

Abstract
Binding of selectins to P-selectin glycoprotein ligand-1 (PSGL-1) mediates tethering and rolling of leukocytes on the endothelium during inflammation. Previous measurements obtained with a flow-chamber assay have shown that mutations of three tyrosines at the PSGL-1 N-terminus (Y46, Y48, and Y51) increase the reverse rates and their sensitivity to the force of bonds with P- and L-selectin. However, the effects of these mutations on the binding affinities and forward rates have not been studied. We quantified these effects by using an adhesion frequency assay to measure two-dimensional affinity and kinetic rates at zero force. Wild-type PSGL-1 has 2.2- to 8.5-fold higher binding affinities for P- and L-selectin than PSGL-1 mutants with two of three tyrosines substituted by phenylalanines, and 9.6- to 49-fold higher affinities than the PSGL-1 mutant with all three tyrosines replaced. In descending order, the affinity decreased from wild-type to Y48/51F, Y46/51F, Y46/48F, and Y46/48/51F. The affinity differences were attributed to major changes in the forward rate and minor changes in the reverse rate, suggesting that these tyrosines regulate the accessibility of PSGL-1 to P- and L-selectin via electrostatic interactions, which is supported by molecular-dynamics simulations. Our results provide insights into the structure-function relationship of receptor-ligand binding at a single-residue level.
Introduction
Binding of selectins to glycoconjugates initiates the first step of leukocyte recruitment to sites of inflammation and injury (1–4). L-selectin, which is expressed on most leukocytes, binds ligands on endothelial cells and other leukocytes. P- and E-selectin, which are expressed on activated platelets and/or endothelial cells, bind ligands on leukocytes and tumor cells. The best-characterized selectin ligand is P-selectin glycoprotein ligand-1 (PSGL-1), a homodimeric leukocyte mucin with two 120-kDa subunits linked by a disulfide bond (5). Each subunit contains an N-terminal portion in which the binding site for P- and L-selectin resides, a long stalk consisting of a series of decameric repeats, a transmembrane domain, and a short cytoplasmic tail. The association and dissociation of selectin and PSGL-1 pair regulate leukocyte tethering and rolling on the endothelium.
Selectin-PSGL-1 interactions depend on the molecular structure, cellular presentation, and mechanochemical microenvironment of the interacting molecules (1). One key factor is sialylation and fucosylation on the branched core-2 O-glycan (6–13). Another crucial requirement is the sulfation of at least one of the three tyrosines at residues 46, 48, and 51. The interactions between P-selectin lectin (Lec) domain and PSGL-1 N-terminal peptide have been demonstrated by the x-ray crystallographic structure of P-selectin lectin and epidermal growth factor (EGF)-like domains (P-LE in short) ligated with a synthesized sulfoglycopeptide of PSGL-1 with three tyrosine sulfate residues (Y46, Y48, and Y51) and an sLeX-modified glycan at T57 (SGP-3 in short). In combination, electrostatic and hydrophobic interactions at the interface form two major contacts: 1), the sLeX-modified glycan, mainly via the fucose (FUC) that interacts with the Ca2+ ion and its nearby residues in lectin domain; and 2), two of the three sulfated tyrosines (Y48 and Y51) that interact with the lectin domain via hydrogen bonds or salt bridges (8). Replacement of all three tyrosines with phenylalanines on transfected cells was shown to eliminate binding to P- and L-selectin (10,12,14–17), and substitution of two of the three tyrosines was shown to affect the dissociation kinetics and their force dependence under shear flow (10,12,14–17). A flow-chamber analysis of tether lifetimes suggested that L-selectin dissociated faster from these double mutants than wild-type (WT) PSGL-1, whereas P-selectin unbound from double mutants with reverse rates similar to those observed for WT PSGL-1 (10). In this analysis, the values were predicted by zero-force extrapolation from the measured force-dependent reverse rates using the Bell model (18). However, the bindings of PSGL-1 with P-selectin (19) and L-selectin (20) show catch-bond behaviors that do not obey the Bell model, raising a question about these extrapolated zero-force reverse rates. Furthermore, the flow-chamber studies did not address how the binding affinities and forward rates are affected by variations of the PSGL-1 N-terminal sulfated tyrosines.
Here, we used an adhesion frequency assay (21–25) to quantify the two-dimensional (2D) binding affinities and zero-force kinetic rates of P- or L-selectin interacting with WT PSGL-1 or mutants with two or all three N-terminal tyrosines replaced by phenylalanines (double or triple mutants). In contrast to surface plasmon resonance, which determines the three-dimensional (3D) kinetics with one of the interacting molecules in solution, 2D kinetics measurements quantify binding with both interacting molecules anchored on the apposing surfaces. This is biologically relevant because cell adhesion molecules are expressed on the cell surface and bind their ligands on another cell or extracellular matrix. We also performed molecular dynamics (MD) simulations to explore the structural impact of N-terminal tyrosines on interactions between P- or L-selectin and PSGL-1. Our data indicate the importance of these tyrosine residues for the forward rate and the accessibility of PSGL-1 for selectin binding.
Materials and Methods
Cells
We used transfected Chinese hamster ovary (CHO) cells stably expressing human WT or mutant PSGL-1 as previously described (10,12). We refer to PSGL-1 with tyrosines (green) replaced by phenylalanines (red) at residues 46, 48, and/or 51 of the N-terminal region as Y46/48F, Y46/51F, and Y48/51F for double mutants, and Y46/48/51F for the triple mutant (cf. Fig. 1 b). CHO cells were transfected with cDNAs for both α1,3-fucosyltransferase VII (FucTVII) and core-2β1,6-N-acetylglucosaminyltransferase (C2GnT). A CHO cell clone expressing high levels of both FucTVII and C2GnT was then transfected with cDNAs for WT or mutant PSGL-1. Cells were selected and cultured in high-glucose, low-salt Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Grand Island, NY), supplemented with 4 mM glutamine, 200 U/ml penicillin, 200 μg/ml streptomycin, 1% 100× HT, 1% 100× nonessential amino acid (Hyclone, Logan, UT), 600 μg/ml geneticin (Gibco), 100 μg/ml Hygromycin B (Boehringer Mannheim, Indianapolis, IN), 10% fetal bovine serum, and 200 μg/ml Zeocin (Invitrogen, Carlsbad, CA), where geneticin, Hygromycin B, and Zeocin were used as selection agents for FucTVII, C2GnT, and PSGL-1, respectively. Control cells were cultured without Zeocin. PSGL-1 expression was periodically checked by flow cytometry (FACSCalibur; BD Biosciences, San Jose, CA). Before micropipette experiments were conducted, the cells were detached by 5 mM PBS/EDTA, washed with HBSS+, and resuspended in culture media.
