

Summary
Background
Osteoarthritis is the most common form of arthritis worldwide and is a major cause of pain and disability in elderly people. The health economic burden of osteoarthritis is increasing commensurate with obesity prevalence and longevity. Osteoarthritis has a strong genetic component but the success of previous genetic studies has been restricted due to insufficient sample sizes and phenotype heterogeneity.
Methods
We undertook a large genome-wide association study (GWAS) in 7410 unrelated and retrospectively and prospectively selected patients with severe osteoarthritis in the arcOGEN study, 80% of whom had undergone total joint replacement, and 11 009 unrelated controls from the UK. We replicated the most promising signals in an independent set of up to 7473 cases and 42 938 controls, from studies in Iceland, Estonia, the Netherlands, and the UK. All patients and controls were of European descent.
Findings
We identified five genome-wide significant loci (binomial test p≤5·0×10−8) for association with osteoarthritis and three loci just below this threshold. The strongest association was on chromosome 3 with rs6976 (odds ratio 1·12 [95% CI 1·08–1·16]; p=7·24×10−11), which is in perfect linkage disequilibrium with rs11177. This SNP encodes a missense polymorphism within the nucleostemin-encoding gene GNL3. Levels of nucleostemin were raised in chondrocytes from patients with osteoarthritis in functional studies. Other significant loci were on chromosome 9 close to ASTN2, chromosome 6 between FILIP1 and SENP6, chromosome 12 close to KLHDC5 and PTHLH, and in another region of chromosome 12 close to CHST11. One of the signals close to genome-wide significance was within the FTO gene, which is involved in regulation of bodyweight—a strong risk factor for osteoarthritis. All risk variants were common in frequency and exerted small effects.
Interpretation
Our findings provide insight into the genetics of arthritis and identify new pathways that might be amenable to future therapeutic intervention.
Funding
arcOGEN was funded by a special purpose grant from Arthritis Research UK.
Introduction
Osteoarthritis is the most common form of arthritis worldwide, affecting about 40% of people older than 70 years.1 It is a complex disease of the musculoskeletal system with both genetic and environmental risk factors.2 From the results of heritability studies in twins, sibling pairs, and families, genetic factors are estimated to account for about 50% of the risk of developing osteoarthritis in the hip or knee, although precise estimates vary according to sex, affected site, and severity of disease.3,4 Despite extensive efforts, only three loci (GDF5, chromosome 7q22, and MCF2L)5–11 have thus far been associated with osteoarthritis at genome-wide significance levels (p≤5·0×10−8) in European populations. This lack of success in osteoarthritis might be attributed to several factors such as insufficient sample sizes in previous studies and disease heterogeneity that might result from different underlying causes, both genetic and environmental, depending on which joints are affected.4 The three established osteoarthritis loci have fairly small effect sizes (allele-wise odds ratios [OR] of about 1·15) and we have previously shown that the genetic basis of osteoarthritis is likely to consist of several signals of similar or smaller magnitude that necessitate large sample sizes for their detection at genome-wide significance levels.12 Phenotype definition in cases and controls is also a likely factor contributing to the dilution of power to detect strong signals. Improved definition of phenotype and reduced misclassification in such a prevalent disorder can enhance power.
To identify additional loci that confer susceptibility to osteoarthritis, we undertook a large well powered genome-wide association study (GWAS) of osteoarthritis within the context of the Arthritis Research UK Osteoarthritis Genetics (arcOGEN) Consortium. In 2011, we reported the results of an interim analysis of 43·8% of the full GWAS sample size, detecting no novel loci for osteoarthritis, but showing the polygenic nature of the disease's underlying genetics.12 Here, we report the results of the full-scale arcOGEN GWAS.
Methods
Study population
We undertook a large case–control genome-wide association study (GWAS) in which 7410 unrelated patients were retrospectively and prospectively selected from hospitals and clinics, with about 80% of patients ascertained for the severe endpoint of total joint replacement [TJR]), and publicly available data were obtained from 11 009 unrelated controls from the UK's Wellcome Trust Case Control Consortium 2 (WTCCC2), Avon Longitudinal Study of Parents and Children, Type 1 Diabetes Genetics Consortium, and People of the British Isles studies (table 1; appendix p 2). We replicated the most promising signals in an independent set of up to 7473 cases and 42 938 controls from studies in Iceland (deCODE), Estonia (Estonian Genome Centre of University of Tartu [EGCUT]), the Netherlands (Genetics OsteoArthritis and Progression [GARP], Rotterdam study RSI and RSII cohorts), and the UK (TwinsUK). All patients and controls were of European descent. For further details, see appendix pp 4–5.
Table 1.
Characteristics of study populations
| Study description | Country of origin | Number of SNPs |
Cases |
Controls |
Effective sample size | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number | Women | Radiography | Joint replacement | Hip | Knee | Hip and knee | Definition | Number | Women | |||||
| arcOGEN | Discovery | UK | 485 491 | 7410 | 4476 (60·4%) | 1606 (21·7%) | 5804 (78·3%) | 3266 (44·1%) | 3498 (47·2%) | 646 (8·7%) | Population* | 11 009 | 5515 (50·1%) | 17 716 |
| arcOGEN | Discovery | UK | 532 234 | 4476 | 4476 (100%) | 1113 (24·9%) | 3363 (75·1%) | 1934 (43·2%) | 2135 (47·7%) | 407 (9·1%) | Osteoarthritis free | 1828 | 1828 (100%) | 5192 |
| deCODE | In silico | Iceland | 128 | 2031 | 1179 (58·1%) | 0 | 2031 (100%) | 1269 (62·5%) | 636 (31·3%) | 126 (6·2%) | Osteoarthritis free | 31 487 | 17 209 (54·7%) | 3055 |
| EGCUT | In silico | Estonia | 80 | 213 | 159 (74·6%) | NA | NA | 64 (30·0%) | 123 (57·7%) | 26 (12·2%) | Population | 2531 | 1426 (56·3%) | 786 |
| GARP | In silico | Netherlands | 128 | 215 | 170 (79·1%) | 169 (78·6%) | 46 (21·4%) | 67 (31·2%) | 109 (50·7%) | 39 (18·1%) | Population | 1670 | 925 (55·4%) | 762 |
| RSI | In silico | Netherlands | 129 | 1950 | 1353 (69·4%) | 1628 (83·5%) | 322 (16·5%) | 458 (23·5%) | 1179 (60·5%) | 313 (16·1%) | KL<2 | 3243 | 1642 (50·6%) | 4871 |
| RSII | In silico | Netherlands | 129 | 485 | 306 (63·1%) | 398 (82·1%) | 87 (17·9%) | 116 (23·9%) | 326 (67·2%) | 43 (8·9%) | KL<2 | 1460 | 752 (51·5%) | 1456 |
| TwinsUK | In silico | UK | 129 | 170 | 170 (100%) | 170 (100%) | 0 | 57 (33·5%) | 102 (60·0%) | 11 (6·5%) | KL<2 | 228 | 228 (100%) | 390 |
| UK replication | De novo | UK | 24 | 2409 | 1453 (60·3%) | 392 (16·3%) | 2017 (83·7%) | 1032 (42·8%) | 1170 (48·6%) | 207 (8·6%) | Population | 2319 | 334 (14·4%) | 4726 |
Data are number (%), unless otherwise indicated. SNP=single nucleotide polymorphism. arcOGEN=Arthritis Research UK Osteoarthritis Genetics. EGCUT=Estonian Genome Centre of University of Tartu. NA=not available. GARP=Genetics OsteoArthritis and Progression. RS=Rotterdam study. KL=Kellgren-Lawrence score.
