model.57 A lower dose of resveratrol (5 mg/kg) also showed therapeutic efficacy by diminishing neural injury, superoxide production, pro-inflammatory signals (TNFalpha, IL-1beta), and Iba1-positive cells when administered 3 hours, but not 6 hours, following 30 minutes of tMCAO in mice.56 Consistent with these results, flavonoids have also been shown to promote functional recovery in association with reducing oxidative stress in both rat and mouse. Mice subjected to 5 minutes of bilateral common carotid artery occlusion (BCCAO) showed improvements in neurological deficits and locomotor activity following the administration of 50 or 100 mg/kg naringin for 10 days, beginning 7 days prior to insult.52 In these experiments, treatments produced alterations in oxidation, lipid peroxidation, and several intermediates of the glutathione pathway. Treatment with 30 mg/kg quercetin (administered in a liposomal preparation due to bioavailability issues) beginning 30 minutes after pMCAO also reduced cell death, behavioral deficits, and reductions in GSH in rats.61 Liposomal luteolin (5 or 20 mg/kg) resulted in similar reductions in neural injury and behavioral deficits that included decreased ROS when administered chronically (14 days) beginning 6 hours after 40-minute tMCAO.49 Additionally, chronic administration of 100 or 200 mg/kg total oligomeric flavonoids (TOFs) to rats beginning 4 days prior to tMCAO (2 hours occlusion) reduced oxidative markers, and neurological deficits; this protection extended to reduced memory impairments and decreased glutamate accumulation.42
In terms of preventative therapy, pretreatment with TOFs mimics consumption of foods containing high concentrations of various oligomeric flavonoids. To more closely mimic dietary consumption of isoflavonoids, experiments were performed in which rats were maintained on a high-soy diet both prior to and after tMCAO (90 minutes). Results indicated that chronic consumption (5 weeks prior to stroke) reduced infarct volume60 and decreased neurological deficits.58 A subsequent study truncated pretreatment duration to 3 weeks and added mechanism, demonstrating reductions in delayed cell death mediators including caspase 3, cleaved spectrin, cell lymphoma 2 (Bcl2), and nuclear localization of apoptosis-inducing factor (AIF). Additionally, high-soy diet increased expression of the anti-apoptotic protein B cell lymphoma-extra large.59 Despite these encouraging data, the degree to which these isoflavonoids are absorbed following oral administration and the bioactivity of their metabolites remain critically important questions to be answered by future studies prior to clinical application.
Consistent with a role for flavonoids in reducing apoptotic-like signaling, studies with baicalein have demonstrated a link between improved neurohistological outcomes, behavioral recovery, and reductions in delayed cell death. For example, rats that received 20 mg/kg baicalein 30 minutes prior to and twice (2 and 4 hours) following 2 hours of tMCAO showed reduced infarct volume and neurological deficits that were accompanied by decreased caspase 3 and TUNEL staining.44 Similar histological and functional benefits resulted from a single administration of 30 mg/kg immediately following stroke in the rat pMCAO model. Data from these experiments also showed reductions in p38 map kinase (p38 MAPK), 12/15-LOX, and cytosolic phospholipase A2β43. Studies in mice have shown similar results. Prophylactic treatment with 300 mg/kg immediately prior to 2 hours of ischemia/reperfusion reduced the protein expression and colocalization of AIF with 12/15-LOX.45 Additionally, the administration of 300 mg/kg baicalein immediately prior to tMCAO (90 minute occlusion) reduced claudin 5 degradation and vasogenic edema. This group also demonstrated similar protection in LOX15−/− mice, indicating a role for 12/ 15-lipoxygenase (12/15-LOX) in stroke-induced vascular disruption.47 Protection by flavones has also been demonstrated with wogonin, where administration of 20 mg/kg 30 minutes prior to and 4 hours after pMCAO reduced infarct volume, and behavioral deficits.48
Interestingly, p38 MAPK has been shown to activate cPLA2 in cultured cells,62,63 and activation of this pathway also contributed to BBB permeability in a rat model of tMCAO.64 Taken together, these studies suggest a mechanism by which MAPK signaling leads to downstream effects including increased lipoxygenase activity, delayed cell death signaling, and BBB permeability, although maybe not directly. Interestingly, proteolysis of claudin 5 and other BBB constituents occurs through several pathological mechanisms, one of which is activation of MMPs. The association of flavonoid-mediated protection, alterations of cytoprotective genes, and downstream MMP activity will be discussed in the following section.
