

Abstract
Loss of PTEN is frequently observed in androgen-independent prostate cancer, resulting in the deregulation of metastatic events. SDF1α activation of CXCR4 induces signaling pathways that have been implicated in prostate metastasis and progression to an advanced disease. The pathways of CXCR4 and PTEN converge, leading to the promotion and regulation of tumorigenesis, respectively. However, loss of PTEN may permit CXCR4 to progress prostate cancer to an advanced disease. In the present study, we investigated the involvement of PTEN in CXCR4-mediated tumorigenesis. When screening advanced metastatic prostate cancer cell lines for PTEN, we observed a loss of expression in PC3 and LNCaP cells, whereas Du145 expressed wildtype PTEN. All three cell lines were positive for surface expression of CXCR4. Reconsitution of PTEN induced a mesenchymal to epithelial-like morphological change and inhibited CXCR4-mediated migration and proliferation in PC3 cells. Downregulation of PTEN by siRNA enhanced the CXCR4-mediated migratory behavior of Du145 cells. By western blot analysis, we observed that PTEN inhibited basal AKT phosphorylation, but not ERK1/2 phosphorylation in PTEN expressing cells. Upon CXCR4 stimulation, PTEN inhibited ERK1/2 phosphorylation, but not phosphorylation of AKT. The CXCR4- mediated migration of PC3 cells was through the ERK1/2 pathway, as confirmed by chemical inhibitors. Based on these studies, we suggest that loss of PTEN permits CXCR4-mediated functions in prostate cancer cells, through the ERK1/2 pathway. Antagonizing CXCR4 and downstream signaling cascades may provide an efficient approach for treating patients with advanced prostate cancer, when hormone therapy fails to the stop the growth and containment of tumors.
Keywords: Prostate cancer, PTEN, CXCR4, metastasis, ERK1/2
INTRODUCTION
The relative 5-year survival rate among prostate cancer survivors is nearly 100%, versus a 15-year relative survival rate of 76% (1). Improved treatments and methods of detection have translated into prostate cancers being found earlier in their development, and being treated more effectively. Despite these advances, prostate cancer still accounts for approximately 10% of cancer-related deaths in men (1). Prostate cancer growth is dependent upon increasing levels of androgens, which stimulate the growth, survival and function of cells that express the androgen receptor. During the early stages of prostate cancer, hormone deprivation therapy proves to be effective. However, advanced prostate tumors grow accustomed to androgen depletion, circumvent restraints on growth and movement, and eventually develop metastatic colonies in the bones, and other distal organs.
Progression to androgen-insensitivity is mediated largely by androgen receptors (AR); however, this process is distinct from metastatic progression, where tumor suppressors fail to regulate signals that influence cell adhesion, anchorage and movement (2). Genomic deletions in the tumor suppressor Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) are common in androgen-insensitive prostate tumors, although the incidence and its downstream effects have not been well elucidated in clinical samples of hormone refractory prostate cancers. Loss of PTEN is well documented in prostate cancer and cancer overall, and appears to act as a permissive event for uncontrolled cell proliferation, invasion and metastasis (3–6). Although PTEN haploinsufficiency is strongly correlated with the conversion of a high-grade prostatic intraepithelial neoplasia (PIN) to an invasive adenocarcinoma, the underlying mechanisms permitting ensuing invasion and metastasis are poorly understood (3, 7).
PTEN functions as a dual-specificity lipid and protein phosphatase that inhibits cell proliferation, survival and growth, predominantly through dephosphorylation of phosphatidylinositol 3,4,5-trisphosphate (PIP3), thus antagonizing phosphatidylinositol 3-kinase (PI3K)/Protein Kinase B (AKT)–mediated signaling events (8). By converting PIP3 into phosphatidylinositol 4,5-bisphosphate (PIP2), PTEN negatively regulates PI3K-AKT signaling and subsequent downstream pathways: (i)apoptosis; (ii)protein synthesis; (iii)metabolism; (iv)cell cycle; (v)proliferation; (vi) invasion; (vii)metastasis; (viii) angiogenesis, and overall survival (9, 10). Regulating the PI3K/AKT/mTOR signaling pathway has been shown to be pivotal to prostate cancer proliferation, and the pathogenesis of an advanced disease (11, 12).
Wallace et al. demonstrated that prostate tumors can carry alleles that contribute to advanced, metastatic stages of prostate cancer; among the genes with elevated expression was CXCR4 (13). The chemokine receptor CXCR4, and its ligand stromal cell-derived factor 1 alpha (SDF1α or CXCL12), play a crucial role in targeting solid tumor metastases to sites outside of the primary tumor. CXCR4 has become a potential target for therapeutic intervention in malignancies that metastasize (14); a study by Akashi et al revealed that CXCR4 expression was higher in malignant prostate tumors than in their normal healthy counterparts, suggesting that its expression level correlated with increased metastasis-associated mortality (15). Positive expression of CXCR4 has become a superior predictor of tumor aggressiveness, poor prognosis and prostate cancer bone metastasis (16, 17). Upon SDF1α binding to CXCR4, the activation of metastasis-associated pathways makes this receptor favorable to tumorigenesis: (i) G-protein coupled receptor (GPCR) signaling, (ii) PI3K/AKT, (iii)MAPK, (iv) JAK/STAT, (v) Src kinase and (vi) HER2 (12, 18, 19). Downstream, CXCR4-initiated signaling leads to cell polarization, an initial step in metastasis, and the transcription of genes involved in migration (14). It has been reported that CXCR4 was expressed on the surface of prostate cancer cells, and was involved in facilitating prostate metastasis (16–18).
Independently, PTEN and CXCR4 have been noted for their involvement in prostate cancer invasion, metastasis and progression. PTEN alterations are strongly implicated in prostate cancer development; placing the tumor suppressor high among the most common genetic alterations in human prostate tissues (8, 20, 21). PTEN deletions and/or mutations are found in up to 30% of primary prostate cancers and 60–63% of metastatic prostate tissues (21–23). Functionally, loss of PTEN developed prostatic neoplasia into an advanced, metastatic state (3), and correlated with increased prostate cancer cell migration towards bone-conditioned medium (24). Conversely, reconstituted PTEN in prostate cancer cells controlled migration (25) and conferred sensitivity to chemotherapy (26). Collectively, these data establishes PTEN as an essential tumor suppressor in the prostate. Therefore, the absence of PTEN may contribute to a tumor environment that is conducive to prostate cancer development and progression.
To date, one link has been established between CXCR4 and PTEN in inflammatory chemotaxis, where PTEN inhibited movement of Jurkat cells stimulated with SDF1α (27). In non-small cell lung cancer, Phillips et al observed that PTEN blocked hypoxia-induced expression of CXCR4 (28). Likewise in prostate cancer, Carver et al observed a correlation in expression between PTEN and CXCR4; however, neither study reported a functional relationship. To our knowledge, a functional relationship between PTEN and CXCR4 has not been established in prostate cancer. Therefore, our aim is to determine whether loss of PTEN in prostate cancer cells provides a “permissive switch” for CXCR4-mediated signaling and functions, as upregulation of CXCR4 is associated with the development of an advanced disease.
MATERIALS AND METHODS
Cell Culture, Antibodies and Reagents Conditions
LNCaP, PC3, Du145 human prostate cancer cell lines and 293T human embryonic kidney cell line were obtained from American Type Culture Collection. C42 human prostate cancer cells were a kind gift from Dr. Leland Chung, Cedars-Sinai Medical Center, Los Angeles, CA. All cells were maintained in RPMI 1640 containing 10% fetal bovine serum (FBS), 1% non-essential amino acids and 1% antibiotic-antimycotic at 37°C in 5% CO2, or starvation media (RPMI). All cells were maintained at 60% to 80% confluency. PD98059 was from Sigma Aldrich; LY294002 was from Cayman Chemicals; cell culture supplies were from MediaTech; SDF1α was from PeproTech. The following human antibodies were from Cell Signaling: anti-PTEN, anti-AKT, anti-phospho-AKT (p-AKT) and anti-phospho-ERK1/2 (p-ERK1/2). Anti-ERK1/2 was from Biosource; β-actin, anti-GFP and Fusin-CXCR4 were from Santa Cruz Biotech.
Plasmid Construct
PTEN (pcDNA3-GFP-PTEN) and GFP (pcDNA3-GFP) constructs were generated as described previously (29). Briefly, GFP was cloned into the 5’ end of pcDNA3 plasmid (Invitrogen) using HindIII and BamHI restriction enzyme sites to generate pcDNA3-GFP. The coding sequence for human PTEN was cloned into BamHI and EcoRI restriction sites of pcDNA3-GFP, to generate pcDNA3-GFP-PTEN.
Western Blot Analysis
Cells were grown on 10 mm dishes, washed with 1X PBS and harvested in lysis buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, 1µg/ml leupeptin and 1mM PMSF (Cell Signaling). Protein concentrations were estimated using Bradford protein assay (BioRad). Equal concentrations of total cell lysate were resolved by 10% SDS-PAGE and transferred to a polyvinylidene fluoride transfer membrane (PVDF). Non-specific binding sites were blocked with 5% nonfat dry milk/0.1% Tween 20/1X TBS, followed by an incubation with primary antibodies for the proteins of interest in 3% BSA-TBST (p-ERK1/2, p-AKT, AKT, PTEN) or 3% nonfat dry milk-TBST (ERK1/2). Membranes were washed in 1X TBST, and incubated in horseradish peroxidase-conjugated anti-rabbit (p-ERK1/2, p-AKT, AKT, PTEN) or anti-mouse secondary antibodies (ERK1/2) (1:10,000; Jackson ImmunoResearch) in 3% BSA-TBST or 3% nonfat dry milk-TBST for 1 hour at room temperature. Immunoblots were visualized using Super Signal West Pico Chemiluminescence reagent (Thermo Scientific).
PCR Amplification
Total RNA was isolated with Total RNA Kit-I (Omega Bio-TeK), as described by the manufacturer. Total RNA was reversed transcribed with M-MLV Transcriptase (Promega) to generate cDNA for PCR amplification. Specific sense and antisense primers for PTEN, and housekeeping gene L19, were synthesized by Integrated DNA Technologies as follows: PTEN (forward GGA CGA ACT GGT GTA ATG ATA TG; reverse TCT ACT GTT TTT GTG AAG TAC AGC) and L19 (forward GAA ATC GCC AAT GCC TC; reverse TCT TA ACC TC GAG CCT CA).
Flow Cytometry Analysis
Cells (1×106) were plated in 100 mm dishes and serum starved overnight for synchronization, prior to treating with SDF1α. Cells were detached with 1X citric saline and fixed in 4% paraformaldehyde at 4°C for 30 minutes. Cells were washed in 1X PBS, followed by centrifugation at 3000rpm. Non-specific binding sites were blocked in blocking buffer (1% donkey serum in 1X PBS) at 4°C for 1 hour, followed by an incubation overnight in CXCR4 antibody (1:100) in blocking buffer at 4°C or without antibody (control). Cells were washed once in 1X PBS, followed by incubation in fluorescein isothiocyanate (FITC) or Indocarbocyanine (Cy3) fluorophore-conjugated secondary antibodies (1:500; Jackson ImmunoResearch) in blocking buffer at 4°C for 45 minutes. Cells were washed, resuspended in 1X PBS and the surface expression of CXCR4 was analyzed by flow cytometry (Accuri C6). Results were quantified using GraphPad Prism 5 statistical software.
Transient Transfection
Transient transfections of pcDNA3-GFP or pcDNA3-GFP-PTEN were performed in PC3 cells using Lipofectamine2000 (Invitrogen), generating the cell lines PC3-GFP and PC3-PTEN, respectively. Briefly, cells (5×105) were plated in 6-well dishes, transfected according to the manufacturer’s instructions and incubated at 37 °C in 5% CO2 for 8 hours, prior to the addition of RPMI/10% FBS overnight. The next day, cells were serum starved or harvested. Transfection efficiency was monitored by measuring the percent of fluorescent cells among 500 total cells using the Cellometer Vision Trio 5 Program (Nexcelom Biosciences).