Drawn from the general population without consideration of their osteoarthritis status.
Procedures
The arcOGEN samples were genotyped by use of Illumina Human 610-Quad BeadChips (Illumina, San Diego, CA, USA; appendix p 2). Most cases had primary osteoarthritis requiring joint replacement of the hip or knee (table 1). We did quality-control checks both at the sample and single nucleotide polymorphism (SNP) levels (appendix p 2). We did a case–control analysis for an overlapping set of 485 491 autosomal SNPs in 7410 arcOGEN cases of osteoarthritis and 11 009 controls (table 1; appendix p 2, p 73). We imputed genotypes for autosomal SNPs by use of the directly typed data and phased genotype data from all HapMap III populations but did not see any additional signals (p<10−5) arising from imputed SNPs that were not captured by the directly genotyped SNP analyses (appendix p 3, p 6). To assess the effect of phenotype misclassification in controls, we also analysed genome-wide genotypes in 4476 female cases of osteoarthritis from arcOGEN compared with 1828 osteoarthritis-free female controls from the TwinsUK dataset. We used a multiplicative model for the analysis of all data and also stratified by site of osteoarthritis, sex, and osteoarthritis severity—ie, only cases with TJR were assessed (for a total of 24 specific phenotypes). We have presented the results for all analyses in the appendix pp 81–98, and for the most significant specific phenotype in the main text. 129 prioritised SNPs (p<10−5) were followed up by in-silico replication in the studies deCODE, EGCUT, GARP, Rotterdam study RSI and RSII cohorts, and TwinsUK (total in-silico replication sample size of 5064 cases of osteoarthritis and 40 619 controls). We subsequently undertook a de-novo replication of the 26 most significant SNPs in an additional independent set of 2409 arcOGEN cases of osteoarthritis and 2319 WTCCC2 population-based controls (UK replication). For our primary analysis, we used a meta-analysis framework to combine results for all data (discovery and replication) by use of a fixed-effects model (appendix p 5).
We used RT-PCR to assess the expression of at least one gene per signal in joint tissues from patients with osteoarthritis who were undergoing TJR, and in cartilage from individuals without clinical osteoarthritis. We investigated the expression patterns of nucleostemin, encoded by the GNL3 gene, through immunohistochemical staining of normal and osteoarthritic cartilage samples and analysed protein expression in cultured chondrocytes from samples of normal and osteoarthritic cartilage by use of western blotting. Full details are available in the appendix (p 6).
Statistical analysis
Statistical and imputational methods and Hardy-Weinberg equilibrium are described in the appendix pp 2–5.
Role of the funding source
The sponsors of the study had no role in the study design, data collection, analysis or interpretation, or writing of the report. The corresponding authors had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
Genome-wide analysis showed an excess of signals compared with the null expectation of no association (genomic control inflation factor λ1000=1·009; appendix p 74). In the main analysis (all cases of osteoarthritis vs population-based controls), 28 SNPs (representing 12 independent signals) had a p value of less than 1·0×10−5 compared with five SNPs according to the null expectation (binomial test p=5·0×10−13; appendix p 12, p 36). We checked for false positives that were attributable to population stratification by adjusting our analyses with the first ten ancestry-informative principal components: our results did not change qualitatively (appendix p 6, p 75). New signals were not detected with HapMap-based imputation across the genome (appendix p 3, p 6, pp 60–61).
After large-scale replication, 71 (55%) of 129 signals taken forward had effects in the same direction as the discovery analysis on the basis of which they had been prioritised (appendix pp 76–81). All 26 SNPs selected for further follow-up had effects in the same direction (binomial p=2·98×10−8). Five of 26 loci had genome-wide significance (p=5·0×10−8) and three were very close to this (table 2). Effect size estimates for seven of eight loci were lower in the replication analysis than in the discovery analysis (table 2), with the exception of one signal (ASTN2).
Table 2.