Figure 1.
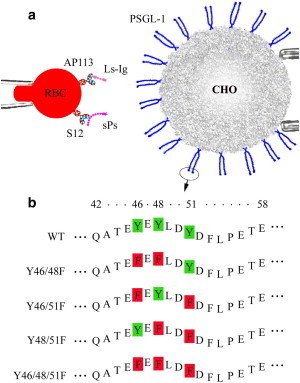
(a) Schematic of the micropipette adhesion frequency assay. Dimeric WT or mutant PSGL-1 was expressed on transfected CHO cells expressing FucTVII and C2nT. Soluble P-selectin or L-selectin-Ig chimera was coupled to an RBC with uniform orientation by capturing antibodies (S12 or AP113) precoated by CrCl3 coupling. (b) PSGL-1 mutants. PSGL-1 was mutated by replacing two of the three (double mutants) or all three (triple mutant) sulfated tyrosines at residues 46, 48, and 51 by phenylalanines. Only the first 17 amino acids of mature PSGL-1 are shown.
Proteins and antibodies
Soluble P-selectin, L-selectin IgG Fc chimera, anti-P-selectin blocking (G1), and capturing (S12) mAbs (26–28), anti-L-selectin blocking (DREG56), and anti-PSGL-1 blocking (PL1) mAbs (all mIgG1) are described elsewhere (19,20). P-selectin IgG Fc chimera was purchased from R&D Systems (Minneapolis, MN). Goat anti-human IgG Fc capturing antibody (AP113; mIgG1) was from Chemicon (Temecula, CA). Fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse polyclonal antibody and irrelevant control mIgG1 were obtained from Sigma Chemical (St. Louis, MO). FITC-labeled Fab fragment of rabbit anti-goat IgG was purchased from Jackson ImmunoResearch (West Grove, PA).
Coating proteins onto red blood cells
Human red blood cells (RBCs) were isolated from fresh blood of healthy volunteers. A chromium chloride (CrCl3) protocol was used to covalently coat capturing mAbs (S12 or AP113), anti-PSGL-1 mAb (PL1), or irrelevant mIgG onto RBC surface (24,29). Coating efficiency was analyzed by flow cytometry (30). Antibody-coated RBCs were incubated with 200 ng/ml of selectin constructs at room temperature for 40 min. This coating procedure resulted in uniform orientation of P- or L-selectin on the RBC surface, which is optimal for micropipette kinetics measurements (22).
Site density determination
Site densities of WT or mutant PSGL-1s on CHO cells were measured by flow cytometry. Cells were incubated with 10 μg/ml anti-PSGL-1 mAb PL1 in FACS buffer (RPMI/5 mM EDTA/1% BSA/0.02% sodium azide) on ice for 40 min and with FITC-labeled goat anti-mouse antibody on ice for another 30 min. After washing, cells were resuspended in PBS and analyzed by flow cytometry. The fluorescent intensities of the cells were quantified by standard fluorescence calibration beads (Quantum 25; Bangs Laboratories, Fishers, IN) to determine the mean number of molecules of equivalent soluble fluorochrome (MESF) per cell. The MESF was divided by the number of fluorochromes per antibody (provided by manufacturer) and then the surface area (assuming a spherical cell of 20 μm in diameter (10)) to convert into PSGL-1 site density, assuming monomeric antibody binding.
The site densities of selectins captured by mAb-coated RBCs were determined by immunoradiometric assay (IRMA) (22,25). RBCs were coated with capturing mAbs at several densities (five to seven levels for each selectin) and then segregated into two sets. One set was incubated with selectins whose site densities were measured by IRMA. The other set was incubated with FITC-labeled secondary antibodies, and the fluorescence intensities were measured by flow cytometry. Plotting selectin-site density against fluorescence intensity generates a calibration curve for calculating the site density of P-selectin (22) or L-selectin (see Fig. S1in the Supporting Material) from the fluorescence intensity of the capturing antibody. Similarly, we measured the site densities of G1 and PL1 using flow cytometry by incubating them with FITC-labeled secondary antibodies.
2D kinetics measurements
The adhesion frequency assay for measuring 2D kinetics was previously described (21,24). Briefly, ∼103 selectin-coated RBCs and ∼102 PSGL-1-expressing CHO cells were added into a chamber solution of 50% HBSS(+) (8 g/L or 68.5 mM NaCl, 0.4 g/L KCl, 1 g/L glucose, 60 mg/L KH2PO4, and 47.5 mg/L Na2HPO4) and 50% H2O to swell the RBC as a mechanical transducer. In some cases, the ionic strength of chamber solution was modified to 6 or 10 g/L NaCl in the stock HBSS solution, resulting in a final ionic concentration of 51.5 or 85.5 mM NaCl, respectively. An RBC and a CHO cell were each aspirated by a micropipette and driven by a computer-controlled piezoelectric translator to go through an approach-contact-withdrawal cycle. The repeated manipulations were performed on a light microscope and monitored with a camera (Fig. 1 a). The suction pressure was kept the same to set the same cell stiffness (25). An adhesion event was determined from the RBC membrane deflection during withdrawal. An adhesion probability, Pa, at a given contact duration, t, was determined from 100 repeated tests. For each molecular pair examined, two Pa versus t curves corresponding to two receptor-site densities (mr) and a single ligand-site density (ml) were obtained from 76–133 cell pairs. Each curve was fitted by a kinetic model (21),
| (1) |
to calculate a zero-force reverse rate, kr0, and effective binding affinity, AcKa0, where Ac is the contact area kept constant in all experiments. Equation 1 is an approximate solution of master equations from a probabilistic model of small system kinetics and has been widely used to determine the binding affinity and kinetic rates for various molecular systems (e.g., selectin, integrin, and antibody binding to their counterpart molecules) (25,31,32). Thus, we obtained two sets of (kr0, AcKa0) values for each selectin-PSGL-1 pair, which allowed us to calculate their mean and standard deviation. The statistical significance of the difference between the 2D affinities (or reverse rates) of WT and mutated PSGL-1 was determined using Student’s t-test.