Migration Assay
Cell migration assays were done using 8 µm pore transwell chambers (Costar). Briefly, cells were grown in starvation media for 24 hours prior to detachment with 1X citric saline. Cells were resuspended in RPMI media, and 2×104 cells/well were added to the upper transwell chamber. RPMI containing 100 ng/mL of SDF1α was added to the lower chamber, and cells were allowed to migrate towards SDF1α in the lower chamber for 4–6 hours at 37°C. Cells that remained in the upper chamber were removed with a cotton swab, fixed and stained with Hemacolor Solution 3 Kit (EMD). Migrated cells were counted using a Zeiss Axiovert 200M light microscope. Results were quantified using GraphPad Prism 5 statistical program.
Proliferation (Viability) Assay
Cell proliferation was assessed by a MTT dye conversion assay at 570 nm following manufacturer’s instructions (Trevigen). In triplicates, 1 × 103 cells/well were seeded in a 96-well flat-bottomed plate. Cells were serum starved for 24 hours prior to treating with 100 ng/mL of SDF1α at 37 °C in 5% CO2. At each time point (24 and 48 hours), cell were incubated with 10 µL of MTT reagent for 2 hours at 37°C, followed by 100 µL of detergent reagent at 37 °C for 2 hours. Proliferation (viability) was measured at 570 nm using a microplate reader (Bio-Tek Synergy HT). Results were quantified using GraphPad Prism 5 statistical program.
Short Interfering RNA Transfection
Transient transfection of PTEN specific siRNA (Cell Signaling) was performed on Du145 cells using Lipofectamine2000. Briefly, cells (5×105) were plated in 100mm dishes and transfected with 100 µM PTEN-siRNA or scramble-siRNA (Santa Cruz) in starvation media at 37 °C in 5% CO2 for 24 hours. Transfected cells were harvested for western blot analysis or a migration assay.
Immunocytochemistry
Cells (3×104) were plated on glass slips in each well of a 10mm plate. Cells were washed with 1X PBS, fixed with 4% paraformaldehyde, washed twice for 5 minutes in Tris-glycine (0.1M glycine, pH 7.4), followed by a wash in 1X PBS. Non-specific binding sites were blocked in blocking buffer (5% normal donkey serum, 1% BSA, 0.1% Tween20 in PBS) for 30 minutes, followed by incubation overnight with CXCR4 antibody (1:100) in blocking buffer at 4°C. Cells were washed thrice in 1X PBS, followed by an incubation in Cy3-conjugated secondary antibody (1:500) in blocking buffer for 45 minutes in the dark at room temperature. Cells were washed thrice in 1X PBS, followed by an incubation in 4',6-diamidino-2-phenylindole (DAPI, Sigma, 1:500 in PBS) for 1 minute. Glass coverslips were washed in 1X PBS prior to mounting on microscope slides with AquaPolymount (PolySciences, Inc). Fluorescence was analyzed by using a Zeiss Axio Imagwe.z1 fluorescence microscope.
[3H]Thymidine Incorporation
Cells were plated at a density of 4×104 cells/well in 24-well culture plates and transfected with pcDNA3-GFP or pcDNA3-GFP-PTEN plasmids as described previously. Cells were serum starved for 24 hours prior to incubating with 100ng/mL of SDF1α at 37 °C in 5% CO2. At each time point (24 and 48 hours), the media was replaced with RPMI containing 1µCi/ml of [3H] thymidine (PerkinElmer) and cells were incubated for 4 h at 37°C. Cells were sonicated and filtered through Whatman DE81 filter paper discs using a vacuum pump, prior to measuring the filter discs for radioactivity incorporation using a LS6500 multi-purpose scintillation counter (Backman Coulter, Brea, CA). Results were quantified using GraphPad Prism 5 statistical program.
Statistics and Quantifications
Data are presented as the mean ± SE of at least three independent experiments. The data was analyzed for twoway ANOVA or Student t-test. All statistical analyses were done, and all graphs generated, using GraphPad Prism 5.0 software (GraphPad).
RESULTS
PTEN was differentially expressed, while reconstitution of PTEN induced morphological changes in prostate cancer cells
Based on the reports that PTEN haplosufficiency is strongly correlated with the conversion of prostate tumors to an invasive adenocarcinoma (3, 7), and the observations that CXCR4 is highly expressed in advanced prostate tissues (17, 30), we surmised that the absence of PTEN permits CXCR4-mediated functions and development of an aggressive phenotype in prostate cancer. We compared the levels of PTEN expression among prostate cancer cell lines (PC3, LNCaP, C42 and Du145) by western blot and RT-PCR assays. PTEN was expressed at both the protein and mRNA levels in androgen-independent Du145 cells and positive control human embryonic kidney cells, 293T (Fig. 1A). Protein (Fig. 1A), but not mRNA (Fig. 1B), PTEN expression was absent in androgen-dependent LNCaP and androgen-independent C42 cell lines (Fig. 1A). Genetic studies have shown that Du145 cells carry one functional PTEN allele, while the other allele is deleted (31). LNCaP cells carry a base-pair deletion found on codon 6 of PTEN, inhibiting translation (31). C42 cells are a metastatic derivative of LNCaP isolated from the bone (32). Finally, PTEN was not detected in androgen-independent PC3 cells at both the protein and mRNA levels, due to homologous deletions of the PTEN gene (Figs. 1A and B) (31). The PTEN expression profiles observed in PC3, LNCaP and Du145 cells is concurrent with previously published data (33).
Figure 1. Expression profiles of CXCR4 and PTEN in PC3 and LNCaP prostate cancer cells.
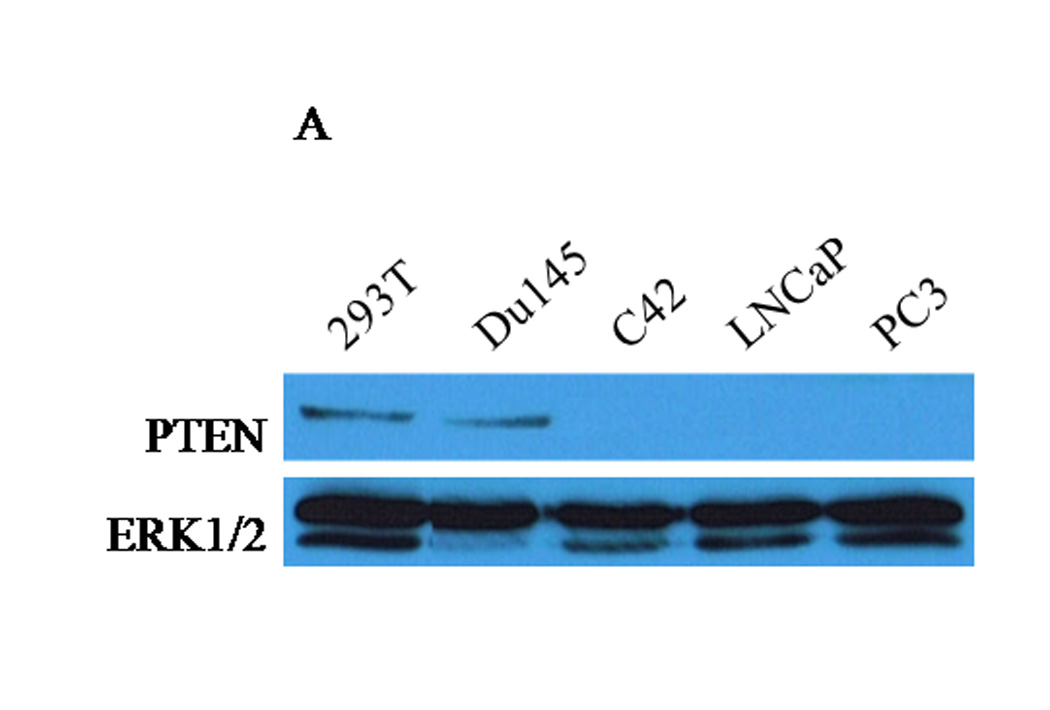
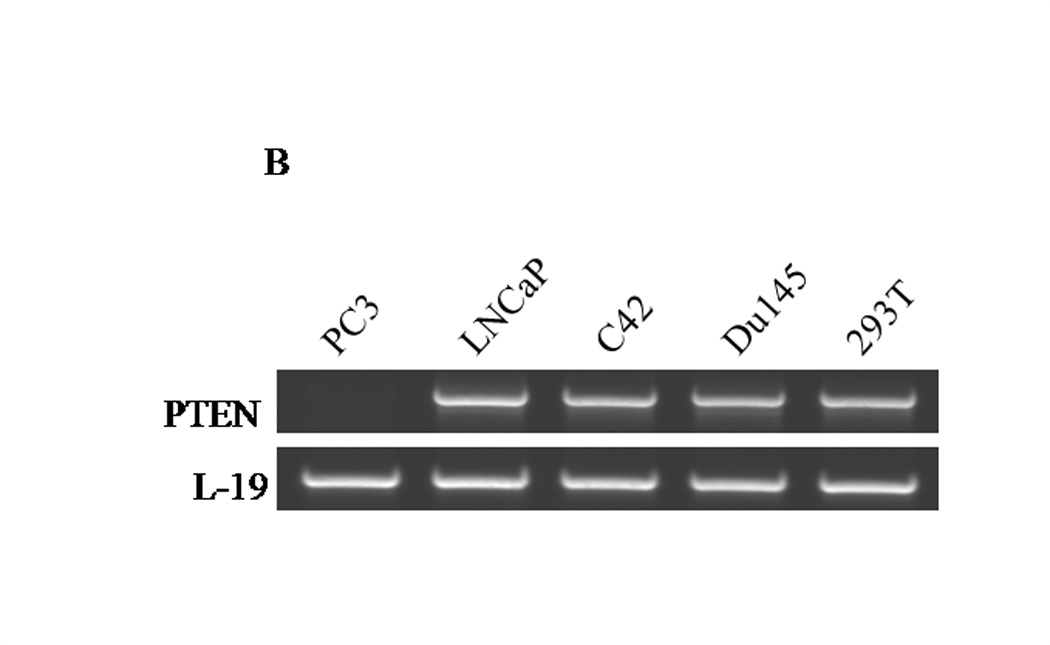
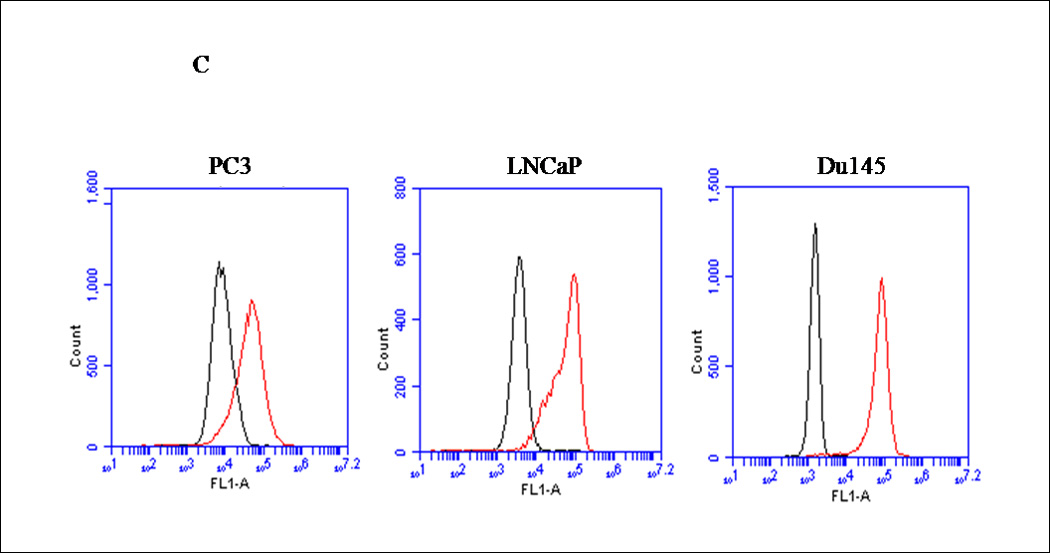

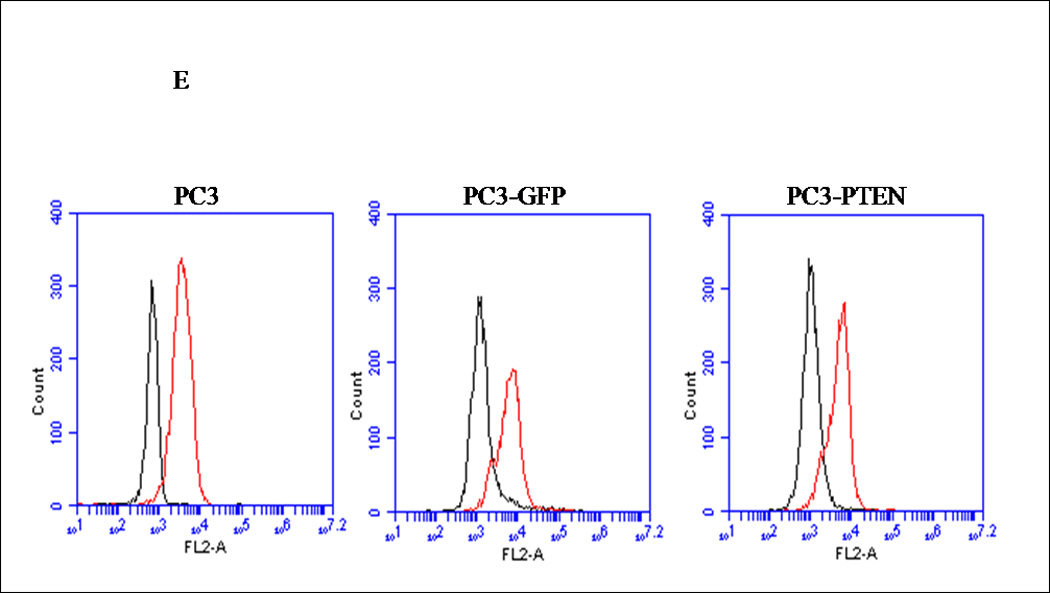

(A) Forty micrograms of total protein were analyzed for PTEN expression by western blot analysis using a PTEN specific antibody. Total ERK1/2 served as a loading control. (B) Two micrograms of total RNA were isolated and reverse transcribed to cDNA for PCR amplification using primers specific for PTEN. L19 served as a loading control. (C) PC3, LNCaP and Du145 cells were fixed in 4% paraformaldehyde, blocked in 1% donkey serum/PBS, and probed with a CXCR4-specific antibody. CXCR4 was detected with a FITC-conjugated donkey anti-mouse antibody. Cells were analyzed for FITC fluorescence intensity using an Accuri C6 flow cytometer. (D) Mean fluorescence was quantified and graphed to represent fold change. Experiments were repeated thrice and the data are presented as fluorescence fold change over control. (E) pcDNA3-GFP (PC3-GFP) and pcDNA3-GFP-PTEN (PC3-PTEN) constructs were transiently transfected into PC3 cells, prior to fixing with 4% paraformaldehyde, blocking in 1% donkey serum/PBS and probing with a CXCR4-specific antibody. CXCR4 was detected with a Cy3-conjugated donkey anti-mouse antibody prior to analysis for Cy3 fluorescence intensity by flow cytometry. (F) Immunofluorescent images of PC3-GFP and PC3-PTEN cells. Cells were probed with a CXCR4 (red) specific antibody. Nuclei were stained with DAPI (blue). (*, P < 0.05).
