

Abstract
We present the unique case of a two-component collagen peptide hydrogel that self-assembles through non-covalent electrostatic interactions. Natural collagen materials, such as those of connective tissue or the basement membrane, assemble in a hierarchic fashion. Similarly, the synthetic peptides presented here proceed from monomer to trimer to fiber, and finally to a hydrogel. By varying stoichiometry and concentration, we are able to dissect the stages of higher order assembly. Insight gained from this study will improve molecular design of biomimetic materials.
Keywords: collagen, fiber, composition, high order structure, hydrogel
Collagen has many favorable properties making it an attractive target for biomaterials: low antigenicity, biodegradability, low toxicity and high biocompatibility 1,2. Currently, animal-derived collagens are utilized in biomedical, cosmetic and food science applications, but they are costly to purify and run the risk of prion contamination 1,3. Synthetic collagens could circumvent some of these issues and potentially allow greater control of material properties.
α-helical or β-hairpin peptides have been successfully designed to form self-assembling materials 4. Although natural collagens are widely used, only a few recent examples of designed collagen peptide materials exist5. A greater diversity of available crosslinking elements will facilitate the design of complex functional biomaterials. Different molecular strategies, such as using sidechain hydrophobic and electrostatic interactions, disulfide or metal mediated cross-linking, have successfully induced higher order assembly of collagen-like peptides6,7,8. The assembled structures ranged from particles and fibers to sheets. Here, we use electrostatic interactions not only to drive fiber formation but also to further induce assembly of a hydrogel, a transparent, highly hydrated polymer network. Hydrogels are a useful phase of biomaterials that have clear applications in tissue regeneration and drug delivery 9,10.
We hypothesize that using heterotrimeric collagen peptides will allow control over the higher order assembly and chemical functionality of biomaterials. Recently, there has been significant progress in the design of heterotrimeric collagen peptides11, and disulfide cross-linking of neutral collagen peptides has been used to design heterotrimers assemble into fibrils8. The two peptides described in this study use electrostatic interactions between acidic and basic amino acids to form heterotrimers that further associate. By varying peptide stoichiometry and total concentration, we are able to modulate the nature of higher order assembly.
In previous work, three Charged Peptides (referred herein as CPA, CPB and CPC) were designed to form a mixture of heterotrimers12. Although it was found that a 2:1 ratio of CPB to CPC formed stable and soluble triple-helices, a 1:2 mixture resulted in rapid aggregation at low temperatures. This discrepancy motivated us to explore the effects of composition on higher order assembly. Below, we demonstrate the complex dependence of assembly on concentration and mixing stoichiometry of CPB and CPC.
The relationship between composition and assembly is best visualized using a phase diagram where the axes are peptide concentration (Figure 1). The relative mixing ratio of the two peptides divides the phase diagram into two domains. The upper domain where [CPB] > [CPC] is characterized solely by soluble monomeric or triple-helical species. The lower domain where [CPB] ≤ [CPC] yields properties that are pertinent for materials development, and mixtures form various higher-order assemblies. In this domain, total concentration determines whether aggregates form an opaque precipitate or a semi-transparent hydrogel. The relative ratio of CPB:CPC influences aggregate morphology, which can range from particles to fibers. We explore the mechanisms of assembly in the lower domain of this phase diagram.
Figure 1.
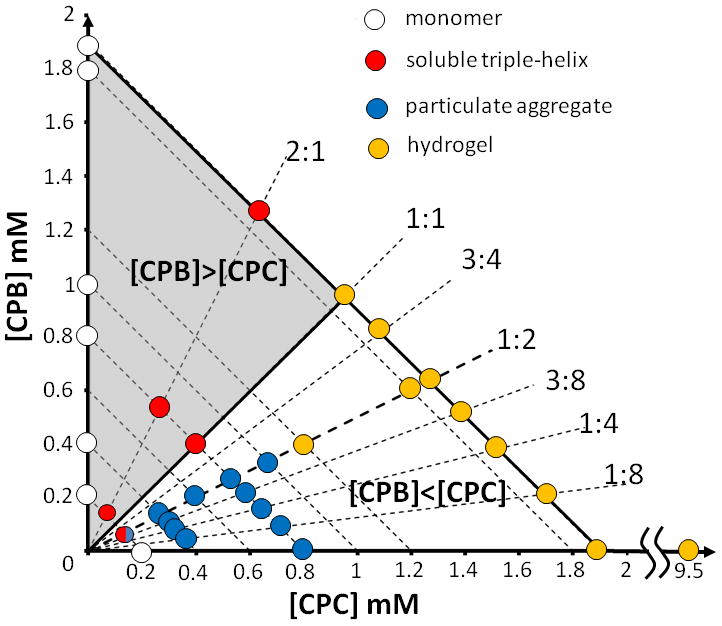
(a) Phase diagram of CPB and CPC combinations at 4 °C. Radial dashed lines constrain the peptide mixing ratio (annotated as CPB:CPC), while parallel lines constrain total peptide concentration. The domain where [CPB]>[CPC] is represented as the. (b) Sequences of CPB and CPC. P = proline, R = arginine, E = glutamic acid, O = hydroxyproline.