MD simulations
To elucidate the structural bases of P-selectin binding to PSGL-1, we performed free MD simulations using the crystal structure of WT P-LE ligated with SGP-3. SGP-3 is a 19 amino acid sulfoglycopeptide of the PSGL-1 N-terminal region that includes three tyrosine sulfate residues (Y46, Y48, and Y51) and an sLex-modified glycan at T57 (PDB code: 1G1S) (8). We built the PSGL-1 double mutants Y46/48F, Y46/51F, and Y48/51F; triple mutant Y46/48/51F; and single mutants Y48F, Y48E, and Y48R by replacing the respective tyrosine residues by phenylalanines, glutamic acid, or arginine. The strontium ion Sr2+ was replaced by calcium ion Ca2+. Every simulation system was established by solvating the target molecule into a water sphere. The complex surface was covered by at least four to five water shells and neutralized with Na+ and Cl− ions to mimic the physiological ionic concentration. Energy minimization of 5000 steps and system heating to 300 K over 25,000 steps were performed for a >3 ns equilibration using a NAMD program (33). A CHARMM22 all-atom force field for protein (34) and a custom-built force field for six sugar and three tyrosine-sulfate residues in SGP-3 ligand (35) were used. The simulations were performed with a time step of 1 fs, a uniform dielectric constant of 1.0, a cutoff of nonbonded interactions with a switching function starting at a distance of 11 Å and reaching zero at 14 Å, and a scaling factor of 1.0 for one to four interactions. During equilibration with a thermal bath of 300 K, the temperature was controlled with the use of Langevin dynamics. The water molecules in the outer 5 Å shell enclosing the system were harmonically restrained to their positions to maintain the water bubble shape, with periodic readjustment every 300 ps. The system was built up and data analyses were performed using the VMD program (36).
Results
Binding controls
We quantified the binding of cells transfected with WT or mutant PSGL-1 (Fig. 1 b) to RBCs bearing selectins using the steady-state adhesion frequency, which we estimated by extrapolating the adhesion frequency versus time curve to infinite contact time (t→∞) to test ligand binding selectivity and nonspecific binding controls. As shown in Fig. 2, a–d (rows 1–3), adhesions of P-selectin-bearing RBCs to CHO cells expressing WT, Y46/48F, Y46/51F, and Y48/51F PSGL-1 (solid-line box) were abolished when the P-selectin capture mAb S12 was substituted with an irrelevant mIgG1. Binding of P-selectin-bearing RBCs to Y46/48/51F-expressing cells was only slightly higher than nonspecific binding (Fig. 2 e), consistent with observations that at least one of the three tyrosines is required for PSGL-1 to bind P-selectin (10,12). Adhesions were also abolished when anti-PSGL-1 blocking mAb PL1, anti-P-selectin blocking mAb G1, or calcium chelator EDTA was present (Fig. 2, a–e, rows 4–6), demonstrating binding specificity. Similar results were obtained for binding of L-selectin-bearing RBCs to WT and mutant PSGL-1 expressing cells (Fig. S2, a–e). Furthermore, the adhesion frequencies of P- and L-selectin-coupled RBCs to cells expressing FucTVII and C2GnT without PSGL-1 were very low (data not shown). These results demonstrate that the adhesions measured were mediated by specific selectin-PSGL-1 interactions.
Figure 2.

(a–e) Binding specificity of PSGL-1 constructs to P-selectin. Adhesion frequency under each condition was obtained by fitting the Pa versus t curves to Eq. 1 and then setting t→∞. The curves were obtained using 6 to 59 cell pairs, each contacting 100 times, to estimate an adhesion frequency for a given contact duration. Data are presented as the mean ± SE. The test of P-selectin captured by S12 has a higher adhesion frequency than any controls and blocking tests (p < 0.01).
Kinetic measurements
Adhesion probabilities were measured using the micropipette assay at contact durations ranging from 0.05 to 12 s for P-selectin (Fig. 3) and L-selectin (Fig. 4) interacting with a WT (panel a), three double-mutant (panels b–d) and one triple-mutant (panel e) PSGL-1 constructs, respectively. Nonspecific adhesion frequency data were fitted by Eq. 1 to obtain a nonspecific binding curve, Pn(t) (dashed lines; data points not shown for clarity). The specific adhesion probability, Pa (solid lines), was obtained by removing Pn from the total adhesion frequency, Pt, according to Pa = (Pt − Pn)/(1 − Pn) (21). Pa followed a simple kinetics, exhibiting a transition phase when it increased with t and a steady phase when it reached equilibrium. The transient phases of the P-selectin curves (Fig. 3) were longer than those of the L-selectin curves (Fig. 4), consistent with previous reports that PSGL-1 dissociates from L-selectin much more rapidly than P-selectin (32). Each Pa versus t data set was fitted by Eq. 1 to obtain two kinetic parameters: the zero-force reverse rate, kr0, and the effective binding affinity, AcKa0, for the selectin density. The theoretical predictions (solid curves, based on the average of two sets of kr0 and AcKa0 values obtained from 76–150 cells and the corresponding selectin-site densities) describe both Pa versus t data sets well for each PSGL-1 construct (Figs. 3 and 4), providing confidence in the validity of the model and the reliability of the kinetic parameters.
Figure 3.

Binding curves of P-selectin for WT and mutant PSGL-1. Adhesion probability was plotted against contact duration for two densities (indicated) of P-selectin interacting with WT (a), Y46/48F (b), Y46/51F (c), Y48/51F (d), or Y46/48/51F (e) PSGL-1 at the indicated densities. Experimental data (points), presented as the mean ± SE at each contact time and obtained from 22–38 cell pairs for each curve, were compared with the predictions (solid lines) calculated from Eq. 1 using the averaged best-fit kinetic parameters and the corresponding mr and ml values. The dashed line represents nonspecific binding, obtained by fitting Eq. 1 to the nonspecific data (for clarity, 30 data points in a–e are not shown).
Figure 4.