CXCR4 is expressed in various cancer models, including prostate (18). We compared the cell surface expression of CXCR4 in PC3, LNCaP and Du145 cells by flow cytometry analysis. PC3, LNCaP and Du145 cells were chosen for this assessment because PC3 and LNCaP are absent for PTEN protein, and Du145 cells express functional PTEN. Since PC3 cells are an androgen-independent model, C42 cells were excluded. Prostate cancer cells were fixed, incubated with a rabbit antibody against PTEN and analyzed for FITC intensity by flow cytometry. We found that CXCR4 was expressed on the cell surface of all three cell lines, as detected by the positive shift in fluorescence (redline) compared to background control (blackline) (Fig. 1C). Quantitatively, these data revealed a 5-fold increase in overall fluorescence intensity of CXCR4 over background (control) in PC3 cells, while LNCaP and Du145 cells revealed a 20- and 15-fold increase, respectively (Fig. 1D). These values were standardized against the values of the background (control), which contained secondary antibody only.
Although a functional relationship has not been elucidated, Carver et al observed a correlation in expression between PTEN and CXCR4 in prostate cancer (7). To address this relationship, PTEN expression was reconstituted in PC3 cells, since they are androgen-independent, highly metastatic and represent an advanced stage of prostate cancer. We generated PC3 clones transiently transfected with PTEN (pcDNA3- GFP -PTEN) or GFP (pcDNA3-GFP). Expression of PTEN did not affect the surface expression of CXCR4 in PC3 cells (Fig. 1E), nor did PTEN expression affect the diffuse subcellular localization of CXCR4, compared to control (Fig. 1F). Interestingly, we observed a morphological change in PC3-PTEN cells compared to PC3-GFP cells, 48 hours post-transfection. Both PC3 and PC3-GFP cells demonstrated a mesenchymal-like morphology, as represented by lamellipodia-like projections (Fig. 2A). However, PC3-PTEN cells demonstrated an epithelial-like morphology compared to PC3 and PC3-GFP cells (Fig. 2A). In agreement with our observations, Kotelevets et al observed that canine kidney epithelial cells transfected with PTEN retained an epithelioid morphotype, compared to fibroblastic-like cells transfected with PTEN mutants (34). To further investigate this morphological transition, we analyzed the expression pattern of vimentin, an EMT marker (Fig. 2D). We found that vimentin expression decreased in PC3-PTEN cells, compared to PC3-GFP cells. Copy DNA constructs were labeled with GFP; therefore, we utilized fluorescence microscopy to confirm that PTEN was expressed in these epithelial-like cells. We detected GFP-PTEN fusion protein at the cell membrane of PC3-PTEN cells, where it primarily functions (Fig. 2B). To ensure that cDNA constructs were expressing the fusion protein, we detected PTEN expression by western blot analysis (Fig. 2C).
Figure 2. Transient transfection of PTEN into PC3 cells induced a mesenchymal to epithelial-like morphological change.
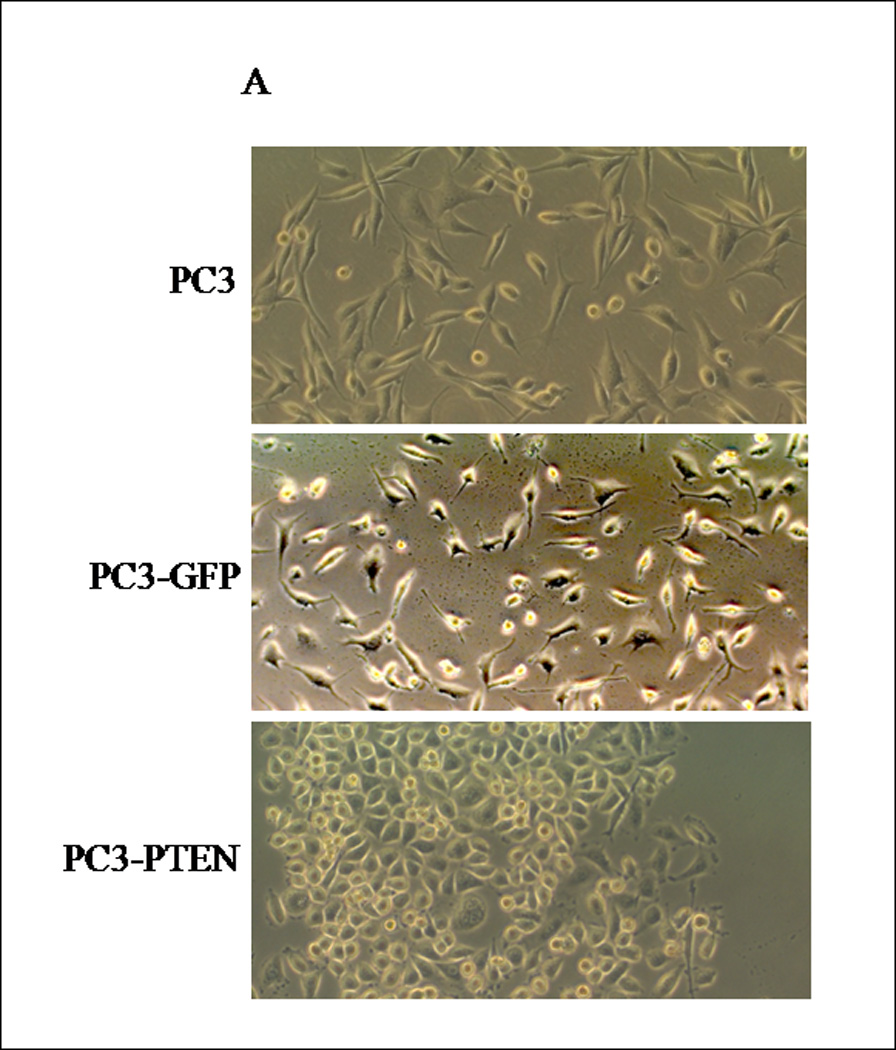
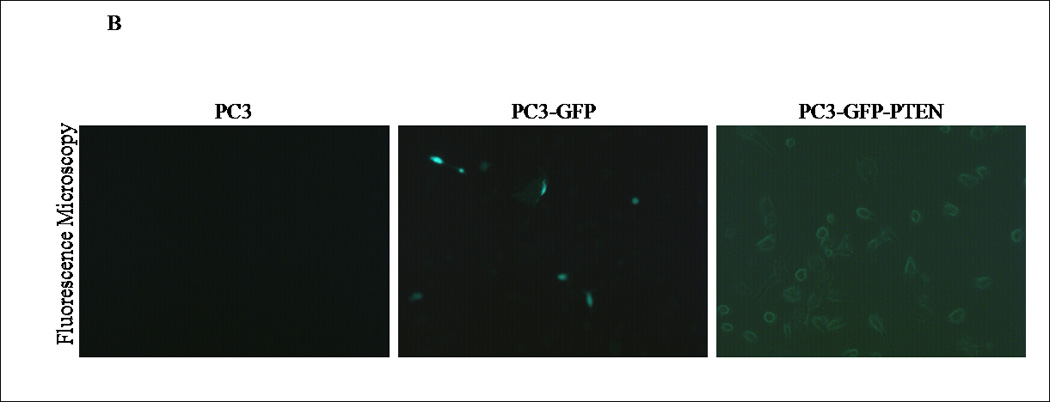