Flavanols protect against stroke and traumatic brain injury: from anti-oxidation to gene regulation
Flavanols are attractive candidates for natural and alternative medicine due to their high abundance ingreen tea, red wine, cocoa, and chocolates. Interestingly, it was found that a relatively small community of Kuna Indians residing in a Panamanian archipelago exhibits low blood pressure and a reduced incidence of cardiovascular disease and stroke. After a comprehensive assessment of genetic and environmental factors, epidemiological studies concluded that these health benefits may result not necessarily from genetic predisposition but likely from a high intake of cocoa extract that was found to be especially enriched in flavanols.65
These observations support the notion that flavanols may provide efficacy as a stroke therapy, and experimental models have further explored this possibility. An early study using the BCCAO approach demonstrated that while 100 mg/kg of either (+)-catechin (CA) or (−)-EC administered approximately 5 minutes prior to occlusion was insufficient to prevent cortical and hippocampal cell death, this dose improved behavioral performance on a passive avoidance task.36 In contrast to these findings, a subsequent study showed that 25 or 50 mg/kg of (−)-epigallocatechin gallate (EGCG) administered immediately following BCCAO reduced CA1 hippocampal pyramidal cell death in gerbils.39 This discrepancy may be due to methodological differences, as the former produced an occlusion for 10 minutes compared with 3 minutes in the latter. Another study supported protection by CAs in female gerbils, demonstrating reductions in infarct volume and improvements in functional recovery after chronic (3 weeks) pretreatment with green tea extract prior to 5 minutes of BCCAO.41 This group also linked these protective effects to reduced hydrogen peroxide, lipid peroxidation (MDA and 4-hydroxynonenal assay), and TUNEL-stained apoptotic-like cell bodies. The protective effects of EGCG were further demonstrated in gerbils using the tMCAO model, where administration of 50 mg/kg 30 minutes prior to and immediately following occlusion reduced infarct volume (60-minute ischemia), lipid peroxidation (60-minute ischemia), and brain water content (60- and 90-minute occlusion).38 Administration of this same dose was later found to promote behavioral recovery in rats subjected to a sticky tape removal test following 2 hours of ischemia/reperfusion brain injury model.40
Although flavanol-mediated protection has most often been investigated in the context of oxidative stress and lipid peroxidation, presumably through direct effects, recent data show that flavanols may exert their protective effects through alterations in gene expression. In a seminal study, Shih et al.66 used rats, Nrf2−/− mice and several different stroke models (tMCAO in rat; permanent distal MCAO, and endothelin-1 in mice) to demonstrate a protective role for Nrf2. Indeed, there is now evidence that Nrf2 mediates the protective effects of EC, possibly through the induction of HO1. Prophylactic (90 minutes before tMCAO) or delayed (3.5 hours after tMCAO) treatment with 30 mg/kg EC reduced infarct volume in wild-type mice subjected to 90-minute ischemia, while protection was lost in mice deficient in either Nrf2 or HO1.35 Another recent study also demonstrated Nrf2-mediated induction of HO1 that occurred concomitantly with neuroprotection from low-dose carbon monoxide after pMCAO in mouse.67 The protective effects of Nrf2 were also demonstrated in a mouse model of hemorrhagic stroke, where Nrf2 knockouts showed decreased infarct volume and neurological deficits.68 Interestingly, these data also showed that Nrf2 deficiency increased infiltration of myeloperoxidase-positive immune cells into the brain parenchyma but did not increase numbers of Iba1-expressing macrophages/microglia. In addition to the rodent stroke model, Nrf2 activation was shown to reduce oxidative injury, cell death, and neurological deficits in rats subjected to traumatic brain injury, and these effects were accompanied by upregulated expression of both HO1 and NAD(P)H quinone oxidoreductase 1.69