Binding curves of L-selectin for WT and mutant PSGL-1. Adhesion probability was plotted against contact duration for two densities (indicated) of L-selectin interacting with WT (a), Y46/48F (b), Y46/51F (c), Y48/51F (d), or Y46/48/51F (e) PSGL-1 at the indicated densities. Experimental data (points), presented as the mean ± SE at each contact time and obtained from 36–59 cell pairs for each curve, were compared with the predictions (solid lines) calculated from Eq. 1 using the averaged best-fit kinetic parameters and the corresponding mr and ml values. The dashed line represents nonspecific binding, obtained by fitting Eq. 1 to the nonspecific data (for clarity, 35 data points in a–e are not shown).
Effects of tyrosine replacements on the binding kinetics of PSGL-1 to P-selectin
The different binding curves of WT and mutated PSGL-1 indicate different kinetics and affinities. To obtain the same level of steady-state adhesion frequency (e.g., Pa (∞) ∼ 0.3), the Y46/51F mutant (ml = 342 μm−2; lower solid curve in Fig. 3 c) required a 4.9-fold higher site density than the WT (ml = 70 μm−2; lower solid curve in Fig. 3 a) PSGL-1. However, they required the same time to reach the half-equilibrium binding level, t1/2. The equilibrium adhesion probability Pa(∞) is related to the effective binding affinity, AcKa0 = −ln[1 − Pa(∞)]/(mr×ml), and the half-time t1/2 is related to the reverse rate, kr0 ≈ 0.5/t1/2 (21). Therefore, the data indicate that the Y46/51F mutant binds to P-selectin with a lower effective binding affinity but a similar reverse rate, as compared with the WT PSGL-1. This was confirmed by the kinetic parameters obtained by fitting Eq. 1 to the Pa versus t data. The zero-force reverse rate for the Y46/51F mutant was similar to that observed for the WT (kr0 = 1.4 ± 0.2 vs. 1.3 ± 0.1 s−1, p > 0.44; Fig. 5 a), and the binding affinity for the Y46/51F mutant was 5.6-fold lower than that for the WT (AcKa0 = (2.71 ± 0.01) × 10−4 vs. (1.52 ± 0.29) × 10−3 μm4, p < 0.026; Fig. 5 b). These results suggest that replacing the two tyrosines at residues 46 and 51 significantly reduced the effective forward rate of PSGL-1 for P-selectin when both molecules were anchored on the apposing cell surfaces. Indeed, the value calculated from Ackf0 = AcKa0 × kr0 for the Y46/51F mutant was 5.0-fold lower than that for the WT (Ackf0 = 3.82 × 10−4 vs. 1.90 × 10−3 μm4·s−1, p < 0.018).
Figure 5.

Kinetic parameters for PSGL-1-P-selectin binding. Data are presented as the mean ± SE of reverse rates, kr0 (a), and effective binding affinities, AcKa0 (b), for P-selectin interacting with WT (solid bars), Y46/48F (open bars), Y46/51F (dotted bars), Y48/51F (leftward-hatched bars), and Y46/48/51F (rightward-hatched bars) PSGL-1. The reverse rate of WT is similar to that of Y46/51F (p > 0.44), Y48/51F (p > 0.14), and Y46/48/51F (p > 0.06), but lower than that of 46/48 (p < 0.001). The binding affinity of WT is higher than that of Y46/48F (p < 0.02), Y46/51F (p < 0.03), Y48/51F (p < 0.032), and Y46/48/51F (p < 0.02).
Similar results were obtained for the Y46/48F and Y48/51F mutants. Compared with the WT PSGL-1, their binding affinities (AcKa0 = (1.79 ± 0.30) × 10−4 and (3.94 ± 0.49) × 10−4 μm4, respectively) were 8.5- and 3.9-fold lower (p < 0.023 and p < 0.032, respectively; Fig. 5 b). The reverse rate for the Y46/48F mutant (2.41 ± 0.01 s−1) was slightly higher than that for the other PSGL-1 constructs (p < 0.028). The forward rates for the Y46/48F and Y48/51F mutants (Ackf0 = 4.31 × 10−4 and 4.07 × 10−4 μm4·s−1, respectively) were similar to that for the Y46/51F mutant but much lower than that for the WT (p < 0.026). Therefore, replacing two of the three PSGL-1 tyrosines by phenylalanines reduces the forward rate for P-selectin.
The P-selectin binding kinetics of the triple mutant (Y46/48/51F), however, was drastically different from those of the WT and three double mutants. Here the equilibrium adhesion frequency was marginally higher than that of nonspecific binding (Pa(∞) ∼ 0.1 in Fig. 3 e). Despite the noisy data, the adhesion frequency curves of the triple mutant provide a 1.2-fold higher reverse rate (kr0 = 1.62 ± 0.13 s−1) but a 49-fold lower binding affinity (AcKa0 = (3.08 ± 0.90) × 10−5 μm4) than those of the WT PSGL-1 (p < 0.06 for kr0 and p < 0.019 for AcKa0, respectively). The forward rate (Ackf0 = 5.00 × 10−5 μm4·s−1) is ∼8-fold lower than those for the double mutants (p < 0.036), and 38-fold lower than that for WT PSGL-1 (p < 0.026). These results confirm previous observations that at least one of the three tyrosine residues is required for PSGL-1 to access its receptors (10,12).
Effects of tyrosine replacements on binding kinetics of PSGL-1 to L-selectin
To further define the functional consequences of tyrosine replacement of PSGL-1, we studied the binding of the PSGL-1 constructs to L-selectin. Soluble L-selectin-Ig was captured by AP113 mAb on RBCs to interact with PSGL-1 constructs on CHO cells. Compared with the WT PSGL-1, the double mutants required 2.4- to 4.5-fold higher combined site densities (mr×ml) to reach comparable equilibrium adhesion frequencies (Pa(∞) ∼ 0.2–0.4), but similar times to reach the half-equilibrium binding level (Fig. 4). Fitting Eq. 1 to the Pa versus t curves yielded comparable reverse rates for the three double mutants (kr0 = 8.4 ± 0.1, 12.1 ± 1.1, and 9.4 ± 1.1 s−1 for Y46/48F, Y46/51F, and Y48/51F, respectively) with WT PSGL-1 (kr0 = 8.2 ± 0.06 s−1; p = 0.445, 0.086, and 0.695, respectively). In contrast, the binding affinities were much lower (5.5-, 3.5-, and 2.2-fold) for the Y46/48F, Y46/51F, and Y48/51F mutants (AcKa0 = (0.72 ± 0.06) × 10−5, (1.16 ± 0.02) × 10−5, and (1.85 ± 0.09) × 10−5 μm4) than that for the WT (AcKa0 = (3.98 ± 0.49) × 10−5 μm4; p < 0.001, 0.002, and 0.005, respectively; Fig. 6). The calculated forward rates were slightly varied in the Y46/48F, Y46/51F, and Y48/51F mutants (Ackf0 = (0.60, 1.39, and 1.73) × 10−4 μm4·s−1, respectively), but all were significantly lower than that for the WT (Ackf0 = 3.26 × 10−4 μm4·s−1; p < 0.002, 0.005, and 0.03, respectively). Substitutions of all three tyrosines, however, resulted in very low binding (Pa(∞) < 0.1) at densities comparable to those obtained for the three double mutants, confirming the lack of specific binding between the triple mutant and L-selectin.
