

Abstract
Aberrant expression and mutations of thyroid hormone receptor genes (TRs) are closely associated with several types of human cancers. To test the hypothesis that TRs could function as tumor suppressors, we took advantage of mice with deletion of all functional TRs (TRα1−/− TRβ−/−mice). As these mice aged, they spontaneously developed follicular thyroid carcinoma with pathological progression from hyperplasia to capsular invasion, vascular invasion, anaplasia and metastasis to the lung, similar to human thyroid cancer. Detailed molecular analysis revealed that known tumor promoters such as pituitary tumor-transforming gene were activated and tumor suppressors such as peroxisome proliferator-activated receptor γ and p53 were suppressed during carcinogenesis. In addition, consistent with the human cancer, AKT–mTOR–p70S6K signaling and vascular growth factor and its receptor were activated to facilitate tumor progression. This report presents in vivo evidence that functional loss of both TRα1 and TRβ genes promotes tumor development and metastasis. Thus, TRs could function as tumor suppressors in a mouse model of metastatic follicular thyroid cancer.
Keywords: thyroid cancer, mouse model, mutations of thyroid hormone receptors
Introduction
Thyroid hormone nuclear receptors (TRs) are ligand-dependent transcription factors critical for growth, development and differentiation. Alternative splicing of the primary transcripts of the two TR genes, α and β, yields three major thyroid hormone (T3) binding isoforms: α1, β1 and β2. TRs regulate expression of target genes through their interaction with thyroid hormone response elements located in the promoter regions. Genes regulated by TRs include growth factors, cell surface receptors, transcription factors, cell-cycle regulators, oncogenes and tumor suppressors (Puzianowska-Kuznicka et al., 2006). Recently, it was also shown that TRs directly modulate the activities of signaling through protein-protein interaction with key effectors in the pathways (Davis et al., 2008; Guigon et al., 2008; Furuya et al., 2009). For example, TRs physically interact with the regulatory p85α subunit of phosphatidylinositol 3-kinase (PI3K) to modulate the downstream AKT-mammalian target of rapamycin (mTOR) and p70S6K and PI3K–integrin-linked kinase (ILK)–matrix metalloproteinase (MMP)-2 signaling pathways (Furuya et al., 2009).
Early evidence suggesting that mutated TR could be involved in carcinogenesis came from the discovery that TRα1 is the cellular counterpart of the retroviral v-erbA that induces acute erythroleukemia and sarcomas in birds (Sap et al., 1986; Weinberger et al., 1986; Thormeyer and Baniahmad, 1999). v-erbA is a highly mutated chicken TRα1 that does not bind T3 and loses the ability to activate gene transcription. It competes with TR for binding to thyroid hormone response elements and interferes with the transcriptional activity of liganded TR on several promoters (Chen and Privalsky, 1993; Yen et al., 1994). Since those early studies, mutated TRs have been reported to associate with several human cancers, including liver (Lin et al., 1999), kidney (Kamiya et al., 2002), pituitary (Safer et al., 2001; Ando et al., 2001a, b) and thyroid (Puzianowska-Kuznicka et al., 2002). Reduced expression of TRβ1 mRNA was also implicated in the carcinogenesis of human kidney cancer (Puzianowska-Kuznicka et al., 2000) and papillary thyroid carcinomas (Puzianowska-Kuznicka et al., 2002; Takano et al., 2003). The silencing of the TRβ gene by hypermethylation and the concurrent reduction of TRβ1 transcripts were shown in breast cancer (Li et al., 2002). In cotransfection experiments, it was shown that both TRα1 and TRβ1 strongly repressed Ha-rasval 12-induced transformation of NIH3T3 fibroblasts, reduced tumor volume and inhibited tumor growth in nude mice (Garcia-Silva and Aranda, 2004). Recently, it was shown that the transfected TRβ1 in hepatocarcinoma and breast cancer cells reduced tumor growth, caused partial mesenchymal-to-epithelial cell transition, and had a striking inhibitory effect on invasiveness, extravasation and metastasis formation in mice (Martinez-Iglesias et al., 2009).
Previously, we created a knockin mutant mouse (TRβPV mouse) by targeting a mutation (denoted PV) to the TRβ gene locus through homologous recombination and the Cre-LoxP system (Kaneshige et al., 2000). TRßPV was derived from a patient, PV, with a genetic disease known as resistance to thyroid hormone (Weiss and Refetoff, 2000). PV has a C-insertion at codon 448, which produces a frame shift of the COOH-terminal 14 amino acids of TRß1. PV has lost T3 binding completely and shows potent dominant negative activity (Meier et al., 1992). As TRβPV/PV mice age, they spontaneously develop follicular thyroid carcinomas with a pathological progression similar to human thyroid cancer (Suzuki et al., 2002; Ying et al., 2003a, b). In subsequent studies, we found that mice with one mutated TRβ allele in the absence of the other wild-type allele (TRβ PV/− mouse) also develop follicular thyroid carcinoma. The pathological progression of TRβ PV/− mice is indistinguishable from that of TRβPV/PVmice. Moreover, there is a striking similarity in the patterns of several altered signaling pathways between TRβ PV/− mice and TRβPV/PV mice during carcinogenesis. Thus, the mutation of one TRβ allele in the absence of the other wild-type allele is sufficient to induce thyroid carcinoma. These findings indicate that the mutations of two TRβ alleles, as well as mutation of one TRβ allele in the absence of the other wild-type allele, lead to the development of metastatic follicular thyroid carcinoma. However, it is not clear whether the loss-of-function or gain-of-function mutation causes carcinogenesis. One possibility is that the mutant TRβPV could act as an oncogene through gain-of-function to promote carcinogenesis. Indeed, it was shown that PV could act through the gain-of-function mode to activate PI3K signaling pathways through physical interaction with the regulatory p85α subunit of PI3K whereas the wild-type TRβ1 has very weak activity (Furuya et al., 2009). Alternatively, PV could, through the loss-of-function mode, act as a dominant negative mutant through the nucleus-initiated transcription to promote thyroid carcinogenesis (Kaneshige et al., 2000; Zhang et al., 2002; Suzuki et al., 2003). Indeed, studies in TRβ PV/− mice and TRβPV/PV mice suggest that PV could act through gain-of-function as well as a loss-of-function mutation (Kato et al., 2006; Guigon et al., 2008; Furuya et al., 2009). However, it is not clear whether total loss of the normal functions of TRs alone is sufficient to lead to thyroid carcinogenesis.
To address this question, we took advantage of available mice that are devoid of all known functional TRs. Remarkably, we found that the TRα1−/−TRβ−/− mice spontaneously developed follicular thyroid cancer as they aged. Histological evaluation showed the progression from hyperplasia to capsular invasion, vascular invasion, anaplasia and metastasis to the lung. Molecular studies revealed that multiple signaling pathways were altered to promote thyroid carcinogenesis. This study provided direct evidence to indicate that TRs could function in vivo as suppressors of follicular thyroid carcinomas.
Results
Spontaneous development of follicular thyroid carcinoma in TRα1−/− TRβ−/− mice
Figure 1 shows the Kaplan-Meier cumulative survival curves for TRα1−/−TRβ−/− mice over 20.9 months. The 50% survival age was 9.3 months (n = 94). In contrast, wild-type TRα1+/+TRβ+/+ mice (WT-mice) were healthy with no deaths during the same observation period.
Figure 1.
