

Abstract
Restrictive cardiomyopathy (RCM) has been linked to mutations in the thin filament regulatory protein cardiac troponin I (cTnI). As the pathogenesis of RCM from genotype to clinical phenotype is not fully understood, transgenic (Tg) mice were generated with cardiac specific expression of an RCM-linked missense mutation (R193H) in cTnI. R193H Tg mouse hearts with 15% stoichiometric replacement had smaller hearts and significantly elevated end diastolic pressures (EDP) in vivo. Using a unique carbon microfiber-based assay, membrane intact R193H adult cardiac myocytes generated higher passive tensions across a range of physiologic sarcomere lengths resulting in significant Ca2+ independent cellular diastolic tone that was manifest in vivo as elevated organ-level EDP. Sarcomere relaxation and Ca2+ decay was uncoupled in isolated R193H Tg adult myocytes due to the increase in myofilament Ca2+ sensitivity of tension, decreased passive compliance of the sarcomere, and adaptive in vivo changes in which phospholamban (PLN) content was decreased. Further evidence of Ca2+ and mechanical uncoupling in R193H Tg myocytes was demonstrated by the biphasic response of relaxation to increased pacing frequency verses the negative staircase seen with Ca2+ decay. In comparison, non-transgenic myocyte relaxation closely paralleled the accelerated Ca2+ decay. Ca2+ transient amplitude was also significantly blunted in R193H Tg myocytes despite normal mechanical shortening resulting in myocyte hypercontractility when compared to non-transgenics. These results identify for the first time that a single point mutation in cTnI, R193H, directly causes elevated EDP due to a myocyte intrinsic loss of compliance independent of Ca2+ cycling or altered cardiac morphology. The compound influence of impaired relaxation and elevated EDP represents a clinically severe form of diastolic dysfunction similar to the hemodynamic state documented in RCM patients.
Keywords: restrictive cardiomyopathy, hypertrophic cardiomyopathy, cardiac troponin I
Introduction
Inherited restrictive cardiomyopathy (RCM) is a malignant heart muscle disease that can transition to heart failure and early morbidity [1, 2]. RCM is relatively rare, evident in roughly 2% of diagnosed inherited cardiomyopathy cases [1, 2]. The clinical hallmark of RCM is near normal systolic function accompanied by marked diastolic dysfunction resulting from impaired ventricular filling and an extremely stiff heart [1, 2]. Ventricular wall thickness in RCM hearts is normal or near normal with variable fibrosis that can range from non-existent to severe [1]. RCM was thought to be idiopathic until the recent identification of six mutations in the gene that encodes cardiac troponin I (cTnI), TNNI3, cosegregated with RCM in human patients [3].
Cardiac TnI is a key component of the thin filament protein complex responsible for regulating cardiac muscle contraction. It functions as a molecular switch that inhibits actin-myosin interaction during diastole. During systole, cTnI moves from its inhibitory position on actin to one in contact with troponin C (cTnC) in response to intracellular elevation in the activating ligand Ca2+ [4]. This TnI switch further facilitates the binding of Ca2+ to cTnC [4]. The cTnI locus is a hotspot for cardiomyopathy mutations, as over twenty different mutations have been linked to either hypertrophic cardiomyopathy (HCM) [5] or those associated with RCM [3]. HCM is clinically heterogeneous disease, distinct from RCM, with an estimated prevalence of 0.2% of the population [2]. In contrast to RCM, the clinical hallmarks of HCM include ventricular hypertrophy with myocyte disarray and fibrosis, normal or hyperdynamic systolic function, and diastolic dysfunction [2, 6–9]. HCM is the most common cause of sudden cardiac death in athletes and adolescents [9]. Approximately 75% of the identified cardiomyopathy mutations in cTnI cluster in the carboxy-terminal domain (C-terminal, amino acids 164-210). TnI’s C-terminus contains a secondary actin-tropomyosin binding domain that is necessary for fully inhibiting cross-bridge cycling during diastole [10–13]. Delineating the mechanistic basis of how mutations in the same functional domain of cTnI can result in the distinct clinical presentations of RCM or HCM has remained elusive.
The primary effects of the highly malignant RCM C-terminal mutant cTnI, R193H, on cardiac myocyte function have been elucidated using acute adenoviral gene transfer to adult cardiac myocytes [14]. In that study, the R193H mutant cTnI caused a novel Ca2+ independent cellular diastolic tone and hypersensitized the myofilaments to Ca2+, which slowed myocyte relaxation and Ca2+ transient decay. Biochemical reconstitution preparations also found that R193H cTnI increased myofilament Ca2+ sensitivity in ATPase and tension assays [15–17]. The R193H mutation has been engineered into the human cTnI sequence and expressed in a published mouse model in which a 25% replacement elicited a small increase in myofilament Ca2+ sensitivity [18] that manifest itself in vivo as diastolic dysfunction beginning around 6 months of age and becoming more prominent with a loss of myocardial compliance at 12 months of age [19, 20]. From these studies it was concluded that myocardial diastolic dysfunction in R193H mice can be ascribed to the following properties measured in isolated myocytes: short end diastolic sarcomere lengths, slow of relaxation, and slow Ca2+ transient decay that occurs in the absence of altered SR-load or Ca2+ handling proteins [18]. These results are similar to those obtained with acute adenoviral gene transfer of rodent R193H cTnI to isolated cardiac myocytes.
Despite these published results several key questions still remain unanswered. (1) What is the physiologic basis for diastolic dysfunction in R193H Tg mice and is cardiac performance altered with physiologic stressors? (2) Given that R193H mutant myocytes have a high Ca2+-independent diastolic tone and altered cellular morphology, do R193H cTnI Tg mice exhibit altered left ventricular end diastolic pressures? (3) If R193H mice have reduced myocardial compliance is it due to cell intrinsic or extrinsic properties? (4) Given the current paradigm that Ca2+ sensitizing cardiomyopathic sarcomeric proteins increase the buffering capacity of the myofilaments, does Ca2+ handling adapt to accommodate an R193H cTnI that elicits a greater change in myofilament Ca2+ sensitivity when expressed in vivo?
To address these important questions we generated independently a cTnI R193H Tg mouse model that, in contrast to the previously published transgenic R193H mouse, was engineered with rodent cTnI and found to elicit a 3-fold increase in myofilament Ca2+ sensitivity despite a low level of replacement. Using real time hemodynamic measurements we uncovered the new finding that R193H cTnI Tg mice have significantly elevated LV end diastolic pressure (EDP) that becomes more severe with increased expression of R193H cTnI. Notably this elevated organ-level EDP could be directly attributed to the significantly reduced compliance in R193H Tg myocytes as demonstrated by their resistance to passively stretch. This poor cellular distensibility, in combination with the high Ca2+-independent diastolic tone described in R193H myocytes [14], suggests that the resistance to passive stretch is due to over-activation of the R193H myofilaments during diastole. Furthermore functional measurements in isolated myocytes directly demonstrate that diastolic dysfunction, elevated LV EDP, and poor compliance are Ca2+-independent as the Ca2+ cycling was uncoupled from myofilament function in isolated R193H Tg myocytes.
Taken together, the results of altered myocardial passive-elastic properties (altered pressure-volume loops), loss of cellular compliance and slowed relaxation provide a basis for the in vivo diastolic dysfunction and poor running performance of R193H Tg mice. Unlike previous reports, the expression of Ca2+ handling proteins were altered in this R193H model which is likely due to homeostatic preservation of Ca2+ handling in the context of highly Ca2+ sensitized and “stiff” myofilaments. Despite the corrected Ca2+ transient decay myocyte relaxation still remained impaired suggesting that the R193H mutant cTnI directly influences cardiac performance. Overall R193H transgenic mice have a unique hybrid cardiomyopathic phenotype characterized by elevated diastolic pressures, diastolic dysfunction, and hypercontractility.
Methods
Engineering transgenic mice carrying R193H mutant cTnI construct
The R193H Tg construct utilized a cardiac specific α-myosin heavy chain (α-MHC) promoter (kindly provided by Dr. Jeffry Robbins, Children’s Hospital, Cincinnati, OH) to drive expression of murine R193H mutant cTnI (Figure 1A). This construct also contained a 3′ flag epitope tag, which has been shown previously not to affect isolated myocyte Ca2+ sensitivity of tension, pH sensitivity, shortening, or Ca2+ cycling when expressed [14, 21, 22]. Previous reports of transgenic animals expressing cTnI with a 5′ flag tag have also been shown to function normally [23–25]. The R193H construct was enzymatically removed from a pDC315 vector (Admax, Microbix) [14] by EcoRI digestion and subcloned into pBluescript 2SK plasmid (Stratagene). A second subcloning step was performed using restriction enzymes XhoI and NotI to transfer the R193H cTnI construct into the plasmid containing the αMHC promoter and SV40 polyadenylation sequence (Figure 1A). The transgenic construct was verified by DNA sequencing and given to the transgenic animal core (University of Michigan) for DNA purification and injection into C57BL/6 X SJL F2 fertilized eggs and surgically transplanted into pseudopregnant females. Ninety-five potentially transgenic mice were PCR screened for the R193H transgenic construct using the following primer pairs: sense 5′ AGACAGATCCCTCCTATCTC 3′ located in the αMHC promoter and antisense 5′ CTTTCGGCCTTCCATTCCACTTAG 3′ located in the cTnI 3′ untranslated region. Eleven PCR positive founders were identified. Three positive founders died within two weeks of obtaining the Tg mice. Five lines were established by backcrossing positive founders to C57BL/6 mice (Jackson Labs). One line produced no detectable transgene expression by Western blot. Another independent line produced pups with 25% replacement; however, transmission of the transgene was extremely low (20% vs. the expected 50%). The founder and 2 additional F1 positive breeders from this line never had more than 3 litters and died of early unknown causes, with no noticeable signs of cardiac altered growth or dilation although the mice were found ~24 hours post-mortem. This line was terminated shortly after determining its replacement given the difficulty of maintaining this line. For all experiments, N2-N3 progeny from line 594 (R193H TG) were studied, and NTG littermates were used as controls. In attempt to increase the level of transgene expression, double transgenic mice were developed by brother-sister matings from line 594 (R193H DTG) in some experiments. All animals were used and cared for according to principles outlined in the NIH Guide for the care and use of laboratory animals (NIH publication no. 85-23. Revised 1985) and protocols reviewed and approved by the Institutional Animal Care and Use Committee of the University of Minnesota.