Association summary statistics for the eight replicating signals
| Chromosome | Position | Nearest gene(s) | Effect allele | Stratum |
Discovery |
Replication |
Discovery and replication |
Discovery and replication for analysis of all cases of osteoarthritis |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Effect allele frequency in controls | Odds ratio (95% CI) | p value | Odds ratio (95% CI) | p value | Odds ratio (95% CI) | p value | Odds ratio (95% CI) | p value | ||||||
| rs6976* | 3 | 52703844 | GLT8D1 | T | TJR | 0·37 | 1·16 (1·11–1·22) | 2·27×10−10 | 1·07 (1·02–1·13) | 7·79×10−03 | 1·12 (1·08–1·16) | 7·24×10−11 | 1·09 (1·06–1·12) | 6·56×10−09 |
| rs11177* | 3 | 52696345 | GNL3 | A | TJR | 0·38 | 1·16 (1·11–1·22) | 2·12×10−10 | 1·07 (1·02–1·13) | 7·70×10−03 | 1·12 (1·08–1·16) | 1·25×10−10 | 1·09 (1·06–1·12) | 5·13×10−09 |
| rs4836732 | 9 | 118306516 | ASTN2 | C | THR–female | 0·47 | 1·19 (1·10–1·28) | 1·19×10−05 | 1·23 (1·12–1·34) | 9·54×10−06 | 1·20 (1·13–1·27) | 6·11×10−10 | 1·04 (1·01–1·07) | 1·56×10−02 |
| rs9350591 | 6 | 76298247 | FILIP1; SENP6 | T | Hip† | 0·11 | 1·20 (1·11–1·30) | 2·49×10−06 | 1·16 (1·07–1·25) | 1·64×10−04 | 1·18 (1·12–1·25) | 2·42×10−09 | 1·09 (1·04–1·13) | 2·78×10−04 |
| rs10492367 | 12 | 27906237 | KLHDC5; PTHLH | T | Hip | 0·19 | 1·18 (1·11–1·27) | 1·20×10−06 | 1·11 (1·04–1·18) | 1·18×10−03 | 1·14 (1·09–1·20) | 1·48×10−08 | 1·06 (1·03–1·10) | 9·02×10−04 |
| rs835487 | 12 | 103584897 | CHST11 | G | THR | 0·34 | 1·15 (1·08–1·22) | 3·26×10−06 | 1·11 (1·04–1·19) | 9·32×10−04 | 1·13 (1·09–1·18) | 1·64×10−08 | 1·05 (1·02–1·08) | 6·22×10−04 |
| rs12107036 | 3 | 191082854 | TP63 | G | TKR–female | 0·52 | 1·23 (1·13–1·35) | 3·03×10−06 | 1·17 (1·05–1·30) | 4·73×10−03 | 1·21 (1·13–1·29) | 6·71×10−08 | 1·05 (1·02–1·08) | 2·15×10−03 |
| rs8044769‡ | 16 | 52396636 | FTO | C | Female | 0·5 | 1·17 (1·10–1·23) | 5·98×10−08 | 1·06 (1·01–1·12) | 2·01×10−02 | 1·11 (1·07–1·15) | 6·85×10−08 | 1·07 (1·04–1·10) | 3·56×10−06 |
| rs10948172 | 6 | 44885669 | SUPT3H; CDC5L | G | Male | 0·29 | 1·17 (1·10–1·26) | 5·02×10−06 | 1·11 (1·04–1·19) | 2·48×10−03 | 1·14 (1·09–1·20) | 7·92×10−08 | 1·08 (1·05–1·12) | 6·14×10−07 |
The details of the individual discovery and replication results of analyses of all cases of osteoarthritis are shown in the appendix p 99. TJR=total joint replacement. THR=total hip replacement. TKR=total knee replacement. arcOGEN=Arthritis Research UK Osteoarthritis Genetics.
Represent the same signal, r2=1; both were prioritised and showed similar p values and effect sizes before and after replication.
Analyses in which a subset of samples with osteoarthritis at the hip and knee are also included in the arcOGEN discovery set and in the UK replication set.
This signal was attenuated after adjustment for body-mass index, suggesting that the FTO locus exerts its effect on osteoarthritis through obesity.
The most significant signal was on chromosome 3 and was followed up by two SNPs in perfect linkage disequilibrium (LD) with each other: rs11177 (allele A; figure A), a missense polymorphism within exon three of GNL3, coding for nucleostemin, and rs6976 (allele T; table 2), situated in the 3′ untranslated region (UTR) of the GLT8D1 gene. It was the only genome-wide significant signal in the discovery GWAS before replication (table 2; appendix p 99) for all osteoarthritis and TJR strata (with stronger evidence for association in the TJR strata).
Figure.
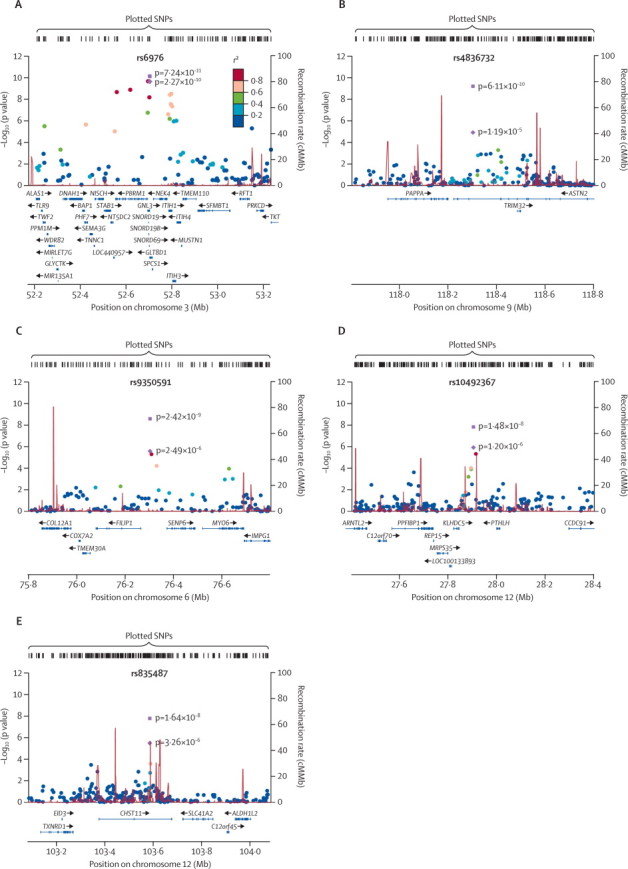
Regional association plots of replicating signals
Case–control association results (–log10 [p value]) for genotyped SNPs in the discovery set are plotted against genomic position (National Center for Biotechnology Information build 36) for the stratum in which the most significant meta-analysis p value occurred. The index SNP is denoted by a purple diamond in the discovery set and by a purple square in the final meta-analysis. The circles indicate association results of genotyped SNPs in the region; the colour reflects the correlation coefficient (r2) of each genotyped SNP with the index SNP estimated with the CEU HapMap II panel. Estimated recombination rates (in cM/Mb) are plotted in red. The region shown in the plots extends to either 500 kb upstream and downstream of the index SNP or until the next recombination hotspot if this lies further than a distance of 500 kb. (A) Chromosome 3 signal centred on rs6976 in total joint replacement. (B) Chromosome 9 signal centred on rs4836732 in female total hip replacement. (C) Chromosome 6 signal centred on rs9350591 in hip osteoarthritis. (D) Chromosome 12 signal centred on rs10492367 in hip osteoarthritis. (E) Chromosome 12 signal centred on rs835487 in total hip replacement. SNP=single nucleotide polymorphism. CEU=Utah residents with ancestry from northern and western Europe.