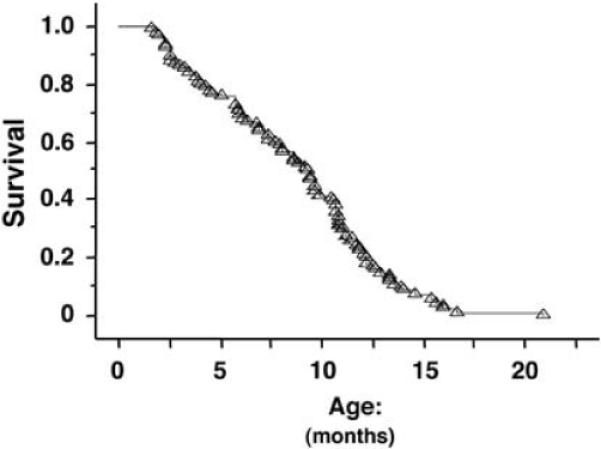
Kaplan–Meier survival curve for TRα1−/− TRβ−/− mice up to 20.9 months of age. The analysis was performed with log-rank (Mantel–Cox) test by using StatView 5.0. The 50% survival age was 9.3 months (n=94).
Detailed characterization was carried out for moribund >TRα1−/−TRβ−/− mice. A common obvious abnormality was the marked enlargement of thyroid glands. The thyroid weights of TRα1−/−TRβ−/− mice were 27.2 ± 2.9 (mean ± s.e.: n = 6), 56.7 ± 4.6 (n = 10) and 115 ± 27.1 (n = 8) mg at the ages of 2–5, 6–9 and 10–13 months, respectively. The average weight of adult thyroids of the wild-type (TRα1+/+TRβ+/+) mice was 4.0 ± 0.3 mg (n = 14) and no significant changes in the thyroid weights of adult wild-type mice were observed as they aged. Thus, this represents a 7-, 14- and 29-fold enlargement in the thyroid glands of the TRα1−/−TRβ−/− mice at the ages of 2−5, 6−9 and 10−13 months, respectively. The enlargement of thyroid glands was progressive as indicated by the increasing ratios of thyroid to body weight from 11.3 ± 1.1 at the age of 2–5 months to 36.8 ± 7.4 at the age of 10–13 months (Figure 2).
Figure 2.
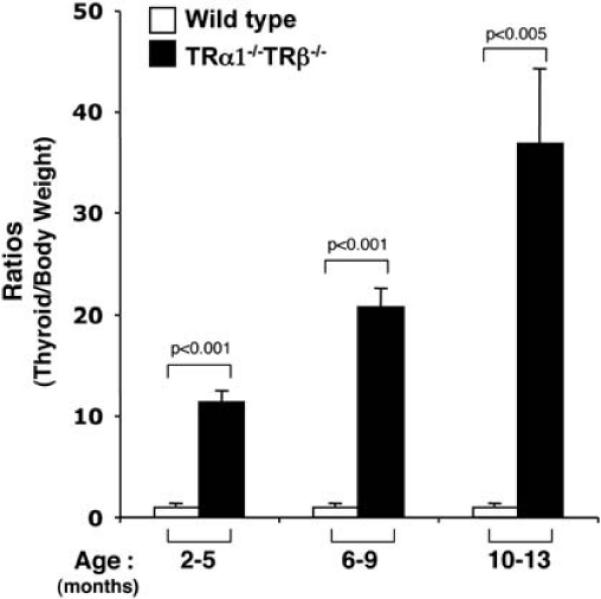
Thyroid weights of TRα1−/− TRβ−/− mice (n=6–25) at the ages of 2–5, 6–9 and 10–13 months. Thyroid glands of TRα1−/− TRβ−/− mice were dissected and weighed. The data are presented as the ratios of thyroid weight to body weight (mg/g).
Histopathological evaluation of the thyroids of TRα1−/−TRβ−/− mice showed grossly enlarged glands, displaying diffuse adenomatous hyperplasia with dense nuclear chromatin and a glandular pattern characteristic of follicular carcinoma. In the 6- to 12-month-old moribund mice, histopathological changes consistent with neoplastic progression of invasion of the thyroid capsule by follicular elements (Figure 3a), invasion of vascular spaces within and adjacent to the thyroid (Figure 3b), focal anaplasia of epithelial cells within the hyperplastic areas (Figure 3c) and finally appearance of distant metastatic lesions in the lung (Figure 3d). The metastatic lesions in the lung showed characteristics of both spindle cell anaplastic and follicular morphology, including focal accumulations of colloid. The nuclear features observed in papillary carcinoma of the thyroid in humans were not observed. This interpretation of histologic type is further supported by a selective pattern of lung metastasis, with no evidence of lymph node involvement, features characteristic of follicular carcinoma of the thyroid in humans. In contrast, the histological patterns of the thyroid glands from 10- to 12-month-old WT-mice showed no detectable hyper-plasia. The quantitative analysis showed progression of these pathological changes as the mice aged. At 2–5 months of age, only one-third of mice (two out of six) were observed to have capsular invasion. At the ages of 10–13 months, 85.7% (12 out of 14) of mice had capsular invasion (Figure 4a). Vascular invasion was not observed in mice younger than 5 months, but was observed only in mice approximately 15% (2 out of 13) aged 6–9 months, and in 35.7% (5 out of 14) aged 10–13 months (Figure 4b). Anaplasia was observed in mice only at the age of 10–13 months (Figure 4c). Approximately, 21% (3 out of 14) of mice had distant lung metastasis at the age of 10–13 months (Figure 4d). This pathological progression is similar to human follicular thyroid carcinoma.
Figure 3.
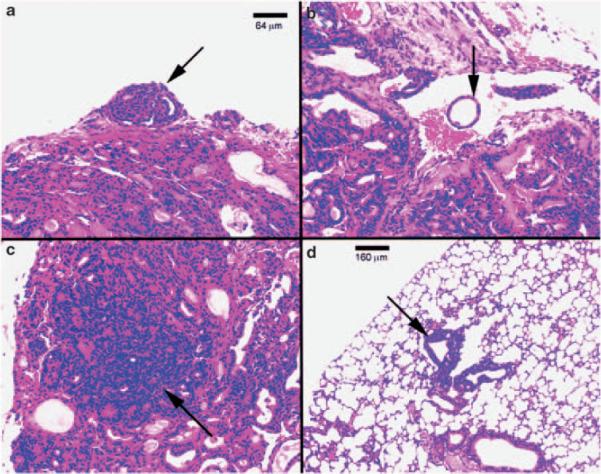
Hematoxylin and eosin (H&E) staining of thyroids and lungs of representative sections from TRα1−/− TRβ−/− mice. Histological sections from tissues of mice showed evidence of capsular invasion in thyroid (a) (arrow), vascular invasion in thyroid (b) (arrow), anaplasia in thyroid (c) and metastatic thyroid carcinoma lesions in lung (d) (arrow).
Figure 4.
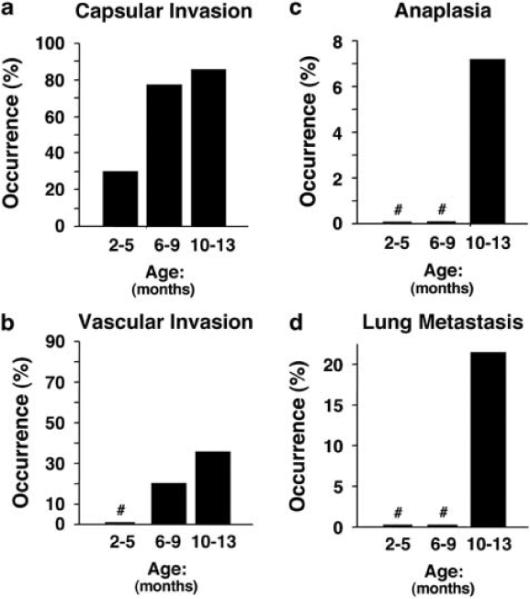
Quantitative analysis of age-dependent occurrence frequency (%) of capsular invasion (a), vascular invasion (b), anaplasia (c) and lung metastasis (d) of TRα1−/− TRβ−/− (n=6–14) mice. Sections of thyroids and lungs from TRα1−/− TRβ−/− mice were stained with Hematoxylin and eosin (H&E) and analyzed for age-dependent pathological progression. The data are expressed as the percentage of occurrence frequency of the mice examined. The designation (#) indicates 0 occurrence frequency (%).