Previously we observed that a 0.2 mM CPB:2CPC mixture aggregated overnight 12. Increasing the concentration above 0.3 mM significantly accelerates aggregation. By 0.8 mM, aggregation is essentially complete within thirty minutes (Figure 2a). The subsequent drop of signal is presumably due to settling of the precipitate.
Figure 2.
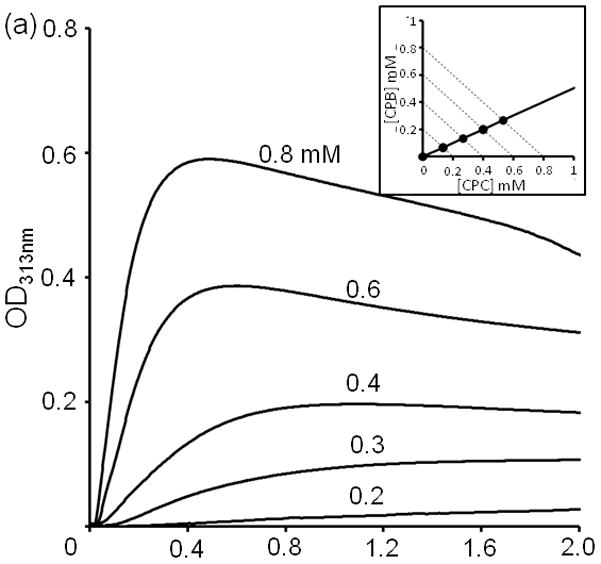
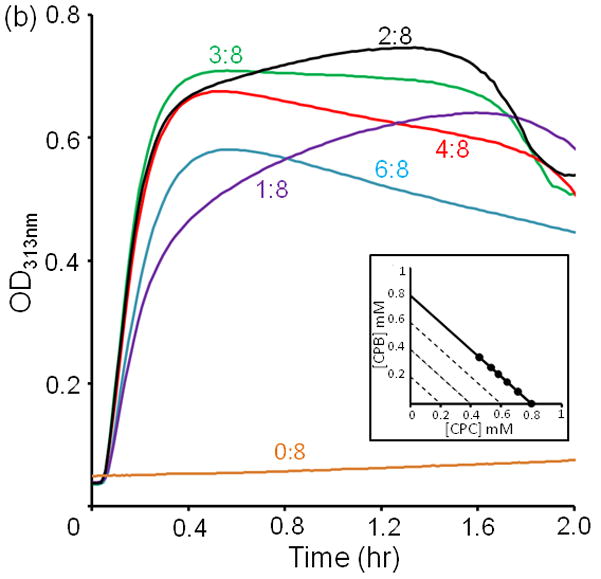
Turbidity kinetics at 4 °C of (a) CPB:2CPC for total concentration from 0.2 to 0.8 mM (b) CPB:CPC with various mixing ratios at a total concentration 0.8 mM.
The relative fraction of CPB and CPC in the mixture also has a significant impact on the rate of aggregation. Assemblies are monitored for a series of mixing ratios, holding the total concentration at 0.8 mM (Figure 2b). CPC alone requires two to three days to form visible precipitate. Aggregation is most efficient at 3:8. This is surprising because one would expect the optimal ratio for collagen peptide assembly to be consistent with formation of a trimeric species, i.e. 3:0, 1:2, 2:1 or 0:3. The increase of turbidity correlates with an increase in particle size as measured by multi-angle static light scattering (Figure S1). Again the particle size increases most rapidly at mixing ratio 3:8 even though the concentration is lower (0.1 mM). As these are kinetics experiments, it is necessary to explore whether these properties are relevant in the equilibrium composition of peptide aggregates.