Figure 1. The expression and replacement of mutant R193H cTnI in transgenic mouse hearts.
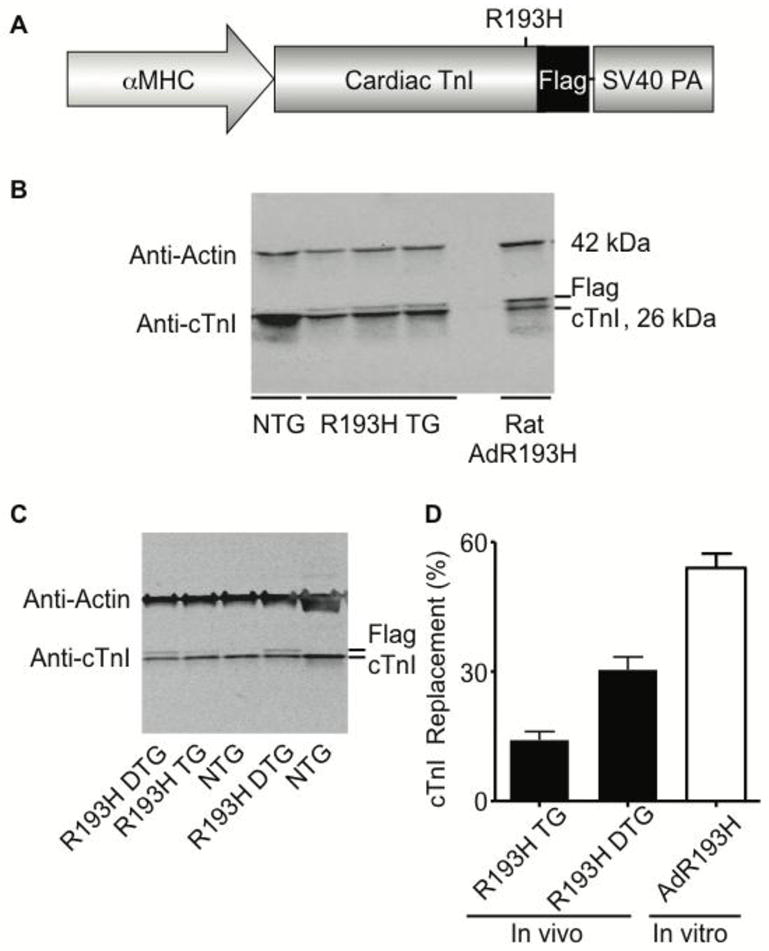
(A) Transgenic construct containing the cardiac specific α-myosin heavy chain (αMHC) promoter driving expression of a C-terminal flag epitope-tagged mutant R193H cTnI with a simian virus 40 (SV40) polyadenylation signal. (B) Representative Western blot of whole heart homogenates from R193H Tg line 594, non-transgenic (NTG), and rat myocytes adenovirally transduced for 2.5 days with R193H cTnI. (C) Representative Western blot of whole heart homogenates from R193H double Tg mice derived from inbreeding line 594 (R193H DTG) and single Tg mice from line 594 (R193H TG). Blots were probed with anti-cTnI and anti-sarcomeric actin specific primary antibodies. Anti-cTnI antibody displays the relative replacement of the native cTnI with the flag epitope-tagged R193H cTnI. There were no detectable changes in other thin filament proteins. (D) A summary of the percent replacement of native cTnI for the R193H mutant cTnI. Values mean ± SEM, n=6.
Mouse Echocardiography
Six to seven-month old NTG and R193H Tg mice were lightly anesthetized by isoflurane and prepared for echocardiography as previously described [22]. M-mode imaging of the parasternal short axis view was used to assess systolic and diastolic dimensions in addition to septal (IV septal thickness) and posterior wall (PW) thickness. M-mode imaging was also used to calculate fractional shortening (FS) and ejection fraction (EF). Pulsed-wave spectral Doppler was used to measure mitral valve inflow, which creates two waves, early (E wave) and late atrial (A wave), from which deceleration time (DT) and isovolumetric relaxation time (IVRT) could be measured. Mitral valve inflow parameters were used as an index of diastolic function [26]. Tissue Doppler imagining provided early (Esa) and late (Asa) tissue velocities of the septal annulus and early (Ela) and late (Ala) tissue velocities of the lateral annulus. Both parameters were used as indices of diastolic tissue velocities [26].
In vivo cardiovascular hemodynamics
Murine cardiovascular hemodynamics were measured using previously described conductance micromanometry [27]. Briefly, mice were anesthetized with 1% isoflurane by tracheal cannulation. A pressure-volume catheter (1.0F, PVR-1045; Millar Instruments Inc.) was inserted into the right carotid artery and advanced into the left ventricle. Baseline pressure-volume loops were measured first and then hemodynamic data was collected during 5 minutes of continuous esmolol (250 μg/kg/min) infusion. Transient inferior vena cava (IVC) occlusions were performed in order to measure end-systolic and end-diastolic pressures.
Tissue histology
NTG and R193H Tg mouse hearts were excised and fixed in 4% paraformaldehyde. Heart tissue was thoroughly washed in phosphate buffered saline (PBS) and prepared for paraffin wax embedding. Four-micron paraffin sections were cut with the entire heart being surveyed. Sections were mounted on slides and stained with hematoxylin and eosin (H&E) or Masson’s trichrome (MTC).
Mouse myocyte isolation and preparation for primary culture
Hearts were removed from heparinized (0.1 cc) and anesthetized adult NTG and R193H Tg mice (Nembutal; 100 mg/kg) and cannulated for retrograde perfusion. Ventricular cardiac myocytes were isolated by liberase blendzyme (Roche) digestion followed by a brief gentle mechanical digestion. Post digestion the myocytes were resuspended in MEM (Modified Eagles Medium) plus 5% FBS (fetal bovine serum). Myocytes were counted and approximately 20,000 cells were plated on laminin-coated coverslips for two hours, and media was subsequently replaced with culturing media consisting of MEM modified with 1% L-glutamine and 10% BSA (bovine serum albumin). Functional testing was performed immediately after the 2 hour incubation period and media exchange.
Measurement of steady-state Ca2+ activated isometric force
Single rod-shaped cardiac myocytes were attached to micropipettes coated in silicone adhesive and permeabilized in 0.2% Triton X-100 as previously described [14, 28]. The isometric tension-pCa relationship was constructed by measuring the calcium activated isometric tension at various Ca2+ concentrations at room temperature as previously described [14, 28]. The pCa (−log[Ca2+]) of the relaxation solution was 9.0 (nominal Ca2+, pCa=9.0) and the pCa of the maximal activating solution was 4.0 (maximal activation, pCa=4.0). A non-linear least squares fitting algorithm was used to determine the Hill coefficient (nH) and pCa50, the Ca2+ concentration at which 50% of maximal isometric tension was developed [14, 28].
Measurement of intact myocyte passive tension-extension relationship
Methods for measuring passive tension during physiologically relevant cell extension (cellular preload assay) from intact isolated cardiac myocytes have been previously described [29, 30]. Briefly, single intact murine cardiac myocytes were placed in a custom chamber containing platinum plated electrodes for stimulating the myocytes at 37°. A single myocyte was attached via micro-carbon fibers to a force transducer (200 mV mg-1, Aurora Scientific) and a piezoelectric actuator (P-173, PI Polytec) for motion control [30]. Sarcomere length and myocyte extension were measured by digitizing video recordings of the cell undergoing length excursions. Passive stretches to physiologic sarcomere lengths ranging from 1.8–2.2μm were performed.
Unloaded dynamic myocyte contractility and Ca2+ transient measurements
Sarcomere length measurements were performed using the Ionoptix system (Ionoptix Co., Milton, MA), which permits the mounting of a coverslip containing cultured murine myocytes to a custom chamber where it is bathed in Tyrode’s solution (1.8mM [Ca2+]) and kept at 37°C. The myocytes can then be viewed through a microscope and supramaximally electrically stimulated at 40 volts via parallel platinum electrodes at a frequency of 0.5 Hz. Sarcomere length and Ca2+ transients were measured in real time and averaged over 10–12 contraction cycles. Ca2+ transients were measured simultaneously using a fluorescent Ca2+ indicator, Fura-2 AM (Molecular Probes, 5 μM) loaded for 4 minutes and measured as previously described [31]. In some experiments provided in supplemental figure 5, pairwise comparisons of myocytes + isoproterenol (10nM, Sigma) were made. Stimulation-frequency experiments were performed utilizing the previously described mechanical and Ca2+ imaging methods under steady-state conditions [14, 32]. Stimulation frequency was varied from 0.5, 0.75, 1.0, 2.0, to 4.0 Hz. At each frequency a minimum of 12 contractions was used for the myocyte to achieve a steady state and 8 subsequent contractions were used for analysis.