Alteration of growth and survival signaling pathways in thyroids of TRα1−/−TRβ−/− mice
Along with others, we have shown that serum TSH levels of TRα1−/−TRβ−/− mice were increased 60- to 160-fold as compared with wild-type mice (Gothe et al., 1999; Furumoto et al., 2005). Serum total T4 and T3 concentrations are increased 18- to 26-fold, respectively, in adult TRα1−/−TRβ−/− mice as compared with WT-mice (Furumoto et al., 2005). Consistent with the elevated TSH levels, TSH signaling was increased as indicated by increased phosphorylation of CREB at ages 5–6 and 11–13 months (two- to threefold; Figure 5Aa). Total CREB protein levels were not significantly affected in mice with same age (Figure 5Ab). The key cell-cycle regulator, cyclin D1, which is a downstream effector of TSH–TSH receptor- phosphorylation of CREB signaling was also increased in the protein level (two- to threefold; Figure 5Ba).
Figure 5.
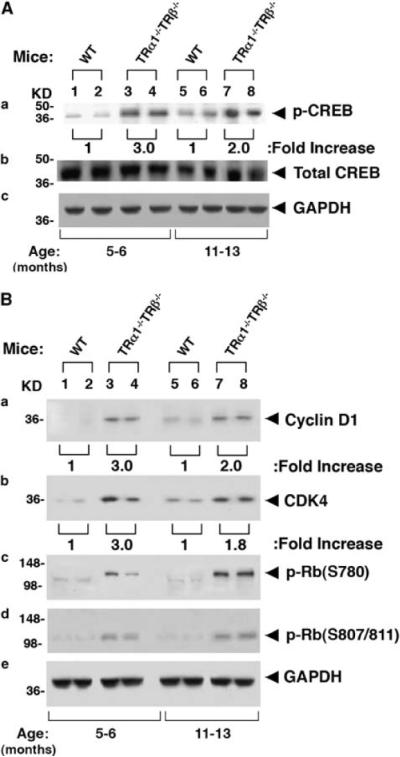
Activation of TSH–TSHR downstream pathway (A) and cyclin-CDK4–Rb pathway (B) in TRα1−/− TRβ−/− mice. For western blot analysis, 30 μg of thyroid extract was used. Two representative results from 4–6 WT (lanes 1, 2, 5 and 6) and TRα1−/− TRβ−/− mice (lanes 3, 4, 7 and 8) are shown for p-CREB and total CREB (A), cyclin D1, CDK4 and p-Rb (B). GAPDH was used as loading controls.
To examine whether increased cyclin D1 led to increased cell-cycle progression, we examined whether the protein level of cyclin-dependent kinase 4 was altered. Indeed, as shown in Figure 5Bb, the protein abundance of cyclin-dependent kinase 4 was increased 1.8- to 3-fold in the thyroids of TRα1−/−TRβ−/− mice (compare lanes 3 and 4 with lanes 1 and 2; 7 and 8 with 5 and 6). Importantly, increased cyclin D1 and cyclin-dependent kinase 4 led to an increase in phospphorylated retinoblastoma protein in the thyroids of TRα1−/−TRβ−/− mice (p-Rb; lanes 3, 4, 7 and 8, S780 in 5Bc and S807/811 in 5Bd). Increased phosphorylation of Rb leads to release the associated-E2F from unphosphorylated Rb-E2F complexes to drive the expression of transcription factors, thereby propelling cells to enter the S-phase to increase cell proliferation (Herwig and Strauss, 1997).
To elucidate the changes in the pathways that have been reported to contribute to thyroid carcinogenesis, we first examined the PI3K–AKT signaling in TRα1−/−TRβ−/− mice. This pathway is critical for tumor proliferation, and its dysregulation in tumors is common (Ringel et al., 2001; Miyakawa et al., 2003). As shown in Figure 6Aa, phosphorylated AKT in TRα1−/−TRβ−/− mice was increased 3- to 3.5-fold (lanes 3, 4, 7 and 8) as compared with the WT-mice (lanes 1, 2, 5 and 6) without significantly affecting the total AKT protein levels (Figure 6Ab). Consistent with the activation of AKT, phosphorylated mTOR at S2448 and at S2481 were increased 1.5- to 2.5-fold in TRα1−/−TRβ−/− mice as compared with WT-mice (Figure 6Ba and b, respectively). A ~twofold increased phosphorylated p70S6K was observed in thyroids of TRα1−/−TRβ−/− mice as compared with WT-mice (Figure 6Bd). No significant changes in the protein abundance of total mTOR (Figure 6Bc) as well as total p70S6K were noted (Figure 6Be). The activation of the AKT–mTOR–p70S6K pathway promotes growth of thyroid tumors.
Figure 6.
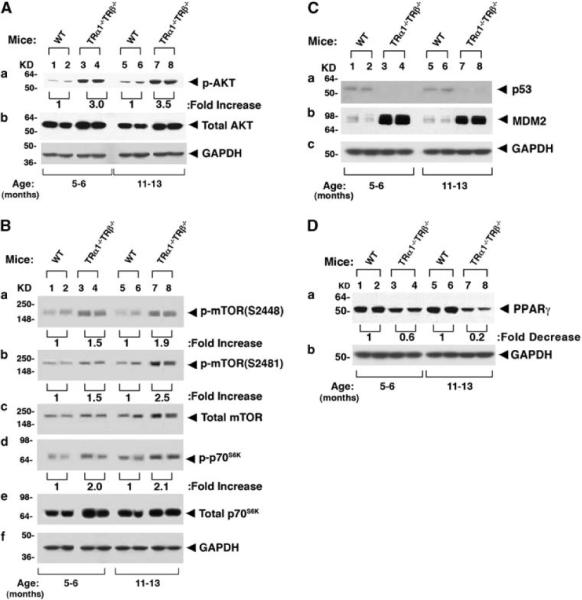
Activation of growth signaling pathways in TRα1−/− TRβ−/− mice. For western blot analysis, 30 μg of thyroid extract was used. Two representative results from 4 to 6 WT (lanes 1, 2, 5 and 6) and TRα1−/− TRβ−/− mice (lanes 3, 4, 7 and 8) are shown for p-AKT and total AKT (A), p-mTOR(S2448), p-mTOR(S2481), total mTOR, p-p70S6K and total 70S6K (B), p53 and MDM2 (C) and PPARγ (D). GAPDH was used as loading controls.
The tumor suppressor p53 induces apoptosis in response to oncogenic transformation (Sharpless and DePinho, 2002). We, therefore, determined whether p53 protein levels were changed to contribute to the growth of thyroid tumors. A weak basal level of p53 was observed in WT-mice aged 5–6 and 11–13 months (lanes 1, 2, 5 and 6, Figure 6Ca). However, no p53 could be detected in the thyroid of TRα1−/−TRβ−/− mice at these two ages (lanes 3, 4, 7 and 8, Figure 6Ca) under the same experimental conditions. The apparent reduced p53 protein level prompted us to examine the expression of its regulator, murine double minute oncogene protein (MDM2). MDM2 acts as a p53 ubiquitin ligase to increase proteasome degradation of p53. Figure 6Cb shows that the MDM2 protein level was markedly elevated eight-to ninefold as compared with that in WT-mice at ages 5–6 and 11–13 months (Figure 6C), indicating that the reduced p53 protein level observed in Figure 6Ca was due at least partially to increase proteasomal degradation of p53. Thus, decreased apoptosis mediated by the lowered p53 level could also contribute to the aberrant thyroid growth of TRα1−/−TRβ−/− mice shown in Figure 2.
