

Abstract
We have successfully designed an A:B:C collagen peptide heterotrimer using an automated computational approach. The algorithm maximizes the energy gap between the target and competing misfolded states while enforcing a minimum target stability. Circular dichroism (CD) measurements confirm that all three peptides are required to form a stable, structured triple helix. This study highlights the power of automated computational design, providing model systems to probe the biophysics of collagen assembly and developing general methods for the design of fibrous proteins.
Keywords: computational protein design, triple helix, negative design, electrostatics, protein folding
Much of computational protein design has focused on coiled-coils and globular folds, leading to a better molecular understanding of stability and structure. Fibrous proteins, such as collagen, have been largely ignored as targets for in silico design, which is surprising given the abundance of collagen in higher animals and its central role in a host of biological processes. Our current study is aimed at providing a greater insight into fibrous protein folding and structure.
A promising strategy for engineering collagen self-assembly is the design of appropriate surface electrostatic interactions 1–6. This approach has been historically successful in controlling the association of natural and model α-helical coiled-coils7–12. Recent progress in increasing the selectivity of intermolecular associations was achieved using computational methods 13–16. Elegant collagen heterotrimer designs have been developed using rational design principles4,6. We demonstrate that similar principles, applied in an automated computational fashion, do promote stable and specific assembly of de novo collagen peptides.
Collagen design is both simple and difficult. Peptides with multiple Gly-X–Y repeats, in which X and Y are often proline (Pro, P) and hydroxyproline (Hyp, O) respectively, can readily form triple-helices. The challenge lies in controlling the specificity of assembly. For example, an equimolar mixture of three collagen-like peptides, denoted A, B and C can potentially associate in 27 ways, e.g. AAA, AAB, ABC, CAB, etc. To achieve a specific target state, one must promote its favorable interchain interactions while simultaneously introducing unfavorable interactions in competing states. Side chain electrostatic interactions have been utilized to promote heterotrimer formation 5,6,17–19. Using this approach, we have computationally designed three peptide sequences that stably and specifically form an A:B:C heterotrimer.
The stability of triple helices was scored by a discrete side chain interaction model19. Only interchain charge-pairs between Y–X and Y–X′ positions were included in the calculation as these are structurally adjacent in the triple helix20,21 (Figure 1a). Interactions between the third and first chains are different than first/second and second/third chain pairs due to the stagger induced by the triple-helical screw; in the third/first case, Y–X′ and Y–X″ interactions are scored as depicted. Intrachain ion pairs are also present in the triple-helix22,23, but their relative energetic contribution to stability of the folded versus unfolded state has yet to be established, which is challenging given the conformational similarity of the triple-helix to an extended chain. Therefore, we focused on intermolecular interactions which exist only in the context of the trimer.
Figure 1.
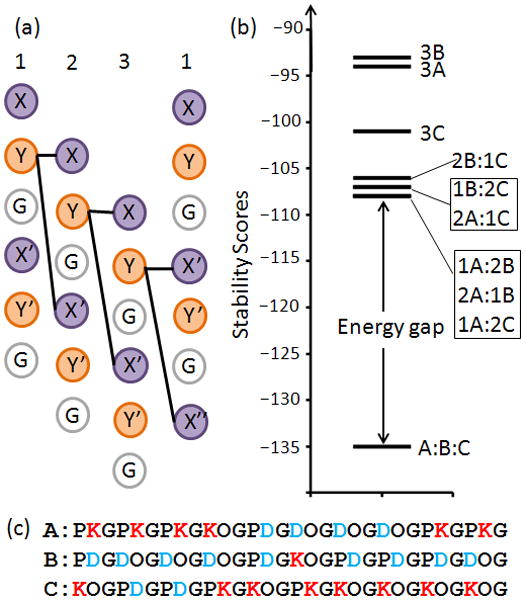
Computational Design. (a) Interactions between peptide chains 1, 2 and 3 included in the computed stability score. (b) Energy landscape of the all association stoichiometries highlights the energy gap between ABC and competing states. (a) Computationally designed sequences of collagen heterotrimers.