Figure 6.
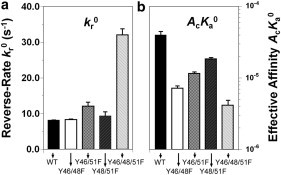
Kinetic parameters for PSGL-1-L-selectin binding. Data are presented as the mean ± SE of reverse rates, kr0 (a), and effective binding affinities, AcKa0 (b), for L-selectin Ig chimera interacting with WT (solid bars), Y46/48F (open bars), Y46/51F (dotted bars), Y48/51F (leftward-hatched bars), and Y46/48/51F (rightward-hatched bars) constructs. The reverse rate of WT is similar to that of Y46/48F (p > 0.44), Y46/51F (p > 0.08), and Y48/51F (p > 0.69), but lower than that of Y46/48/51F (p < 0.003). The binding affinity of WT is higher than that of Y46/48F (p < 0.001), Y46/51F (p < 0.002), Y48/51F (p < 0.005), and Y46/48/51F (p < 0.001).
We compared the impact of PSGL-1 mutations on the binding kinetics between L- and P-selectin. As expected from the more rapid dissociation kinetics of WT PSGL-1 from L-selectin compared with P-selectin, the PSGL-1 double mutants had 3.5- to 9.1-fold higher reverse rates (p < 0.006), 21.3- to 24.9-fold lower binding affinities (p < 0.015), and 2.4- to 7.2-fold lower forward rates (p = 0.018, 0.036, and 0.085 for Y46/48F, Y46/51F, and Y46/48/51F, respectively) for L-selectin than for P-selectin (Figs. 5 and 6).
Effects of electrostatic interactions and molecular orientation of PSGL-1 on selectin binding
To test whether the binding difference between WT and mutated PSGL-1 constructs resulted from modifying the electrostatic interactions by replacing a negatively charged tyrosine with a neutral phenylalanine, and/or from varying the molecular orientation by capturing P-selectin with S12 and the L-selectin Fc chimera with AP113, we performed two additional sets of measurements. In the first set, P-selectin Fc chimera captured by AP113 mAb was tested for interaction with the double mutant Y48/51F. The final ionic strength of the chamber solution was modified from 68.5 mM to 51.5 and 85.5 mM NaCl, respectively. In the single set of site densities of mr = 14 μm−2 for P-selectin and ml = 1,740 μm−2 for PSGL-1, binding reached a higher plateau but had a similar transition phase when the ionic strength was reduced from 68.5 (triangles) to 51.5 (squares) mM (Fig. 7). Fitting these data using Eq. 1 (lines) returned an increased binding affinity (AcKa0 = 1.42 × 10−5 and 1.95 × 10−5 μm4) and a similar reverse rate (kr0 = 0.8 and 1.0 s−1) for 68.5 and 51.5 mM, respectively. By contrast, a similar equilibrium plateau and a fast transition were observed when the ionic strength was increased to 85.5 (cycles) mM, which yielded a similar binding affinity (1.44 × 10−5 μm4) but a faster reverse rate (3.0 s−1; inset in Fig. 7). These results indicate that electrostatic interactions between the interacting molecules regulate their binding kinetics.
Figure 7.
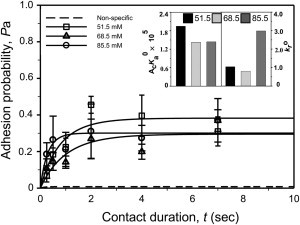
Binding of P-selectin Fc chimera captured by AP113 mAb to Y48/51F PSGL-1 in a final ionic concentration of 51.5 (squares), 68.5 (triangles), and 85.5 (cycles) mM NaCl. Data are presented as the mean ± SE for three to four cell pairs at each contact time and compared with the predictions (lines) calculated from Eq. 1 using the averaged best-fit kinetic parameters (inset) and the corresponding mr and ml values. The dashed line represents nonspecific binding, obtained by fitting Eq. 1 to the nonspecific data from a total of 18 cell pairs (for clarity, data points are not shown).
In the second set, we tested the impact of molecular orientation by comparing P-selectin captured by S12 mAb with P-selectin Fc chimera captured by AP113 mAb. As indicated in Figs. 5 and 7, we found a remarkable difference in binding affinity (AcKa0 = 3.94 × 10−4 and 1.42 × 10−5 μm4) but the reverse rate remained unchanged (kr0 = 1.0 and 0.8 s−1) for binding of Y48/51F to S12-captured P-selectin and AP113-captured P-selectin Fc chimera, respectively. This is consistent with our previous observation that the molecular orientation regulates the binding affinity by altering the forward rate but not the reverse rate (22).
Structural basis of tyrosine replacement on PSGL-1 binding to P-selectin
To elucidate the structural basis at the atomic level, we performed equilibration MD simulations. The SGP-3 peptide displayed significant conformational differences among the WT (cyan) and the three single mutants; the three double mutants of Y46/48F (blue), Y46/51F (red), and Y48/51F (orange); and the triple mutant of Y46/48/51F (green), whereas the SGP-3 O-glycan showed similar conformations except for the two residues linking to T57 of the peptide (Fig. 8 a). We compared the conformational differences using electrostatic (black) and total nonbonded (gray) interactions between the SGP-3 peptide and P-selectin lectin domain (Fig. 8 b). The total nonbonded interactions were further illustrated in all systems with similar orientations of key binding sites between FUC of SGP-3 O-glycan and the Ca2+ ion (Fig. 8 c), as well as comparable distances between Ca2+ and the mass center of O3 and O4 atoms of FUC (Fig. 8 d). These results indicate that the mutations of sulfated tyrosine residues affect the conformation of the peptide but not the glycan of PSGL-1 ligand.