We next determined whether the expression of the peroxisome proliferator-activated receptor γ (PPARγ) was altered in the thyroid of TRα1−/−TRβ−/− mice during thyroid carcinogenesis. PPARγ has been suggested as a tumor suppressor in human follicular thyroid cancer (Shen and Chung, 2005; Teresi and Waite, 2008). PPARγ was shown to suppress proliferation and increase apoptosis of thyroid tumor cells by activating nuclear factor-κB signaling (Kato et al., 2006). Western blot analyses showed that the protein abundance of PPARγ in the thyroid of ageing TRα1−/−TRβ−/− mice was progressively reduced as compared with WT-mice (Figure 6D). Approximately, 40 and 80% reductions of PPARγ protein level were detected at ages 5–6 and 11–13 months, respectively (Figure 6Da). These results indicate that during thyroid carcinogenesis, the expression of PPARγ protein remains low, consistent with the notion that PPARγ could function as a tumor suppressor during thyroid carcinogenesis.
Activation of signaling pathway to increase cell invasion and angiogenesis in thyroids of TRα1−/−TRβ−/− mice
To determine the changes in the signaling pathways leading to increase cell invasion and migration, we analyzed the expression of key effectors in ILK–MMP-2 signaling pathway. ILK is a ubiquitously expressed protein serine-threonine kinase that binds to integrins. The kinase activity of ILK is stimulated by integrins or growth factors in a PI3K–AKT-dependent manner (Persad and Dedhar, 2003; Troussard et al., 2003). Increased ILK expression and activity result in invasive and metastatic phenotypes (Troussard et al., 2000). MMP-2 is critically involved in degradation of extra-cellular matrix (Brinckerhoff and Matrisian, 2002; Turpeenniemi-Hujanen, 2005).
The AKT signaling that was activated in the thyroid of TRα1−/−TRβ−/− mice prompted us to determine whether ILK–MMP-2 was overexpressed. Figure 7A shows that 1.5- and 3-fold increases in the activation of ILK were detected in TRα1−/−TRβ−/− mice at 5–6 and 11–13 months, respectively (Figure 7Aa). The abundance of MMP-2 protein, the direct downstream target of ILK, was increased 1.6- to 2-fold for TRα1−/−TRβ−/− mice at 5–6 and 11–13 months, respectively (Figure 7Ab). These results indicate an increased activation of ILK–MMP-2 signaling as carcinogenesis progressed, thus, contributing to invasion and metastasis of thyroid tumor cells in TRα1−/−TRβ−/− mice.
Figure 7.
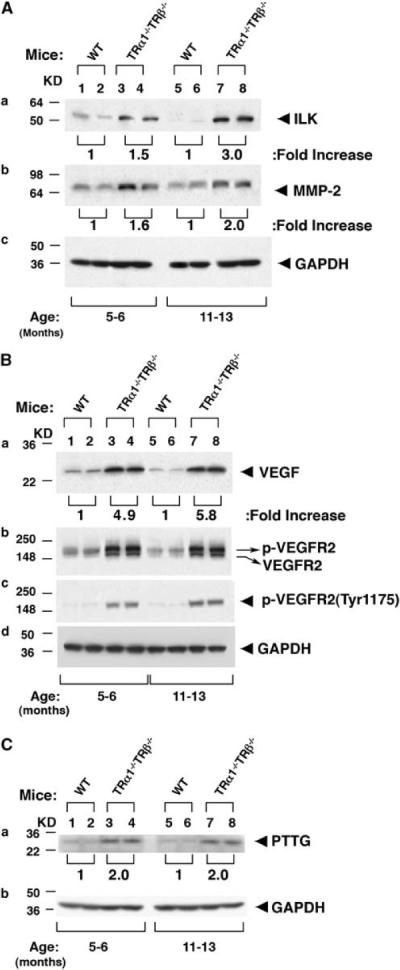
Activation of pathways involved in invasion and angiogenesis. Thyroid extract (30 μg) was used in the western blot analysis, as described in Materials and methods section. Two representative results from 5 to 7 WT (lanes 1, 2, 5 and 6), TRα1−/− TRβ−/− mice (lanes 3, 4, 7 and 8) are shown for ILK and MMP-2 (A), VEGF, VEGFR2 and p-VEGFR2 (B), and PTTG (C). GAPDH was used as loading controls.
Angiogenesis is critical for the growth and metastatic spread of tumors. Vascular endothelial growth factor (VEGF) is the most potent inducer of neovasculature, and its increased expression has been associated with the development of metastases (Weidner et al., 1991) and reduced survival (Maeda et al., 1995). We, therefore, studied the expression of VEGF and its receptor, VEGFR2, in the thyroid of TRα1−/−TRβ−/− mice. Figure 7Ba shows that the abundance of VEGF was 4.9- and 5.8-fold higher than in WT-mice at 5–6 and 11–13 months, respectively. A higher expression of VEGF at an advanced stage of carcinogenesis is consistent with an increase in the occurrence frequency of vascular invasion as mice aged (see Figure 4B).
Using antibodies against VEGFR2 protein, we detected two bands with slightly different molecular weights in the thyroid of TRα1−/−TRβ−/− mice (Figure 7Bb), indicative of the phosphorylated VEGFR2 with a higher molecular weight and the non-phosphorylated form with a lower molecular weight (lanes 3, 4, 7 and 8). In contrast, only the lower non-phosphorylated form of VEGFR2 was detected in the WT-mice (lanes 1, 2, 5 and 6, Figure 7Bb). That VEGFR2 protein with a higher molecular weight band was the activated phosphorylated form was further confirmed by using antibody specific for the phosphorylated VEGFR2 protein with which only the higher molecular weight band was detected in TRα1−/−TRβ−/− mice, but not in WT-mice (Figure 7Bc). All together, the activated VEGF–VEGFR2 signaling promotes the angiogenesis and invasion of tumor cells.
The pituitary tumor-transformation gene (PTTG), originally isolated from GH4 pituitary cells, has been shown to cause cell transformation in vitro and to induce tumor formation in vivo (Pei and Melmed, 1997). Its overexpression occurs in a wide variety of non-endocrine and endocrine tumors including thyroid cancer (Heaney et al., 2001; Kim et al., 2003). PTTG is involved in multiple cellular pathways, including proliferation, DNA repair, transformation, angiogenesis induction, invasion and the induction of genetic instability. Recently, overexpression of the PTTG protein has been shown to induce cell-cycle abnormalities and aneuploidy in thyroid follicular cells (Ying et al., 2003a, 2006; Zimonjic et al., 2005) and to promote angiogenesis in thyroid tumor cells (Kim et al., 2007). In thyroid carcinomas, PTTG expression is a marker of invasiveness. We, therefore, examined the protein abundance of PTTG in the thyroid of TRα1−/−TRβ−/− mice during thyroid carcinogenesis. Figure 7C shows that, similar to human thyroid cancer, PTTG protein abundance was elevated approximately twofold in the thyroid tumors of TRα1−/−TRβ−/− mice at ages 5–6 months (lanes 3 and 4) and 11–13 months (lanes 7 and 8) as compared with WT-mice at the corresponding ages (lanes 1 and 2, and 5 and 6). These results suggest that overexpressed PTTG could increase angiogenesis and invasion to promote tumor progression.