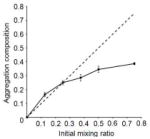
The observed non-canonical 3:8 mixing ratio raises the question whether CPB is physically part of the aggregate, or is somehow serving a catalytic role in nucleating assembly. To determine this, the peptide content of precipitates are determined by HPLC for several initial mixing ratios at 0.8 mM. Peptide CPB is found in the precipitate indicating that it was in fact physically part of the aggregate. When the starting ratio of CPB to CPC was 1:8, or 1:4, the resulting ratio of peptides in the precipitate matches the starting ratio (Table 1). However, as the relative amount CPB increased, we observe a limiting compositional ratio of approximately 3:8. This unusual property suggests that composition may be influenced by long range interactions at the level of higher order assembly rather than triple helix formation.
Table 1.
Initial mixing ratio vs. compositional ratio of aggregates of CPB:CPC obtained by HPLC. Peak areas of CPB and CPC were used to obtain final ratios. Final composition deviates significantly from the initial conditions for mixing ratios at 3:8 or above.
|
|
|
|
| Initial Mixing Ratios | Composition ratios of aggregates | |
|
| ||
| 1:8 | 1.32±0.13:8 | |
| 2:8 | 2.00±0.13:8 | |
| 3:8 | 2.29±0.33:8 | |
| 4:8 | 2.76±0.18:8 | |
| 6:8 | 3.10±0.07:8 | |
|
| ||
If long-range forces, presumably electrostatics, are responsible for mediating assembly, then screening charged sidechain interactions would prevent aggregation. As the salt concentration was increased, the aggregation rate and total extent of aggregate were reduced (Figure 3).
Figure 3.
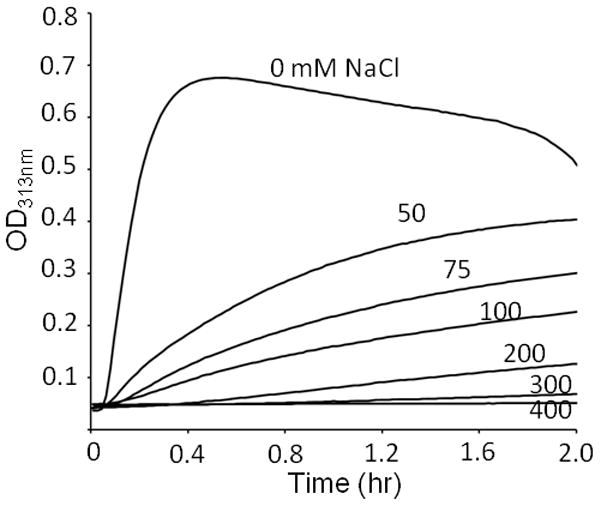
Salt effects on the turbidity of mixture 1CPB:2CPC at the total concentration 0.8 mM. Turbidity was monitored at 4 °C.
Although it was previously determined that mixtures of CPB and CPC formed triple helices, the 3:8 composition of the aggregate calls into question whether secondary structure is required for higher order assembly. Perhaps the zwitterionic nature of CPC is sufficient to drive assembly through nonspecific electrostatic interactions. The following two observations supports a secondary structure prerequisite for assembly: (1) the rate and extent of aggregation decreases with increasing temperature and (2) directly inhibiting triple-helix formation by mutating core glycines prevents aggregation.
If the assembly of CPB and CPC requires a folded intermediate, then raising the temperature should denature the intermediate, preventing or slowing the rate of aggregation at a specific concentration. A 0.8 mM 1:2 mixture was rapidly cooled from a temperature well above the observed melting transition (Figure S2). Aggregation was most efficient at 4°C. No aggregation was observed at 15 °C, even after several hours.
If aggregation is mediated by nonspecific electrostatic interactions, replacing CPC with a modified sequence that has a similar charge distribution but cannot form triple-helices might still promote aggregation. To test this hypothesis, we synthesized CPC-G/P, in which all ten glycines in CPC were replaced by prolines. A circular dichroism (CD) spectrum of CPC-G/P indicates a polyproline II random-coil state (Figure S3a) instead of a triple helix. A 1:2 mixture of CPB:CPC-G/P also lacked secondary structure as measured by CD. Small-angle X-ray scattering (SAXS) measurements are best fit with circular cylinder model at Guinier region to ~15 × 90 Å for an individual rod of CPB:2CPC 13, which is reasonable for a thirty amino-acid long triple helix (Figure S4(d)). Fitted SAXS data for CPB:2CPC-G/P is consistent with a random coil configuration (Figure S4). More importantly, mutating CPC prevented aggregation (Figure S3(b)). A triple-helical intermediate rather than an unfolded state is necessary for the formation of higher-order structure.