As Nrf2 is a potent activator of Phase II detoxifying enzymes and other cytoprotective proteins through binding to AREs, Nrf2 translocation likely serves as a catalyst for relatively low concentrations of flavanols to exert potent actions on pro-oxidative and inflammatory mediators (see Fig. 2). For example, it is well known that ROS are potent activators of MMPs. The administration of 50 mg/kg EGCG immediately prior to tMCAO (80-minute ischemia) resulted in reduced infarct volume that was accompanied by decreased MMP activity, as measured by gelatin and in situ zymography.37 Importantly, these mechanisms are not limited to flavanols. Experiments with resveratrol, the well-studied stilbene present in concentrated amounts in red wine, have demonstrated that its protective effects are also linked to HO1 and MMP activity. The ability of acute (2 hours) pretreatment with 20 mg/kg resveratrol to reduce reperfusion injury (90-minute tMCAO) was lost in HO1−/−.55 Additionally, chronic (7 days) pretreatment with 50 mg/kg resveratrol was effective in blocking upregulated MMP-9 expression and activity,54 while acute (5 minute) pretreatment with the same dose decreased mRNA and protein levels of MMP-2 and vascular endothelial growth factor53 following 2 hours of ischemia and reperfusion. Notably, each of these studies showed efficacy in reducing infarct volume and the latter expanded to demonstrate decreased neurological deficits. Similarly, kaempferol, another red wine constituent, was effective in decreasing infarct volume, MMP activity, and laminin degradation whenadministered at a dose of 100 μM/l both 30 minutes prior to and immediately following tMCAO (1 hour occlusion).51 These effects were accompanied by reduced levels of poly (ADP-ribose) polymerase (PARP) protein, and caspase 9 activity in ischemic tissue homogenates. Finally, administration of 25 μmol/kg quercetin beginning 1 hour after photothrombotic vessel occlusion decreased BBB permeability (as indicated by Evans Blue extravasation), brain water content, total MMP-9 proteolytic activity, neuronal MMP-9 immunoreactivity, and behavioral deficits; neural injury was not assessed.50
Figure 2.
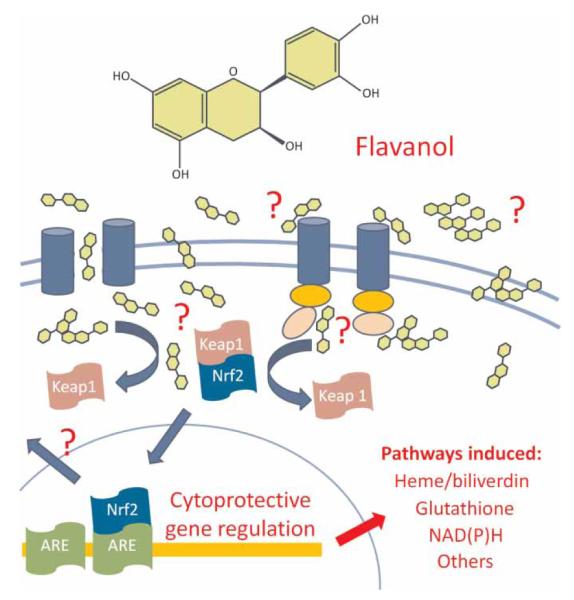
Schematic representation depicting the potential mechanisms by which flavanol-mediated Nrf2 induction leads to activation of cytoprotective pathways after stroke, traumatic brain injury, and/or other neurodegenerative diseases. Flavanols may induce Nrf2 through binding to receptors seated on the plasma membrane and subsequent initiation of intracellular signaling cascades. Alternatively, passive diffusion or active transport through the plasma membrane may permit direct cytosolic dissociation of the Keap1/Nrf2 complex or activation of second messengers that regulate Nrf2 translocation into the nucleus. Upon nuclear translocation, Nrf2 binds to AREs on the promoter regions of cytoprotective genes to regulate heme/biliverdin, glutathione, NAD(P)H, and/or other protective pathways.
To date, the precise mechanism of MMP inhibition remains unknown. The fact that both enzyme activity and protein expression are reduced suggests a transcriptional mechanism rather than a direct interaction with the enzyme itself. Notably, the administration of baicalein to rats immediately following 2 hours of ischemia/reperfusion resulted in a dose-dependent reduction in infarct volume that was accompanied by decreased expression of the NFkB p65 subunit.46 MMP gene transcription is heavily regulated by nuclear factor-κB (NFκB) and AP1 transcription factors,70 and thus Nrf2-mediated repression of these transcriptional activation pathways may represent the mechanism of MMP inhibition.