Discussion
This study shows that TRα1−/−TRβ−/− mice spontaneously developed metastatic FTC as they aged. Extensive molecular analyses showed that complex alterations of multiple signaling pathways contribute to the development and progression of metastatic carcinoma (see Figure 8). Thyroid growth was stimulated by multiple proliferation signals, such as the TSH–TSHR–pCREB–cyclin D1 pathway and AKT–mTOR–p70S6K signaling. Several pathways known to promote cell invasion and motility, such as ILK–MMP-2 and PTTG, were also activated in the thyroid of TRα1−/−TRβ−/− mice. Consistent with reports by others (McCabe et al., 2002; Kim et al., 2006), we also found that an elevated PTTG was accompanied by induction of VEGF and VEGFR, thereby increasing the angiogenesis of tumor cells to further promote tumor progression (Figure 8). The decreased p53 is known to decrease apoptosis. The end result of these multiple alterations is development and progression of thyroid cancer.
Figure 8.
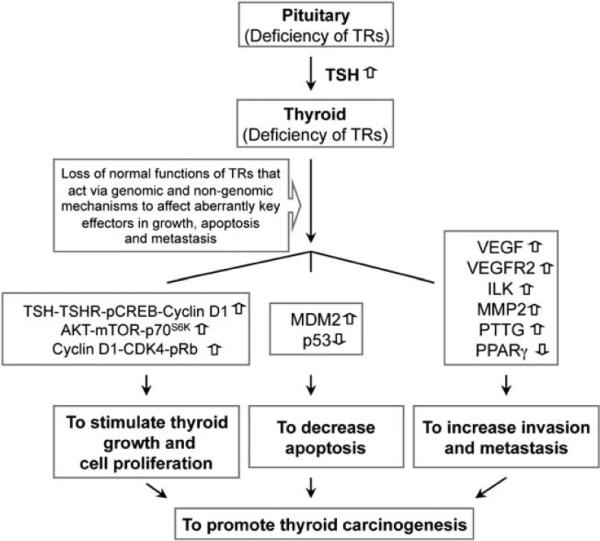
The loss of normal TR functions results in complex alterations of multiple signaling pathways, thereby contributing to thyroid carcinogenesis in TRα1−/− TRβ−/− mice. TRs act through genomic and nongenomic actions to regulate key cellular effectors to maintain normal cellular functions in growth, proliferation, cell motility and migration. TRs through T3 repress the expression of TSHα and TSHβ genes in the pituitary. Deficiency of TRs in the pituitary results in the loss of negative regulation and thus increases expression of TSH. Although little is known regarding the precise mechanisms by which TRs regulate other key effectors, such as VEGFR2 and others indicated in this figure (see also Discussion section), the loss of functional TRs results in disarrays of multiple signaling pathways to contribute to thyroid carcinogenesis. The phenotypic manifestation of tumor development and progression because of the loss of normal functions of TR is consistent with the notion that TRs have key tumor suppressor roles.
Currently, the molecular details by which the loss of both functional TRs mediates the changes of the key effectors affecting the cellular signaling in growth, invasion and angiogenesis remain to be fully elucidated. However, TRs are known to act (directly and indirectly) through genomic and nongenomic actions to regulate target genes expression and activities. Thus, through genomic actions, the loss of TRs increases the expression of negatively regulated genes. Examples are the increased expression of TSHα and TSHβ genes (TR direct target genes) in the pituitary to stimulate thyroid growth and the increased expression of cyclin D1 in the thyroid (through indirect regulation) to increase tumor cell proliferation through cyclin-CDK4–pRb signaling (Furumoto et al., 2005). However, understanding of how the loss of TRs results in altered expression and activities of other key regulators and in the activation of phosphorylation cascades (for example, AKT–mTOR–p70S6K) awaits further studies. Importantly, however, the phenotypic alterations resulting in carcinogenesis because of the loss of normal functions of TRs are consistent with the hypothesis that TRs could function as tumor suppressors in the development of thyroid cancer in TRα1−/−TRβ−/− mice.
TSH has long been known as a major stimulator of thyrocyte proliferation. Whether it is an initiator of thyroid carcinogenesis, however, remains to be clarified. Approximately, 30% of rats fed an iodine-deficient diet developed follicular carcinomas by 18 months, but no metastatic tumors were found (Ward and Ohshima, 1986). Several studies also showed a more prevalent occurrence of follicular carcinomas in patients in iodine-deficient regions (Lawal et al., 2001). These findings suggest the possibility that TSH could initiate thyroid carcinogenesis, but recent studies provide compelling evidence to the contrary. Transgenic mice with thyroid-specific expression of the A2 adenosine receptor (Ledent et al., 1992), a mutated Gsα (Michiels et al., 1994) or cholera toxin A1 (Zeiger et al., 1997) develop thyroid hyperplasia and hyperthyroidism, but not carcinomas. In addition, patients with Graves' disease or with congenital hyperthyroidism because of germline mutations of the TSH receptor do not appear to have a higher rate of thyroid malignancy compared with persons with normal TSH levels (Fagin, 2002). These studies suggest that growth signals provided by TSH are necessary for thyrocyte proliferation, but not sufficient for metastatic carcinoma to occur. Additional genetic changes would need to occur for the transformation of the hyperproliferative thyroid cells to cancer cells. In TRα1−/−TRβ−/− mice, the loss of both functional TR isoforms results in total loss of negative regulation of the pituitary-thyroid axis, thereby elevating and sustaining high serum TSH levels to provide proliferation signals to the follicular cells (60- to 160-fold higher than in WT-mice; (Gothe et al., 1999; Furumoto et al., 2005)). In addition, the regulatory activities of TRs necessary to maintain normal cellular functions are lost, leading to disarrayed signaling to develop metastatic tumors. Indeed, TRα1−/−TRβ−/− mice showed extensive hyperplasia as early as 2–3 months of age, preceding the occurrence of capsular and vascular invasion and distant metastasis at a later stage of cancer progression (see Figure 4). These observations suggest that growth and metastasis are two related, but independent processes. The former is a prerequisite for the latter to occur. However, without additional genetic alterations to enable the latter to occur, there would be no metastatic carcinoma.
This molecular model of thyroid carcinogenesis is supported by the phenotypic expression of several genetically engineered mice. So far, no thyroid cancer has been reported for mice deficient in TRβ (TRβ−/− mice) or TRα1 (TRα1−/− mice) alone. The TRβ−/− mice show mild resistance to thyroid hormone with elevation of two- to threefold of thyroid hormones accompanied by a moderately increased TSH (two- to threefold as compared with WT-mice; (Forrest et al., 1996b; Forrest and Vennstrom, 2000). Mice lacking TRα1 show a mild hypothyroidism with nearly normal TSH levels (Wikstrom et al., 1998). Therefore, in these two mutant mice, there is no strong TSH proliferation signal to sustain long-term aberrant growth in spite of the loss of the suppressor functions of either TRβ or TRα1. TRβPV/PV mice (Suzuki et al., 2002) and TRβPV/− mice (a mutated TRβ gene together with a loss of a wild-type TRβ gene) are known to spontaneously develop metastatic FTC (Kato et al., 2004). TRβPV/PV and TRβPV/− mice show similarly highly elevated TSH levels to drive the growth of follicular cells (Suzuki et al., 2002; Kato et al., 2004). The loss of the suppressor functions of TRβ because of mutations of the two TRβ alleles, as in TRβPV/PV mice, or the mutation of one allele of the TRβ gene together with the loss of the other allele, as in TRβPV/− mice, empowers the hyperplastic follicular cells to progress to metastatic carcinoma.