The discrete interaction scores were based on a similar α-helical coiled-coil design14. To enforce the established role of imino acids in promoting triple-helix stability24, a compositional constraint was used where all triplets contained at least one Pro or Hyp (KOG, PKG, DOG, PDG, POG). Additionally, we used a sequence energy term that scaled with the total number of Pro and Hyp to favor inclusion of the POG triplet.
The algorithm used for sequence selection had a positive and negative design component. In positive design, the target state is stabilized while in negative design, competing states are destabilized. Combining positive and negative design results in an energy gap that promotes both stability and specificity of the target state (Figure 1b)25. Stability of the target state ABC was constrained to be 26 °C or above, as calculated by a previously determined empirical relationship between the energy score and melting temperature, Tm 19:
| Eq. (1) |
Specificity was concurrently enforced by optimizing the energy gap, ΔEgap:
| Eq. (2) |
where min(Ecompeting) was the stability of the best competing stoichiometry. The sequence space of ~1020 possible solutions was searched using a Monte Carlo Simulated Annealing (MCSA) protocol26 (see supplementary methods for details).
The final sequences (Figure 1c) were composed exclusively of charge containing triplets despite the additional stability contributions of POG in the scoring function, potentially due to an inherent stability-specificity tradeoff of this system where the magnitude of the optimized energy gap was inversely related to the number of POG triplets incorporated27. ABC had a predicted Tm of 33.8 °C, with more than twice the num- ber of favorable charge pairs as the next best competing state and no repulsive charge pairs (Figure 2, Table S1). Competing species were highly populated by predicted repulsive interactions. Therefore, we expected that a 1:1:1 mixture of A, B, and C would form the most stable triple helix.
Figure 2.

(left) Twenty one predicted favorable ion-pairs in an ABC heterotrimer. (right) example of a competing state: the AAA homotrimer with eight repulsive interactions and no favorable +/− ion pairs.
To evaluate the design, peptides were synthesized and purified by HPLC. Ten combinations of the peptides in phosphate buffer pH 7.0, (A, B, C, A:2B, 2A:B, A:2C, 2A:C, B:2C, 2B:C and A:B:C) were studied for triple-helix structure and thermal stability by CD. A:B:C formed a stable triple helix with a Tm of 29 °C, less than 5 °C below the computed melting temperature (Figure 3 and Table S2). A:B:C showed a mean residue ellipiticity (MRE) comparable to [(POG)10]3, with a modest two nm shift in the peak wavelength consistent with the lower imino-acid content of the design (Figure S2).
Figure 3.
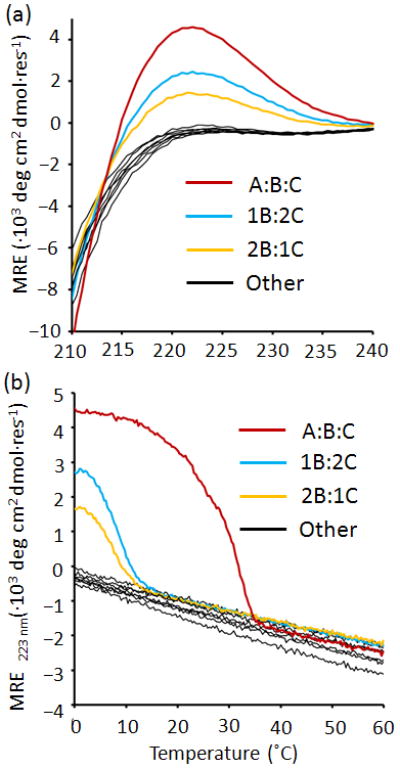
Experimental characterization of the A:B:C heterotrimer. (a) CD spectra at 4 °C and (b) thermal denaturation monitored at 223 nm of the ten mixtures of A, B, C.