All the remaining four genome-wide significant signals emanated from hip-specific strata (table 2): rs4836732 (allele C; female total hip replacement [THR]) located within intron 18 of the ASTN2 gene (figure B; table 2); rs9350591 (allele T; hip osteoarthritis) located 38 kb upstream of FILIP1 and 70 kb upstream of SENP6 (figure C; table 2); rs10492367 (allele T; hip osteoarthritis) 59 kb downstream of KLHDC5 and 96 kb downstream of PTHLH (figure D; table 2); and rs835487 (allele G; THR) located within intron two of CHST11 (figure E; table 2).
Three additional signals lacked genome-wide significance and therefore need to be interpreted with caution. Rs12107036 was associated with osteoarthritis in women with total knee replacement (TKR; allele G) and is located within intron 12 of TP63 (appendix p 70; table 2). Association with rs8044769 was strongest in the female osteoarthritis stratum (allele C), within intron 1 of FTO (appendix p 71; table 2). The final signal was seen in the male osteoarthritis stratum at rs10948172 (allele G), situated in the vicinity of the SUPT3H gene (appendix p 72; table 2).
We investigated the effect of the definition of disease phenotype on the strength of association of the replicating signals (table 2) by comparing the results of analyses of TJR-only cases (severe endpoint of disease) and all cases (TJR and radiography combined) for the stratum that had the lowest meta-analysis p value. We noted that, after large-scale replication, four signals (rs6976, rs4836732, rs835487, and rs12107036) were stronger in the TJR meta-analysis, one signal (rs9350591) was stronger in the meta-analysis of TJR and radiographic cases, and three signals (rs10492367, rs8044769, and rs10948172) were little changed (appendix p 100).
In the discovery set analyses of two types of controls—population-based controls unselected for osteoarthritis (n=11 009) and osteoarthritis-free controls (n=1828)—we compared the strength of association of the three previously established osteoarthritis loci (appendix p 7, p 101) and the eight replicating signals (table 2). We noted that one signal (GDF5) was stronger in the analysis of osteoarthritis-free controls, whereas the other ten were stronger or similar in the analyses of population-based controls (appendix p 7, p 101, p 103). In the meta-analysis of discovery and replication sets, effect size estimates for the replicating loci were the same or smaller for all variants except one (CHST11) when we used disease-free controls, and all signals were statistically stronger in the analyses of population-based controls (appendix p 7, p 104).
Because obesity is an established risk factor for osteoarthritis, we investigated whether any of the 129 prioritised signals were attenuated after adjustment for body-mass index (BMI) with arcOGEN cases and disease-free TwinsUK control data (BMI data were not available for the population-based controls). Two SNPs (rs11107957 and rs2626053) showed greater than 100-times increase in their p value (ie, became less significant) after adjustment for BMI (appendix p 5, p 105). Rs804476 in FTO was the only replicating signal to be attenuated after BMI adjustment in the discovery GWAS, but we cannot exclude the possibility that BMI adjustment might have had an effect on the other variants in the replication cohorts. However, in the discovery scan, the association p value at the four remaining loci that had genome-wide significance and the three signals just below this threshold were not affected by more than one order of magnitude after adjustment for BMI.
All the studied genes were expressed within osteoarthritic or control fracture neck-of-femur joint tissues (appendix p 6, p 108).
Immunohistochemical staining showed strong nucleolar expression of nucleostemin in cytospins of primary cultured chondrocytes from human articular cartilage derived from patients with osteoarthritis (appendix p 69), and a similar pattern of staining was seen in chondrocytes from frozen sections of osteoarthritic cartilage (appendix p 69). We measured levels of nucleostemin protein expression in cultured articular chondrocytes from five controls and five patients with osteoarthritis. Nucleostemin was barely detectable in cultured chondrocytes from controls but was clearly detectable in cultured chondrocytes from patients with osteoarthritis (appendix p 69). When nucleostemin expression was corrected for expression of the housekeeping gene β-actin, the upregulation of nucleostemin protein levels in osteoarthritis chondrocytes versus control chondrocytes was significant (p=0·003; appendix p 69).
Discussion
We have identified eight novel loci that are associated with the risk of developing osteoarthritis, five of which surpassed genome-wide significance and three that were just less than the threshold (panel). With the addition of previously established associations on chromosome 7q22, and in the GDF5 and MCF2L genes,6,7,9,11 the current total of osteoarthritis susceptibility loci in European populations is 11.
Panel. Research in context.
Systematic review
We searched PubMed for articles in English with a combination of the terms “osteoarthritis” and “genetic” to identify reports of loci encoding susceptibility for osteoarthritis. We then focused on those in which genetic associations had been identified with a compelling level of significance (p≤5×10−8) and with replication. This gave three loci: GDF5, 7q22, and MCF2L. These formed the basis by which we put our GWAS results into context. In our search we also took advantage of the extensive knowledge of several of the arcOGEN investigators who have published many reviews about the genetics of osteoarthritis.
Interpretation
We identified eight novel genetic risk loci for osteoarthritis. Two are close to functional candidate genes that suggest clinical implications for osteoarthritis. CHST11 codes for an enzyme that modulates cartilage proteoglycan, with proteoglycan modulation being an active area of osteoarthritis therapeutic development; nutraceutical compounds, such as chondroitin sulfate, have been extensively studied. Although the evidence of efficacy of such compounds in relieving signs and symptoms of osteoarthritis is weak, our results suggest that alternate therapeutic approaches acting on this same pathway could be clinically beneficial. Novel anabolic treatments for osteoporosis have been developed with peptide fragments based on parathyroid hormone, and our results suggest that investigating these compounds in the treatment of osteoarthritis, or assessing endpoints of osteoarthritis in previous clinical trials of parathyroid hormone, would now be sensible. Furthermore, the association with FTO draws attention to the interplay between bodyweight and osteoarthritis, and emphasises existing clinical advice that loss of excess bodyweight is a clinical recommendation for symptom relief and avoidance of osteoarthritis. Our demonstration that osteoarthritis risk loci are on the whole specific to either the hip or the knee informs the clinician that the pathophysiological process of this chronic disease is likely to have joint-specific components that need to be borne in mind when developing treatment regimens.