SDS-gel electrophoresis and Western blots
To assess expression and incorporation of the R193H mutant cTnI transgene into the sarcomere, whole heart homogenates were made in Laemmli sample buffer modified with protease inhibitors and prepared for gel electrophoresis by boiling and sonication [14, 22]. In some instances, isolated myocytes were scraped into Laemmli sample buffer and prepared for gel electrophoresis as previously described [14]. Prior to Western blotting, a dose-response curve (amount of protein X signal) was performed and 20 micrograms was at the optimal detection range. Proteins were separated on a 4–20% precast SDS-page gel (Criterion, Biorad) and prepared for Western blot analysis by the Odyssey system (Licor Biosciences). The following primary antibodies and dilutions were used for immunodection: MAB directed against the flag epitope (M2, 1:1000, Sigma) and a polyclonal antibody directed against cTnI (A1627, 1:1000, Chemicon). Secondary antibodies were fluorophore conjugated goat anti-rabbit and goat anti-mouse (1:5000, Molecular Probes, Invitrogen) for detection by Odyssey. For Ca2+ handling protein expression the following antibodies were used: Serca2 (C-20, 1:500, Santa Cruz), Phospholamban (clone A1, 1:1000, Upstate), Phospho-phospholamban (ser16, 1:1000, Upstate), Sodium-calcium exchanger (R3F1, 1:1000, Swant), and Calsequestrin (1:1000, Upstate).
Results
Limited tolerance for R193H cTnI sarcomeric incorporation in vivo
The 5.6 kb murine α-myosin heavy chain promoter (αMHC) and rodent cTnI cDNA with a single amino acid conversion of arginine (R) to histidine (H) at codon 193 was used to generate multiple R193H cTnI transgenic lines (Figure 1A). The transgene expression cassette contained a 3′ flag epitope to distinguish the transgene product from endogenous cTnI. The cTnI flag tag was previously shown not to alter myocyte function [14]. Five lines were PCR positive for the transgene and were further analyzed for the percent replacement of native cTnI by mutant R193H cTnI. Representative Western blots probed with anti-cTnI (Figure 1B, bottom panel) and anti-sarcomeric actin demonstrate the expression of the transgene in R193H Tg line 594 and expression compared to isolated adult cardiac myocytes that were adenovirally transduced for 2.5 days with the R193H mutant cTnI construct. Cardiac TnI replacement was determined by measuring the ratio of the higher molecular weight cTnI, which represents the flag-tagged R193H cTnI, to total cTnI expression by densitometry. Total cTnI was normalized to silver stain as a loading control as before [33–35]. Thin filament protein stoichiometry did not change in R193H Tg hearts, as assessed by determining the relative expression of other thin filament proteins such as actin and tropomyosin (data not shown) [23, 36]. All of the Tg lines had low levels of stoichiometric replacement ranging from 7 to 25%, but the highest expressing line (25%) proved difficult to breed, as the average litter size was 3 ± 0.3 pups per birth (across multiple breeding pairs). Therefore, the second highest expressing line, 594 (TG), with 15% replacement (Figure 1C and D) was studied. In order to examine the functional consequences of higher levels of replacement, double Tg mice (DTG) were developed by brother-sister matings of the 15% replacement line, and from this cross pups with ~30% replacement were obtained (Figure 1C and D). The lack of a high replacement line, on par for example for the significantly higher levels of R193H incorporation obtained in vitro with adenoviral gene transfer to isolated adult rat cardiac myocytes shown here (~60% day 2.5 post gene transfer; Figure 1B,D) and previously (~90% incorporation at day 4 post gene transfer) [14], is evidence that the R193H transgene is not well tolerated in the working heart in vivo. No alterations in mortality rates, compared to Ntg, were observed in R193H Tg or DTG mice, at least over the one year period that this was examined.
RCM mutant cTnI significantly alters diastolic function in vivo independent of marked changes in cardiac morphology
Survival of R193H TG and DTG mice was not different from NTG littermates up to at least one year of age. Histological analysis of longitudinal-sections and cross-sections of hearts from R193H Tg mice 6–7 months of age revealed no significant changes in cardiac fibrosis, inflammation, or myofibrillar disarray relative to NTG littermates (Supplement Figure 1). Heart-to-body weight ratios (Table 1) and left ventricular (LV) end diastolic chamber volume (EDV, Figure 2A), as determined by M-mode echocardiography of 6–7 month old mice, were significantly smaller in R193H Tg relative to NTG mice. A significant increase in circumferential shortening (Vcf) was also detected in both R193H TG and R193H DTG mice (Figure 2B). The R193H DTG cohort demonstrated a significant increase in shortening rate beyond that of the lower replacement R193H TG mice (Figure 2B) and evidence of a R193H gene dosage effect on some cardiovascular functions in vivo.
Table 1.
Baseline echocardiographic measurements of R193H and non-transgenic mice
| NTG (n=21) | R193H TG (n=30) | R193H DTG (n=11) | |
|---|---|---|---|
| M-Mode Echocardiography | |||
| Posterior Wall Thickness (mm) | 0.45±0.04 | 0.56±0.03 | 0.58±0.04 |
| LV End Diastolic Dimension (mm) | 4.04±0.07 | 3.78±0.06* | 3.57±0.06* |
| LV Systolic Dimension (mm) | 2.90±0.08 | 2.39±0.07* | 2.16±0.08* |
| Fractional Shortening (%) | 28±1 | 37±1* | 41 ±1* |
| Ejection Fraction (%) | 63±2 | 74±1* | 79±1* |
| Heart Weight/Body Weight (g) | 4.2±0.2 | 3.7±0.1* | 3.4±0.1* |
| Doppler Echocardiography | |||
| Mitral Valve E wave (mm/s) | 1249±43 | 1033±41* | 1081±43* |
| Mitral Valve E/A wave (mm/s) | 1.78±0.12 | 1.45±0.07* | 1.45±0.08* |
| Isovolumetric Relaxation Time (ms) | 18.14 ±0.66 | 19.53±0.68 | 20.56±2.92 |
| Tissue Doppler Echocardiography | |||
| Septal Ea/Aa wave (mm/s) | 1.2±0.1 | 1.2±0.1 | 1.1±0.4 |
| Lateral Ela/Ala wave (mm/s) | 1.23±0.08 | 0.99±0.06* | 0.84±0.07* |
| Lateral annulus Ela wave (mm/s) | 25.9±1.4 | 20.6±1.1* | 18.2±1.9* |
| Emitral valve/Ela (E/Ela, mm/s) | 48.2±2.7 | 50.1±2.4 | 67.2±6.5*+ |
Data are presented as mean ± SEM.
One-way ANOVA, Newman Keuls post-tests were used for statistical testing with * P<0.05 and NS = not significant. There were no significant interactions found for any of the described parameters.
Key: IV = intraventricular, LV = left ventricle, E = early wave, A = late wave (atrial contraction).
Figure 2. Echocardiographic measurements of R193H TG and NTG mice and the effects of R193H-dependent cardiac dysfunction on voluntary running.

(A) Summary of left ventricular end diastolic dimension. (B) Summary of circumferential shortening (Vcf). (C) Summary of tissue Doppler lateral annulus Ela/Ala ratio. (D) Summary of the ratio between the mitral inflow early wave (E′) and the lateral annulus early wave (Ela) which serves as an index of diastolic pressure. Data are presented as mean ± SEM. One-way ANOVA, Newman Keuls post-hoc tests were used for statistical testing, P<0.05, n>10 (see Table 1). Correlation between average running bouts per night time active period and echocardiography parameters (E) ejection fraction, (F) E′/Ela, and (G) Ela/Ala average. There was a significant population effect in which R193H Tg mice with heightened ejection fraction (E) and poor diastolic function (F and G) ran fewer bouts per night when compared to NTG littermates with healthy cardiac function.
As summarized in Table 1, functional measurements were also obtained from Doppler and tissue Doppler echocardiography while the mice were kept under light isoflurane anesthesia. The average heart rate of NTG and R193H Tg mice during these measurements was 481± 20 beats/minute (BPM). Tissue Doppler measurements of the lateral mitral annulus, which provides early LV relaxation velocity (Ela) and late LV relaxation velocity during atrial contraction (Ala), showed a significant reduction in LV relaxation velocity and by default a decreased Ela/Ala ratio in R193H Tg mouse hearts (Figure 2C). The significant transgene effect on Ela/Ala ratio is evidence of diastolic dysfunction, a clinical hallmark of both RCM and HCM. Interestingly, the E′/Ela ratio, which correlates to pulmonary wedge pressures [26], was also significantly increased in R193H Tg mouse hearts suggesting these mice have elevated left ventricular diastolic pressure (Figure 2D). There were no significant age-dependent functional differences in transgenic mice spanning 5–10 months of age and diastolic dysfunction was already apparent by 3–4 months of age (data not shown). In separate isolated hanging heart (Langendorff) studies, R193H TG heart contractility (supplement Figure 2A) and relaxation derivatives (supplement Figure 2B) were not statistically different from NTG hearts.