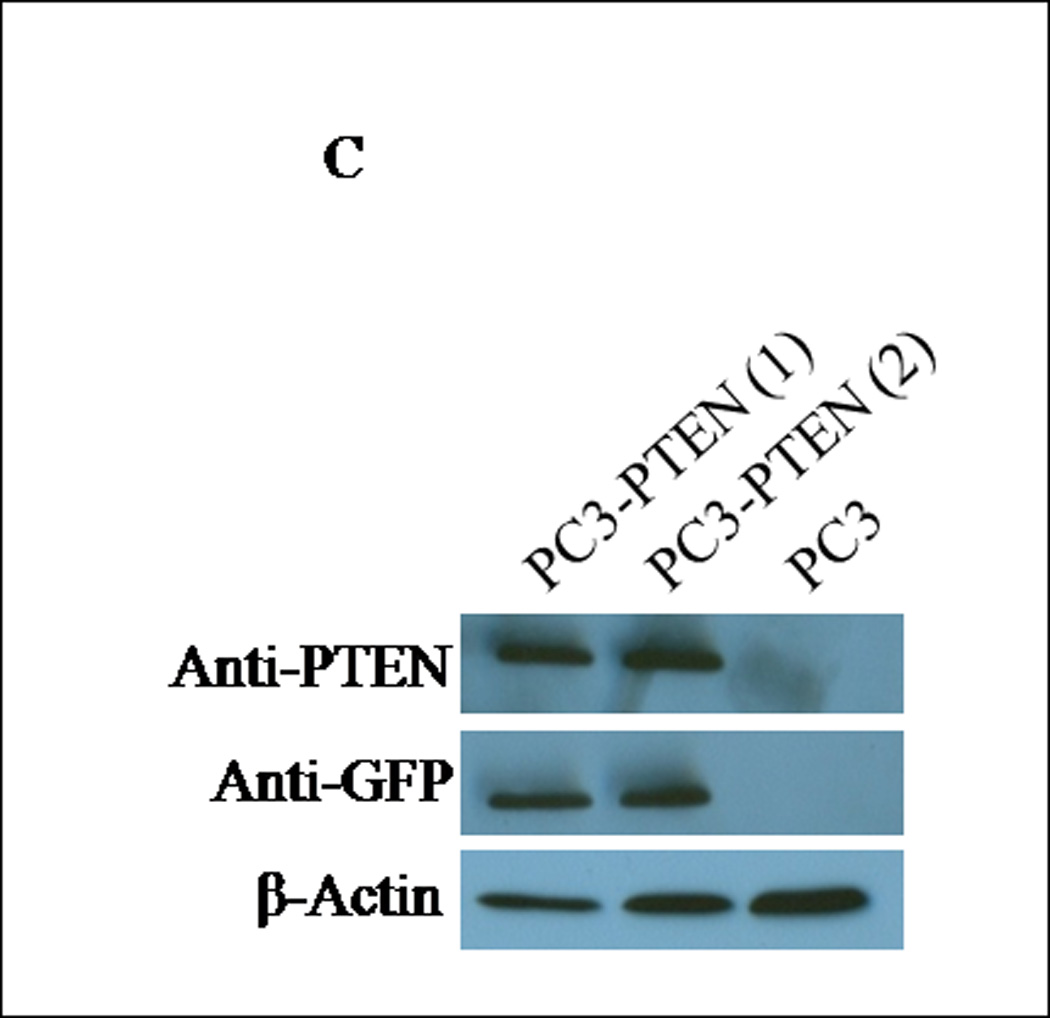
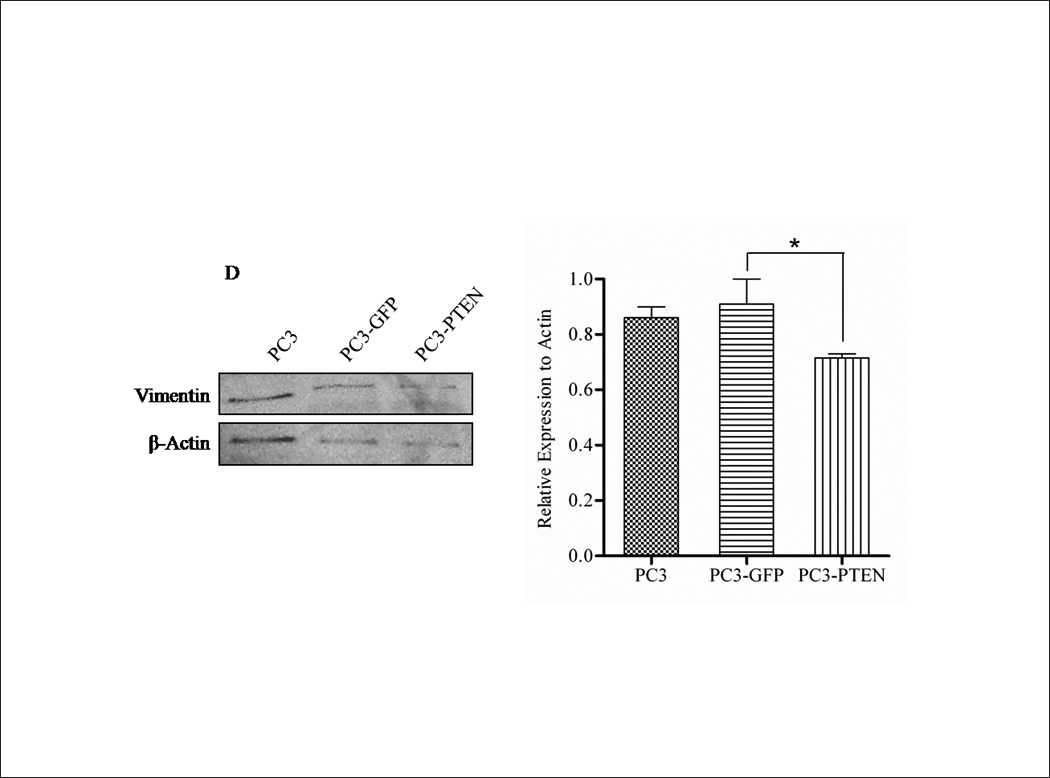
(A) Cells were transiently transfected with pcDNA3-GFP and pcDNA3-GFP-PTEN constructs. Light micrographs were taken 48h post-transfection at 10X magnification using a Zeiss Axiovert 200M fluorescence microscope. (B) Transiently transfected cells were analyzed for GFP fluorescence 48h post-transfection. Cells were excited at 495 nm and micrographs were taken at 10X magnification using a Zeiss Axiovert 200M fluorescence microscope. (C) Thirty micrograms of total protein was extracted from two clones of transfected cells and analyzed by western blot analysis using anti-PTEN and anti-GFP specific antibodies. β-actin served as a loading control. (D) Transiently transfected cells were lysed and 30 µg of protein were analyzed for vimentin. β-actin served as a loading control. Graph represents the relative expression of vimentin compared to control. (*, P < 0.05).
PTEN expression inhibited CXCR-mediated migration and proliferation of prostate cancer cells
Prostate cancer tends to spread to the bones. The CXCR4/SDF1α signaling axis was shown to play a pivotal role in triggering prostate bone metastasis (19), while Wu et al observed that PTEN inhibited C42 cell migration toward calvaria-conditioned medium (24). To examine whether PTEN negatively regulates CXCR4-mediated migration and proliferation, respective assays were performed with PC3, PC3-GFP or PC3-PTEN cells upon CXCR4 stimulation with its ligand, SDF1α. By transwell assay, we observed an increase in cell migration of PC3 and PC3-GFP cells towards SDF1α in the bottom chamber (Fig. 3A). However, SDF1α failed to stimulate movement of PC3-PTEN cells (Fig. 3A), resulting in a significant reduction in cell migration compared to PC3 and PC3-GFP cells (Fig. 3B).
Figure 3. PTEN inhibited CXCR4-mediated migration and proliferation.
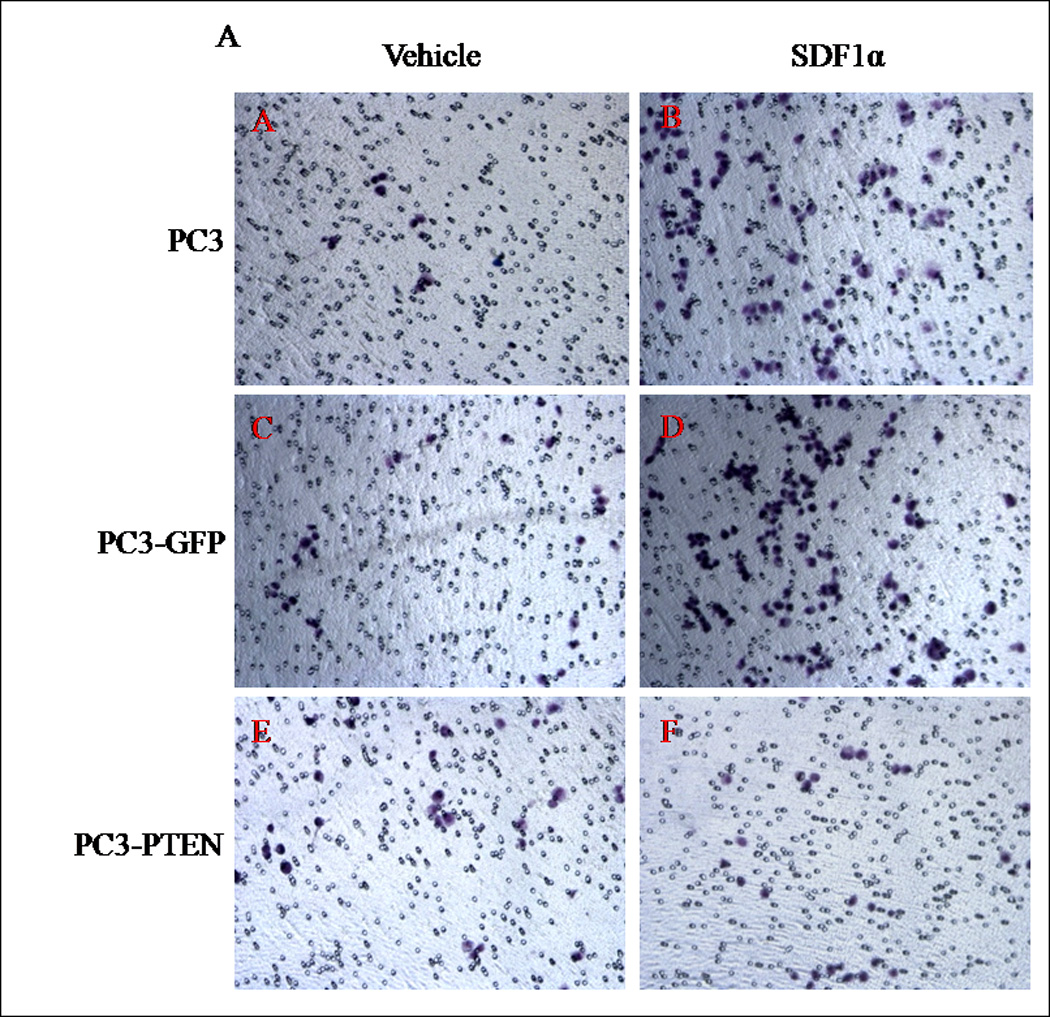


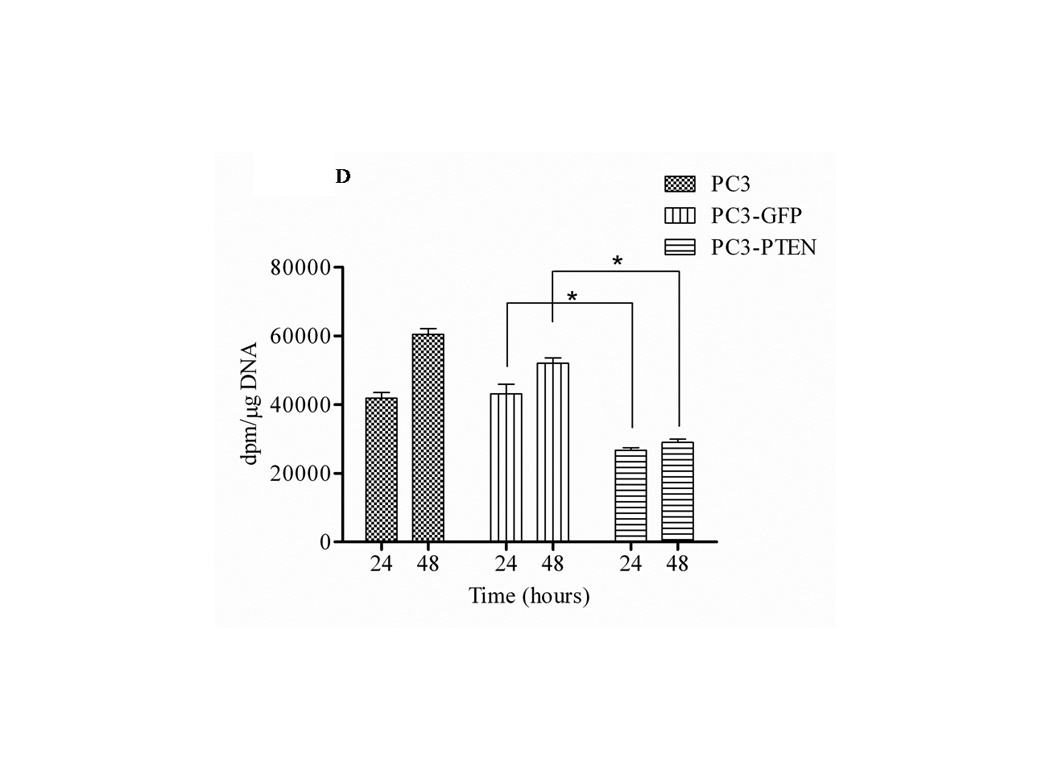
(A) Cells were transiently transfected with 4 µg of pcDNA3-GFP or pcDNA3-GFP-PTEN constructs, prior to serum-starvation for 24 h. Cells (2×104) were added to the upper transwell chamber and allowed to migrate towards 100 ng/mL of SDF1α in the lower wells for 4 h at 37°C. Five fields of each transwell insert were randomly selected and counted for migrated cells at 10X magnification. Micrographs of migrated cells were taken at 10X magnification using a Zeiss Axiovert 200M light microscope. (B) A graphical representation of total migrated cells. Experiments were repeated thrice and data represents the averages of three independent experiments. (C) Cells were transiently transfected and serum starved, prior to treatment with 100ng/mL of SDF1α at the indicated time points. MTT assays were performed after each time point following the manufacturer’s protocol. Experiments were repeated thrice in triplicate and data represents the averages of three independent experiments. (D) Cells were transiently transfected and serum starved, prior to treatment with 100ng/mL of SDF1α at the indicated time points. Proliferation was measured by [3H] thymidine uptake/DNA synthesis assay. (*, P < 0.05).