Our most significant finding was in a gene-rich region of chromosome 3p21·1. Rs6976 lies within the 3′ UTR of GLT8D1 (glycosyltransferase 8 domain containing 1; figure A). Alterations in the expression of GLT8D1 could affect the glycosylation of cartilage proteins. Rs11177, in perfect LD with rs6976, encodes a missense variant (Arg→Gln) in the third exon of GNL3 (guanine nucleotide binding protein-like 3, or nucleostemin; figure A). GNL3 is expressed in mesenchymal stem cells, from which chondrocytes are derived, and regulates the G1–S phase transition in stem cells.13–15 The function of GNL3 in bone and cartilage is not known because mice homozygous for deletion of GNL3 die on embryonic day 4.16 However, of interest is that nucleostemin protein levels were substantially increased in cultured chondrocytes from patients with osteoarthritis compared with controls, raising the possibility that this gene might be functionally important in the pathogenesis of osteoarthritis.
Rs4836732 lies within the ASTN2 gene (astrotactin 2; figure B), which is highly expressed in the developing and adult brain. ASTN2 is a membrane protein that regulates surface levels of ASTN1 during neuronal migration.17 Rs9350591 lies between the FILIP1 (filamin A interacting protein 1) and SENP6 (sentrin specific peptidase 6) genes (figure C). COL12A1 (collagen, type XII, alpha 1), the product of which is found in articular cartilage,18 lies at a distance of about 326 kb. Rs10492367 is located between the KLHDC5 (Kelch domain containing 5) and PTHLH (parathyroid hormone-like hormone) genes (figure D). Parathyroid hormone-related protein (PTHrP) regulates endochondral bone development. Results from studies of Pthrp–/– mice showed that animals who survived gestation had accelerated differentiation of chondrocytes in bone.19
Rs835487 is located in the CHST11 (carbohydrate sulfotransferase 11) gene (figure E), which encodes a Golgi enzyme that catalyses the transfer of sulphate groups to the 4-O position of chondroitin and dermatan sulphate, important components of cartilage proteoglycans.20 CHST11 has a role in skeletal development, signalling pathways, and cancer progression.21 It has substantially higher expression in osteoarthritic cartilage than in normal articular cartilage22 and mice that are null for this gene have a disorganised growth plate, accelerated chondrocyte differentiation, altered patterns of signalling with transforming growth factor β and bone morphogenetic protein, and a fibrillated cartilage extracellular matrix that is characteristic of osteoarthritic cartilage.23 Chondroitin sulfate is used as a symptomatic, slow-acting drug for osteoarthritis, recommended by the latest Osteoarthritis Research Society International treatment guidelines, but evidence for its effectiveness remains controversial.24
Rs12107036 lies within intron 12 of the TP63 gene (encoding tumour protein p63; appendix p 70). p63 null mice have serious defects in their limb, craniofacial, and epithelial development.25,26 Rs8044769 lies within FTO (appendix p 71), a well established locus that is associated with fat mass and obesity27 and is in partial LD (r2>0·6) with the reported index BMI-associated SNPs. This signal was attenuated after adjustment for BMI, suggesting that the FTO gene exerts its effect on osteoarthritis through obesity. Rs10948172 occurs between CDC5L (CDC5 cell division cycle 5-like) and SUPT3H genes (suppressor of Ty3 homologue; appendix p 72). At a distance of 500 kb, but possibly containing SNPs correlated with rs10948172, is the RUNX2 (runt-related transcription factor 2) gene, which encodes a protein that is essential for osteoblast differentiation28 and skeletal morphogenesis.29 RUNX2 was identified in peripheral blood expression profiles as a possible biomarker for bone metabolism in arthritis.30
The results of our study draw attention to three important issues with respect to design and analysis that are of relevance to GWAS investigations of complex disease: definition of significant association in view of several correlated stratified analyses; effect of phenotype homogeneity and misclassification; and allowing for power constraints when seeking replication of previously identified loci. First, we analysed several specific non-independent phenotypes in addition to the main analysis of all osteoarthritis cases versus controls so as to increase power to detect signals specific for joint, sex, or disease severity. Compelling evidence suggests that joint-specific genetic factors play a part in the pathogenesis of osteoarthritis31 and that the familial concordance for hip and knee osteoarthritis is greater in surgically defined than in radiographically defined disease.32–34 The results of these reports, in addition to the significant differences reported in the prevalence of osteoarthritis between skeletal sites, the sexes, and radiographic versus symptomatic disease,35 underpin our decision to stratify our data by site of osteoarthritis (hip or knee), sex, sex with site, and the two modes of osteoarthritis definition (radiographic and joint replacement) that we have used. That most of the loci we have identified are associated with osteoarthritis at a particular joint site provides genetic evidence that the aetiopathogenesis of osteoarthritis is not uniform, but that joint-specific risk factors are in operation. A conservative Bonferroni correction, assuming the independence of these highly correlated specific phenotypes, would artificially move the p-value threshold of declaring genome-wide significant association to 2·08×10−9.36 In this work, we draw attention to eight novel associations with osteoarthritis based on a combination of statistical evidence of association, corroborative evidence from large-scale replication, and biological insights afforded by the replicating loci that we noted (eg, PTHLH and CHST11, both excellent biological candidates, would not have surpassed the more stringent threshold).
Second, we accrue evidence that, in general, analyses of patients ascertained by the more stringent criterion of arthroplasty have power advantages over those including radiographically diagnosed cases, emphasising the value of patient phenotype homogeneity (appendix p 100). All eight loci reported in this study were prioritised on the basis of analyses of osteoarthritis-status-agnostic controls, indicating that the large increase in sample size afforded by the use of population-based controls might have power advantages despite potential misclassification (appendix pp 101–04).
Third, we assessed evidence for association at the three previously reported osteoarthritis loci in our discovery GWAS. We detected signals with p=0·002 to p=6×10−4 for the chromosome 7 and MCF2L loci, but little evidence for association at GDF5. These results are in keeping with findings in other complex disease discovery GWAS37 and can be ascribed to a combination of power, stochastic variation in allele frequency estimates, and the absence of the index variant from the genotyping platform (appendix p 7, p 101).
We have studied common variants and therefore the findings of our report cannot be used to estimate the effect that rarer variants might have on causing osteoarthritis. Further, although our discovery sample size of 7410 cases is the largest yet used for osteoarthritis, the sample was stratified; an analysis of larger stratified subsets might show additional associated loci. In our study, we focused on Europeans and it will be of great interest to assess whether these loci have global relevance to osteoarthritis risk through similar studies of non-European cohorts with osteoarthritis.