Of the nine competing stoichiometries, only B:2C and 2B:C showed any indication of triple-helical structure and cooperative unfolding upon melting. Both states were only marginally stable and were fully unfolded above 15 °C.
To confirm that favorable charge pair interactions promoted assembly of A:B:C, structure and stability of the peptides were measured under high salt concentrations. At 100 mM NaCl, stability and structure of A:B:C was reduced (Figure S3, Table S3). At 1.0 M, no secondary structure was observed.
The thermal denaturation transition of A:B:C (Figure 3B) was broad, indicative of multiple species, potentially corresponding to permutations BCA and CAB which had near-target stability scores. In 100 mM NaCl, a level folded-state baseline was observed, suggesting that less stable species were not able to form at intermediate ionic strengths due to screening of charge-pair interactions.
Previous collagen heterotrimer designs used arginine and glutamic acid as charge-pair promoting residues. An equivalent set of peptides were synthesized using the same charge pattern, with all Lys mutated to Arg and all Asp mutated to Glu. The R/E version of A:B:C was the most stable, but ex- hibited a lower Tm than the K/D counterpart (Table S2). Sev- eral competing states also formed triple-helical structures with melting temperatures between 12–15°C. In cases where both R/E and K/D competing stoichiometries folded, the R/E species were more stable.
Comparing the experimental outcome of K/D and R/E peptides provides insight into contributions of sidechain electrostatics to stability. Gauba and Hartgerink showed that replacing Arg with Lys and Glu with Asp improved the stability of an A:B:C heterotrimer by 9 °C 5. Despite significant differences in total composition, our results mirror theirs, with the K/D peptides 7°C more stable than equivalent R/E sequences. This was previously attributed to stronger sidechain interactions between Lys and Asp1. In contrast, competing states are more stable in the R/E peptide mixtures than their K/D counterpart. This may be due to the higher backbone stability of Arg and Glu, particularly at the Y-position in the triple helix28. The net result of these effects is a larger energy gap in the K/D versus the R/E peptide system. Reconciling the relative contributions of amino acid content and intermolecular electrostatics will be useful in modulating the stability and specificity of future designs.
This is the first successful computational design of an A:B:C collagen heterotrimer. The design is robust, where the target is thermodynamically more stable than competing states in the ensemble. The outcome is impressive given the relatively simple scoring function used, and highlights the importance of surface electrostatics in driving stability and specificity of collagen self-assembly.
A key challenge in any molecular engineering endeavor is selecting the appropriate level of chemical accuracy in the simulation to achieve the target design goals. Here, intermolecular charge pairs were scored using discrete energies based on amino acid identity. For the sake of improving future designs, it is worth discussing molecular aspects of collagen stability that not included in the final scoring function.
We did not consider energetic differences in amino acid substitutions at X and Y positions; i.e. in the absence of intermolecular charge pair effects, the effect on stability of PKG and KOG are considered identical, although host peptides containing either of these triplets can differ in stability by ~ 5°C 28. Similar position-dependent stabilities are present for Asp, Glu and Arg substitutions. Optimizing positional preferences during sequence design would cause at least two issues. With the exception of arginine, substitutions at the X position are more favorable than at Y, but limiting mutations to the X position would preclude the attractive and repulsive charge-pair interactions that mediate specificity. Second, such optimization would stabilize all states of the ensemble, narrowing the energy gap and pushing competing states across the threshold stability required for folding27.
Discrepancies between predicted stability and observed Tm are due both to limitations of the empirical model and the fact that melting temperature will not correlate linearly with thermodynamic stability. Although the stabilities of natural collagens are most strongly correlated with hydroxyproline content29, our recent scoring function developed on a set of synthetic peptides does not differentiate between proline at X and hydroxyproline at Y. The presence of weak heterotrimers in binary mixtures of B:C in this study may be due to the higher hydroxyproline content of peptides B and C relative to A. The observed difference between K/D and R/E designs is certainly due to both amino acid propensities as well as intermolecular electrostatics. We expect that empirical predictions of collagen stability will be improved by appropriately considering position specific amino acid preferences.