Figure 8.
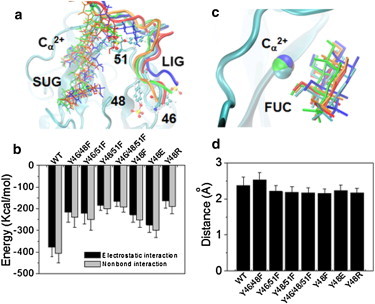
Structural features of P-selectin binding to PSGL-1 mutants. (a) Superposition of interaction interfaces (3-ns snapshot) of P-selectin lectin domain with WT, Y46/48F, Y46/51F, Y48/51F, and Y46/48/51F by aligning the rigid regions of P-selectin lectin domain. Calcium (Cα2+), O-glycan (SUG), and ligand peptide (LIG) are presented as VDW, bonds, and newcartoon, respectively. For clarity, only the P-LE domains and three sulfated tyrosine residues of 46, 48, and 51 in WT PSGL-1 are shown as newcartoon and CPK, respectively. (b) Electrostatic (black) and all nonbonded (gray) interactions between the SGP-3 peptide and P-selectin lectin domain for the eight systems. (c) Key interactions between Cα2+ and the O-glycan FUC. (d) Distance between Cα2+ and the mass center of O3 and O4 atoms of FUC. Data are shown as the mean ± SD of the last 1 ns equilibration process. For clarity, the Y48E, Y48F, and Y48R systems are not shown in panels a and c.
Discussion
The presence of three sulfated tyrosines at the PSGL-1 N-terminal region is crucial for the biological function of PSGL-1, and tyrosine replacement affects its binding to selectins (10,12,14–17). Here, using a micropipette adhesion frequency assay, we further quantified the 2D kinetics of interactions of WT as well as double and triple tyrosine mutants of PSGL-1 with P- and L-selectin. Our results confirm that at least one of the three tyrosine residues at the N-terminus of PSGL-1 is necessary to support binding to P- and L-selectin (10,12). Our zero-force reverse-rate data for WT PSGL-1 and P- or L-selectin are comparable to those previously measured by flow-chamber and atomic force microscopy techniques (10,20,22,25). In fact, the mean values of zero-force reverse rate in the previous flow-chamber study, extrapolated using the Bell model from cell-rolling dynamics at various shear stresses, yield 1.1–1.8 s−1 for P-selectin and 8.6–18.3 s−1 for L-selectin (10,12), which are close to the data obtained here (1.0–1.4 s−1 for P-selectin and 8.2–12.1 s−1 for L-selectin), as summarized in Table S1. The different mean values of the rates for double mutants measured by the two techniques are within a factor of 2. The discrepancy may be due to the distinct surface presentations of selectin molecules: transfected murine L1-2 preB cells expressing human P-selectin or L-selectin were used in the flow-chamber study, whereas P- and L-selectin were presented with the use of capturing mAbs on RBCs in our micropipette assay. The difference may also come from our limited temporal resolution (22,25), which produces less precise data, as reported in Figs. 5 a and 6 a.
Of more importance, in this work we measured 2D binding affinities and forward rates using a micropipette adhesion frequency assay, which revealed information that would be unobtainable with the previous flow-chamber technique. For example, compared with WT PSGL-1, the double mutants reduced the binding affinities for P- and L-selectin by one or two orders of magnitude, and reduced the forward rates accordingly. The differences may explain the different rolling dynamics of the PSGL-1 expressing cells under applied shear stresses in the flow-chamber study (10), because cell rolling is a dynamic process that involves bond formation in the front and bond rupture in the rear, and bond formation is governed by the forward rate whereas bond rupture is controlled by the reverse rate. The triple mutant has even lower forward rates and binding affinities to P- and L-selectin. It should be pointed out that tyrosine sulfation is also important for PSGL-1 binding. Although robust rolling adhesion on P-selectin was found to be mediated sufficiently by PSGL-1 mimics with tyrosine sulfation (SGP3) and without tyrosine sulfation (GP1) (37), the resulting rolling velocity was significantly lower for SGP3 than for GP1. These results imply that the binding capacity is higher for the former than for the latter, which would be consistent with the results of the work presented here, in which we removed not only the tyrosine sulfation but also the tyrosines themselves.
The influence of electrostatic interactions on protein-ligand binding was predicted by theoretical calculations (38–40) and demonstrated by experiments with different molecules (41,42). The crystal structure of P-selectin and an N-terminal glycosulfopeptide fragment of PSGL-1 reveals electrostatic interactions between selectin Lec domain residues and the sulfate esters on tyrosines 48 and 51 but not tyrosine 46 (8). It is conceivable that a significant conformational change in the binding pocket of P-selectin (e.g., core-2 glycan; Figs. 7 and 8) forms electrostatic interactions with the sulfate ester on tyrosine 46 that the crystal structure does not capture. Our results suggest that these interactions increase binding affinity primarily by augmenting the forward rate. Substitution of charged tyrosine(s) with neutralized phenylalanines may prevent electrostatic interactions with P-selectin. This explains the reduction in the forward rate and in turn the affinity for P-selectin. As reported in our previous studies (22,25), the forward rate strongly correlates with the molecular accessibility to its counterpart molecule. In the work presented here, we observed via MD simulations a significant reduction in the electrostatic interactions between PSGL-1 and the P-selectin lectin domain when the negatively charged tyrosines were replaced by the neutral phenylalanine (Fig. 8 c). This confirms that the reduction in forward rate most likely stems from the defective accessibility of these PSGL-1 mutants.