All eight identified loci are represented by common SNPs (minor allelic frequency 0·11–0·50) and have small effect sizes (allelic ORs from 1·11 to 1·21). In keeping with other common complex trait studies, these findings indicate that common SNPs with large effect sizes are not likely to have a role in the pathogenesis of osteoarthritis. Association at the index SNPs reported here does not imply causality. Functional studies will be necessary to pinpoint the precise functional variants. The eight signals identified in this study have a combined sibling recurrence risk (λs) estimate of 1·028 and account for only a small fraction of familial clustering of osteoarthritis. This outcome leads us to advocate larger-scale sample sizes, typically achieved through GWAS meta-analysis efforts in complex traits. The arcOGEN GWAS is the largest study of osteoarthritis genetics so far and has sufficient power to detect slight effects at common loci at the genome-wide significance level (eg, 80% power to detect an allele with frequency 0·35 and allelic OR of 1·15). We have established novel loci represented by common SNPs that confer a slight risk for osteoarthritis and are associated with the clinically important phenotype of TJR. These results provide a basis for functional studies to identify the underlying causative variants, biological networks, and molecular cause of osteoarthritis.
Correspondence to: Dr Eleftheria Zeggini, Wellcome Trust Sanger Institute, Morgan Building, Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1HH, UK eleftheria@sanger.ac.uk
Prof John Loughlin, Newcastle University, Institute of Cellular Medicine, 4th Floor Catherine Cookson Building, Newcastle University, Medical School, Framlington Place, Newcastle upon Tyne NE2 4HH, UK john.loughlin@ncl.ac.uk
Acknowledgments
Acknowledgments
arcOGEN was funded by a special purpose grant from Arthritis Research UK (grant 18030). A full list of acknowledgements and funding sources are provided in the appendix p 9.
Contributors
EZ, KP, LS, GAW, JMW, and JL participated in the writing group. EZ, KC, PD, WERO, GAW, NiA, AC, MD, AMcC, JMW, SHR, AMV, TDS, and JL participated in the arcOGEN study design. EZ, KP, LS, NWR, AGD-W, MCL, VB, CB, KSE, SYS, VKY, GZ, MM, and SaM participated in data analysis and interpretation. NiA, AC, MD, AMcC, JMW, SHR, TDS, JL, FB, AR, EA, NaA, P-KB, IC, SD, AG, JJ, RK, NCK, ShM, FON, EP, MRR, DiS, KW, BW, and MW participated in arcOGEN patient and data collection. WERO, KSh, and KD participated in sample DNA processing. SuB and PD participated in genotyping for the arcOGEN study. HB, SEH, and SCP provided informatics support for the arcOGEN study. TI, IJ, HJ, GT, KSt, and US participated in data replication from the deCODE study. TE and AM participated in data replication from the EGCUT study. JJH-D, MK, StB, PES, and IM participated in data replication from the GARP study. AH, HJMK, FR, AGU, and JBvM participated in data replication from the Rotterdam study. EE, MM, SaM, JPAI, AMV, and TDS participated in data replication from the TwinsUK study. SHR, EVAR, MaR, MiR, and DoS participated in functional studies.
arcOGEN Consortium and arcOGEN Collaborators
Eleftheria Zeggini*†, Kalliope Panoutsopoulou*, Lorraine Southam*, Nigel W Rayner, Aaron G Day-Williams, Margarida C Lopes, Vesna Boraska, Tonu Esko, Evangelos Evangelou, Albert Hofman, Jeanine J Houwing-Duistermaat, Thorvaldur Ingvarsson, Ingileif Jonsdottir, Helgi Jonsson, Hanneke J M Kerkhof, Margreet Kloppenburg, Steffan D Bos, Massimo Mangino, Sarah Metrustry, P Eline Slagboom, Gudmar Thorleifsson, Emma V A Raine, Madhushika Ratnayake, Michelle Ricketts, Claude Beazley, Hannah Blackburn, Suzannah Bumpstead, Katherine S Elliott, Sarah E Hunt, Simon C Potter, So-Youn Shin, Vijay K Yadav, Guangju Zhai, Kate Sherburn, Kate Dixon, Elizabeth Arden, Nadim Aslam, Phillippa-Kate Battley, Ian Carluke, Sally Doherty, Andrew Gordon, John Joseph, Richard Keen, Nicola C Koller, Sheryl Mitchell, Fiona O'Neill, Ellen Paling, Mike R Reed, Fernando Rivadeneira, Diane Swift, Kirsten Walker, Bridget Watkins, Maggie Wheeler, Fraser Birrell, John P A Ioannidis, Ingrid Meulenbelt, Andres Metspalu, Ashok Rai, Donald Salter, Kari Stefansson, Unnur Styrkarsdottir, André G Uitterlinden, Joyce B J van Meurs, arcOGEN Consortium: Kay Chapman, Panos Deloukas, William E R Ollier, Gillian A Wallis, Nigel Arden, Andrew Carr, Michael Doherty, Andrew McCaskie, J Mark Wilkinson, Stuart H Ralston, Ana M Valdes, Tim D Spector, John Loughlin†. *Joint first authors. †Joint corresponding authors.