The deleterious effects of mutations or the loss of the TRβ gene in the thyroid is evident in the TRβPV/PV mice (Suzuki et al., 2002) and TRβPV/− mice (Kato et al., 2004). Less clear are the consequences of loss of TRα1 in the thyroid during carcinogenesis. It is known that the major TR isoform in the thyroid is TRβ (Ying et al., 2003b). Therefore, it is possible that in TRβPV/PV mice and TRβPV/− mice, normal functions of TRα1 are also inhibited by TRβPV through the dominant negative effect as similarly as shown in the liver (Zhang et al., 2002). Thus, the normal activities of TRα1 are lost in the thyroids of TRβPV/PV and TRβPV/− mice as in those of TRα1−/−TRβ−/− mice, although the underlying mechanisms are different. In TRβPV/PV and TRβPV/− mice, the activities of TRα1 are inactivated because of the potent dominant negative activity of TRβPV, whereas in TRα1−/−TRβ−/− mice, it is the deletion of both of the alleles of the TRα gene. These results would indicate that both TRs are critically important to maintain normal functions of the thyroid. Their loss by mutations of the TRβ gene or deletion of the TRβ and the TRα genes would propel hyperplastic thyroid cells to become metastatic cancer cells. Thus, this study has revealed a novel tumor suppressor role of TRs in thyroid carcinogenesis.
Materials and methods
Experimental animals
Animal experiments were performed according to the protocols approved by the Animal Care and Use Committee at the National Cancer Institute. Mice deficient in TRα1 and TRβ genes were genotyped as described (Forrest et al., 1996a; Wikstrom et al., 1998). Heterozygous TRα1- and TRβ- deficient mice were intercrossed to generate WT- and TRα1−/−TRβ−/− mice. Thyroids and other tissues were harvested from TRα1−/−TRβ−/− mice and WT-mice littermates for weighing, histological analyses and biochemical studies.
Western blot analysis
Thyroids dissected from TRα1−/−TRβ−/− mice and wild-type siblings were washed with phosphate-buffered saline and homogenized in a solution with 50 mM Tris buffer, 150 mM NaCl, 1 mM ethylene-diaminetetraacetic acid, 1% NP40 and proteinase/phosphatase inhibitors. The western blot analysis was carried out as described by Furumoto et al. (Furumoto et al., 2005). Primary antibodies for phosphorylated-S473 AKT (#9271), total AKT (#9272), phosphorylated-T421/S424 p70S6K (#9204), total p70S6K (#9202), CREB (#9197), phospho-CREB (#9198), phospho-mTOR (Ser2448) (#2971), phospho-mTOR (Ser2481) (#2974), cyclin-dependent kinase-4 (DCS156) (#2906), retinoblastoma protein (Rb:S807/811, #9307S; S780, #9308S), phospho-VEGFR2 (Tyr1175) (#2478S) and GAPDH (#2118) were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-MMP2 (SC-10736), MDM2 (sc-965), VEGF (sc-507), VEGFR2(C17) (sc-316), PPARγ (sc-7196), p53 (sc-6243), Cyclin D1 (sc-450) and ILK (sc-13075) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-PTTG-1 (34–1500) was purchased from Zymed Laboratories Inc. (San Francisco, CA, USA). Anti-mTOR antibody (T2949) was purchased from Sigma-Aldrich (St Louis, MO, USA). Antibodies were used at a concentration recommended by manufacturers. For control of protein loading, the blots were probed with the antibodies against GAPDH.
Histological analysis
Thyroids and lungs were dissected and embedded in paraffin. Five-micrometer-thick sections were prepared and stained with hematoxylin and eosin. For each animal, single random sections through the thyroid (usually both lobes), lung and heart were examined. For thyroids, single section hyperplasia, capsular invasion, vascular invasion and anaplasia were routinely evaluated and scored. Hyperplasia was generally diffuse throughout the gland. Evidence of any of these changes in any section was counted as positive for that change. On average, in those cases with capsular invasion and/or vascular invasion, these morphological changes were observed in multiple locations (usually two or three) in any one single thyroid section. The presence of a single microscopic focus of metastatic follicular carcinoma in the lung was counted as positive for metastasis in that animal.
Statistical analysis
Data are expressed as means ± standard errors. Statistical analysis was performed with the use of analysis of variance, and P < 0.05 was considered significant unless otherwise specified. StatView 5.0 (SAS Institute Inc., Cary, NC, USA) was used to perform Kaplan-Meier cumulative survival analysis, and Student's t-test using odds ratios and Fisher's exact probability test were used to analyze the data of pathological progression. PRISM 4.0a (GraphPad Software, San Diego, CA, USA) was used for log-rank testing for statistical significance.
Acknowledgements
This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health. We thank Drs H Ying and Y Kato for the analyses of survival curves and thyroid growth in the early phase of the present work.
Footnotes
Conflict of interest The authors declare no conflict of interest.
References
- Ando S, Sarlis NJ, Krishnan J, Feng X, Refetoff S, Zhang MQ, et al. Aberrant alternative splicing of thyroid hormone receptor in a TSH-secreting pituitary tumor is a mechanism for hormone resistance. Mol Endocrinol. 2001a;15:1529–1538. doi: 10.1210/mend.15.9.0687. [DOI] [PubMed] [Google Scholar]
- Ando S, Sarlis NJ, Oldfield EH, Yen PM. Somatic mutation of TRbeta can cause a defect in negative regulation of TSH in a TSH-secreting pituitary tumor. J Clin Endocrinol Metab. 2001b;86:5572–5576. doi: 10.1210/jcem.86.11.7984. [DOI] [PubMed] [Google Scholar]
- Brinckerhoff CE, Matrisian LM. Matrix metalloproteinases: a tail of a frog that became a prince. Nat Rev Mol Cell Biol. 2002;3:207–214. doi: 10.1038/nrm763. [DOI] [PubMed] [Google Scholar]
- Chen HW, Privalsky ML. The erbA oncogene represses the actions of both retinoid X and retinoid A receptors but does so by distinct mechanisms. Mol Cell Biol. 1993;13:5970–5980. doi: 10.1128/mcb.13.10.5970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis PJ, Leonard JL, Davis FB. Mechanisms of nongenomic actions of thyroid hormone. Front Neuroendocrinol. 2008;29:211–218. doi: 10.1016/j.yfrne.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Fagin JA. Minireview: branded from the start-distinct oncogenic initiating events may determine tumor fate in the thyroid. Mol Endocrinol. 2002;16:903–911. doi: 10.1210/mend.16.5.0838. [DOI] [PubMed] [Google Scholar]
- Forrest D, Erway LC, Ng L, Altschuler R, Curran T. Thyroid hormone receptor beta is essential for development of auditory function. Nat Genet. 1996a;13:354–357. doi: 10.1038/ng0796-354. [DOI] [PubMed] [Google Scholar]
- Forrest D, Hanebuth E, Smeyne RJ, Everds N, Stewart CL, Wehner JM, et al. Recessive resistance to thyroid hormone in mice lacking thyroid hormone receptor beta: evidence for tissue-specific modulation of receptor function. EMBO J. 1996b;15:3006–3015. [PMC free article] [PubMed] [Google Scholar]