In parallel studies, the voluntary running behavior of Tg mice was examined to determine if cardiac dysfunction would translate to alterations in whole animal exercise fitness. Compared to NTG mice, R193H mice had a significant reduction in the number of running bouts per day, while the average distance run/day was not altered (Supplement Figure 3). In this study subset, echocardiography analysis revealed a significant segregation of genotypes (NTG v TG) when ejection fraction, E/Ela and Ela/Ala were plotted vs. running bouts per day (Figure 2E–G). We speculate that the reduction in running bouts per day is due to the impaired cardiac performance of R193H mice, which deleteriously affects their natural behavior to run.
To further examine the effect of the R193H cTnI transgene on global cardiac function a cohort of 8–9 month old double transgenic (R193H DTG) and single transgenic (R193H TG) mice were studied using Millar catheter implantation for real-time acquisition of in vivo hemodynamics as shown in the representative pressure-volume loops in Figure 3A. This age was selected as echocardiography demonstrated no significant age-dependent functional differences in transgenic mice spanning 5–10 months of age. At baseline both single and double Tg R193H (DTG) mice had compromised diastolic performance as revealed in the pressure-volume loops (Figure 3B), elevated end diastolic pressures (EDP, Figure 3C), and increased rate constant for isovolumetric relaxation (Tau, Figure 3D). Notably, both R193H TG and DTG cohorts had significantly elevated EDP (Figure 3C) similar to the relaxation parameters measured by tissue Doppler imaging (Figure 2D). Elevated EDP is a phenotype characteristic of RCM in patient populations [1]. The baseline hemodynamic dysfunction of R193H Tg mice was independent of the level of transgene replacement as there was no significant difference between the single and double transgenic (R193H TG and R193H DTG) cohorts despite the different levels of cTnI replacement (15% and 30%).
Figure 3. In vivo hemodynamic function of 8–9 month old R193H Tg and NTG mice by conductance micromanometry.

(A) Representative raw pressure-volume traces from R193H double Tg (R193H DTG, n=6), R193H single Tg (R193H TG, n=9), and NTG mice (n=8). (B) Representative pressure-volume loops from R193H double Tg (R193H DTG), R193H single Tg (R193H TG), and NTG mice. Summary of (C) end diastolic pressure (EDP) and (D) the relaxation constant Tau. Data are presented as mean ± SEM. One-way ANOVA, Newman Keuls post-hoc tests were used for statistical testing, P<0.05. The effects of manipulating β-blockade on (E) stroke volume (SV), (F) end diastolic pressure (EDP), and (G) the relaxation constant Tau. Data are presented as mean ± SEM. Two-way ANOVA, Bonferroni post-hoc tests were used for statistical testing, P<0.05.
To address how R193H cTnI affects cardiac performance in vivo under conditions in which cardiac demands are altered, hemodynamic function was assessed during an acute bout of β-blockade using esmolol infusion. Esmolol treatment markedly reduced stroke volume (SV) in NTG mice; whereas, R193H Tg mice were unaffected (Figure 3E). Similarly, esmolol significantly increased EDP (Figure 3F) and the relaxation constant Tau (Figure 3G) in NTG mice, whereas these parameters in R193H Tg mice were not significantly changed by β-blockade.
R193H mutant cTnI results in a Ca2+ independent passive tension at physiologic sarcomere lengths
For all in vitro single myocyte measurements, single adult Tg R193H (TG) myocytes were used, as there were no statistically significant differences between 15% (R193H TG) and 30% (R193H DTG) cTnI replacement on these contractile parameters at the cellular level. To provide insight into the molecular basis for the high EDP and small chamber volumes of R193H Tg hearts, single adult cardiac myocytes were acutely isolated from 3–4 month old R193H Tg and NTG hearts. Membrane intact cardiac myocytes were then attached to a force transducer and piezoelectric device via micro-carbon fibers for measuring passive tension-extension across a range of physiologically relevant sarcomere lengths (SL,1.8–2.2 μm) [29]. At diastolic SL (1.9 μm) and over a series of extensions to longer sarcomere lengths, R193H myocytes generated markedly higher passive tension relative to NTG myocytes (Figure 4A). As such, R193H myocytes had a lower maximum tolerated sarcomere length of ~2.05 μm while the NTG myocyte could be extended over a normal range to ~2.2 μm (Figure 4B). Together these data demonstrate that R193H myocytes are extremely stiff, thus hindering the passive diastolic extension range of the cell. The unloaded resting SL of R193H Tg myocytes was 35% shorter (1.75 μm vs. 1.9 μm, Figure 4C) than NTG in the absence of detected changes in diastolic Ca2+ (Figure 4D). Taken together, these finding suggest that R193H myocytes generate a significant Ca2+ independent diastolic tone, similar to previous reports from acute gene transfer of R193H mutant cTnI to isolated rat cardiac myocytes [14]. Notably, the elevated diastolic tone provides a cellular correlate to the elevated in vivo EDP measured in R193H Tg mice.
Figure 4. The effects of the R193H cTnI transgene on intact myocyte passive tension and diastolic [Ca2+] over a range of sarcomere lengths.

(A) R193H single Tg myocytes generate significantly higher passive tensions over sarcomere lengths (SL) that range from 1.9–2.1μm at diastolic [Ca2+]. (B) The maximal sarcomere length extension tolerated by R193H single Tg myocytes (~2.07μm) is significantly less than the extensions tolerated by NTG (>2.2μm). (C) The R193H transgene causes a significantly smaller myocyte resting SL relative to NTG myocytes despite having (D) similar diastolic [Ca2+]. Data is presented as the mean ± SEM, * P<0.05 significantly different, t-test, n=10 myocytes in passive tension-extension assays and n=40 myocytes in resting SL and diastolic [Ca2+] assays.
R193H mutant cTnI enhances Ca2+ activated tension causing a myofilament-based diastolic dysfunction
To further understand the cellular basis for the diastolic dysfunction measured in R193H Tg mice in vivo, Ca2+ activated isometric tension development was measured in membrane permeabilized acutely isolated myocytes. Maximal isometric tension (P0) development was not different between R193H and NTG myocytes (31.8±3.2 and. 26.5±2.3 kN/m2 respectively). Only 15% replacement of native cTnI with R193H cTnI was necessary to alter myofilament cooperativity (R193H Tg nH=1.87±0.11 vs. NTG nH=2.97±0.25) and to produce a significant leftward shift of the tension-pCa relationship in R193H mutant myocytes (Figure 5). This left shift in the Ca2+-tension relationship resulted in a pCa50 that was 0.31 units higher (ΔpCa50=0.31) in R193H relative to NTG myocytes (Figure 5B), indicating that low levels of R193H cTnI replacement markedly enhance myofilament sensitivity to Ca2+.
Figure 5. Ca2+ activated tension in single permeabilized R193H Tg cardiac myocytes.

(A) Representative tension-pCa relationships for R193H single Tg (TG) and non-transgenic (NTG) permeabilized single myocytes. (B) Effects of non-transgenic (NTG) and R193H Tg (TG) on Ca2+ sensitivity (pCa50) of tension generation in adult cardiac myocytes at pH 7.0. Maximum tension generation (P0) was not different, but the Hill coefficient (nH) was significantly lower in R193H Tg myocytes (nH=1.87±0.11) versus NTG myocytes (nH=2.97±0.25). Data represents mean ± SEM, * P<0.001, n=9.
Given the heightened myofilament Ca2+ sensitivity in R193H myocytes, baseline contractile function and Ca2+ transients were simultaneously measured at 37°C in unloaded intact R193H Tg and NTG myocytes isolated from mice 3–4 months of age and summarized in Table 2. Figure 6A displays a representative SL shortening transient from NTG and R193H Tg myocytes, normalized to peak shortening amplitude in order to illustrate the effects of the R193H mutant cTnI on slowing sarcomere relaxation. Preliminary studies on R193H Tg myocytes isolated from mice 8 weeks of age indicated these myocytes already had a mechanical relaxation defect. When quantified, R193H myocytes from 3–4 month old mice were nearly twice as slow to relax relative to NTG (Figure 6B). R193H cTnI myocytes did not elicit significant changes in Ca2+ transient decay time (Figure 6C). This result is opposite to the effects of acute adenoviral expression of R193H cTnI in which both mechanical relaxation and the decay of the Ca2+ transient were tightly coupled and markedly slowed [14]. The uncoupling of mechanical relaxation from the Ca2+ transient in R193H Tg myocytes suggests that Ca2+ handling mechanisms have adapted in order to compensate for the gain in myofilament Ca2+ sensitivity in vivo.
Table 2.