Taken together, these data demonstrate the therapeutic potential for flavonoids in the treatment of stroke. Activation of Nrf2 results in the induction of various gene transcripts capable of altering levels of oxidation and lipid peroxidation, inflammatory signaling, and proteolytic activity that contributes to BBB disruption and vascular permeability. Future studies are needed to identify the net effects of Nrf2 induction, as well as the specific downstream targets that will be most helpful in combating neurological injury after stroke.
Conclusion
The health benefits of polyphenolic food sources are well documented. In particular, flavanols are strong candidates for stroke prevention and treatment due to their dietary origin and reproducible efficacy in experimental models. Of interest, the previous conception attributing these effects to direct anti-oxidation is slowly giving way to a novel mechanism involving the modulation of neuroprotective gene expression. Accumulated evidence suggests that the pleiotropic effects resulting from flavonoid-mediated Nrf2 induction may lead to more promising therapies for the treatment of stroke and other acute and/or chronic neurodegenerative disorders.
Acknowledgment
This work is supported in part by the National Institute of Health research grants (AT005085, AT005246). Note that this review highlights studies based upon relevance to the topic, and was not intended to provide an exhaustive summary of all flavonoid studies.
References
- 1.Heiss C, Keen CL, Kelm M. Flavanols and cardiovascular disease prevention. Eur Heart J. 2010;31(21):2583–92. doi: 10.1093/eurheartj/ehq332. [DOI] [PubMed] [Google Scholar]
- 2.Arab L, Liebeskind DS. Tea, flavonoids and stroke in man and mouse. Arch Biochem Biophys. 2010;501(1):31–6. doi: 10.1016/j.abb.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 3.Arts ICW, Hollman PCH. Polyphenols and disease risk in epidemiological studies. Am J Clin Nutr. 2005;81(suppl):317S–25S. doi: 10.1093/ajcn/81.1.317S. [DOI] [PubMed] [Google Scholar]
- 4.Mann GE, Bonacasa B, Ishii T, Siow RC. Targeting the redox sensitive Nrf2-Keap1 defense pathway in cardiovascular disease: protection afforded by dietary isoflavones. Curr Opin Pharmacol. 2009;9(2):139–45. doi: 10.1016/j.coph.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 5.Alfieri A, Srivastava S, Siow RC, Modo M, Fraser PA, Mann GE. Targeting the Nrf2-Keap1 antioxidant defence pathway for neurovascular protection in stroke. J Physiol. 2011 Jun 6; doi: 10.1113/jphysiol.2011.210294. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perez-Vizcaino F, Duarte J. Flavonols and cardiovascular disease. Mol Aspects Med. 2010;31(6):478–94. doi: 10.1016/j.mam.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 7.Serafini M, Peluso I, Raguzzini A. Flavonoids as anti-inflammatory agents. Proc Nutr Soc. 2010;69(3):273–8. doi: 10.1017/S002966511000162X. [DOI] [PubMed] [Google Scholar]
- 8.Holt RR, Lazarus SA, Sullards MC, Zhu QY, Schramm DD, Hammerstone JF, et al. Procyanidin dimer B2 [epicatechin-(4beta-8)-epicatechin] in human plasma after the consumption of a flavanol-rich cocoa. Am J Clin Nutr. 2002;76(4):798–804. doi: 10.1093/ajcn/76.4.798. [DOI] [PubMed] [Google Scholar]
- 9.Spencer JP, Chaudry F, Pannala AS, Srai SK, Debnam E, Rice-Evans C. Decomposition of cocoa procyanidins in the gastric milieu. Biochem Biophys Res Commun. 2000;272(1):236–41. doi: 10.1006/bbrc.2000.2749. [DOI] [PubMed] [Google Scholar]
- 10.Manach C, Scalbert A, Morand C, Remesy C, Jimenez L. Polyphenols: food sources and bioavailability. Am J Clin Nutr. 2004;79(5):727–47. doi: 10.1093/ajcn/79.5.727. [DOI] [PubMed] [Google Scholar]