Transmission Electron Microscopy (TEM) images of the CPB/CPC assemblies reveals a transition in aggregate morphology when the composition is 3:8 (Figure 4). CPC alone requires high concentrations to get reasonable aggregation densities for EM imaging and are observed to form curly, gnarled fibers. Addition of small amount of CPB where CPB:CPC ≤ 3:8, results in elongated, granular particles. In contrast, peptide compositions above 3:8 formed fibers of uniform thickness up to 1 micrometer in length, often found in bundles where the individual fibers are clearly visible. Below 3:8, many of the images show heterogeneous mixtures of structures, including the coexistence of granular particles and long-thin fibers. Above 3:8, the fiber morphologies are consistently observed in multiple preparations (Figure S5).
Figure 4.
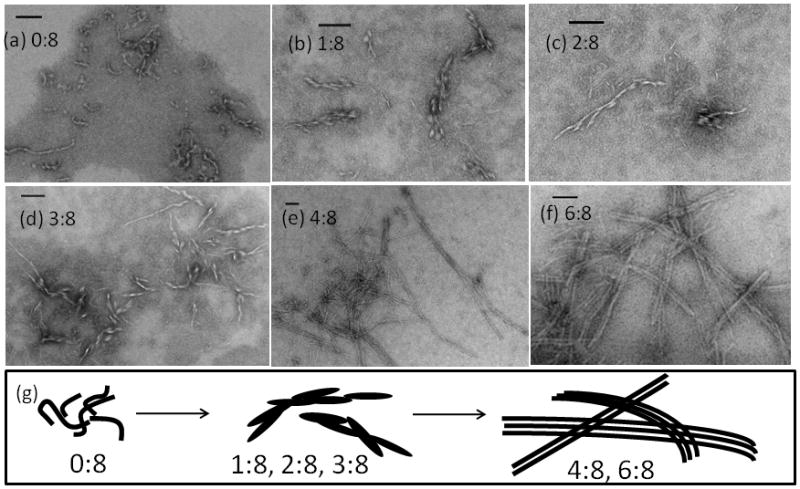
Electron Microscopy images of negatively stained CPB:CPC samples at various mixing ratios (a) 0:8, (b) 1:8, (c) 2:8, (d) 3:8, (e) 4:8, and (f) 6:8 in pH 7, 10 mM phosphate buffer. (g) Schematic sketch of fiber morphology under various mixing ratio. The scale bar is 100 nm. Except for CPC alone or 0:8 at 0.8 mM, all the samples were prepared at the same total concentration, 0.4 mM, and incubated at 4 °C for ~72 hrs.
The thickness of individual fibers is uniformly ~12 nm, near the length of a thirty-residue triple-helix, suggesting that individual peptides may be aligned perpendicular to the fiber axis. The addition of two amino acids at the N-terminus of CPB did not disrupt aggregation or fiber morphology (Table S1 and Figure S6). In other synthetic peptides and some natural collagens, periodic banding along the fiber axis is caused by coherent gaps between adjacent chains 7,14. Such banding is not observed in this system, and would not be expected if peptides are associating perpendicular to the fiber axis.
At concentrations above 0.8 mM, CPB:2CPC mixtures are semi-transparent rather than opaque. Samples do not flow when the tube containing them is inverted, suggesting formation of a peptide hydrogel. This is confirmed using micro-rheological measurements. The trajectories of 1.0 μm fluorescent polystyrene beads embedded in samples are videotaped and analyzed to determine the mean square displacement as a function of time 15. At total concentrations of less than 0.8 mM, the mixture behaves as a viscous solution where G″ > G′ at all frequencies (Figure 5 and S7). Above this concentration, G′ exceeds G″, indicating formation of a soft hydrogel. CD of a 2 mM CPB:2CPC sample in a thin cuvette showed the presence of collagen triple helix in the hydrogel (Figure S8). Although others have synthesized collagen peptide hydrogels using disulfide crosslinking, the peptide concentrations required for gelation are ten to twenty-fold higher (~10% versus 0.6% weight/volume).