Unloaded R193H and NTG myocyte contractile and Ca2+ transient parameters
| Sarcomere Shortening Parameters
| ||||||
|---|---|---|---|---|---|---|
| Resting SL (um) | Shortening Amplitude (um) | Dep Velocity (%amp) | 50% Relaxation Time (s) | 75% Relaxation Time (s) | 90% Relaxation Time (s) | |
| NTG | 1.835±0.008 | 0.103±0.009 | 19.69±0.49 | 0.0689±0.002 | 0.107±0.004 | 0.18±0.01 |
| R193H | 1.675±0.01* | 0.091±0.004 | 20.85±0.49 | 0.104±0.004* | 0.209±0.008* | 0.39±0.015* |
| Ca2+ Transient Parameters
| ||||||
|---|---|---|---|---|---|---|
| Diastolic Ca2+ (360/380) | Ca2+ Amplitude (360/380) | Rise Velocity (%amp) | 50% Decay Time (s) | 75% Decay Time (s) | 90% Decay Time (s) | |
| NTG | 1.14+0.02 | 0.115+0.012 | 87.7+8.6 | 0.141+0.004 | 0.266+0.012 | 0.42+0.03 |
| R193H | 0.09+0.02 | 0.055+0.004* | 105.2+10.5 | 0.139+0.005 | 0.250+0.011 | 0.40+0.02 |
Departure and Ca2+ transient rise velocity is normalized to shortening and Ca2+ amplitude. Relaxation and Ca2+ decay time is calculated from peak shortening to 50, 75, or 90% of relaxation.
Values are represented as the mean ± SEM,
P<0.05 significantly different, t-test, n = 30 myocytes from 5 preparations.
Key: SL=Sarcomere Length, Dep Velocity = sarcomere length shortening velocity, and Rise Velocity = rate of rise of the Ca2+ transient.
Figure 6. Intact single R193H Tg myocyte sarcomere length shortening and Ca2+ transients.

(A) Representative raw sarcomere shortening trace from intact myocytes isolated from single R193H Tg and non-transgenic (NTG, solid line). (B) Sarcomere shortening amplitude and (C) the corresponding Ca2+ transient amplitude. (D) Sarcomere relaxation time and (E) and the corresponding Ca2+ transient decay time. Relaxation and decay times were determined by calculating the difference between peak shortening and 75% relaxation. Data represent the mean ± SEM, * P<0.05 significantly different, t-test, n = 40 myocytes from 5 different preparations.
Further evidence of an uncoupling of Ca2+ cycling from mechanical performance is also demonstrated in cellular fractional shortening measurements in which shortening amplitude did not differ between R193H and NTG myocyte (Figure 6D), but the Ca2+ transient amplitude was significantly reduced in R193H myocytes (Figure 6E). This gain in shortening amplitude with less Ca2+ release suggests that R193H myocytes are hypercontractile similar to the heightened in vivo systolic function observed by M-mode echocardiography (Figure 2B). The enhancement of mechanical shortening for a given [Ca2+] in R193H myocytes could be explained by the heightened Ca2+ sensitivity of the myofilaments (Figure 5).
The effects of the β-adrenergic agonist, isoproterenol (Iso), on R193H Tg myocyte contractile function was also examined using pairwise comparisons. It is known that cTnI is a primary substrate for direct covalent modification via β-adrenergic mediated PKA (protein kinase A) phosphorylation (supplement figure 5) that causes a desensitization of the myofilaments to Ca2+. Acute exposure to isoproterenol revealed a significant positive lusitropic effect on myocytes in both groups (supplement figure 5). Notably, R193H Tg myocytes in the presence of isoproterenol were still unable to attain WT isoproterenol-mediated relaxation times, but the magnitude of inotropic and lusitropic responses were similar between NTG and R193H Tg myocytes, which concurs with previous reports [14, 18]. Likely the inability of R193H Tg myocytes to achieve the same relaxation rates as NTG would be expected to be detrimental during bouts of stress. In keeping with this point, R193H Tg mice performed less well at voluntary running (Figure 2E–G).
Enhanced pacing frequency uncouples cellular mechanical relaxation and Ca2+ transient decay
To further assess the uncoupling of mechanical function and Ca2+ handling at the cellular level, isolated R193H and NTG myocytes were subjected to progressive escalations in pacing frequency [14]. R193H Tg myocytes demonstrated a frequency-dependent uncoupling of mechanical function and Ca2+ handling that was not present in NTG myocytes (Figure 7). During escalations in pacing frequency, R193H Tg myocytes retained shorter resting sarcomere lengths and reduced diastolic [Ca2+] (Figure 7A and B). Resting sarcomere length had a frequency-dependent decline in diastolic sarcomere length due mostly to poor relaxation at higher frequencies. Both NTG and R193H Tg myocytes displayed typical negative staircase patterns [37] in which shortening amplitude decreased as a function of frequency (Figure 7C). Shortening amplitudes were not different among groups at any frequency, but the corresponding Ca2+ amplitudes were significantly reduced in R193H Tg myocytes (Figure 7D).
Figure 7. The effects of escalations in pacing frequency on R193H Tg myocyte mechanical and Ca2+ transient function.
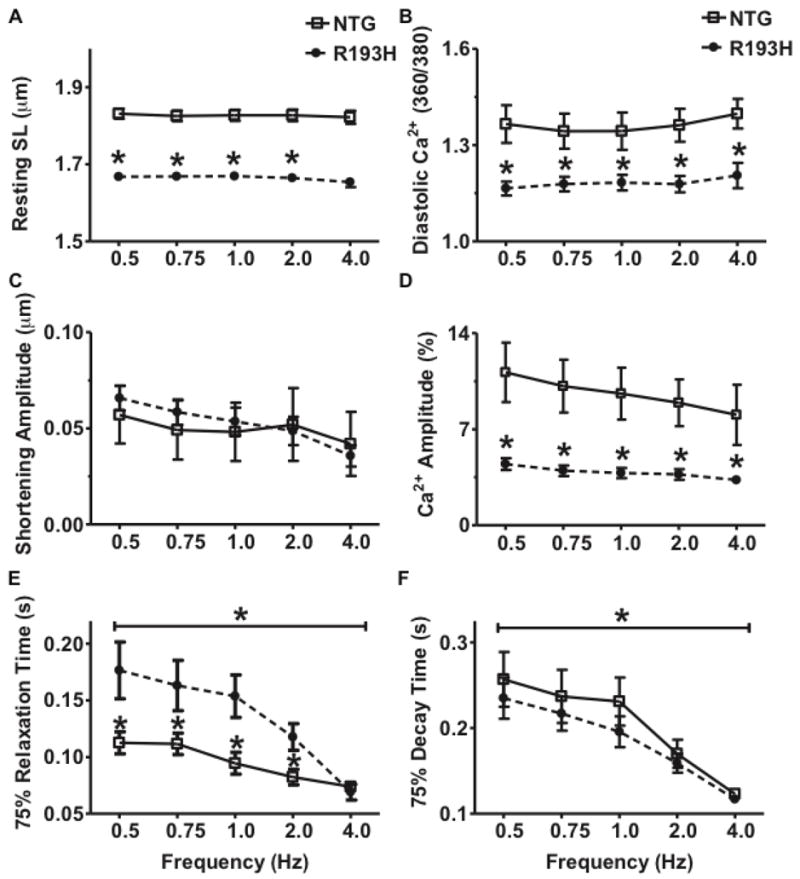
Two-way ANOVA, Bonferroni post-tests were used for statistical testing with two experimental factors: transgene effects (R193H vs. NTG) and frequency (0.5 – 4.0 Hz). Data are presented as mean ± SEM, n=20. (A) Summary of resting sarcomere length (SL) as a function of frequency with a significant transgene main effects (P<0.01). (B) Summary of diastolic Ca2+ ratios as a function of frequency. (C) Summary of shortening amplitude as a function of frequency. (D) Summary of Ca2+ amplitude as a function of frequency with a significant transgene main effect (P<0.01). (E) Summary of relaxation time as a function of frequency. There are significant transgene and frequency main effects (P<0.01). (F) Summary of Ca2+ decay time as a function of frequency with a significant frequency main effect (P<0.01).
Escalation in pacing frequency also caused a biphasic response to decrease myocyte relaxation in R193H Tg myocytes (Figure 7E). Ca2+ transient decay time decreased in a negative staircase pattern in both Tg and NTG myocytes (Figure 7F). R193H Tg myocytes had a much steeper mechanical response at higher frequencies (1.0–4.0 Hz, Figure 7E) even with a linear decline in Ca2+ transient decay time (Figure 7F). R193H Tg and NTG myocytes had similar Ca2+ transient decay times at each frequency (Figure 7F, significant frequency effect, Two-way ANOVA, P<0.01). By contrast, the R193H mutant cTnI had a significant transgene effect to slow relaxation across all frequencies with the exception of 4 Hz, contributing to the R193H Tg myocyte biphasic relaxation response (Figure 5–7F, Two-way ANOVA, P<0.01). NTG myocytes had a much shallower relationship between relaxation time and frequency. Taken together, these data suggest that the Ca2+ handling in R193H Tg myocytes is functioning like NTG, but mechanical relaxation has in part uncoupled from the Ca2+ transient at lower frequencies.