To further investigate the regulatory role of PTEN in CXCR4-mediated functions, PC3, PC3-GFP and PC3-PTEN cells were analyzed for proliferation and viability. By MTT assay, we observed increases in the viability of both PC3 and PC3-GFP cells 48 hours post treatment with SDF1α. However, the viability of PC3-PTEN cells was significantly reduced compared to PC3-GFP cells at both 24 and 48 hours post SDF1α treatment (Fig. 3C). By [3H] thymidine incorporation assay, we observed increases in proliferation in both PC3 and PC3-GFP cells 48 hours post ligand treatment; whereas the proliferation of PC3-PTEN cells was significantly reduced compared to PC3-GFP cells up to 48 hours post SDF1α treatment (Fig. 3C).
Suppression of ERK1/2 phosphorylation inhibited CXCR4-mediated migration of PC3 cells
PTEN functions as a dual protein and lipid phosphatase. The major known substrate of PTEN is the lipid second messenger phosphatidylinositol 3,4,5-trisphosphate (PIP3), which activates downstream signaling components, most notably the protein kinase AKT (35). The subsequent activation of CXCR4/SDF1α involves classical pathways of cell survival: (i) PI3K/AKT, (ii) the MAPK cascade and (iii) PLC-β (36). Many reports have observed AKT activation in response to SDF1α, while others have observed that ERK1/2 activity is required for GPCR-mediated migration (37, 38). When we investigated the basal levels of ERK1/2 and AKT in PC3-GFP and PC3-PTEN cells, we observed a decrease in phospho-AKT expression in PC3-PTEN cells compared to PC3-GFP cells (Fig. 4A). Phospho-ERK1/2 levels did not change (Fig. 4A). Treatment of serum starved PC3-GFP and PC3-PTEN cells with SDF1α resulted in ERK1/2 phosphorylation in a biphasic manner, while no changes in AKT phosphorylation were observed compared to control (Fig. 4B). Phospho-ERK1/2 was detected in PC3-GFP cells upon SDF1α stimulation, but not in PC3-PTEN cells under the same conditions (Fig. 4B).
Figure 4. PTEN inhibited CXCR4-mediated migration by inhibiting ERK1/2 phosphorylation.
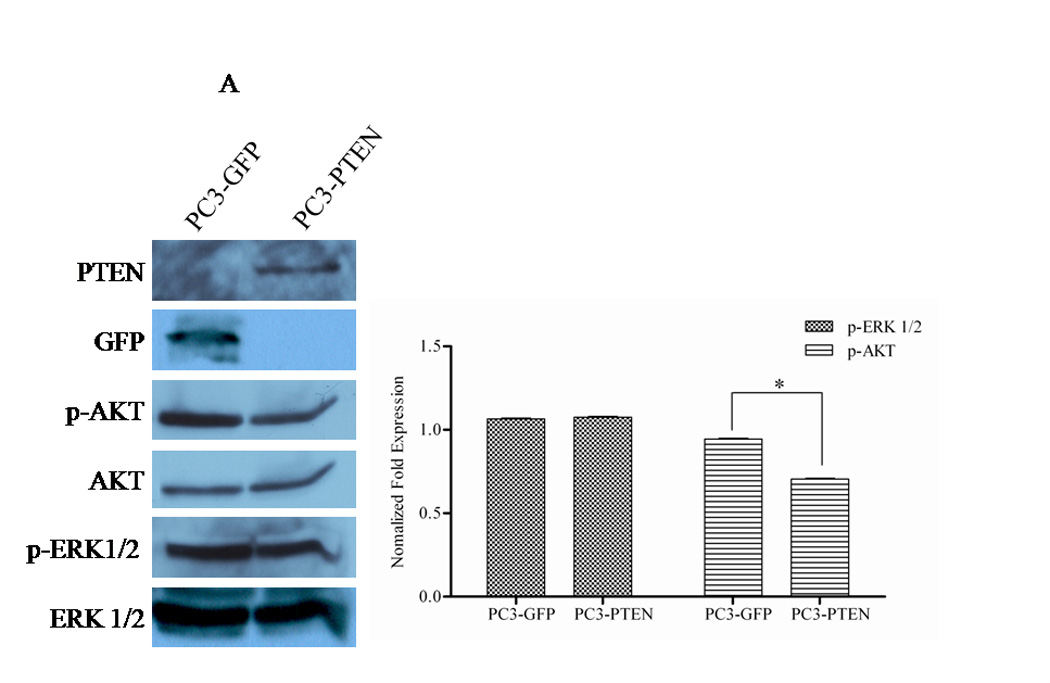
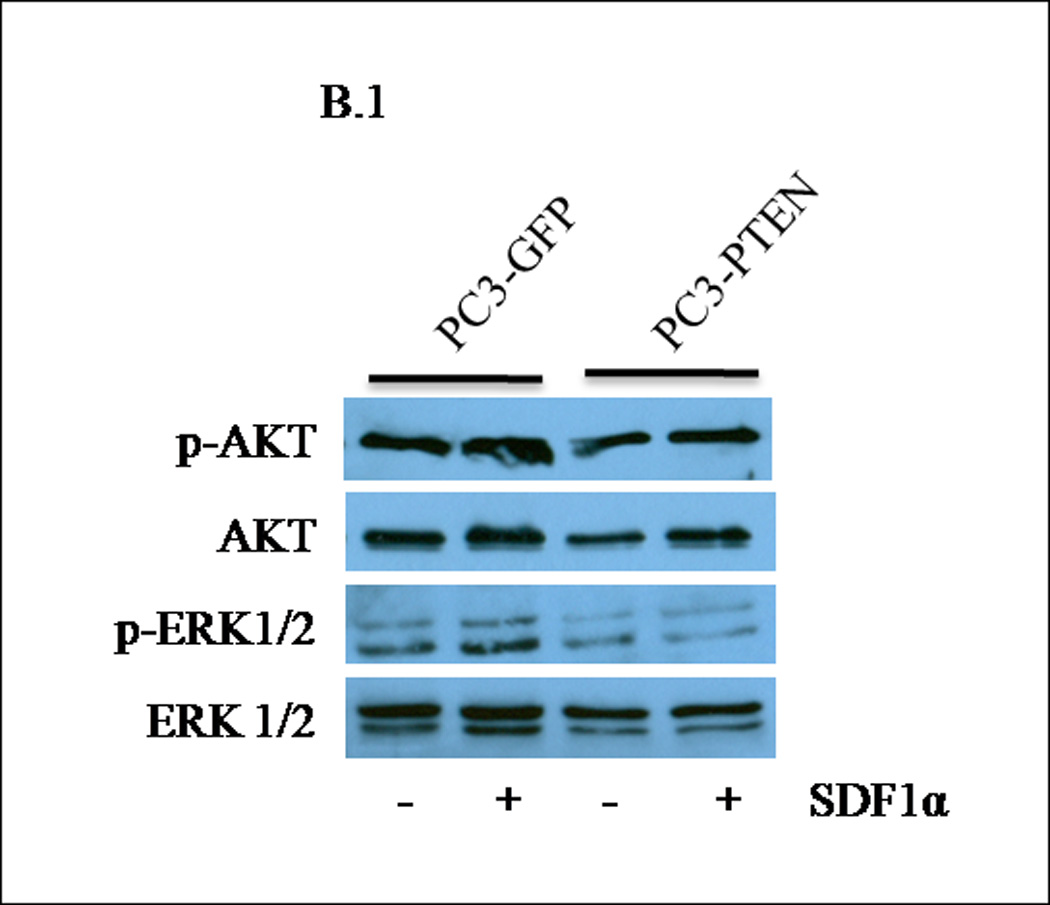
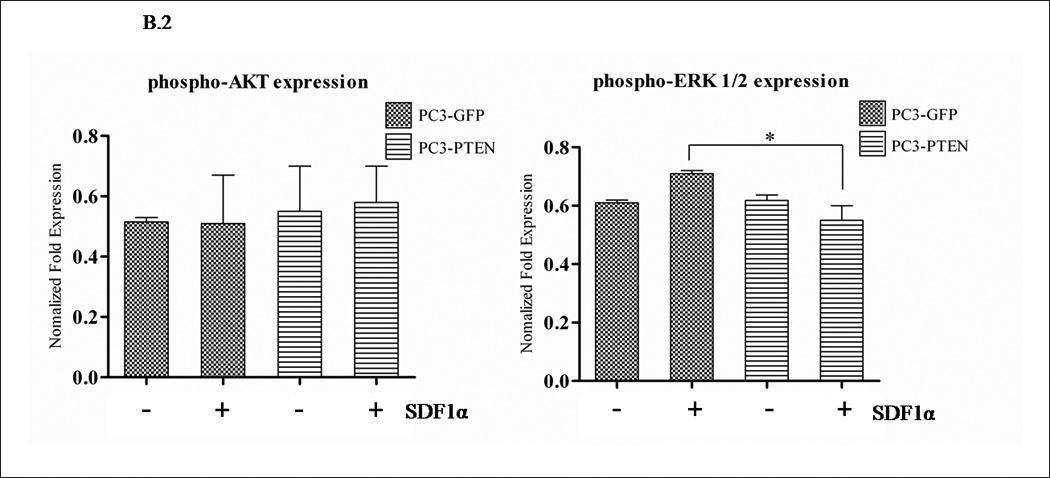
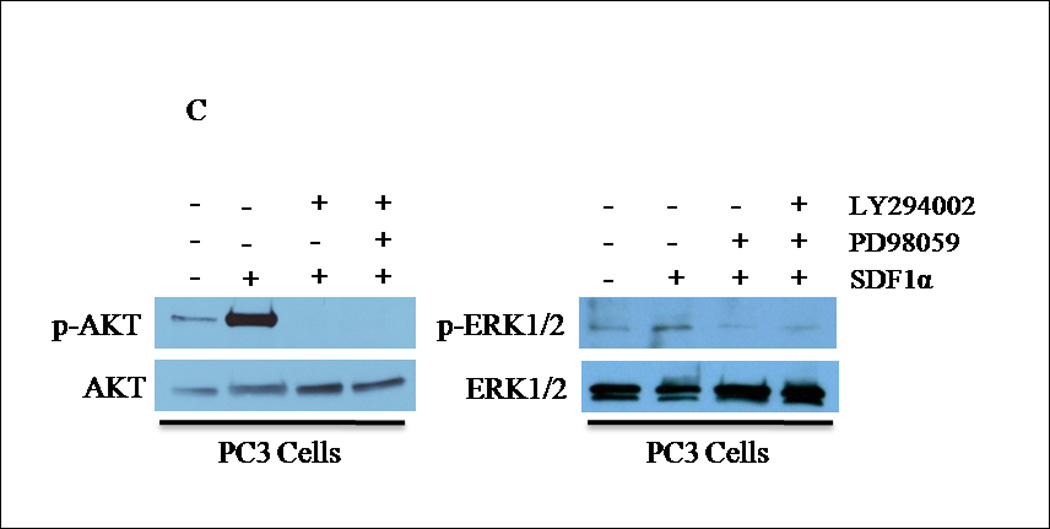
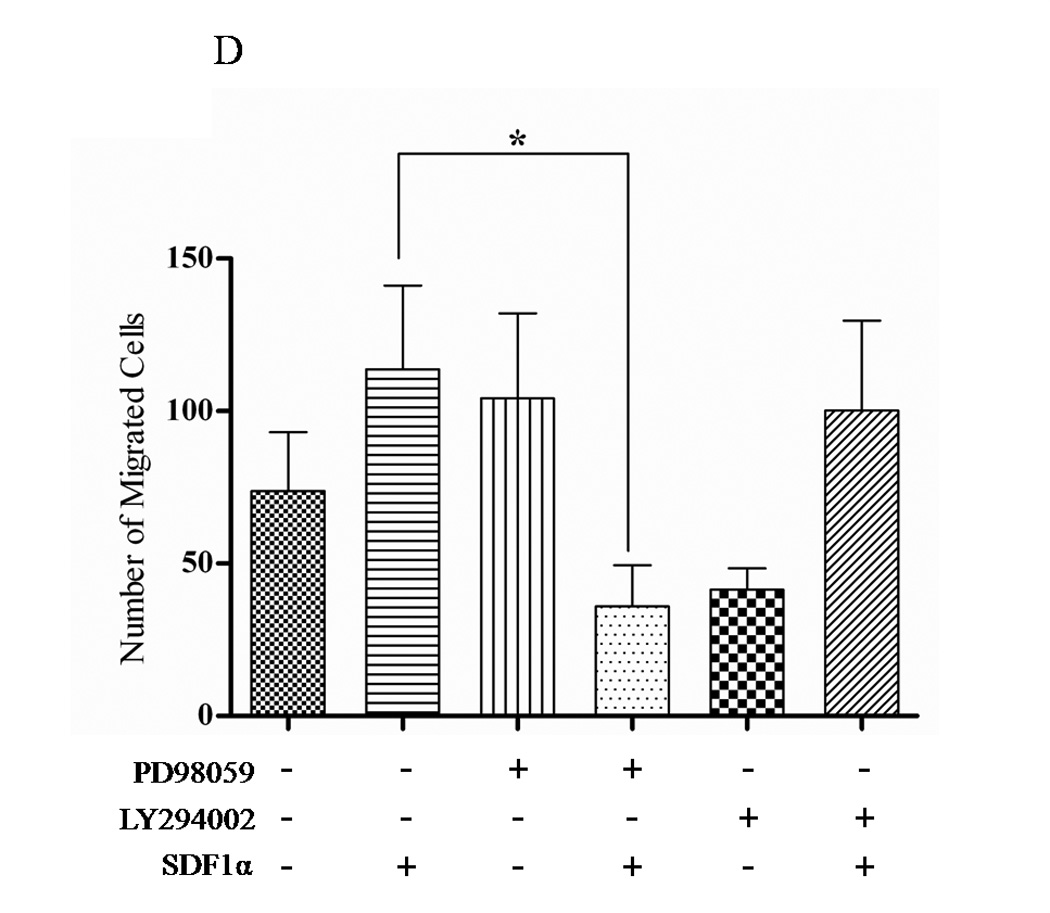
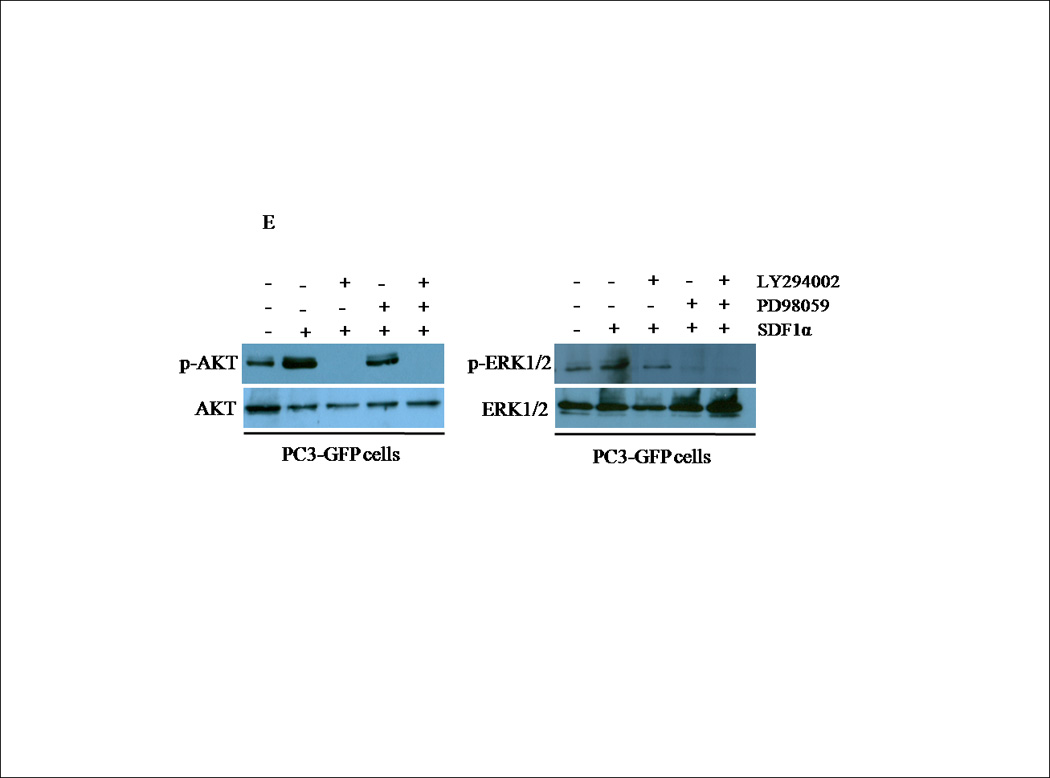

(A) Transiently transfected cells were lysed and 30 µg of protein were analyzed for PTEN, p-AKT, total AKT, p-ERK1/2 and total ERK1/2 expression by western blot analysis using specific antibodies. Graph represents a densitometric analysis for the relative expression of p-AKT and p-ERK1/2 compared to controls. (B) Transiently transfected cells were treated with 100 ng/mL of SDF1α for 10 minutes followed by western blot analysis of 30 µg of protein using p-AKT, total AKT, p-ERK1/2 and total ERK1/2 specific antibodies. Graph represents a densitometric analysis for the relative expression of p-AKT and p-ERK1/2 compared to controls. (C) PC3 and (E) PC3-GFP cells were serum starved prior to treatment with 50mM of PD98059 and/or 10mM of LY294002 for 1 h at 37°C. Cells were then stimulated with 100 ng/mL of SDF1α for 10 minutes followed by western blot analysis of 30 µg of protein using p-AKT, total AKT, p-ERK and total ERK1/2 specific antibodies. (D) PC3 and (F) PC3-GFP cells were serum starved and treated with 50 µM of PD98059 and/or 10 µM of LY294002 for 1 h at 37°C. Cells (2×104) were added to the upper transwell chamber and allowed to migrate towards 100 ng/mL of SDF1α in the lower wells for 6 h at 37°C. Five fields were randomly selected and counted for migrated cells at 10X magnification. Experiments were repeated thrice and data represents the averages of three independent experiments. (*, P < 0.05).