Figure 5.
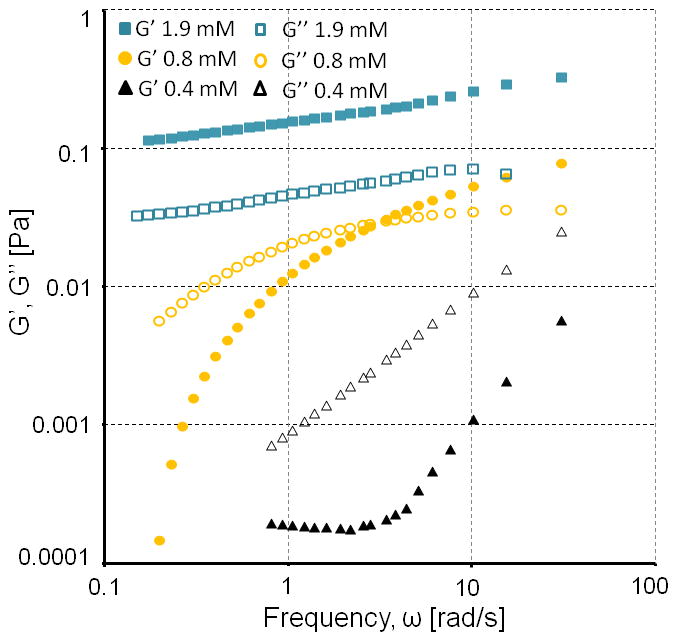
Storage and loss moduli G′ and G″ of CPB:2CPC at 4°C.
It is possible to control the nature of the assembly and material properties of the two-component system by varying the mixing ratio and total concentration of the charged peptides. The advantage of the two-component system is the ability to generate a library of compositions with tunable material properties. We have only begun to characterize the rich behavior of these peptides, and there are still significant unexplored areas of this phase diagram that may present unique behaviors in terms of aggregate morphology or material rheology. Multi-component systems might be used to generate spatial gradients within a material such as discrete or gradual changes in mechanical properties or density of ligand functionalization. Such applications would require further optimization of material, perhaps by replacing arginine with lysine and glutmate with aspartate, thus forming more stable ion pairs16, or by including structured hydrophobic domains10 could improve the practical utility of this system.
The disconnect between the optimal peptide ratio for aggregation and the canonical stoichiometry of a collagen triple-helix highlights a key challenge in rational design of peptide biomaterials. The current design process is focused on optimizing atomic-scale interactions such as protein topology or intermolecular association. The ability of the assembling fiber to constrain the peptide composition to ~ 3 CPB : 8 CPC suggests that long-range forces are playing a significant role in specifying the structure of the final assembly. We suspect that for ratios greater than 3:8 the charge density of cationic CPB is too high to allow efficient aggregation, and above 1:1 no higher-order assembly of any kind is observed. As such, the assembly is able to maintain the ratio of charged to neutral peptides. The repulsive interactions may also direct morphology by favoring a uniform fiber over granular particles10,17. These types of interactions represented uncharted territory for computational protein design, and will require the development of new mutliscale computational methods that treat the structure of fibrous proteins atomistically and consider protein fibers as polyelectrolytes in higher order association processes18.