Compensatory changes in Ca2+ handling proteins in R193H myocytes
To understand the basis for the uncoupling between myocyte mechanical performance and Ca2+ cycling, Ca2+ handling proteins from R193H and NTG cellular lysates taken at the same time the isolated myocyte experiments were performed and were examined by Western Blot. Serca2A protein levels were unchanged (Supplement Figure 4); however, there was a significant reduction in PLN expression in R193H myocytes (Figure 8A), which increased the SERCA/PLN ratio (Figure 8B) providing an adaptation that differs from conventional heart failure. In addition there was an increase of phosphorylated PLN (Ser16, Figure 8C) in R193H myocytes (Figure 8C). The altered Ca2+ handling proteins in R193H Tg myocytes differs from previous reports of myocytes acutely engineered with R193H mutant cTnI in which Ca2+ handling protein expression was not directly altered [14]. In this previous gene transfer study adult myocytes were quiescent and cultured in serum-free conditions in the absence of load or constant contractile cycles. Here replacement of the native cTnI with R193H mutant cTnI was 25% at day 2 and rose to ~90% replacement at day 4. In vitro, adaptations to the acutely Ca2+ sensitized myofilaments were minimal and showed no alterations in gene expression. By contrast, transgenic expression of R193H cTnI in the Tg mouse model represents a system in which physiologic loads and Ca2+ cycling are in place to influence homeostasis and thus leaves open the possibility of adaptation to the primary effects of the R193H cTnI transgene. Indeed, low levels of R193H cTnI replacement in vivo combined with physiologic or pathophysiologic signals caused a decrease in PLN expression and increased PLN phosphorylation. This alteration in a key Ca2+ handling protein is likely a compensatory response to the enhanced Ca2+ sensitivity of the myofilaments and is in a direction that would speed Ca2+ decay. This could partially explain the uncoupling of the Ca2+ transient from mechanical performance (Figure 6D and E).
Figure 8. The effects of the R193H cTnI transgene on Ca2+ handling protein expression.
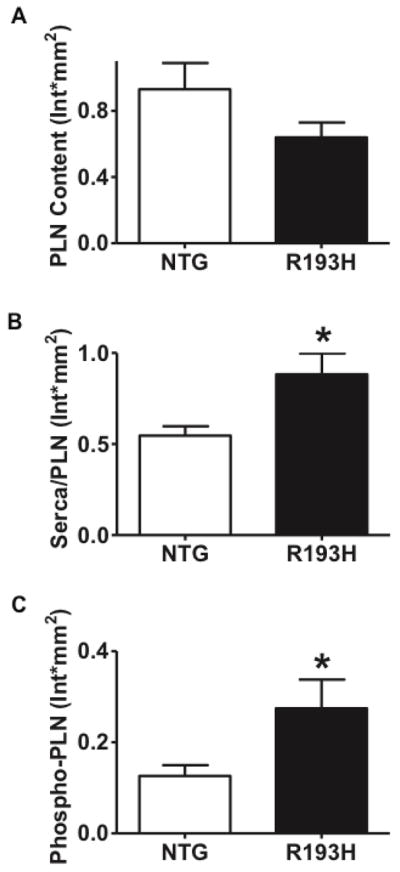
Summary of (A) phospholamban (PLN) content (B) the ratio of Serca2A to PLN content (Serca/PLN) and (C) phospho-phospholamban content from whole heart homogenates of 3–4 month old R193H single Tg and NTG mice. Data represents mean ± SEM, * P<0.05, n=5 hearts per group were examined. Representative Western blots and SR load data are presented in Supplement Figure 4.
Discussion
We report a hybrid hemodynamic phenotype in transgenic mice harboring the restrictive cardiomyopathy-associated mutant allele cTnI R193H. This RCM mouse model is characterized by poor myocardial compliance, elevated LV end diastolic pressure and incomplete relaxation, which combine to create severe diastolic dysfunction all in the absence of fibrotic lesions or altered morphology. With the use of a unique micro-carbon fiber assay on intact myocytes we uncovered the new finding that R193H cTnI Tg myocytes produce heightened passive tension without changes in intracellular Ca2+ during physiologically relevant extensions in sarcomere length. This poor myocyte distensibility was manifest at the organ level as elevated end diastolic pressure and poor ventricular filling and can be directly linked to the Ca2+-independent diastolic tone in R193H myocytes. As Ca2+ cycling was uncoupled from mechanical function in isolated R193H Tg myocytes these data demonstrate that the main pathologic feature of this R193H mouse model, diastolic dysfunction, is indeed Ca2+-independent and due to cTnI disinhibition. This underscores cell intrinsic heightened resistance to distension as a newly defined contributing factor to the clinical presentation of restrictive cardiomyopathy.
Restrictive cardiomyopathy (RCM) is a rare form of cardiomyopathy [1, 3]. In RCM the walls of the ventricles are abnormally stiff and resistant to normal ventricular chamber expansion in diastole as the heart refills with blood. The systolic function of the heart is typically normal. A full understanding of the molecular basis of the restrictive phenotype is lacking. To gain insight into cellular mechanisms we employed a novel micro-carbon fiber assay enabling the mechanical tethering of membrane intact, single adult cardiac myocytes while simultaneously monitoring tension, intracellular Ca2+ and sarcomere length during controlled excursions in overall myocyte length [29]. Compared to controls, cTnI R193H Tg myocytes display abrupt increases in passive tension over discrete SL extensions in the 1.90–2.05 um physiological range. During diastole in vivo, the ventricular chamber is distended as the chamber is re-primed with blood. Accordingly, individual myocytes in the heart are passively extended over the same sarcomere lengths used in the carbon fiber single myocyte assay [29]. In this context, it is informative that cTnI R193H adult cardiac myocytes had marked resistance to small SL excursions leading to a steep passive tension-SL extension relationship compared to controls. This occurs in the absence of detected changes in intracellular Ca2+ during length extension suggesting a mechanical basis for this loss in myocyte compliance. We hypothesize that heightened myofilament Ca2+ sensitivity by cTnI R193H tuned the sarcomeres to hyper-responsiveness as SL lengths are passively extended causing greater resistance to these physiological length steps. This mechanism is consistent with our prior work showing cTnI R193H renders the myocyte in a quasi “pre-contractile state” [14]. We propose this cTnI R193H fundamental defect of enhanced myofilament activation underlies the mechanism of heightened LV end diastolic pressure in R193H Tg mice in vivo.
Here, invasive hemodynamic measurements reveal poor ventricular filling and loss of compliance due to the increased stiffness of R193H cardiac myocytes. These outcomes parallel the clinical definition of RCM in which there is a compromised hemodynamic state of ventricular filling and loss of myocardial compliance [1, 38]. As tail cuff blood pressures (data not shown), gross cardiac anatomy, and Ca2+ dynamics were unchanged we can ascribe the loss of compliance directly to cTnI. This interpretation has potentially important clinical impact as it identifies a primary biophysical defect underpinning restrictive physiology, a “stiff” LV chamber, and pronounced diastolic dysfunction associated with inherited RCM patients. This is in contrast to an alternate hypothesis of restrictive physiology that develops due to secondary causes such as structurally abnormalities, fibrosis, or amylodoidosis. Our findings of heightened myocyte passive tension-extension in the absence of structural defects of fibrotic lesions implicates the dysfunctional myocyte as a key and cell intrinsic source of the restrictive organ level phenotype defined by the R193H cTnI-dependent high pressures and reduced end diastolic volumes.
Transgenic mice provide an important animal model to probe clinically relevant human health questions. Independent lines of Tg mice harboring RCM associated cTnI R193H could tolerate a maximum 15–30% stoichiometric replacement of native cTnI in the sarcomere in vivo. In keeping with this finding, attempts to create full replacement of WT cTnI with cTnI R193H in vivo leads to early death following cardiac transgene activation in vivo [18]. In vitro isolated cardiac myocytes can attain much greater levels of replacement (60–90%) by adenoviral gene transfer indicating tolerance in this case is not dictated by some fundamental limitation in sarcomere replacement but rather due to physiological intolerance at the whole organ level in vivo. This is further born out in other cTnI transgenic mice using a histidine button engineered cTnI that can tolerate at least 90% cTnI replacement in vivo [22, 39]. In vivo intolerance for cardiomyopathic sarcomeric proteins has been demonstrated for other thin filament regulatory proteins including cTnT [40]. In this cTnT truncation model, lines expressing greater than 5% of the transgene were lethal just after birth [40]. Collectively, transgenic cardiac expression profiles provide an in vivo functional tolerance test offering insight into disease processes. We speculate that in single allele RCM cTnI R193H patients express at most 50%, and likely much less, content of R193H residing in the sarcomere.
We posit that the increased in vivo diastolic pressure in R193H hearts results from the dominant role of R193H cTnI to elicit a significant Ca2+ independent diastolic tone, stiffened myocyte and heightened myofilament Ca2+ sensitivity. In addition, R193H cTnI causes a distinct secondary outcome of excitation-contraction uncoupling (EC-uncoupling) in which mechanical relaxation is significantly slowed with no change in Ca2+ transient decay rates. The adaptive decrease in phospholamban (PLN) expression and increase in phospho-PLN levels helps partially explain the normal Ca2+ decay despite a heightened myofilament Ca2+ buffering by R193H cTnI. Interestingly, a 6.5-fold greater replacement of native cTnI with R193H mutant cTnI shown with acute adenoviral gene transfer [14] versus the replacement reported here in R193H Tg myocytes exhibits the same magnitude of gain in myofilament Ca2+ sensitivity tension suggesting that very little mutant R193H cTnI is required to elicit significant pathophysiology. Together these data support the hypothesis that R193H Tg myocytes have adapted Ca2+ handling mechanisms that are insufficient to overcome the dominant effect of mutant myofilaments on mechanical performance. Likely the property of cooperativity in the thin filament regulation [41] permits small levels of mutant cTnI replacement to have large changes in Ca2+ sensitivity. This has implications when considering inherited cardiomyopathies as they are primarily autosomal dominant disorders in which the mutant sarcomeric allele acts as a dominant negative protein [7, 8], suggesting that very little mutant protein needs to be expressed in order to have large physiologic ramifications.