Extending the empirical model to a three-dimensional, atomistic representation of the design would allow explicit treatment of molecular forces. Intrachain ion pairs23, interchain networks of sidechain interactions30–32, and positional preferences of amino acids33 have been modeled using all-atom methods. The challenge at this level of computation for protein design is one of sufficient conformational sampling. The unfolded and folded triple-helical states of collagen peptides are very similar in terms of extended conformation and degree of solvation. As a result, distinguishing sometimes subtle effects of amino acid substitutions on these two states requires significant simulation times and computational resources. This computational cost is exacerbated in the case where an ensemble of states must be simulated to calculate an energy gap. State of the art approaches to the computational design of globular proteins address this problem by judiciously combining knowledge-based terms (such as positional specific preferences of amino acids) with atomic level calculations34. A similar hybrid approach will probably be the most effective route in improving collagen designs.
Automated computational design can overcome limitations of rational methods to produce complex, unintuitive solutions. Through this work, we have gained a better understanding of intermolecular forces important for the design of fibrous proteins.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by the NIH DP2 OD006478–01 and NSF DMR-0907273.
ABBREVIATIONS
- MRE
molar residue ellipticity
- CD
circular dichroism
- HPLC
high pressure liquid chromatography
- MCSA
Monte Carlo Simulated Annealing
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. Details on computational design, experimental methods and additional figures and tables are provided in the supplemental information. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Persikov AV, Ramshaw JAM, Kirkpatrick A, Brodsky B. Biochemistry. 2005;44:1414. doi: 10.1021/bi048216r. [DOI] [PubMed] [Google Scholar]
- 2.Chan VC, Ramshaw JAM, Kirkpatrick A, Beck K, Brodsky B. J Biol Chem. 1997;272:31441. doi: 10.1074/jbc.272.50.31441. [DOI] [PubMed] [Google Scholar]
- 3.Venugopal MG, Ramshar JAM, Braswell E, Zhu D, Brodsky B. Biochemistry. 1994;33:7948. doi: 10.1021/bi00191a023. [DOI] [PubMed] [Google Scholar]
- 4.O’Leary LER, Fallas JA, Hartgerink JD. J Amer Chem Soc. 2011;133:5432. doi: 10.1021/ja111239r. [DOI] [PubMed] [Google Scholar]
- 5.Gauba V, Hartgerink JD. J Am Chem Soc. 2007;129:15034. doi: 10.1021/ja075854z. [DOI] [PubMed] [Google Scholar]
- 6.Gauba V, Hartgerink JD. J Am Chem Soc. 2007;129:2683. doi: 10.1021/ja0683640. [DOI] [PubMed] [Google Scholar]
- 7.Bryson JW, Desjarlais JR, Handel TM, DeGrado WF. Protein Sci. 1998;7:1404. doi: 10.1002/pro.5560070617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oakley MG, Kim PS. Biochemistry. 1997;36:2544. doi: 10.1021/bi962391t. [DOI] [PubMed] [Google Scholar]
- 9.Oshea EK, Klemm JD, Kim PS, Alber T. Science. 1991;254:539. doi: 10.1126/science.1948029. [DOI] [PubMed] [Google Scholar]
- 10.Phelan P, Gorfe AA, Jelesarov I, Marti DN, Warwicker J, Bosshard HR. Biochemistry. 2002;41:2998. doi: 10.1021/bi011920c. [DOI] [PubMed] [Google Scholar]