- Forrest D, Vennstrom B. Functions of thyroid hormone receptors in mice. Thyroid. 2000;10:41–52. doi: 10.1089/thy.2000.10.41. [DOI] [PubMed] [Google Scholar]
- Furumoto H, Ying H, Chandramouli GV, Zhao L, Walker RL, Meltzer PS, et al. An unliganded thyroid hormone beta receptor activates the cyclin D1/cyclin-dependent kinase/retinoblastoma/E2F pathway and induces pituitary tumorigenesis. Mol Cell Biol. 2005;25:124–135. doi: 10.1128/MCB.25.1.124-135.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya F, Lu C, Guigon CJ, Cheng SY. Nongenomic activation of phosphatidylinositol 3-kinase signaling by thyroid hormone receptors. Steroids. 2009;74:628–634. doi: 10.1016/j.steroids.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Silva S, Aranda A. The thyroid hormone receptor is a suppressor of ras-mediated transcription, proliferation, and transformation. Mol Cell Biol. 2004;24:7514–7523. doi: 10.1128/MCB.24.17.7514-7523.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gothe S, Wang Z, Ng L, Kindblom JM, Barros AC, Ohlsson C, et al. Mice devoid of all known thyroid hormone receptors are viable but exhibit disorders of the pituitary-thyroid axis, growth, and bone maturation. Genes Dev. 1999;13:1329–1341. doi: 10.1101/gad.13.10.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigon CJ, Zhao L, Lu C, Willingham MC, Cheng SY. Regulation of beta-catenin by a novel nongenomic action of thyroid hormone beta receptor. Mol Cell Biol. 2008;28:4598–4608. doi: 10.1128/MCB.02192-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaney AP, Nelson V, Fernando M, Horwitz G. Transforming events in thyroid tumorigenesis and their association with follicular lesions. J Clin Endocrinol Metab. 2001;86:5025–5032. doi: 10.1210/jcem.86.10.7886. [DOI] [PubMed] [Google Scholar]
- Herwig S, Strauss M. The retinoblastoma protein: a master regulator of cell cycle, differentiation and apoptosis. Eur J Biochem. 1997;246:581–601. doi: 10.1111/j.1432-1033.1997.t01-2-00581.x. [DOI] [PubMed] [Google Scholar]
- Kamiya Y, Puzianowska-Kuznicka M, McPhie P, Nauman J, Cheng SY, Nauman A. Expression of mutant thyroid hormone nuclear receptors is associated with human renal clear cell carcinoma. Carcinogenesis. 2002;23:25–33. doi: 10.1093/carcin/23.1.25. [DOI] [PubMed] [Google Scholar]
- Kaneshige M, Kaneshige K, Zhu X, Dace A, Garrett L, Carter TA, et al. Mice with a targeted mutation in the thyroid hormone beta receptor gene exhibit impaired growth and resistance to thyroid hormone. Proc Natl Acad Sci USA. 2000;97:13209–13214. doi: 10.1073/pnas.230285997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Ying H, Willingham MC, Cheng SY. A tumor suppressor role for thyroid hormone beta receptor in a mouse model of thyroid carcinogenesis. Endocrinology. 2004;145:4430–4438. doi: 10.1210/en.2004-0612. [DOI] [PubMed] [Google Scholar]
- Kato Y, Ying H, Zhao L, Furuya F, Araki O, Willingham MC, et al. PPARgamma insufficiency promotes follicular thyroid carcinogenesis via activation of the nuclear factor-kappaB signaling pathway. Oncogene. 2006;25:2736–2747. doi: 10.1038/sj.onc.1209299. [DOI] [PubMed] [Google Scholar]
- Kim CS, Ying H, Willingham MC, Cheng SY. The pituitary tumor-transforming gene promotes angiogenesis in a mouse model of follicular thyroid cancer. Carcinogenesis. 2007;28:932–939. doi: 10.1093/carcin/bgl231. [DOI] [PubMed] [Google Scholar]
- Kim DS, Franklyn JA, Boelaert K, Eggo MC, Watkinson JC, McCabe CJ. Pituitary tumor transforming gene (PTTG) stimulates thyroid cell proliferation via a vascular endothelial growth factor/kinase insert domain receptor/inhibitor of DNA binding-3 autocrine pathway. J Clin Endocrinol Metab. 2006;91:4603–4611. doi: 10.1210/jc.2006-1291. [DOI] [PubMed] [Google Scholar]
- Kim DS, McCabe CJ, Buchanan MA, Watkinson JC. Oncogenes in thyroid cancer. Clin Otolaryngol Allied Sci. 2003;28:386–395. doi: 10.1046/j.1365-2273.2003.00732.x. [DOI] [PubMed] [Google Scholar]
- Lawal O, Agbakwuru A, Olayinka OS, Adelusola K. Thyroid malignancy in endemic nodular goitres: prevalence, pattern and treatment. Eur J Surg Oncol. 2001;27:157–161. doi: 10.1053/ejso.2000.1085. [DOI] [PubMed] [Google Scholar]
- Ledent C, Dumont JE, Vassart G, Parmentier M. Thyroid expression of an A2 adenosine receptor transgene induces thyroid hyperplasia and hyperthyroidism. EMBO J. 1992;11:537–542. doi: 10.1002/j.1460-2075.1992.tb05084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Meng ZH, Chandrasekaran R, Kuo WL, Collins CC, Gray JW, et al. Biallelic inactivation of the thyroid hormone receptor beta1 gene in early stage breast cancer. Cancer Res. 2002;62:1939–1943. [PubMed] [Google Scholar]
- Lin KH, Shieh HY, Chen SL, Hsu HC. Expression of mutant thyroid hormone nuclear receptors in human hepatocellular carcinoma cells. Mol Carcinog. 1999;26:53–61. doi: 10.1002/(sici)1098-2744(199909)26:1<53::aid-mc7>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Maeda K, Chung YS, Takatsuka S, Ogawa Y, Sawada T, Yamashita Y, et al. Tumor angiogenesis as a predictor of recurrence in gastric carcinoma. J Clin Oncol. 1995;13:477–481. doi: 10.1200/JCO.1995.13.2.477. [DOI] [PubMed] [Google Scholar]
- Martinez-Iglesias O, Garcia-Silva S, Tenbaum SP, Regadera J, Larcher F, Paramio JM, et al. Thyroid hormone receptor beta1 acts as a potent suppressor of tumor invasiveness and metastasis. Cancer Res. 2009;69:501–509. doi: 10.1158/0008-5472.CAN-08-2198. [DOI] [PubMed] [Google Scholar]
- McCabe CJ, Boelaert K, Tannahill LA, Heaney AP, Stratford AL, Khaira JS, et al. Vascular endothelial growth factor, its receptor KDR/Flk-1, and pituitary tumor transforming gene in pituitary tumors. J Clin Endocrinol Metab. 2002;87:4238–4244. doi: 10.1210/jc.2002-020309. [DOI] [PubMed] [Google Scholar]
- Meier CA, Dickstein BM, Ashizawa K, McClaskey JH, Muchmore P, Ransom SC, et al. Variable transcriptional activity and ligand binding of mutant beta 1 3,5,3′-triiodothyronine receptors from four families with generalized resistance to thyroid hormone. Mol Endocrinol. 1992;6:248–258. doi: 10.1210/mend.6.2.1569968. [DOI] [PubMed] [Google Scholar]
- Michiels FM, Caillou B, Talbot M, Dessarps-Freichey F, Maunoury MT, Schlumberger M, et al. Oncogenic potential of guanine nucleotide stimulatory factor alpha subunit in thyroid glands of transgenic mice. Proc Natl Acad Sci USA. 1994;91:10488–10492. doi: 10.1073/pnas.91.22.10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakawa M, Tsushima T, Murakami H, Wakai K, Isozaki O, Takano K. Increased expression of phosphorylated p70S6 kinase and Akt in papillary thyroid cancer tissues. Endocr J. 2003;50:77–83. doi: 10.1507/endocrj.50.77. [DOI] [PubMed] [Google Scholar]
- Pei L, Melmed S. Isolation and characterization of a pituitary tumor-transforming gene (PTTG) Mol Endocrinol. 1997;11:433–441. doi: 10.1210/mend.11.4.9911. [DOI] [PubMed] [Google Scholar]