To determine whether PTEN-mediated inhibition of ERK1/2 phosphorylation was responsible for the decreased CXCR4-mediated cell migration of PC3-PTEN cells, we used PD98059, a small molecule MEK inhibitor to suppress ERK1/2 phosphorylation, and LY294002, a small molecule PI3K inhibitor to suppress AKT phosphorylation. Pretreatment with PD98059 for 1hour abrogated SDF1α-induced phosphorylation of ERK1/2, while LY294002 abrogated phosphorylation of AKT (Fig. 4C). Since we observed that PTEN blocked SDF1α-induced phosphorylation of ERK1/2, we then determined whether ERK1/2 abrogation inhibited CXCR4-mediated migration of PC3 cells. We monitored transwell cell migration of PC3 and PC3-GFP cells towards SDF1α in the presence and absence of LY294002 and PD98059. Pretreatment with PD98059 significantly inhibited PC3 and PC3-GFP migration, which was not inhibited by LY294002 (Figs. 4D and 4F). PD98059, nor LY294002, was cytotoxic to the cells (data not shown).
Downregulation of PTEN expression enhanced CXCR4-mediated migration of Du145 cells
PTEN inactivation correlates with invasiveness and metastasis in prostate cancers. Loss of PTEN is common in prostate cancers that have transitioned to an advanced disease. Du145 cells have low-to-moderate metastatic potential and express a functional PTEN allele. Therefore, we tested whether downregulation of PTEN could serve as the permissive switch for CXCR4-mediated migration. We utilized small interfering RNA (siRNA) to downregulate the expression of PTEN in Du145 cells. Cells transfected with a fluoresceinconjugated siRNA targeted for PTEN or control (scramble siRNA) were subjected to a transwell migration assays towards SDF1α. Clones transfected with PTEN siRNA exhibited decreased PTEN expression by Western blot analysis (Fig. 5A) and a significant increase in migratory activity towards SDF1α (Fig. 5B). Migratory activity towards SDF1α was not observed in control transfected cells. These finding suggest that there is a reciprocal relationship between CXCR4 activity and PTEN expression. Taken together, these results support the idea that the loss of PTEN regulation permits the advancement of prostate cancer through CXCR4, and its concomitant pathways.
Figure 5. PTEN downregulation enhanced CXCR4-mediated migration of Du145 cells.


(A) Du145 cells were transfected with PTEN specific siRNA or scramble-siRNA using Lipofectamine2000. Fourty-eight hours posttransfection, cells were lysed and 30 µg of protein were analyzed for PTEN expression by western blot analysis. β-actin served as a loading control. (B) Cells were transfected with PTEN specific siRNA or scramble-siRNA and serum starved for 24 h. Cells (2×104) were seeded into the upper chamber of transwell inserts and allowed to migrate towards 100ng/mL of SDF1α for 6 h at 37°C. Five fields were randomly selected and counted for migrated cells at 10X magnification. Experiments were repeated thrice and data represents the averages of three independent experiments. (*, P < 0.05).
DISCUSSION
A physical interaction between PTEN and CXCR4 has not been elucidated. Upon SDF1α binding to CXCR4, tumorgenic-associated pathways are activated: (i) G-protein coupled receptor (GPCR) signaling, (ii) PI3K/AKT, (iii)MAPK, (iv)JAK/STAT, (v) Src kinase and (vi) HER2 (12, 18, 19). Downstream, CXCR4-initiated signaling leads to the transcription of genes involved in migration and tumorigenesis (14). Traditionally, PTEN functions as a dual-specificity lipid and protein phosphatase that inhibits tumorigenic events through dephosphorylation of PIP3, thus antagonizing PI3K/AKT–mediated signaling (8). By converting PIP3 into PIP2, PTEN negatively regulates PI3K/AKT signaling and subsequent downstream pathways. PTEN and CXCR4 converge at the PI3K/AKT and/or MAPK signaling level. This is supported by our research and others, that PI3K/AKT and/or ERK inhibitors mimicked PTEN’s effect of negatively regulating CXCR4 (27). Therefore, our research describes that the loss of PTEN expression provides a “permissive switch” for CXCR4-mediated signaling and functions (Fig. 6).
Figure 6. Proposed mechanism for the role of PTEN in CXCR4-mediated signaling.