The novel defect of EC-uncoupling is a contributing factor to the distinctly different tolerance for and phenotype produced in R193H cTnI Tg myocytes compared to the primary effects of R193H cTnI in adenovirally transduced rat myocytes [14] and from other HCM-linked mouse models. Myocytes acutely engineered with R193H cTnI have a tightly coupled slowing of mechanical relaxation and Ca2+ decay despite a similar increase in myofilament Ca2+ sensitivity [14]. The basis for this acute prolongation of Ca2+ decay was ascribed to the heightened Ca2+ buffering capacity of the R193H mutant myofilaments, which has been implicated in other HCM models [14, 42, 43]. A comparison between the primary outcomes previously measured in an R193H acute gene transfer [14] model versus results from the R193H Tg myocytes here suggests that R193H Tg mice have adapted Ca2+ handling processes to accommodate the increased buffering capacity of the R193H myofilaments. This adaptation is through changes in the SERCA/PLN ratio and in the phosphorylation status of PLN (Figure 8, Supplement Figure 4). The modification to PLN content and enhanced phosphorylation should increase SERCA2a pump function but in this case only enough to normalize the R193H cTnI-dependent changes to the Ca2+ transient decay. Together this can explain the normal Ca2+ decay despite the increase Ca2+ buffering by the myofilaments and SERCA2a/PLN ratio. This adaptation is in contrast to isolated mouse myocytes from other HCM transgenic models [42–45] or non-disease linked cTnI variants [22, 46] with heightened myofilament Ca2+ sensitivity that still retain a tight coupling between the slow mechanical function and slowed Ca2+ transient decay suggesting that this RCM-linked R193H Tg model is adapting differently to the primary defects of heightened myofilament Ca2+ sensitivity and elevated diastolic tone.
In comparing this R193H mouse model with a previously published model [18–20] both show no cardiac hypertrophy, each have a loss of myocardial compliance, and each have impaired relaxation in vivo and in vitro. Beyond this the models diverge significantly. Here we demonstrate that the loss of myocardial compliance is due to non-compliant R193H myocytes, which directly impacts the end diastolic pressure and resistance to filling measured hemodynamically in R193H mice. Furthermore, the differences in Ca2+ handling, compliance, and diastolic function between the two R193H models are likely due to the magnitude of change in Ca2+ sensitivity (ΔpCa50=0.3 shown here verses 0.1 [18]). The R193H Tg mouse model presented here has diastolic impairment already at 3 verses 6 months of age [18–20]. Ca2+ handling is also different. The Ca2+ transient closely followed mechanical function with no change in SERCA/PLN ratios in the previously published R193H model [18]. Here we demonstrate an uncoupling between Ca2+ cycling and mechanical function due to changes in expression of Ca2+ handling proteins. In addition our R193H model has high EDPs that were not reported in previous models. The basis for these differential results is not known. The R193H cTnI transgene contains a 3′ epitope tag; however, in multiple previous studies we have not detected any flag epitope-dependent effects [14, 21, 22].
The high EDP and diastolic dysfunction in R193H Tg mice should compromise cardiac function. During β-blockade, R193H Tg mice had an increased functional capacity (Figure 3E) and diastolic dysfunction was no longer evident (Figure 3F & G). This is surprising as rodents have a high sympathetic tone that can mask diastolic dysfunction, particularly in models with heightened myofilament Ca2+ sensitivity [47]. Paradoxically, the R193H Tg mice have impaired relaxation at baseline rather than under β-blockade. The basis for this apparent gain in contractile function and improved relaxation is not known but could in principle relate to prolonged diastole and time for passive filling; thus, the beneficial change in R193H diastolic function similar to the effects of β-blockade in another mouse model of HCM [20]. β-blockers are a common treatment for cardiomyopathies and in the case of inherited RCM patients it may be beneficial in terms of lessening impaired relaxation but maintaining stroke volume.
In conclusion, RCM-linked R193H mutant cTnI results in a hybrid RCM/HCM phenotype with the distinct features of an elevated Ca2+ -independent diastolic tone, a myofilament based loss of myocardial compliance and the secondary adaptive outcome of EC-uncoupling at the cellular and organ level. This mixed phenotype falls in line with the latest report on the contemporary classifications of inherited cardiomyopathy which highlight phenotypic overlap as a common clinical presentation of cardiomyopathy subtypes like HCM and RCM that were formally perceived as distinct clinical entities [3]. In addition, reports on a human patient with the R193H cTnI allele indicate a transition from RCM to HCM at the time of death [3]. Importantly, a low level of native cTnI replacement was all that was required to cause the prominent diastolic dysfunction that could not be overcome by the adaptive changes in Ca2+ handling genes that established a “normal” Ca2+ decay rate. These results underscore the prominent influence of mutant myofilaments on cardiac and organismal function. Collectively, this study points to factors outside of Ca2+ sensitivity, including compensatory adaptation and allele specific functional effects as being critical for determining the threshold for functional deficits tolerated by an organism. Finding these intersecting pathways will be a key step toward understanding the pathogenic progression of the inherited cardiomyopathies.
Supplementary Material
Highlights.
The RCM mutation R193H in troponin I decreased myocardial and myocyte compliance.
R193H troponin I elevated end diastolic pressures.
Sarcomere relaxation and Ca2+ decay was uncoupled in isolated R193H myocytes.
Altered Ca2+ handling proteins compensated for R193H Ca2+ sensitive myofilaments.
Acknowledgments
This work was supported by grants from the NIH (JMM) and American Heart Association (JD).
Footnotes
Disclosures
The authors have no potential conflicts of interest to disclose.
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Kushwaha SS, Fallon JT, Fuster V. Restrictive cardiomyopathy. N Engl J Med. 1997;336:267–76. doi: 10.1056/NEJM199701233360407. [DOI] [PubMed] [Google Scholar]
- 2.Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–16. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 3.Mogensen J, Kubo T, Duque M, Uribe W, Shaw A, Murphy R, et al. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J Clin Invest. 2003;111:209–16. doi: 10.1172/JCI16336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Metzger JM, Westfall MV. Covalent and noncovalent modification of thin filament action: the essential role of troponin in cardiac muscle regulation. Circ Res. 2004;94:146–58. doi: 10.1161/01.RES.0000110083.17024.60. [DOI] [PubMed] [Google Scholar]
- 5.Mogensen J, Murphy RT, Kubo T, Bahl A, Moon JC, Klausen IC, et al. Frequency and clinical expression of cardiac troponin I mutations in 748 consecutive families with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:2315–25. doi: 10.1016/j.jacc.2004.05.088. [DOI] [PubMed] [Google Scholar]
- 6.Braunwald E, Seidman CE, Sigwart U. Contemporary evaluation and management of hypertrophic cardiomyopathy. Circulation. 2002;106:1312–6. doi: 10.1161/01.cir.0000030314.11999.6a. [DOI] [PubMed] [Google Scholar]
- 7.Fatkin D, Graham RM. Molecular mechanisms of inherited cardiomyopathies. Physiol Rev. 2002;82:945–80. doi: 10.1152/physrev.00012.2002. [DOI] [PubMed] [Google Scholar]
- 8.Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, et al. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. doi: 10.1016/0092-8674(90)90274-i. [DOI] [PubMed] [Google Scholar]
- 9.Maron BJ, Shirani J, Poliac LC, Mathenge R, Roberts WC, Mueller FO. Sudden death in young competitive athletes. Clinical, demographic, and pathological profiles. Jama. 1996;276:199–204. [PubMed] [Google Scholar]
- 10.Blumenschein TM, Stone DB, Fletterick RJ, Mendelson RA, Sykes BD. Dynamics of the C-terminal region of TnI in the troponin complex in solution. Biophys J. 2006;90:2436–44. doi: 10.1529/biophysj.105.076216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gomes AV, Potter JD. Cellular and molecular aspects of familial hypertrophic cardiomyopathy caused by mutations in the cardiac troponin I gene. Mol Cell Biochem. 2004;263:99–114. doi: 10.1023/B:MCBI.0000041852.42291.aa. [DOI] [PubMed] [Google Scholar]
- 12.Pirani A, Vinogradova MV, Curmi PM, King WA, Fletterick RJ, Craig R, et al. An atomic model of the thin filament in the relaxed and Ca2+-activated states. J Mol Biol. 2006;357:707–17. doi: 10.1016/j.jmb.2005.12.050. [DOI] [PubMed] [Google Scholar]
- 13.Solaro RJ, Van Eyk J. Altered interactions among thin filament proteins modulate cardiac function. J Mol Cell Cardiol. 1996;28:217–30. doi: 10.1006/jmcc.1996.0021. [DOI] [PubMed] [Google Scholar]