- 11.Manach C, Williamson G, Morand C, Scalbert A, Remesy C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am J Clin Nutr. 2005;81(1 Suppl):230S–42S. doi: 10.1093/ajcn/81.1.230S. [DOI] [PubMed] [Google Scholar]
- 12.D’Archivio M, Filesi C, Di Benedetto R, Gargiulo R, Giovannini C, Masella R. Polyphenols, dietary sources and bioavailability. Ann Ist Super Sanita. 2007;43(4):348–61. [PubMed] [Google Scholar]
- 13.Dajas F, Rivera F, Blasina F, Arredondo F, Echeverry C, Lafon L, et al. Cell culture protection and in vivo neuroprotective capacity of flavonoids. Neurotox Res. 2003;5(6):425–32. doi: 10.1007/BF03033172. [DOI] [PubMed] [Google Scholar]
- 14.Rivera F, Urbanavicius J, Gervaz E, Morquio A, Dajas F. Some aspects of the in vivo neuroprotective capacity of flavonoids: bioavailability and structure-activity relationship. Neurotox Res. 2004;6(7-8):543–53. doi: 10.1007/BF03033450. [DOI] [PubMed] [Google Scholar]
- 15.Pannala AS, Rice-Evans CA, Halliwell B, Singh S. Inhibition of peroxynitrite-mediated tyrosine nitration by catechin polyphenols. Biochem Biophys Res Commun. 1997;232(1):164–8. doi: 10.1006/bbrc.1997.6254. [DOI] [PubMed] [Google Scholar]
- 16.Rice-Evans CA, Miller NJ, Paganga G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic Biol Med. 1996;20(7):933–56. doi: 10.1016/0891-5849(95)02227-9. [DOI] [PubMed] [Google Scholar]
- 17.Maffei Facino R, Carini M, Aldini G, Bombardelli E, Morazzoni P, Morelli R. Free radicals scavenging action and anti-enzyme activities of procyanidines from Vitis vinifera. A mechanism for their capillary protective action. Arzneimittelforschung. 1994;44(5):592–601. [PubMed] [Google Scholar]
- 18.Spencer JP. Beyond antioxidants: the cellular and molecular interactions of flavonoids and how these underpin their actions on the brain. Proc Nutr Soc. 2010;69(2):244–60. doi: 10.1017/S0029665110000054. [DOI] [PubMed] [Google Scholar]
- 19.Schroeter H, Heiss C, Balzer J, Kleinbongard P, Keen CL, Hollenberg NK, et al. (−)-Epicatechin mediates beneficial effects of flavanol-rich cocoa on vascular function in humans. Proc Natl Acad Sci USA. 2006;103(4):1024–9. doi: 10.1073/pnas.0510168103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79(4):1431–568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- 21.del Zoppo GJ, Milner R, Mabuchi T, Hung S, Wang X, Berg GI, et al. Microglial activation and matrix protease generation during focal cerebral ischemia. Stroke. 2007;38(2 Suppl):646–51. doi: 10.1161/01.STR.0000254477.34231.cb. [DOI] [PubMed] [Google Scholar]
- 22.Rosell A, Lo EH. Multiphasic roles for matrix metalloproteinases after stroke. Curr Opin Pharmacol. 2008;8(1):82–9. doi: 10.1016/j.coph.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 23.Jean WC, Spellman SR, Nussbaum ES, Low WC. Reperfusion injury after focal cerebral ischemia: the role of inflammation and the therapeutic horizon. Neurosurgery. 1998;43(6):1382–96. doi: 10.1097/00006123-199812000-00076. [DOI] [PubMed] [Google Scholar]
- 24.Matsuo Y, Onodera H, Shiga Y, Nakamura M, Ninomiya M, Kihara T, et al. Correlation between myeloperoxidase-quantified neutrophil accumulation and ischemic brain injury in the rat. Effects of neutrophil depletion. Stroke. 1994;25(7):1469–75. doi: 10.1161/01.str.25.7.1469. [DOI] [PubMed] [Google Scholar]
- 25.Schwarting S, Litwak S, Hao W, Bahr M, Weise J, Neumann H. Hematopoietic stem cells reduce postischemic inflammation and ameliorate ischemic brain injury. Stroke. 2008;39(10):2867–75. doi: 10.1161/STROKEAHA.108.513978. [DOI] [PubMed] [Google Scholar]
- 26.Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab. 2005;26(5):654–65. doi: 10.1038/sj.jcbfm.9600217. [DOI] [PubMed] [Google Scholar]