arcOGEN Investigators
Sanger Institute, Hinxton, UK (E Zeggini PhD, K Panoutsopoulou PhD, L Southam BSc, A G Day-Williams PhD, M C Lopes PhD, V Boraska PhD, C Beazley PhD, H Blackburn MSc, S Bumpstead BSc, S E Hunt BSc, S C Potter PhD, S-Y Shin PhD, V K Yadav PhD, P Deloukas PhD); Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford, UK (L Southam BSc, N W Rayner PhD, M Lopes PhD, K S Elliott PhD); Oxford Centre for Diabetes, Endocrinology and Metabolism, University of Oxford, Oxford, UK (N W Rayner PhD); University of Split School of Medicine, Split, Croatia (V Boraska PhD); Estonian Genome Centre and Centre of Translational Genomics, University of Tartu, Tartu, Estonia (T Esko MSc, Prof A Metspalu PhD); Institute of Molecular and Cell Biology, University of Tartu, Tartu, Estonia (T Esko MSc, Prof A Metspalu MD); Estonian Biocenter, Tartu, Estonia (T Esko MSc, Prof A Metspalu MD); University of Ioannina School of Medicine, Ioannina, Greece (E Evangelou PhD, Prof J P A Ioannidis MD); Erasmus Medical Centre, Rotterdam, Netherlands (Prof A Hofman MD, H J M Kerkhof MD, F Rivadeneira MD, Prof A G Uitterlinden PhD, J B J van Meurs PhD); Leiden University Medical Centre, Leiden, Netherlands (Prof J J Houwing-Duistermaat PhD, M Kloppenburg MD, S D Bos PhD, Prof P E Slagboom PhD, I Meulenbelt PhD); University of Akureyri, Akureyri, Iceland (Prof T Ingvarsson MD); University of Iceland, Reykjavik, Iceland (Prof T Ingvarsson MD, Prof I Jonsdottir FilD, Prof H Jonsson MD, Prof K Stefansson MD); deCODE Genetics, Reykjavik, Iceland (Prof I Jonsdottir, G Thorleifsson PhD, Prof K Stefansson MD, U Styrkarsdottir PhD); King's College London, London, UK (M Mangino PhD, S Metrustry MSc, G Zhai PhD, F O'Neill MSc, A M Valdes PhD, Prof T D Spector MD); Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, UK (E V A Raine MSc, M Ratnayake BSc, F Birrell PhD, Prof A McCaskie MBChB, Prof John Loughlin PhD); Institute of Genetics and Molecular Medicine, University of Edinburgh, Edinburgh, UK (M Ricketts BSc, N C Koller RN, Prof D Salter MD, Prof S H Ralston MD); Memorial University of Newfoundland, St John's, NF, Canada (G Zhai PhD); Centre for Integrated Genomic Medical Research, University of Manchester, Manchester, UK (K Sherburn BSc, K Dixon PhD, Prof W E R Ollier PhD); Wellcome Trust Clinical Research Facility, Southampton General Hospital, Southampton, UK (E Arden RGN, P-K Battley DipHE); Worcestershire Acute Hospitals NHS Trust, Worcester, UK (N Aslam FRCSOrth, J Joseph DipHE, A Rai MBChB); Wansbeck General Hospital, Ashington, UK (I Carluke MBChB, M R Reed MD, K Walker RN, F Birrell MBBChir); Academic Rheumatology, University of Nottingham, Nottingham, UK (S Doherty SRN, M Wheeler BA, Prof M Doherty MD); Academic Unit of Bone Metabolism, Department of Human Metabolism, University of Sheffield, Sheffield, UK (A Gordon PhD, E Paling BMedSci, D Swift BA, J M Wilkinson PhD); Royal National Orthopaedic Hospital, Stanmore, UK (R Keen MD); Freeman Hospital, Newcastle upon Tyne, UK (S Mitchell BSc, Prof A McCaskie MBChB); Sheffield National Institute for Health Research Musculoskeletal Biomedical Research Unit, Northern General Hospital, Sheffield, UK (E Paling BMedSci, D Swift BA, J M Wilkinson PhD); Nuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Sciences, University of Oxford, Oxford, UK (B Watkins RN, K Chapman PhD, Prof N Arden MD, Prof A Carr FMedSci); Tufts Clinical and Translational Science Institute and Tufts University School of Medicine, Boston, USA (Prof J P A Ioannidis MD); Stanford Prevention Research Centre, Stanford University School of Medicine, Stanford, USA (Prof J P A Ioannidis MD); Wellcome Trust Centre for Cell-Matrix Research, University of Manchester, Manchester, UK (Prof G A Wallis PhD); Medical Research Council Epidemiology Resource Centre, University of Southampton, Southampton, UK (Prof N Arden MD).
Conflicts of interest
IJ, GT, KSt, and US are employed by deCODE. The other authors declare that they have no conflicts of interest.
Supplementary Material
References
- 1.Dieppe PA, Lohmander LS. Pathogenesis and management of pain in osteoarthritis. Lancet. 2005;365:965–973. doi: 10.1016/S0140-6736(05)71086-2. [DOI] [PubMed] [Google Scholar]
- 2.Valdes AM, Spector TD. Genetic epidemiology of hip and knee osteoarthritis. Nat Rev Rheumatol. 2011;7:23–32. doi: 10.1038/nrrheum.2010.191. [DOI] [PubMed] [Google Scholar]
- 3.Loughlin J. The genetic epidemiology of human primary osteoarthritis: current status. Expert Rev Mol Med. 2005;7:1–12. doi: 10.1017/S1462399405009257. [DOI] [PubMed] [Google Scholar]
- 4.Valdes AM, McWilliams D, Arden NK. Involvement of different risk factors in clinically severe large joint osteoarthritis according to the presence of hand interphalangeal nodes. Arthritis Rheum. 2010;62:2688–2695. doi: 10.1002/art.27574. [DOI] [PubMed] [Google Scholar]
- 5.Chapman K, Takahashi A, Meulenbelt I. A meta-analysis of European and Asian cohorts reveals a global role of a functional SNP in the 5′ UTR of GDF5 with osteoarthritis susceptibility. Hum Mol Genet. 2008;17:1497–1504. doi: 10.1093/hmg/ddn038. [DOI] [PubMed] [Google Scholar]
- 6.Day-Williams AG, Southam L, Panoutsopoulou K. A variant in MCF2L is associated with osteoarthritis. Am J Hum Genet. 2011;89:446–450. doi: 10.1016/j.ajhg.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evangelou E, Chapman K, Meulenbelt I. Large-scale analysis of association between GDF5 and FRZB variants and osteoarthritis of the hip, knee, and hand. Arthritis Rheum. 2009;60:1710–1721. doi: 10.1002/art.24524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evangelou E, Valdes AM, Kerkhof HJ. Meta-analysis of genome-wide association studies confirms a susceptibility locus for knee osteoarthritis on chromosome 7q22. Ann Rheum Dis. 2011;70:349–355. doi: 10.1136/ard.2010.132787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kerkhof HJ, Lories RJ, Meulenbelt I. A genome-wide association study identifies an osteoarthritis susceptibility locus on chromosome 7q22. Arthritis Rheum. 2010;62:499–510. doi: 10.1002/art.27184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miyamoto Y, Mabuchi A, Shi D. A functional polymorphism in the 5′ UTR of GDF5 is associated with susceptibility to osteoarthritis. Nat Genet. 2007;39:529–533. doi: 10.1038/2005. [DOI] [PubMed] [Google Scholar]