- 14.Davis J, Wen H, Edwards T, Metzger JM. Thin filament disinhibition by restrictive cardiomyopathy mutant R193H troponin I induces Ca2+-independent mechanical tone and acute myocyte remodeling. Circ Res. 2007;100:1494–502. doi: 10.1161/01.RES.0000268412.34364.50. [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi T, Solaro RJ. Increased Ca2+ affinity of cardiac thin filaments reconstituted with cardiomyopathy-related mutant cardiac troponin I. J Biol Chem. 2006;281:13471–7. doi: 10.1074/jbc.M509561200. [DOI] [PubMed] [Google Scholar]
- 16.Gomes AV, Liang J, Potter JD. Mutations in human cardiac troponin I that are associated with restrictive cardiomyopathy affect basal ATPase activity and the calcium sensitivity of force development. J Biol Chem. 2005;280:30909–15. doi: 10.1074/jbc.M500287200. [DOI] [PubMed] [Google Scholar]
- 17.Yumoto F, Lu QW, Morimoto S, Tanaka H, Kono N, Nagata K, et al. Drastic Ca2+ sensitization of myofilament associated with a small structural change in troponin I in inherited restrictive cardiomyopathy. Biochem Biophys Res Commun. 2005;338:1519–26. doi: 10.1016/j.bbrc.2005.10.116. [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Charles PY, Nan C, Pinto JR, Wang Y, Liang J, et al. Correcting diastolic dysfunction by Ca2+ desensitizing troponin in a transgenic mouse model of restrictive cardiomyopathy. J Mol Cell Cardiol. 2010;49:402–11. doi: 10.1016/j.yjmcc.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Du J, Liu J, Feng HZ, Hossain MM, Gobara N, Zhang C, et al. Impaired relaxation is the main manifestation in transgenic mice expressing a restrictive cardiomyopathy mutation, R193H, in cardiac TnI. Am J Physiol Heart Circ Physiol. 2008;294:H2604–13. doi: 10.1152/ajpheart.91506.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Du J, Zhang C, Liu J, Sidky C, Huang XP. A point mutation (R192H) in the C-terminus of human cardiac troponin I causes diastolic dysfunction in transgenic mice. Arch Biochem Biophys. 2006;456:143–50. doi: 10.1016/j.abb.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 21.Davis J, Wen H, Edwards T, Metzger JM. Allele and species dependent contractile defects by restrictive and hypertrophic cardiomyopathy-linked troponin I mutants. J Mol Cell Cardiol. 2008;44:891–904. doi: 10.1016/j.yjmcc.2008.02.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Day SM, Westfall MV, Fomicheva EV, Hoyer K, Yasuda S, La Cross NC, et al. Histidine button engineered into cardiac troponin I protects the ischemic and failing heart. Nat Med. 2006;12:181–9. doi: 10.1038/nm1346. [DOI] [PubMed] [Google Scholar]
- 23.James J, Zhang Y, Osinska H, Sanbe A, Klevitsky R, Hewett TE, et al. Transgenic modeling of a cardiac troponin I mutation linked to familial hypertrophic cardiomyopathy. Circ Res. 2000;87:805–11. doi: 10.1161/01.res.87.9.805. [DOI] [PubMed] [Google Scholar]
- 24.Sanbe A, James J, Tuzcu V, Nas S, Martin L, Gulick J, et al. Transgenic rabbit model for human troponin I-based hypertrophic cardiomyopathy. Circulation. 2005;111:2330–8. doi: 10.1161/01.CIR.0000164234.24957.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yasuda S, Coutu P, Sadayappan S, Robbins J, Metzger JM. Cardiac transgenic and gene transfer strategies converge to support an important role for troponin I in regulating relaxation in cardiac myocytes. Circ Res. 2007;101:377–86. doi: 10.1161/CIRCRESAHA.106.145557. [DOI] [PubMed] [Google Scholar]
- 26.Oh JK, Hatle L, Tajik AJ, Little WC. Diastolic heart failure can be diagnosed by comprehensive two-dimensional and Doppler echocardiography. J Am Coll Cardiol. 2006;47:500–6. doi: 10.1016/j.jacc.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 27.Michele DE, Gomez CA, Hong KE, Westfall MV, Metzger JM. Cardiac dysfunction in hypertrophic cardiomyopathy mutant tropomyosin mice is transgene-dependent, hypertrophy-independent, and improved by beta-blockade. Circ Res. 2002;91:255–62. doi: 10.1161/01.res.0000027530.58419.82. [DOI] [PubMed] [Google Scholar]
- 28.Westfall MV, Albayya FP, Turner II, Metzger JM. Chimera analysis of troponin I domains that influence Ca(2+)-activated myofilament tension in adult cardiac myocytes. Circ Res. 2000;86:470–7. doi: 10.1161/01.res.86.4.470. [DOI] [PubMed] [Google Scholar]
- 29.Yasuda S, Townsend D, Michele DE, Favre EG, Day SM, Metzger JM. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature. 2005;436:1025–9. doi: 10.1038/nature03844. [DOI] [PubMed] [Google Scholar]
- 30.Yasuda SI, Sugiura S, Kobayakawa N, Fujita H, Yamashita H, Katoh K, et al. A novel method to study contraction characteristics of a single cardiac myocyte using carbon fibers. Am J Physiol Heart Circ Physiol. 2001;281:H1442–6. doi: 10.1152/ajpheart.2001.281.3.H1442. [DOI] [PubMed] [Google Scholar]
- 31.Coutu P, Bennett CN, Favre EG, Day SM, Metzger JM. Parvalbumin corrects slowed relaxation in adult cardiac myocytes expressing hypertrophic cardiomyopathy-linked alpha-tropomyosin mutations. Circ Res. 2004;94:1235–41. doi: 10.1161/01.RES.0000126923.46786.FD. [DOI] [PubMed] [Google Scholar]
- 32.Coutu P, Metzger JM. Genetic manipulation of calcium-handling proteins in cardiac myocytes. II. Mathematical modeling studies. Am J Physiol Heart Circ Physiol. 2005;288:H613–31. doi: 10.1152/ajpheart.00425.2004. [DOI] [PubMed] [Google Scholar]
- 33.Metzger JM, Michele DE, Rust EM, Borton AR, Westfall MV. Sarcomere thin filament regulatory isoforms. Evidence of a dominant effect of slow skeletal troponin I on cardiac contraction. J Biol Chem. 2003;278:13118–23. doi: 10.1074/jbc.M212601200. [DOI] [PubMed] [Google Scholar]
- 34.Westfall MV, Borton AR, Albayya FP, Metzger JM. Myofilament calcium sensitivity and cardiac disease: insights from troponin I isoforms and mutants. Circ Res. 2002;91:525–31. doi: 10.1161/01.res.0000034710.46739.c0. [DOI] [PubMed] [Google Scholar]
- 35.Westfall MV, Rust EM, Metzger JM. Slow skeletal troponin I gene transfer, expression, and myofilament incorporation enhances adult cardiac myocyte contractile function. Proc Natl Acad Sci U S A. 1997;94:5444–9. doi: 10.1073/pnas.94.10.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsoutsman T, Chung J, Doolan A, Nguyen L, Williams IA, Tu E, et al. Molecular insights from a novel cardiac troponin I mouse model of familial hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2006;41:623–32. doi: 10.1016/j.yjmcc.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 37.Bers DM. Excitation-contraction coupling and cardiac contractile force. 2. Dordrecht; Boston: Kluwer Academic Publishers; 2001. [Google Scholar]
- 38.Sen-Chowdhry S, Syrris P, McKenna WJ. Genetics of restrictive cardiomyopathy. Heart Fail Clin. 2010;6:179–86. doi: 10.1016/j.hfc.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 39.Palpant NJ, Day SM, Herron TJ, Converso KL, Metzger JM. Single histidine-substituted cardiac troponin I confers protection from age-related systolic and diastolic dysfunction. Cardiovasc Res. 2008;80:209–18. doi: 10.1093/cvr/cvn198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tardiff JC, Factor SM, Tompkins BD, Hewett TE, Palmer BM, Moore RL, et al. A truncated cardiac troponin T molecule in transgenic mice suggests multiple cellular mechanisms for familial hypertrophic cardiomyopathy. J Clin Invest. 1998;101:2800–11. doi: 10.1172/JCI2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 42.Semsarian C, Ahmad I, Giewat M, Georgakopoulos D, Schmitt JP, McConnell BK, et al. The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J Clin Invest. 2002;109:1013–20. doi: 10.1172/JCI14677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Semsarian C, Healey MJ, Fatkin D, Giewat M, Duffy C, Seidman CE, et al. A polymorphic modifier gene alters the hypertrophic response in a murine model of familial hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2001;33:2055–60. doi: 10.1006/jmcc.2001.1466. [DOI] [PubMed] [Google Scholar]
- 44.Fatkin D, McConnell BK, Mudd JO, Semsarian C, Moskowitz IG, Schoen FJ, et al. An abnormal Ca(2+) response in mutant sarcomere protein-mediated familial hypertrophic cardiomyopathy. J Clin Invest. 2000;106:1351–9. doi: 10.1172/JCI11093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Knollmann BC, Potter JD. Altered regulation of cardiac muscle contraction by troponin T mutations that cause familial hypertrophic cardiomyopathy. Trends Cardiovasc Med. 2001;11:206–12. doi: 10.1016/s1050-1738(01)00115-3. [DOI] [PubMed] [Google Scholar]
- 46.Fentzke RC, Buck SH, Patel JR, Lin H, Wolska BM, Stojanovic MO, et al. Impaired cardiomyocyte relaxation and diastolic function in transgenic mice expressing slow skeletal troponin I in the heart. J Physiol. 1999;517 ( Pt 1):143–57. doi: 10.1111/j.1469-7793.1999.0143z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cho MC, Rapacciuolo A, Koch WJ, Kobayashi Y, Jones LR, Rockman HA. Defective beta-adrenergic receptor signaling precedes the development of dilated cardiomyopathy in transgenic mice with calsequestrin overexpression. J Biol Chem. 1999;274:22251–6. doi: 10.1074/jbc.274.32.22251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.