- 27.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22(9):391–7. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 28.Chen XL, Kunsch C. Induction of cytoprotective genes through Nrf2/antioxidant response element pathway: a new therapeutic approach for the treatment of inflammatory diseases. Curr Pharm Des. 2004;10(8):879–91. doi: 10.2174/1381612043452901. [DOI] [PubMed] [Google Scholar]
- 29.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 30.Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114(9):1248–59. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niture SK, Jaiswal AK. Hsp90 interaction with INrf2(Keap1) mediates stress-induced Nrf2 activation. J Biol Chem. 2010;285(47):36865–75. doi: 10.1074/jbc.M110.175802. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116(4):984–95. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, et al. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med. 2005;202(1):47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagai N, Thimmulappa RK, Cano M, Fujihara M, Izumi-Nagai K, Kong X, et al. Nrf2 is a critical modulator of the innate immune response in a model of uveitis. Free Radic Biol Med. 2009;47(3):300–6. doi: 10.1016/j.freeradbiomed.2009.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shah ZA, Li RC, Ahmad AS, Kensler TW, Yamamoto M, Biswal S, et al. The flavanol (−)-epicatechin prevents stroke damage through the Nrf2/HO1 pathway. J Cereb Blood Flow Metab. 2010;30(12):1951–61. doi: 10.1038/jcbfm.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsuoka Y, Hasegawa H, Okuda S, Muraki T, Uruno T, Kubota K. Ameliorative effects of tea catechins on active oxygen-related nerve cell injuries. J Pharmacol Exp Ther. 1995;274(2):602–8. [PubMed] [Google Scholar]
- 37.Park JW, Hong JS, Lee KS, Kim HY, Lee JJ, Lee SR. Green tea polyphenol (−)-epigallocatechin gallate reduces matrix metalloproteinase-9 activity following transient focal cerebral ischemia. J Nutr Biochem. 2010;21(11):1038–44. doi: 10.1016/j.jnutbio.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 38.Lee H, Bae JH, Lee SR. Protective effect of green tea polyphenol EGCG against neuronal damage and brain edema after unilateral cerebral ischemia in gerbils. J Neurosci Res. 2004;77(6):892–900. doi: 10.1002/jnr.20193. [DOI] [PubMed] [Google Scholar]
- 39.Lee S, Suh S, Kim S. Protective effects of the green tea polyphenol (−)-epigallocatechin gallate against hippocampal neuronal damage after transient global ischemia in gerbils. Neurosci Lett. 2000;287(3):191–4. doi: 10.1016/s0304-3940(00)01159-9. [DOI] [PubMed] [Google Scholar]
- 40.Lim SH, Kim HS, Kim YK, Kim TM, Im S, Chung ME, et al. The functional effect of epigallocatechin gallate on ischemic stroke in rats. Acta Neurobiol Exp (Wars) 2010;70(1):40–6. doi: 10.55782/ane-2010-1772. [DOI] [PubMed] [Google Scholar]
- 41.Hong JT, Ryu SR, Kim HJ, Lee JK, Lee SH, Yun YP, et al. Protective effect of green tea extract on ischemia/reperfusion-induced brain injury in Mongolian gerbils. Brain Res. 2001;888(1):11–8. doi: 10.1016/s0006-8993(00)02935-8. [DOI] [PubMed] [Google Scholar]
- 42.Sunil AG, Kesavanarayanan KS, Kalaivani P, Sathiya S, Ranju V, Priya RJ, et al. Total oligomeric flavonoids of Cyperus rotundus ameliorates neurological deficits, excitotoxicity and behavioral alterations induced by cerebral ischemic-reperfusion injury in rats. Brain Res Bull. 2011;84(6):394–405. doi: 10.1016/j.brainresbull.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 43.Cui L, Zhang X, Yang R, Liu L, Wang L, Li M, et al. Baicalein is neuroprotective in rat MCAO model: role of 12/15-lipoxygenase, mitogen-activated protein kinase and cytosolic phospholipase A2. Pharmacol Biochem Behav. 2010;96(4):469–75. doi: 10.1016/j.pbb.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 44.Liu C, Wu J, Xu K, Cai F, Gu J, Ma L, et al. Neuroprotection by baicalein in ischemic brain injury involves PTEN/AKT pathway. J Neurochem. 2010;112(6):1500–12. doi: 10.1111/j.1471-4159.2009.06561.x. [DOI] [PubMed] [Google Scholar]