The 2D affinity of WT PSGL-1 for P-selectin (AcKa = 1.52 × 10−3 μm4) observed here is close to the value previously obtained from a micropipette assay (25), suggesting that the carrier property for transfected cells expressing FucTVII and C2GnT is similar to that for HL-60 cells. The affinity value is also consistent with reported in a previous work (mlAcKa = 0.25 μm2) (22), which yields AcKa = 4.0 × 10−3 μm4 calculated from the 63 μm−2 PSGL-1 site density on the HL-60 cells (25). Considering that the reverse rates are close to 1 s−1, these three reports show similar forward rates for P-selectin binding to WT PSGL-1. However, our P-selectin and L-selectin forward rates (Ackf0 = 1.90 × 10−3 and 3.26 × 10−4 μm4 s−1, respectively) are higher than those reported in a biomembrane force probe (BFP) study (Ackf0 = 1.4 × 10−4 and 5.9 × 10−5 μm4 s−1, respectively) that employed monomeric PSGL-1 constructs and a thermal fluctuation assay (32). Because the monomeric PSGL-1 construct was found to bind selectin with an affinity similar to that obtained for native, dimeric PSGL-1 from human neutrophils (11,43), the discrepancies may result from the contact area, Ac, which acts as a multiplying factor in the effective forward rate, Ackf0. The thermal fluctuation assay used two small rigid beads to present selectin and PSGL-1, respectively, yielding a contact area 5–10 times smaller than that obtained with our micropipette assay, which would explain the lower effective forward rates observed in the previous study.
In summary, our results demonstrate that specific tyrosine residues in PSGL-1 make major contributions to the 2D forward rates for interactions with P- and L-selectin.
Acknowledgments
We thank Cindy Carter, Tadayuki Yago, Juan Chen, Jun Huang, Yuxin Gao, Baoxia Li, and Changliang Fu for technical assistance.
This work was supported by the National Natural Science Foundation of China (grants 30730032, 11072251, and 10902117), the National Key Basic Research Foundation of China (grants 2011CB710904 and 2006CB910303), the Strategic Priority Research Program (grants XDA01030102 and XDA04020219), the Knowledge Innovation Program of the Chinese Academy of Sciences (grant 2005-1-16 to M.L.), and the National Institutes of Health (grants AI77343 to C.Z. and R.P.M., HL090923 to R.P.M., and TW 05774-01 to C.Z. and M.L.).
Footnotes
Botao Xiao’s present address is Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, and Children’s Hospital, Boston, Massachusetts
Contributor Information
Cheng Zhu, Email: cheng.zhu@bme.gatech.edu.
Mian Long, Email: mlong@imech.ac.cn.
Supporting Material
References
- 1.McEver R.P., Zhu C. Rolling cell adhesion. Annu. Rev. Cell Dev. Biol. 2010;26:363–396. doi: 10.1146/annurev.cellbio.042308.113238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu C., Yago T., McEver R.P. Mechanisms for flow-enhanced cell adhesion. Ann. Biomed. Eng. 2008;36:604–621. doi: 10.1007/s10439-008-9464-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wagner D.D., Frenette P.S. The vessel wall and its interactions. Blood. 2008;111:5271–5281. doi: 10.1182/blood-2008-01-078204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu C., Long M., Bongrand P. Measuring receptor/ligand interaction at the single-bond level: experimental and interpretative issues. Ann. Biomed. Eng. 2002;30:305–314. doi: 10.1114/1.1467923. [DOI] [PubMed] [Google Scholar]
- 5.Moore K.L., Stults N.L., McEver R.P. Identification of a specific glycoprotein ligand for P-selectin (CD62) on myeloid cells. J. Cell Biol. 1992;118:445–456. doi: 10.1083/jcb.118.2.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez M., Joffraud M., Spertini O. Regulation of PSGL-1 interactions with L-selectin, P-selectin, and E-selectin: role of human fucosyltransferase-IV and -VII. J. Biol. Chem. 2005;280:5378–5390. doi: 10.1074/jbc.M410899200. [DOI] [PubMed] [Google Scholar]
- 7.Leppänen A., Yago T., Cummings R.D. Model glycosulfopeptides from P-selectin glycoprotein ligand-1 require tyrosine sulfation and a core 2-branched O-glycan to bind to L-selectin. J. Biol. Chem. 2003;278:26391–26400. doi: 10.1074/jbc.M303551200. [DOI] [PubMed] [Google Scholar]
- 8.Somers W.S., Tang J., Camphausen R.T. Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to SLe(X) and PSGL-1. Cell. 2000;103:467–479. doi: 10.1016/s0092-8674(00)00138-0. [DOI] [PubMed] [Google Scholar]
- 9.Leppänen A., White S.P., Cummings R.D. Binding of glycosulfopeptides to P-selectin requires stereospecific contributions of individual tyrosine sulfate and sugar residues. J. Biol. Chem. 2000;275:39569–39578. doi: 10.1074/jbc.M005005200. [DOI] [PubMed] [Google Scholar]
- 10.Ramachandran V., Nollert M.U., McEver R.P. Tyrosine replacement in P-selectin glycoprotein ligand-1 affects distinct kinetic and mechanical properties of bonds with P- and L-selectin. Proc. Natl. Acad. Sci. USA. 1999;96:13771–13776. doi: 10.1073/pnas.96.24.13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leppänen A., Mehta P., Cummings R.D. A novel glycosulfopeptide binds to P-selectin and inhibits leukocyte adhesion to P-selectin. J. Biol. Chem. 1999;274:24838–24848. doi: 10.1074/jbc.274.35.24838. [DOI] [PubMed] [Google Scholar]
- 12.Liu W., Ramachandran V., McEver R.P. Identification of N-terminal residues on P-selectin glycoprotein ligand-1 required for binding to P-selectin. J. Biol. Chem. 1998;273:7078–7087. doi: 10.1074/jbc.273.12.7078. [DOI] [PubMed] [Google Scholar]
- 13.Li F., Wilkins P.P., McEver R.P. Post-translational modifications of recombinant P-selectin glycoprotein ligand-1 required for binding to P- and E-selectin. J. Biol. Chem. 1996;271:3255–3264. [PubMed] [Google Scholar]
- 14.Pouyani T., Seed B. PSGL-1 recognition of P-selectin is controlled by a tyrosine sulfation consensus at the PSGL-1 amino terminus. Cell. 1995;83:333–343. doi: 10.1016/0092-8674(95)90174-4. [DOI] [PubMed] [Google Scholar]