Supplementary Material
Acknowledgments
Funding Sources
FX, VN - NIH DP2 OD006478-01, NSF DMR-0907273; JL, QH - USDA National Research Initiative (#2009-35603-05071); RT, VJ - NSF DMR-1006407
ABBREVIATIONS
- CPB,CPC
charged peptide B, C
- CD
circular dichroism spectroscopy
- TEM
transmission electron microscopy
- SAXS
small-angle X-ray scattering
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. Methods, data analysis, additional figures and tables. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lee CH, Singla A, Lee Y. Intl J Phar. 2001;221:1. doi: 10.1016/s0378-5173(01)00691-3. [DOI] [PubMed] [Google Scholar]
- 2.Friess W. Eur J Phar Biopharm. 1998;45:113. doi: 10.1016/s0939-6411(98)00017-4. [DOI] [PubMed] [Google Scholar]; Rao KP. J Biomater Sci. 1995;7:623. doi: 10.1163/156856295x00526. [DOI] [PubMed] [Google Scholar]
- 3.O’Grady JE, Bordon DM. Adv Drug Deliv Rev. 2003;55:1699. doi: 10.1016/j.addr.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 4.Pandya MJ, Spooner GM, Sunde M, Thorpe JR, Rodger A, Woolfson DN. Biochemistry. 2000;39:8728. doi: 10.1021/bi000246g. [DOI] [PubMed] [Google Scholar]; Pochan DJ, Schneider JP, Kretsinger J, Ozbas B, Rajagopal K, Haines L. J Am Chem Soc. 2003;125:11802. doi: 10.1021/ja0353154. [DOI] [PubMed] [Google Scholar]
- 5.O’Leary LER, Fallas JA, Bakota EL, Kang MK, Hartgerink JD. Nat Chem. 2011;3:821. doi: 10.1038/nchem.1123. [DOI] [PubMed] [Google Scholar]
- 6.Kar K, Ibrar S, Nanda V, Getz TM, Kunapuli SP, Brodsky B. Biochemistry. 2009;48:7959. doi: 10.1021/bi900496m. [DOI] [PMC free article] [PubMed] [Google Scholar]; Przybyla DE, Chmielewski J. J Am Chem Soc. 2008;130:12610. doi: 10.1021/ja804942w. [DOI] [PubMed] [Google Scholar]; Cejas MA, Kinney WA, Chen C, Leo GC, Tounge BA, Vinter JG, Joshi PP, Maryanoff BE. J Am Chem Soc. 2007;129:2202. doi: 10.1021/ja066986f. [DOI] [PubMed] [Google Scholar]
- 7.Rele S, Song Y, Apkarian RP, Qu Z, Conticello VP, Chaikof EL. J Am Chem Soc. 2007;129:14780. doi: 10.1021/ja0758990. [DOI] [PubMed] [Google Scholar]
- 8.Kotch FW, Raines RT. Pro Natl Acad Sci USA. 2006;103:3028. doi: 10.1073/pnas.0508783103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petka WA, Harden JL, McGrath KP, Wirtz D, Tirrell DA. Science. 1998;281:389. doi: 10.1126/science.281.5375.389. [DOI] [PubMed] [Google Scholar]; Nagarkar RP, Hule RA, Pochan DJ, Schneider JP. J Am Chem Soc. 2008;130:4466. doi: 10.1021/ja710295t. [DOI] [PubMed] [Google Scholar]
- 10.Nowak AP, Breedveld V, Pakstis L, Ozbas B, Pine DJ, Pochan D, Deming TJ. Nature. 2002;417:424. doi: 10.1038/417424a. [DOI] [PubMed] [Google Scholar]
- 11.Gauba V, Hartgerink JD. J Am Chem Soc. 2007;129:2683. doi: 10.1021/ja0683640. [DOI] [PubMed] [Google Scholar]
- 12.Xu F, Zhang L, Koder RL, Nanda V. Biochemistry. 2010;49:2307. doi: 10.1021/bi902077d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ilavsky J, Jemian PR. J App Cryst. 2009;42:347. [Google Scholar]; Roe RJ. Methods of X-ray and Neutron Scattering in Polymer Sciences. Oxford University Press; 2000. [Google Scholar]
- 14.Papapostolou D, Smith AM, Atkins EDT, Oliver SJ, Ryadnov MG, Serpell LC, Woolfson DN. Proc Natl Acad Sci USA. 2007;104:10853. doi: 10.1073/pnas.0700801104. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kadler KE, Hojima Y, Prockop DJ. Biochem J. 1990;268:339. doi: 10.1042/bj2680339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mason TG, Weitz DA. Phys Rev Lett. 1995;74:1250. doi: 10.1103/PhysRevLett.74.1250. [DOI] [PubMed] [Google Scholar]; Zimenkov Y, Dublin SN, Ni R, Tu RS, Breedveld V, Apkarian RP, Conticello VP. J Am Chem Soc. 2006;128:6770. doi: 10.1021/ja0605974. [DOI] [PubMed] [Google Scholar]
- 16.Gauba V, Hartgerink JD. J Am Chem Soc. 2007;129:15034. doi: 10.1021/ja075854z. [DOI] [PubMed] [Google Scholar]
- 17.Nowak AP, Breedveld V, Pine DJ, Deming TJ. J Am Chem Soc. 2003;125:15666. doi: 10.1021/ja0381050. [DOI] [PubMed] [Google Scholar]
- 18.Dobrynin AV, Rubinstein M. Progress in Polymer Science. 2005;30:1049. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.