- 11.Valdes AM, Evangelou E, Kerkhof HJ. The GDF5 rs143383 polymorphism is associated with osteoarthritis of the knee with genome-wide statistical significance. Ann Rheum Dis. 2011;70:873–875. doi: 10.1136/ard.2010.134155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Panoutsopoulou K, Southam L, Elliott KS. Insights into the genetic architecture of osteoarthritis from stage 1 of the arcOGEN study. Ann Rheum Dis. 2011;70:864–867. doi: 10.1136/ard.2010.141473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baddoo M, Hill K, Wilkinson R. Characterization of mesenchymal stem cells isolated from murine bone marrow by negative selection. J Cell Biochem. 2003;89:1235–1249. doi: 10.1002/jcb.10594. [DOI] [PubMed] [Google Scholar]
- 14.Han C, Zhang X, Xu W, Wang W, Qian H, Chen Y. Cloning of the nucleostemin gene and its function in transforming human embryonic bone marrow mesenchymal stem cells into F6 tumor cells. Int J Mol Med. 2005;16:205–213. [PubMed] [Google Scholar]
- 15.Ma H, Pederson T. Nucleostemin: a multiplex regulator of cell-cycle progression. Trends Cell Biol. 2008;18:575–579. doi: 10.1016/j.tcb.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 16.Beekman C, Nichane M, De Clercq S. Evolutionarily conserved role of nucleostemin: controlling proliferation of stem/progenitor cells during early vertebrate development. Mol Cell Biol. 2006;26:9291–9301. doi: 10.1128/MCB.01183-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson PM, Fryer RH, Fang Y, Hatten ME. Astn2, a novel member of the astrotactin gene family, regulates the trafficking of ASTN1 during glial-guided neuronal migration. J Neurosci. 2010;30:8529–8540. doi: 10.1523/JNEUROSCI.0032-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gregory KE, Keene DR, Tufa SF, Lunstrum GP, Morris NP. Developmental distribution of collagen type XII in cartilage: association with articular cartilage and the growth plate. J Bone Miner Res. 2001;16:2005–2016. doi: 10.1359/jbmr.2001.16.11.2005. [DOI] [PubMed] [Google Scholar]
- 19.Lanske B, Karaplis AC, Lee K. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science. 1996;273:663–666. doi: 10.1126/science.273.5275.663. [DOI] [PubMed] [Google Scholar]
- 20.Heinegard D. Proteoglycans and more—from molecules to biology. Int J Exp Pathol. 2009;90:575–586. doi: 10.1111/j.1365-2613.2009.00695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kluppel M. The roles of chondroitin-4-sulfotransferase-1 in development and disease. Prog Mol Biol Transl Sci. 2010;93:113–132. doi: 10.1016/S1877-1173(10)93006-8. [DOI] [PubMed] [Google Scholar]
- 22.Karlsson C, Dehne T, Lindahl A. Genome-wide expression profiling reveals new candidate genes associated with osteoarthritis. Osteoarthritis Cartilage. 2010;18:581–592. doi: 10.1016/j.joca.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 23.Kluppel M, Wight TN, Chan C, Hinek A, Wrana JL. Maintenance of chondroitin sulfation balance by chondroitin-4-sulfotransferase 1 is required for chondrocyte development and growth factor signaling during cartilage morphogenesis. Development. 2005;132:3989–4003. doi: 10.1242/dev.01948. [DOI] [PubMed] [Google Scholar]
- 24.Zhang W, Nuki G, Moskowitz RW. OARSI recommendations for the management of hip and knee osteoarthritis: part III: Changes in evidence following systematic cumulative update of research published through January 2009. Osteoarthritis Cartilage. 2010;18:476–499. doi: 10.1016/j.joca.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 25.Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature. 1999;398:708–713. doi: 10.1038/19531. [DOI] [PubMed] [Google Scholar]
- 26.Yang A, Schweitzer R, Sun D. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398:714–718. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- 27.Frayling TM, Timpson NJ, Weedon MN. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 29.Stein GS, Lian JB, van Wijnen AJ. Runx2 control of organization, assembly and activity of the regulatory machinery for skeletal gene expression. Oncogene. 2004;23:4315–4329. doi: 10.1038/sj.onc.1207676. [DOI] [PubMed] [Google Scholar]
- 30.Grcevic D, Jajic Z, Kovacic N. Peripheral blood expression profiles of bone morphogenetic proteins, tumor necrosis factor-superfamily molecules, and transcription factor Runx2 could be used as markers of the form of arthritis, disease activity, and therapeutic responsiveness. J Rheumatol. 2010;37:246–256. doi: 10.3899/jrheum.090167. [DOI] [PubMed] [Google Scholar]
- 31.MacGregor AJ, Li Q, Spector TD, Williams FM. The genetic influence on radiographic osteoarthritis is site specific at the hand, hip and knee. Rheumatology (Oxford) 2009;48:277–280. doi: 10.1093/rheumatology/ken475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chitnavis J, Sinsheimer JS, Clipsham K. Genetic influences in end-stage osteoarthritis. Sibling risks of hip and knee replacement for idiopathic osteoarthritis. J Bone Joint Surg Br. 1997;79:660–664. doi: 10.1302/0301-620x.79b4.7437. [DOI] [PubMed] [Google Scholar]
- 33.Lanyon P, Muir K, Doherty S, Doherty M. Assessment of a genetic contribution to osteoarthritis of the hip: sibling study. BMJ. 2000;321:1179–1183. doi: 10.1136/bmj.321.7270.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neame RL, Muir K, Doherty S, Doherty M. Genetic risk of knee osteoarthritis: a sibling study. Ann Rheum Dis. 2004;63:1022–1027. doi: 10.1136/ard.2003.014498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pereira D, Peleteiro B, Araujo J, Branco J, Santos RA, Ramos E. The effect of osteoarthritis definition on prevalence and incidence estimates: a systematic review. Osteoarthritis Cartilage. 2011;19:1270–1285. doi: 10.1016/j.joca.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 36.Conneely KN, Boehnke M. So many correlated tests, so little time! Rapid adjustment of p values for multiple correlated tests. Am J Hum Genet. 2007;81:1158–1168. doi: 10.1086/522036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wellcome Trust Case Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.