- Persad S, Dedhar S. The role of integrin-linked kinase (ILK) in cancer progression. Cancer Metastasis Rev. 2003;22:375–384. doi: 10.1023/a:1023777013659. [DOI] [PubMed] [Google Scholar]
- Puzianowska-Kuznicka M, Krystyniak A, Madej A, Cheng SY, Nauman J. Functionally impaired TR mutants are present in thyroid papillary cancer. J Clin Endocrinol Metab. 2002;87:1120–1128. doi: 10.1210/jcem.87.3.8296. [DOI] [PubMed] [Google Scholar]
- Puzianowska-Kuznicka M, Nauman A, Madej A, Tanski Z, Cheng S, Nauman J. Expression of thyroid hormone receptors is disturbed in human renal clear cell carcinoma. Cancer Lett. 2000;155:145–152. doi: 10.1016/s0304-3835(00)00416-x. [DOI] [PubMed] [Google Scholar]
- Puzianowska-Kuznicka M, Pietrzak M, Turowska O, Nauman A. Thyroid hormones and their receptors in the regulation of cell proliferation. Acta Biochim Pol. 2006;53:641–650. [PubMed] [Google Scholar]
- Ringel MD, Hayre N, Saito J, Saunier B, Schuppert F, Burch H, et al. Overexpression and overactivation of Akt in thyroid carcinoma. Cancer Res. 2001;61:6105–6111. [PubMed] [Google Scholar]
- Safer JD, Colan SD, Fraser LM, Wondisford FE. A pituitary tumor in a patient with thyroid hormone resistance: a diagnostic dilemma. Thyroid. 2001;11:281–291. doi: 10.1089/105072501750159750. [DOI] [PubMed] [Google Scholar]
- Sap J, Munoz A, Damm K, Goldberg Y, Ghysdael J, Leutz A, et al. The c-erb-A protein is a high-affinity receptor for thyroid hormone. Nature. 1986;324:635–640. doi: 10.1038/324635a0. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, DePinho RA. p53: good cop/bad cop. Cell. 2002;110:9–12. doi: 10.1016/s0092-8674(02)00818-8. [DOI] [PubMed] [Google Scholar]
- Shen WT, Chung WY. Treatment of thyroid cancer with histone deacetylase inhibitors and peroxisome proliferator-activated receptor-gamma agonists. Thyroid. 2005;15:594–599. doi: 10.1089/thy.2005.15.594. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Willingham MC, Cheng SY. Mice with a mutation in the thyroid hormone receptor beta gene spontaneously develop thyroid carcinoma: a mouse model of thyroid carcinogenesis. Thyroid. 2002;12:963–969. doi: 10.1089/105072502320908295. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Zhang XY, Forrest D, Willingham MC, Cheng SY. Marked potentiation of the dominant negative action of a mutant thyroid hormone receptor beta in mice by the ablation of one wild-type beta allele. Mol Endocrinol. 2003;17:895–907. doi: 10.1210/me.2002-0326. [DOI] [PubMed] [Google Scholar]
- Takano T, Miyauchi A, Yoshida H, Nakata Y, Kuma K, Amino N. Expression of TRbeta1 mRNAs with functionally impaired mutations is rare in thyroid papillary carcinoma. J Clin Endocrinol Metab. 2003;88:3447–3449. doi: 10.1210/jc.2003-030012. [DOI] [PubMed] [Google Scholar]
- Teresi RE, Waite KA. PPARgamma, PTEN, and the fight against cancer. PPAR Res. 2008;2008:932632. doi: 10.1155/2008/932632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thormeyer D, Baniahmad A. The v-erbA oncogene (review) Int J Mol Med. 1999;4:351–358. [PubMed] [Google Scholar]
- Troussard AA, Costello P, Yoganathan TN, Kumagai S, Roskelley CD, Dedhar S. The integrin linked kinase (ILK) induces an invasive phenotype via AP-1 transcription factor-dependent upregulation of matrix metalloproteinase 9 (MMP-9) Oncogene. 2000;19:5444–5452. doi: 10.1038/sj.onc.1203928. [DOI] [PubMed] [Google Scholar]
- Troussard AA, Mawji NM, Ong C, Mui A, St -Arnaud R, Dedhar S. Conditional knock-out of integrin-linked kinase demonstrates an essential role in protein kinase B/Akt activation. J Biol Chem. 2003;278:22374–22378. doi: 10.1074/jbc.M303083200. [DOI] [PubMed] [Google Scholar]
- Turpeenniemi-Hujanen T. Gelatinases (MMP-2 and -9) and their natural inhibitors as prognostic indicators in solid cancers. Biochimie. 2005;87:287–297. doi: 10.1016/j.biochi.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Ward JM, Ohshima M. The role of iodine in carcinogenesis. Adv Exp Med Biol. 1986;206:529–542. doi: 10.1007/978-1-4613-1835-4_37. [DOI] [PubMed] [Google Scholar]
- Weidner N, Semple JP, Welch WR, Folkman J. Tumor angiogenesis and metastasis-correlation in invasive breast carcinoma. N Engl J Med. 1991;324:1–8. doi: 10.1056/NEJM199101033240101. [DOI] [PubMed] [Google Scholar]
- Weinberger C, Thompson CC, Ong ES, Lebo R, Gruol DJ, Evans RM. The c-erb-A gene encodes a thyroid hormone receptor. Nature. 1986;324:641–646. doi: 10.1038/324641a0. [DOI] [PubMed] [Google Scholar]
- Weiss RE, Refetoff S. Resistance to thyroid hormone. Rev Endocr Metab Disord. 2000;1:97–108. doi: 10.1023/a:1010072605757. [DOI] [PubMed] [Google Scholar]
- Wikstrom L, Johansson C, Salto C, Barlow C, Campos Barros A, Baas F, et al. Abnormal heart rate and body temperature in mice lacking thyroid hormone receptor alpha 1. EMBO J. 1998;17:455–461. doi: 10.1093/emboj/17.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen PM, Ikeda M, Brubaker JH, Forgione M, Sugawara A, Chin WW. Roles of v-erbA homodimers and heterodimers in mediating dominant negative activity by v-erbA. J Biol Chem. 1994;269:903–909. [PubMed] [Google Scholar]
- Ying H, Furuya F, Zhao L, Araki O, West BL, Hanover JA, et al. Aberrant accumulation of PTTG1 induced by a mutated thyroid hormone beta receptor inhibits mitotic progression. J Clin Invest. 2006;116:2972–2984. doi: 10.1172/JCI28598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Suzuki H, Furumoto H, Walker R, Meltzer P, Willingham MC, et al. Alterations in genomic profiles during tumor progression in a mouse model of follicular thyroid carcinoma. Carcinogenesis. 2003a;24:1467–1479. doi: 10.1093/carcin/bgg111. [DOI] [PubMed] [Google Scholar]
- Ying H, Suzuki H, Zhao L, Willingham MC, Meltzer P, Cheng SY. Mutant thyroid hormone receptor beta represses the expression and transcriptional activity of peroxisome proliferator-activated receptor gamma during thyroid carcinogenesis. Cancer Res. 2003b;63:5274–5280. [PubMed] [Google Scholar]
- Zeiger MA, Saji M, Gusev Y, Westra WH, Takiyama Y, Dooley WC, et al. Thyroid-specific expression of cholera toxin A1 subunit causes thyroid hyperplasia and hyperthyroidism in transgenic mice. Endocrinology. 1997;138:3133–3140. doi: 10.1210/endo.138.8.5347. [DOI] [PubMed] [Google Scholar]
- Zhang XY, Kaneshige M, Kamiya Y, Kaneshige K, McPhie P, Cheng SY. Differential expression of thyroid hormone receptor isoforms dictates the dominant negative activity of mutant Beta receptor. Mol Endocrinol. 2002;16:2077–2092. doi: 10.1210/me.2002-0080. [DOI] [PubMed] [Google Scholar]
- Zimonjic DB, Kato Y, Ying H, Popescu NC, Cheng SY. Chromosomal aberrations in cell lines derived from thyroid tumors spontaneously developed in TRbetaPV/PV mice. Cancer Genet Cytogenet. 2005;161:104–109. doi: 10.1016/j.cancergencyto.2005.02.007. [DOI] [PubMed] [Google Scholar]