- 45.Pallast S, Arai K, Pekcec A, Yigitkanli K, Yu Z, Wang X, et al. Increased nuclear apoptosis-inducing factor after transient focal ischemia: a 12/15-lipoxygenase-dependent organelle damage pathway. J Cereb Blood Flow Metab. 2010;30(6):1157–67. doi: 10.1038/jcbfm.2009.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xue X, Qu XJ, Yang Y, Sheng XH, Cheng F, Jiang EN, et al. Baicalin attenuates focal cerebral ischemic reperfusion injury through inhibition of nuclear factor kappaB p65 activation. Biochem Biophys Res Commun. 2010;403(3-4):398–404. doi: 10.1016/j.bbrc.2010.11.042. [DOI] [PubMed] [Google Scholar]
- 47.Jin G, Arai K, Murata Y, Wang S, Stins MF, Lo EH, et al. Protecting against cerebrovascular injury: contributions of 12/ 15-lipoxygenase to edema formation after transient focal ischemia. Stroke. 2008;39(9):2538–43. doi: 10.1161/STROKEAHA.108.514927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cho J, Lee HK. Wogonin inhibits ischemic brain injury in a rat model of permanent middle cerebral artery occlusion. Biol Pharm Bull. 2004;27(10):1561–4. doi: 10.1248/bpb.27.1561. [DOI] [PubMed] [Google Scholar]
- 49.Zhao G, Zang SY, Jiang ZH, Chen YY, Ji XH, Lu BF, et al. Postischemic administration of liposome-encapsulated luteolin prevents against ischemia-reperfusion injury in a rat middle cerebral artery occlusion model. J Nutr Biochem. 2010 Dec 27; doi: 10.1016/j.jnutbio.2010.07.014. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 50.Lee JK, Kwak HJ, Piao MS, Jang JW, Kim SH, Kim HS. Quercetin reduces the elevated matrix metalloproteinases-9 level and improves functional outcome after cerebral focal ischemia in rats. Acta Neurochir (Wien) 2011;153(6):1321–9. doi: 10.1007/s00701-010-0889-x. discussion 9. [DOI] [PubMed] [Google Scholar]
- 51.Lopez-Sanchez C, Martin-Romero FJ, Sun F, Luis L, Samhan-Arias AK, Garcia-Martinez V, et al. Blood micromolar concentrations of kaempferol afford protection against ischemia/reperfusion-induced damage in rat brain. Brain Res. 2007;1182:123–37. doi: 10.1016/j.brainres.2007.08.087. [DOI] [PubMed] [Google Scholar]
- 52.Aggarwal A, Gaur V, Kumar A. Nitric oxide mechanism in the protective effect of naringin against post-stroke depression (PSD) in mice. Life Sci. 2010;86(25-26):928–35. doi: 10.1016/j.lfs.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 53.Dong W, Li N, Gao D, Zhen H, Zhang X, Li F. Resveratrol attenuates ischemic brain damage in the delayed phase afterstroke and induces messenger RNA and protein express for angiogenic factors. J Vasc Surg. 2008;48(3):709–14. doi: 10.1016/j.jvs.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 54.Gao D, Zhang X, Jiang X, Peng Y, Huang W, Cheng G, et al. Resveratrol reduces the elevated level of MMP-9 induced by cerebral ischemia-reperfusion in mice. Life Sci. 2006;78(22):2564–70. doi: 10.1016/j.lfs.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 55.Sakata Y, Zhuang H, Kwansa H, Koehler RC, Dore S. Resveratrol protects against experimental stroke: putative neuroprotective role of heme oxygenase 1. Exp Neurol. 2010;224(1):325–9. doi: 10.1016/j.expneurol.2010.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shin JA, Lee H, Lim YK, Koh Y, Choi JH, Park EM. Therapeutic effects of resveratrol during acute periods following experimental ischemic stroke. J Neuroimmunol. 2010;227(1-2):93–100. doi: 10.1016/j.jneuroim.2010.06.017. [DOI] [PubMed] [Google Scholar]