- 15.Wilkins P.P., Moore K.L., Cummings R.D. Tyrosine sulfation of P-selectin glycoprotein ligand-1 is required for high affinity binding to P-selectin. J. Biol. Chem. 1995;270:22677–22680. doi: 10.1074/jbc.270.39.22677. [DOI] [PubMed] [Google Scholar]
- 16.Snapp K.R., Ding H., Kansas G.S. A novel P-selectin glycoprotein ligand-1 monoclonal antibody recognizes an epitope within the tyrosine sulfate motif of human PSGL-1 and blocks recognition of both P- and L-selectin. Blood. 1998;91:154–164. [PubMed] [Google Scholar]
- 17.Bernimoulin M.P., Zeng X.L., Spertini O. Molecular basis of leukocyte rolling on PSGL-1. Predominant role of core-2 O-glycans and of tyrosine sulfate residue 51. J. Biol. Chem. 2003;278:37–47. doi: 10.1074/jbc.M204360200. [DOI] [PubMed] [Google Scholar]
- 18.Bell G.I. Models for the specific adhesion of cells to cells. Science. 1978;200:618–627. doi: 10.1126/science.347575. [DOI] [PubMed] [Google Scholar]
- 19.Marshall B.T., Long M., Zhu C. Direct observation of catch bonds involving cell-adhesion molecules. Nature. 2003;423:190–193. doi: 10.1038/nature01605. [DOI] [PubMed] [Google Scholar]
- 20.Sarangapani K.K., Yago T., Zhu C. Low force decelerates L-selectin dissociation from P-selectin glycoprotein ligand-1 and endoglycan. J. Biol. Chem. 2004;279:2291–2298. doi: 10.1074/jbc.M310396200. [DOI] [PubMed] [Google Scholar]
- 21.Chesla S.E., Selvaraj P., Zhu C. Measuring two-dimensional receptor-ligand binding kinetics by micropipette. Biophys. J. 1998;75:1553–1572. doi: 10.1016/S0006-3495(98)74074-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang J., Chen J., Long M. Quantifying the effects of molecular orientation and length on two-dimensional receptor-ligand binding kinetics. J. Biol. Chem. 2004;279:44915–44923. doi: 10.1074/jbc.M407039200. [DOI] [PubMed] [Google Scholar]
- 23.Huang J., Zarnitsyna V.I., Zhu C. The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature. 2010;464:932–936. doi: 10.1038/nature08944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Long M., Zhao H., Zhu C. Kinetic measurements of cell surface E-selectin/carbohydrate ligand interactions. Ann. Biomed. Eng. 2001;29:935–946. doi: 10.1114/1.1415529. [DOI] [PubMed] [Google Scholar]
- 25.Wu L., Xiao B.X., Long M. Impact of carrier stiffness and microtopology on two-dimensional kinetics of P-selectin and P-selectin glycoprotein ligand-1 (PSGL-1) interactions. J. Biol. Chem. 2007;282:9846–9854. doi: 10.1074/jbc.M609219200. [DOI] [PubMed] [Google Scholar]
- 26.Geng J.G., Bevilacqua M.P., McEver R.P. Rapid neutrophil adhesion to activated endothelium mediated by GMP-140. Nature. 1990;343:757–760. doi: 10.1038/343757a0. [DOI] [PubMed] [Google Scholar]
- 27.Mehta P., Patel K.D., McEver R.P. Soluble monomeric P-selectin containing only the lectin and epidermal growth factor domains binds to P-selectin glycoprotein ligand-1 on leukocytes. Blood. 1997;90:2381–2389. [PubMed] [Google Scholar]
- 28.Patel K.D., Nollert M.U., McEver R.P. P-selectin must extend a sufficient length from the plasma membrane to mediate rolling of neutrophils. J. Cell Biol. 1995;131:1893–1902. doi: 10.1083/jcb.131.6.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kofler R., Wick G. Some methodologic aspects of the chromium chloride method for coupling antigen to erythrocytes. J. Immunol. Methods. 1977;16:201–209. doi: 10.1016/0022-1759(77)90198-3. [DOI] [PubMed] [Google Scholar]
- 30.Selvaraj P., Plunkett M.L., Springer T.A. The T lymphocyte glycoprotein CD2 binds the cell surface ligand LFA-3. Nature. 1987;326:400–403. doi: 10.1038/326400a0. [DOI] [PubMed] [Google Scholar]
- 31.Zhu C. Kinetics and mechanics of cell adhesion. J. Biomech. 2000;33:23–33. doi: 10.1016/s0021-9290(99)00163-3. [DOI] [PubMed] [Google Scholar]
- 32.Chen W., Evans E.A., Zhu C. Monitoring receptor-ligand interactions between surfaces by thermal fluctuations. Biophys. J. 2008;94:694–701. doi: 10.1529/biophysj.107.117895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 35.Lü S.Q., Long M. Forced dissociation of selectin-ligand complexes using steered molecular dynamics simulation. Mol. Cell. Biomech. 2005;2:161–177. [PubMed] [Google Scholar]
- 36.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 37.Rodgers S.D., Camphausen R.T., Hammer D.A. Tyrosine sulfation enhances but is not required for PSGL-1 rolling adhesion on P-selectin. Biophys. J. 2001;81:2001–2009. doi: 10.1016/S0006-3495(01)75850-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baker N.A., Sept D., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hendsch Z.S., Tidor B. Do salt bridges stabilize proteins? A continuum electrostatic analysis. Protein Sci. 1994;3:211–226. doi: 10.1002/pro.5560030206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vijayakumar M., Wong K.Y., Zhou H.X. Electrostatic enhancement of diffusion-controlled protein-protein association: comparison of theory and experiment on barnase and barstar. J. Mol. Biol. 1998;278:1015–1024. doi: 10.1006/jmbi.1998.1747. [DOI] [PubMed] [Google Scholar]
- 41.McLaughlin S., Wang J., Murray D. PIP(2) and proteins: interactions, organization, and information flow. Annu. Rev. Biophys. Biomol. Struct. 2002;31:151–175. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- 42.Schreiber G., Fersht A.R. Energetics of protein-protein interactions: analysis of the barnase-barstar interface by single mutations and double mutant cycles. J. Mol. Biol. 1995;248:478–486. doi: 10.1016/s0022-2836(95)80064-6. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Y., Sun G.Y., Long M. Low spring constant regulates P-selectin-PSGL-1 bond rupture. Biophys. J. 2008;95:5439–5448. doi: 10.1529/biophysj.108.137141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.