- 11.Burkhard P, Ivaninskii S, Lustig A. J Mol Biol. 2002;318:901. doi: 10.1016/S0022-2836(02)00114-6. [DOI] [PubMed] [Google Scholar]
- 12.Krylov D, Barchi J, Vinson C. J Mol Biol. 1998;279:959. doi: 10.1006/jmbi.1998.1762. [DOI] [PubMed] [Google Scholar]
- 13.Harvranek JJ, Harbury PB. Nature Struct Bio. 2003;10:45. doi: 10.1038/nsb877. [DOI] [PubMed] [Google Scholar]
- 14.Summa CM, Rosenblatt MM, Hong J–K, Lear JD, DeGrado WF. J Mol Biol. 2002;321:923. doi: 10.1016/s0022-2836(02)00589-2. [DOI] [PubMed] [Google Scholar]
- 15.Reinke AW, Grant RA, Keating AE. J Amer Chem Soc. 2010;132:6025. doi: 10.1021/ja907617a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nautiyal S, Woolfson DN, King DS, Alber T. Biochemistry. 1995;34:11645. doi: 10.1021/bi00037a001. [DOI] [PubMed] [Google Scholar]
- 17.Heino J. BioEssays. 2007;29:1001. doi: 10.1002/bies.20636. [DOI] [PubMed] [Google Scholar]
- 18.Gauba V, Hartgerink JD. Journal of the American Chemical Society. 2008;130:7509. doi: 10.1021/ja801670v. [DOI] [PubMed] [Google Scholar]
- 19.Xu F, Zhang L, Koder RL, Nanda V. Biochemistry. 2010;49:2307. doi: 10.1021/bi902077d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salem G, Traub W. FEBS Lett. 1975;51:94. doi: 10.1016/0014-5793(75)80861-1. [DOI] [PubMed] [Google Scholar]
- 21.Bella J, Eaton M, Brodsky B, Berman HM. Science. 1994;266:75. doi: 10.1126/science.7695699. [DOI] [PubMed] [Google Scholar]
- 22.Persikov AV, Ramshaw JA, Brodsky B. J Biol Chem. 2005;280:19343. doi: 10.1074/jbc.M501657200. [DOI] [PubMed] [Google Scholar]
- 23.Katz EP, David CW. Biopolymers. 1990;29:791. doi: 10.1002/bip.360290413. [DOI] [PubMed] [Google Scholar]
- 24.Engel J, Chen HT, Prockop DJ, Klump H. Biopolymers. 1977;16:601. doi: 10.1002/bip.1977.360160310. [DOI] [PubMed] [Google Scholar]
- 25.Shakhnovich EI, Gutin AM. Protein Eng. 1993;6:793. doi: 10.1093/protein/6.8.793. [DOI] [PubMed] [Google Scholar]
- 26.Hellinga HW, Richards FM. P Natl Acad Sci USA. 1994;91:5803. doi: 10.1073/pnas.91.13.5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nanda V, Zahid S, Xu F, Levine D. Methods Enzymol. 2011;487:575. doi: 10.1016/B978-0-12-381270-4.00020-2. [DOI] [PubMed] [Google Scholar]
- 28.Persikov AV, Ramshaw JAM, Brodsky B. J Biol Chem. 2005;280:19343. doi: 10.1074/jbc.M501657200. [DOI] [PubMed] [Google Scholar]
- 29.Burjanadze T. Biopolymers. 1979;18:931. doi: 10.1002/bip.1979.360180413. [DOI] [PubMed] [Google Scholar]
- 30.Vitagliano L, Nemethy G, Zagari A, Scheraga HA. J Mol Biol. 1995;247:69. doi: 10.1006/jmbi.1994.0123. [DOI] [PubMed] [Google Scholar]
- 31.Vitagliano L, Nemethy G, Zagari A, Scheraga HA. Biochemistry. 1993;32:7354. doi: 10.1021/bi00080a004. [DOI] [PubMed] [Google Scholar]
- 32.Gurry T, Nerenberg PS, Stultz CM. Biophys J. 2010;98:2634. doi: 10.1016/j.bpj.2010.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raman SS, Gopalakrishnan R, Wade RC, Subramanian V. J Phys Chem B. 2011;115:2593. doi: 10.1021/jp109133v. [DOI] [PubMed] [Google Scholar]
- 34.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Science. 2003;302:1364. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.