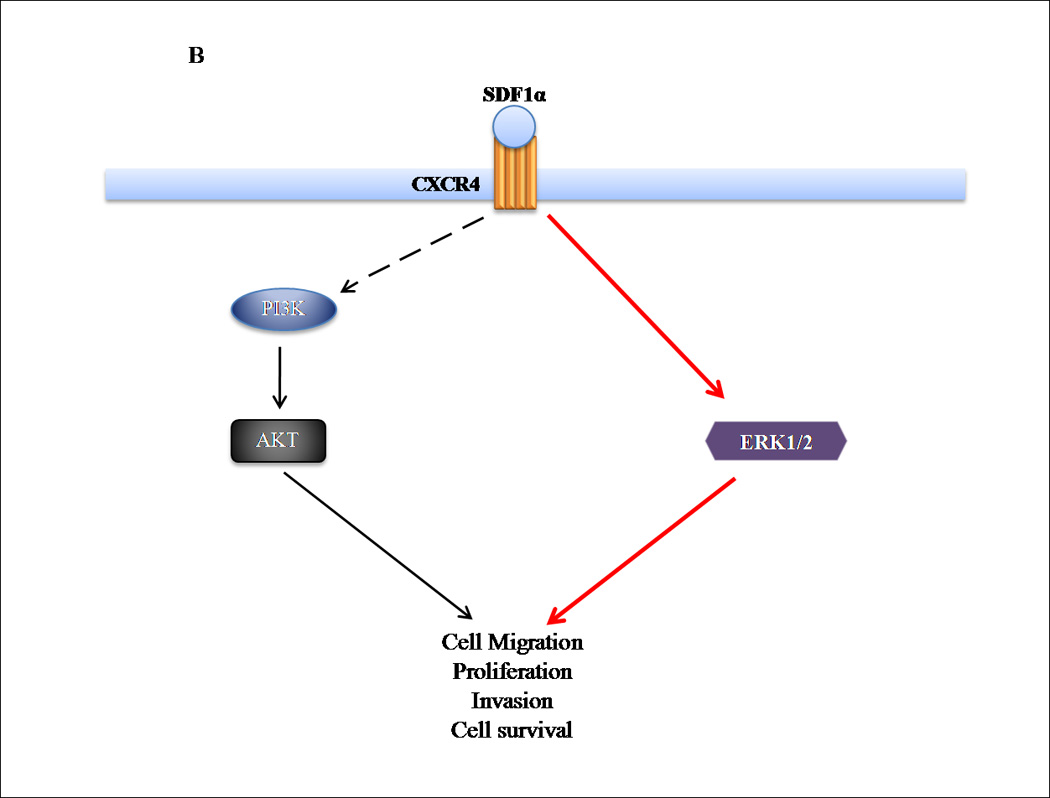
(A) Ligand activation of CXCR4 stimulates several pathways, including PI3K/AKT and ERK1/2, resulting in tumorigenic events. PTEN negatively regulates both pathways, by acting as a protein and lipid phosphatase. (B) Loss of PTEN expression results in a loss of regulation of CXCR4-mediated events, permitting activation of signaling pathways that enhance tumorigenesis.
In this report, we investigated PTEN in null human PC3 and wildtype human Du145 prostate cancer cell lines to characterize the involvement of PTEN in CXCR4-mediated functions. Our findings were as follows: (1) PTEN-null PC3 cells exhibited potent CXCR4-mediated migration, indicating that PTEN was not essential for the movement of prostate cancer cells; (2) By transient transfection, we found that reconstitution of PTEN expression in PC3 cells induced morphological changes and downregulated CXCR4-mediated migration and proliferation; (3) PTEN reconstitution regulated phospho-ERK1/2, but not phospho-AKT in CXCR4-mediated functions. This was further supported by the observation that MAPK inhibitors mimicked PTEN's effect of negatively regulating CXCR4-mediated migration; (4) Expression of PTEN did not affect cell surface expression of CXCR4, suggesting that inhibition of CXCR4-mediated migration and proliferation was at the level of signaling; and (5) Downregulation of wildtype PTEN by siRNA in Du145 cells increased CXCR4-mediated migration. Collectively, these findings indicate that loss of PTEN expression in prostate cancer cells provides the loss of a critical inhibitory function in the signal cascade(s) that lead to cell migration, and may provide the permissive switch to CXCR4-mediate tumorigenesis and advanced stages of prostate cancer.
Prostate cancers have the ability to localize to tissue sites throughout the body. Loss of tumor suppressors, specific chemoattractants and migration-promoting signaling pathways may influence sites of specific distant tumor formation. The long-held view is that metastasis occurs by a multistep process requiring intravasation, cell survival in the blood stream, extravasation, initiation of micrometastasis and the establishment of new blood vessels (39). It has been proposed that the expression, or absence, of particular genes in primary tumors may directly predispose cancer cell growth and metastatic development. Aberrant expression of key genes, including CXCR4 and PTEN, have been established to collectively facilitate cell invasion, bone metastasis, cell adhesion and angiogenesis (12, 16, 22, 40–43). CXCR4 and PTEN are independently identified as gene expression signatures, which reflect the activation status of oncogenic pathways, and in turn provide clinically relevant associations with disease outcomes (4, 44).
The potential prognostic role of the combined alterations in CXCR4 and PTEN in prostate cancer is not well established. A gene expression signature for immunohistochemistry (IHC)-detectable PTEN loss has been developed for breast cancer, which correlates to poor patient outcome in independent data sets of breast, bladder, and prostate carcinoma (45). Primary prostate cancers often show genetic loss or mutation of at least one PTEN allele in approximately 30% to 70% of advanced (locally advanced or metastatic) cases, primarily at the level of transcription (46, 47). Conversely, in Du145, LNCaP and PC3 prostate cancer cells, CXCR4 mRNA expression was approximately 1000, 400 and 21 times, respectively, that of primary and normal prostate cancer cell lines1542 NPTX, Pre 2.8 and 1542 CPT3X (48). In the same study, migration of the metastatic cell lines PC3 and Du145 was enhanced by SDF1α ligand and inhibited by an antibody to CXCR4, indicating that migration of metastatic prostate cancer cells was facilitated and enhanced by the SDF1α/CXCR4 signaling axis, which did not influence the migration of the normal prostate epithelial cells (48). Considering the converging pathways that CXCR4 and PTEN activate and inhibit, respectively, loss of PTEN could provide one of the critical events in human prostate cancer that cooperates to promote tumor development and progression through CXCR4.
There have been few studies on the role of PTEN in the metastatic events of prostate cancer. PC3 cells transfected with wildtype PTEN reverted the invasive phenotype and invasion of collagen type I (34). Wu et al proposed that PTEN loss upregulated cell cycle genes, cdc6 and cyclin E2, which in turn lead to metastatic colonization at distant sites (4). In defining a relationship with PTEN, Carver et al observed that cancer specimens containing chromosomal translocations involving the ERG locus were concomitant with the loss of PTEN expression and upregulation of CXCR4 in prostate cancer (7). Phillips et al reported that overexpression of wild type PTEN in non-small cell lung cancer cells inhibited hypoxia-induced CXCR4 expression (28).
Outside of prostate cancer, one group has studied the role of PTEN and CXCR4 in the chemotatic movement of PTEN-null Jurkat cells, where enhanced chemotaxis was observed. Induction of PTEN expression downregulated Jurkat cell chemotaxis towards SDF1α, which correlated with the reconstitution of PTEN expression. In the studies by Gao et al, they observed that the lipid phosphatase activity of PTEN was essential for the role of PTEN as a negative regulator of chemotaxis, suggesting that PI3K pathway was involved, and that PTEN antagonized PI3K to inhibit chemotaxis (49). In agreement with Gao et al, we observed downregulation of CXCR4-mediated migration and proliferation upon reconstitution of PTEN into PC3 cells. We also observed an increase in CXCR4-mediated migration of poorly migratory Du145 cells, upon downregulation of PTEN by siRNA.
PTEN functions as a dual-functional protein and lipid phosphatase. Physiologically, phosphatidylinositol 3,4,5-trisphosphate (PIP3) is the substrate of PTEN, whereby PTEN dephosphosphorylates PIP3, indirectly inhibiting AKT activation. We found that phospho-ERK1/2, but not phospho-AKT demonstrated biphasic expression in PC3-PTEN cells, following SDF1α stimulation. We further examined the roles of the ERK1/2 and AKT pathways in CXCR4-mediated migration by chemical impairment with PD98059 (MEK inhibitor) and LY294002 (PI3K inhibitor). PD98059 inhibited CXCR4-mediated migration, unlike LY294002, further implicating a role for ERK1/2 in CXCR4-mediated metastasis. Corroborating our studies, Sun et al observed that CXCR4-mediated chondrosarcoma cell invasion was inhibited by the CXCR4 inhibitor AMD3100, as well as with ERK1/2 inhibitor U0126 and ERK1/2 siRNA (50). Likewise, similar results have been demonstrated in human osteosarcoma cells (51), laryngeal and hypopharyngeal squamous cell carcinoma metastasis (52, 53). Classically, the ERK1/2 signaling cascade is not a target of PTEN; however, Thomas et al reported that PTEN reconstitution in SPARC (secreted protein acidic and rich in cysteine) suppressed the SHC-RAF-ERK signaling pathway in SPARC-expressing cells (54). Moreover, restoration of wildtype PTEN induced apoptosis in Jun(+/+) cells undergoing cellular transformation by oncogenic Ras (55).
In summary, metastatic signature genes (e.g. chemokine and growth factor receptor/ligand axes) have been implicated to facilitate the cell autonomous transforming functions of a primary disease to a malignant disease, throughout the course of tumor progression. Since loss of PTEN activity in advanced, metastatic prostate cancers leads to increased cell proliferation, reduced cell death and metastasis, it is not implausible to suggest that loss of PTEN activity in prostate cancer provides the permissive switch to a CXCR4-mediated advanced, metastatic disease. Current management options for prostate cancer are hormonal therapy for early stage tumors and chemotherapy, which is often reserved for diseases that have spread beyond the prostate. Radiation therapy may be used for some advanced tumors. Overall, the treatment options for advanced, metastatic prostate cancers become slim, often relying on general chemotherapy and radiation. During advanced stages of prostate cancer, these treatment options are geared toward easing symptoms rather than slowing the disease. Deregulated CXCR4, PI3K/AKT and/or MAPK signaling pathways substantially contribute to the pathogenesis of prostate cancers. Likewise, PTEN inactivation is associated with a hormone-refractory disease (22). Understanding the relationship between PTEN and CXCR4 will lead to new treatment options, especially for aggressive, androgen-insensitive cancers that express low or diminished levels of PTEN and high levels of CXCR4. Thus, when hormone therapy is no longer an option, antagonists against CXCR4, PI3K/AKT and/or MAPK signaling could prove to be beneficial in regulating tumor progression once PTEN expression and function is lost. We suggest that targeted therapies against these critical and frequent events (loss of PTEN; upregulation of CXCR4 expression in advanced prostate cancers) should be tested in the future, in combination with current chemotherapeutic agents.
ACKNOWLEDGEMENTS
Limitations of space preclude extensive citation of literature; we apologize to those whose work is not mentioned herein. Research in this lab is supported, in part, by National Institutes of Health Grants 5R25GM060414 (MBRS-RISE)(MAC), 1P20MD002285 (P20)(VOM) and 2G12RR003062-22 (RCMI)(CVH). We would like to thank Dr. Alonzo Ross (UMass, Worcester) for the pcDNA3-GFP and pcDNA3-GFP-PTEN constructs. We also thank Brittany T. Jones, Amber L. Taylor and T’Shane C. Williams (CAU) for technical support.