- 57.Sinha K, Chaudhary G, Gupta YK. Protective effect of resveratrol against oxidative stress in middle cerebral artery occlusion model of stroke in rats. Life Sci. 2002;71(6):655–65. doi: 10.1016/s0024-3205(02)01691-0. [DOI] [PubMed] [Google Scholar]
- 58.Burguete MC, Torregrosa G, Perez-Asensio FJ, Castello-Ruiz M, Salom JB, Gil JV, et al. Dietary phytoestrogens improve stroke outcome after transient focal cerebral ischemia in rats. Eur J Neurosci. 2006;23(3):703–10. doi: 10.1111/j.1460-9568.2006.04599.x. [DOI] [PubMed] [Google Scholar]
- 59.Lovekamp-Swan T, Glendenning M, Schreihofer DA. A high soy diet reduces programmed cell death and enhances bcl-xL expression in experimental stroke. Neuroscience. 2007;148(3):644–52. doi: 10.1016/j.neuroscience.2007.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schreihofer DA, Do KD, Schreihofer AM. High-soy diet decreases infarct size after permanent middle cerebral artery occlusion in female rats. Am J Physiol Regul Integr Comp Physiol. 2005;289(1):R103–8. doi: 10.1152/ajpregu.00642.2004. [DOI] [PubMed] [Google Scholar]
- 61.Rivera F, Costa G, Abin A, Urbanavicius J, Arruti C, Casanova G, et al. Reduction of ischemic brain damage and increase of glutathione by a liposomal preparation of quercetin in permanent focal ischemia in rats. Neurotox Res. 2008;13(2):105–14. doi: 10.1007/BF03033562. [DOI] [PubMed] [Google Scholar]
- 62.Coulon L, Calzada C, Moulin P, Vericel E, Lagarde M. Activation of p38 mitogen-activated protein kinase/cytosolic phospholipase A2 cascade in hydroperoxide-stressed platelets. Free Radic Biol Med. 2003;35(6):616–25. doi: 10.1016/s0891-5849(03)00386-1. [DOI] [PubMed] [Google Scholar]
- 63.Lin LL, Wartmann M, Lin AY, Knopf JL, Seth A, Davis RJ. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993;72(2):269–78. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- 64.Nito C, Kamada H, Endo H, Niizuma K, Myer DJ, Chan PH. Role of the p38 mitogen-activated protein kinase/cytosolic phospholipase A2 signaling pathway in blood-brain barrier disruption after focal cerebral ischemia and reperfusion. J Cereb Blood Flow Metab. 2008;28(10):1686–96. doi: 10.1038/jcbfm.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hollenberg NK, Fisher ND, McCullough ML. Flavanols, the Kuna, cocoa consumption, and nitric oxide. J Am Soc Hypertens. 2009;3(2):105–12. doi: 10.1016/j.jash.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shih AY, Li P, Murphy TH. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci. 2005;25(44):10321–35. doi: 10.1523/JNEUROSCI.4014-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang B, Cao W, Biswal S, Dore S. Carbon monoxide-activated Nrf2 pathway leads to protection against permanent focal cerebral ischemia. Stroke. 2011;42(9):2605–10. doi: 10.1161/STROKEAHA.110.607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang J, Fields J, Zhao C, Langer J, Thimmulappa RK, Kensler TW, et al. Role of Nrf2 in protection against intracerebral hemorrhage injury in mice. Free Radic Biol Med. 2007;43(3):408–14. doi: 10.1016/j.freeradbiomed.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hong Y, Yan W, Chen S, Sun CR, Zhang JM. The role of Nrf2 signaling in the regulation of antioxidants and detoxifying enzymes after traumatic brain injury in rats and mice. Acta Pharmacol Sin. 2010;31(11):1421–30. doi: 10.1038/aps.2010.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vincenti MP, Brinckerhoff CE. Signal transduction and cell-type specific regulation of matrix metalloproteinase gene expression: can MMPs be good for you? J Cell Physiol. 2007;213(2):355–64. doi: 10.1002/jcp.21208. [DOI] [PubMed] [Google Scholar]