REFERENCES
- 1.Association AC. What Are the Key Statistics About Prostate Cancer? 2010 www.cancer.org. [Google Scholar]
- 2.Sircar K, Yoshimoto M, Monzon FA, et al. PTEN genomic deletion is associated with p-Akt and AR signalling in poorer outcome, hormone refractory prostate cancer. J Pathol. 2009;218:505–513. doi: 10.1002/path.2559. [DOI] [PubMed] [Google Scholar]
- 3.Wang S, Gao J, Lei Q, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209–221. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 4.Wu Z, Cho H, Hampton GM, Theodorescu D. Cdc6 and cyclin E2 are PTEN-regulated genes associated with human prostate cancer metastasis. Neoplasia. 2009;11:66–76. doi: 10.1593/neo.81048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ratnacaram CK, Teletin M, Jiang M, Meng X, Chambon P, Metzger D. Temporally controlled ablation of PTEN in adult mouse prostate epithelium generates a model of invasive prostatic adenocarcinoma. Proc Natl Acad Sci U S A. 2008;105:2521–2526. doi: 10.1073/pnas.0712021105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whang YE, Wu X, Suzuki H, et al. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc Natl Acad Sci U S A. 1998;95:5246–5250. doi: 10.1073/pnas.95.9.5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carver BS, Tran J, Gopalan A, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet. 2009;41:619–624. doi: 10.1038/ng.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dahia PL. PTEN, a unique tumor suppressor gene. Endocr Relat Cancer. 2000;7:115–129. doi: 10.1677/erc.0.0070115. [DOI] [PubMed] [Google Scholar]
- 9.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 10.Fang J, Ding M, Yang L, Liu LZ, Jiang BH. PI3K/PTEN/AKT signaling regulates prostate tumor angiogenesis. Cell Signal. 2007;19:2487–2497. doi: 10.1016/j.cellsig.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang W, Zhu J, Efferson CL, et al. Inhibition of tumor growth progression by antiandrogens and mTOR inhibitor in a Pten-deficient mouse model of prostate cancer. Cancer Res. 2009;69:7466–7472. doi: 10.1158/0008-5472.CAN-08-4385. [DOI] [PubMed] [Google Scholar]
- 12.Chinni SR, Yamamoto H, Dong Z, Sabbota A, Bonfil RD, Cher ML. CXCL12/CXCR4 transactivates HER2 in lipid rafts of prostate cancer cells and promotes growth of metastatic deposits in bone. Mol Cancer Res. 2008;6:446–457. doi: 10.1158/1541-7786.MCR-07-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wallace TA, Prueitt RL, Yi M, et al. Tumor immunobiological differences in prostate cancer between African-American and European-American men. Cancer Res. 2008;68:927–936. doi: 10.1158/0008-5472.CAN-07-2608. [DOI] [PubMed] [Google Scholar]
- 14.Wong D, Korz W. Translating an Antagonist of Chemokine Receptor CXCR4: from bench to bedside. Clin Cancer Res. 2008;14:7975–7980. doi: 10.1158/1078-0432.CCR-07-4846. [DOI] [PubMed] [Google Scholar]
- 15.Akashi T, Koizumi K, Tsuneyama K, Saiki I, Takano Y, Fuse H. Chemokine receptor CXCR4 expression and prognosis in patients with metastatic prostate cancer. Cancer Sci. 2008;99:539–542. doi: 10.1111/j.1349-7006.2007.00712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002;62:1832–1837. [PubMed] [Google Scholar]
- 17.Sun YX, Wang J, Shelburne CE, et al. Expression of CXCR4 and CXCL12 (SDF-1) in human prostate cancers (PCa) in vivo. J Cell Biochem. 2003;89:462–473. doi: 10.1002/jcb.10522. [DOI] [PubMed] [Google Scholar]
- 18.Chinni SR, Sivalogan S, Dong Z, et al. CXCL12/CXCR4 signaling activates Akt-1 and MMP-9 expression in prostate cancer cells: the role of bone microenvironment-associated CXCL12. Prostate. 2006;66:32–48. doi: 10.1002/pros.20318. [DOI] [PubMed] [Google Scholar]
- 19.Kukreja P, Abdel-Mageed AB, Mondal D, Liu K, Agrawal KC. Up-regulation of CXCR4 expression in PC-3 cells by stromal-derived factor-1alpha (CXCL12) increases endothelial adhesion and transendothelial migration: role of MEK/ERK signaling pathway-dependent NF-kappaB activation. Cancer Res. 2005;65:9891–9898. doi: 10.1158/0008-5472.CAN-05-1293. [DOI] [PubMed] [Google Scholar]
- 20.Sellers WR, Sawyers CL. Somatic Genetics of Prostate Cancer: Oncogenes and Tumore Suppressors. Philadelphia: Lippincott Williams and Wilkins; 2002. [Google Scholar]
- 21.Suzuki H, Freije D, Nusskern DR, et al. Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple metastatic prostate cancer tissues. Cancer Res. 1998;58:204–209. [PubMed] [Google Scholar]
- 22.Shen MM, Abate-Shen C. Pten inactivation and the emergence of androgen-independent prostate cancer. Cancer Res. 2007;67:6535–6538. doi: 10.1158/0008-5472.CAN-07-1271. [DOI] [PubMed] [Google Scholar]
- 23.Vlietstra RJ, van Alewijk DC, Hermans KG, van Steenbrugge GJ, Trapman J. Frequent inactivation of PTEN in prostate cancer cell lines and xenografts. Cancer Res. 1998;58:2720–2723. [PubMed] [Google Scholar]
- 24.Wu Z, McRoberts KS, Theodorescu D. The role of PTEN in prostate cancer cell tropism to the bone micro-environment. Carcinogenesis. 2007;28:1393–1400. doi: 10.1093/carcin/bgm050. [DOI] [PubMed] [Google Scholar]
- 25.Leslie NR, Yang X, Downes CP, Weijer CJ. PtdIns(3,4,5)P(3)-dependent and -independent roles for PTEN in the control of cell migration. Curr Biol. 2007;17:115–125. doi: 10.1016/j.cub.2006.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grunwald V, DeGraffenried L, Russel D, Friedrichs WE, Ray RB, Hidalgo M. Inhibitors of mTOR reverse doxorubicin resistance conferred by PTEN status in prostate cancer cells. Cancer Res. 2002;62:6141–6145. [PubMed] [Google Scholar]
- 27.Gao PWR, Zhang N, Oppenheim JJ, Howard OM. Negative regulation of CXCR4-mediated chemotaxis by the lipid phosphatase activity of tumor suppressor PTEN. Blood. 2005;106:2619–2669. doi: 10.1182/blood-2004-08-3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phillips RJ, Mestas J, Gharaee-Kermani M, et al. Epidermal growth factor and hypoxia-induced expression of CXC chemokine receptor 4 on non-small cell lung cancer cells is regulated by the phosphatidylinositol 3-kinase/PTEN/AKT/mammalian target of rapamycin signaling pathway and activation of hypoxia inducible factor-1alpha. J Biol Chem. 2005;280:22473–22481. doi: 10.1074/jbc.M500963200. [DOI] [PubMed] [Google Scholar]
- 29.Liu F, Wagner S, Campbell RB, Nickerson JA, Schiffer CA, Ross AH. PTEN enters the nucleus by diffusion. J Cell Biochem. 2005;96:221–234. doi: 10.1002/jcb.20525. [DOI] [PubMed] [Google Scholar]
- 30.Darash-Yahana M, Pikarsky E, Abramovitch R, et al. Role of high expression levels of CXCR4 in tumor growth, vascularization, and metastasis. FASEB J. 2004;18:1240–1242. doi: 10.1096/fj.03-0935fje. [DOI] [PubMed] [Google Scholar]
- 31.Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 32.Bosland MC, Chung LWK, Greenberg NM, Ho S, Isaacs JT, Lane K, Peehl DM, Thompson TC, van Steenbrugge GJ, van Weerden WM. Recent advances in the development of animal and cell culture models for prostate cancer research. Urologic Oncology. 1996;2:99–128. doi: 10.1016/s1078-1439(96)00077-4. [DOI] [PubMed] [Google Scholar]
- 33.Bastola DR, Pahwa GS, Lin MF, Cheng PW. Downregulation of PTEN/MMAC/TEP1 expression in human prostate cancer cell line DU145 by growth stimuli. Mol Cell Biochem. 2002;236:75–81. doi: 10.1023/a:1016191913274. [DOI] [PubMed] [Google Scholar]
- 34.Kotelevets L, van Hengel J, Bruyneel E, Mareel M, van Roy F, Chastre E. The lipid phosphatase activity of PTEN is critical for stabilizing intercellular junctions and reverting invasiveness. J Cell Biol. 2001;155:1129–1135. doi: 10.1083/jcb.200105109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sarker D, Reid AH, Yap TA, de Bono JS. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin Cancer Res. 2009;15:4799–4805. doi: 10.1158/1078-0432.CCR-08-0125. [DOI] [PubMed] [Google Scholar]
- 36.Hinton CV, Avraham S, Avraham HK. Contributions of integrin-linked kinase to breast cancer metastasis and tumourigenesis. J Cell Mol Med. 2008;12:1517–1526. doi: 10.1111/j.1582-4934.2008.00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat Rev Cancer. 2004;4:937–947. doi: 10.1038/nrc1503. [DOI] [PubMed] [Google Scholar]
- 38.Ahn S, Shenoy SK, Wei H, Lefkowitz RJ. Differential kinetic and spatial patterns of beta-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J Biol Chem. 2004;279:35518–35525. doi: 10.1074/jbc.M405878200. [DOI] [PubMed] [Google Scholar]
- 39.Hinton CV, Avraham S, Avraham HK. Role of the CXCR4/CXCL12 signaling axis in breast cancer metastasis to the brain. Clin Exp Metastasis. 27:97–105. doi: 10.1007/s10585-008-9210-2. [DOI] [PubMed] [Google Scholar]
- 40.Liang Z, Brooks J, Willard M, et al. CXCR4/CXCL12 axis promotes VEGF-mediated tumor angiogenesis through Akt signaling pathway. Biochem Biophys Res Commun. 2007;359:716–722. doi: 10.1016/j.bbrc.2007.05.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh S, Singh UP, Grizzle WE, Lillard JW., Jr CXCL12-CXCR4 interactions modulate prostate cancer cell migration, metalloproteinase expression and invasion. Lab Invest. 2004;84:1666–1676. doi: 10.1038/labinvest.3700181. [DOI] [PubMed] [Google Scholar]
- 42.Engl T, Relja B, Marian D, et al. CXCR4 chemokine receptor mediates prostate tumor cell adhesion through alpha5 and beta3 integrins. Neoplasia. 2006;8:290–301. doi: 10.1593/neo.05694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang S, Qi L, Li M, et al. Chemokine CXCL12 and its receptor CXCR4 expression are associated with perineural invasion of prostate cancer. J Exp Clin Cancer Res. 2008;27:62. doi: 10.1186/1756-9966-27-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bild AH, Yao G, Chang JT, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–357. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 45.Saal LH, Johansson P, Holm K, et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc Natl Acad Sci U S A. 2007;104:7564–7569. doi: 10.1073/pnas.0702507104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reiter RE, Gu Z, Watabe T, et al. Prostate stem cell antigen: a cell surface marker overexpressed in prostate cancer. Proc Natl Acad Sci U S A. 1998;95:1735–1740. doi: 10.1073/pnas.95.4.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McMenamin ME, Soung P, Perera S, Kaplan I, Loda M, Sellers WR. Loss of PTEN expression in paraffin-embedded primary prostate cancer correlates with high Gleason score and advanced stage. Cancer Res. 1999;59:4291–4296. [PubMed] [Google Scholar]
- 48.Arya M, Patel HR, McGurk C, et al. The importance of the CXCL12-CXCR4 chemokine ligand-receptor interaction in prostate cancer metastasis. J Exp Ther Oncol. 2004;4:291–303. [PubMed] [Google Scholar]
- 49.Gao P, Wange RL, Zhang N, Oppenheim JJ, Howard OM. Negative regulation of CXCR4-mediated chemotaxis by the lipid phosphatase activity of tumor suppressor PTEN. Blood. 2005;106:2619–2626. doi: 10.1182/blood-2004-08-3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun X, Wei L, Chen Q, Terek RM. CXCR4/SDF1 mediate hypoxia induced chondrosarcoma cell invasion through ERK signaling and increased MMP1 expression. Mol Cancer. 9:17. doi: 10.1186/1476-4598-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang CY, Lee CY, Chen MY, et al. Stromal cell-derived factor-1/CXCR4 enhanced motility of human osteosarcoma cells involves MEK1/2, ERK and NF-kappaB-dependent pathways. J Cell Physiol. 2009;221:204–212. doi: 10.1002/jcp.21846. [DOI] [PubMed] [Google Scholar]
- 52.Tan CT, Chu CY, Lu YC, et al. CXCL12/CXCR4 promotes laryngeal and hypopharyngeal squamous cell carcinoma metastasis through MMP-13-dependent invasion via the ERK1/2/AP-1 pathway. Carcinogenesis. 2008;29:1519–1527. doi: 10.1093/carcin/bgn108. [DOI] [PubMed] [Google Scholar]
- 53.Ghosh S, Preet A, Groopman JE, Ganju RK. Cannabinoid receptor CB2 modulates the CXCL12/CXCR4-mediated chemotaxis of T lymphocytes. Mol Immunol. 2006;43:2169–2179. doi: 10.1016/j.molimm.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 54.Thomas SL, Alam R, Lemke N, Schultz LR, Gutierrez JA, Rempel SA. PTEN augments SPARC suppression of proliferation and inhibits SPARC-induced migration by suppressing SHC-RAF-ERK and AKT signaling. Neuro Oncol. doi: 10.1093/neuonc/noq048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vasudevan KM, Burikhanov R, Goswami A, Rangnekar VM. Suppression of PTEN expression is essential for antiapoptosis and cellular transformation by oncogenic Ras. Cancer Res. 2007;67:10343–10350. doi: 10.1158/0008-5472.CAN-07-1827. [DOI] [PubMed] [Google Scholar]