

Abstract
Tumors of the gastrointestinal tract are among the most frequent human malignancies and account for approximately 30% of cancer-related deaths worldwide. Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors that control diverse cellular functions such as proliferation, differentiation, and cell death. Owing to their involvement in so many processes, they play crucial roles also in the development and physiology of the gastrointestinal tract. Consistently, PPARs deregulation has been implicated in several pathophysiological conditions, including chronic inflammation and cancer development. This paper summarizes the current knowledge on the role that the various PPAR isoforms play in the pathogenesis of the esophageal, gastric, and intestinal cancer. Elucidation of the molecular mechanisms underlying PPARs' signaling pathways will provide insights into their possible use as predictive biomarkers in the initial stages of the process. In addition, this understanding will provide the basis for new molecular targets in cancer therapy and chemoprevention.
1. Introduction
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors belonging to the nuclear hormone receptor superfamily. Three subtypes, PPARα, PPARβ/δ, and PPARγ, have been identified so far. PPARα is expressed in the liver, kidney, small intestine, heart, and muscle, where it activates fatty acid catabolism and controls lipoprotein assembly in response to long-chain unsaturated fatty acids, eicosanoids, and hypolipidemic drugs (e.g., fenofibrate) [1, 2]. PPARβ/δ is more broadly expressed and is implicated in fatty acid oxidation, keratinocyte differentiation, wound healing, and macrophage response to VLDL metabolism. This isoform has been implicated in transcriptional-repression functions and has been shown to repress the activity of PPARα or PPARγ target genes [2–7]. PPARγ1 and γ2 are generated from a single-gene PPARG by differential promoter usage and alternative splicing [8–12]. PPARγ1 is expressed in colon, immune system (e.g., monocytes and macrophages), and other tissues where it participates in the modulation of inflammation, cell proliferation, and differentiation. PPARγ2 contains 28 additional amino acids at the N-terminus, as compared to PPARγ1, and is expressed in adipose tissue where it plays a pivotal role in adipocyte differentiation, lipid storage, and energy dissipation [12]. PPARG3 and PPARG4 are splicing variants of PPARG1 mRNA and give rise to the same PPARγ1 protein [8, 9, 12]. Since PPARγ is also involved in glucose metabolism improving insulin sensitivity, selective ligands such as the thiazolidinediones (TZD) are used as insulin-sensitizing drugs in type 2 diabetes [2, 4, 5].
As all nuclear receptors, PPARs share a modular structure with four distinct domains [13, 14]. The A/B domain at the N-terminus is the key determinant of isotype-selective gene function and harbors a ligand-independent transcriptional activating function (AF-1) motif. The C domain is the DNA binding domain, with the typical two-zinc-finger structure with which the receptor binds the major groove of the double helix DNA of the peroxisome proliferator response elements (PPREs). They are formed by direct repeats (DRs) of the core sequence AGG(A/T)CA, separated by one or two nucleotides (DR1 and DR2, resp.). The D domain or hinge region allows receptor dimerization and DNA binding. The E/F domain contains the ligand-binding domain (LBD), a large binding pocket in which a variety of natural and synthetic ligands, such as fatty acids, eicosanoids, linoleic acid derivatives, as well as oxidized and nitrated fatty acids, accommodate. In addition, this domain exhibits the ligand-dependent transcriptional-activating function (AF2) motif on the C-terminus helix 12 [13]. Both the D and E/F domains are required to the dimerization with the 9-cis retinoic acid receptor (RXR) with which PPARs form permissive heterodimers bound to their cognate PPREs. Several genes involved in lipid metabolism and energy homeostasis, as well as genes modulating cell proliferation, differentiation, and survival, have functional PPREs in their regulatory regions [1, 2, 13, 15].
PPARs regulate gene expression through distinct mechanisms: ligand-dependent transactivation, ligand-independent repression, and ligand-dependent transrepression (Figure 1) [16, 17]. Ligand-dependent transactivation is considered the “classical mode of action” of PPARs: upon ligand binding, the helix 12 of the LBD folds back exposing the AF2 motif that governs the recruitment of transcriptional coactivators. These, in turn, facilitate the assembly of the general transcriptional machinery at PPRE-containing promoters [16, 17]. In the absence of ligand, PPARs repress transcription of target genes by recruiting corepressor complexes (e.g., NCoR and SMRT). Finally, recent studies have disclosed an additional “nongenomic” mode of action defined “transrepression” that involves gene repression in a ligand-dependent manner through protein-protein interactions with NFκB, AP1, Smads, STATs, and NFATs [17–19]. In contrast to transcriptional activation and repression, transrepression does not involve binding to PPREs but is attained through the recruitment and stabilization of the corepressor complexes on the promoters of pro-inflammatory genes. This mechanism might explain the anti-inflammatory properties of PPARs [17–20].
Figure 1.
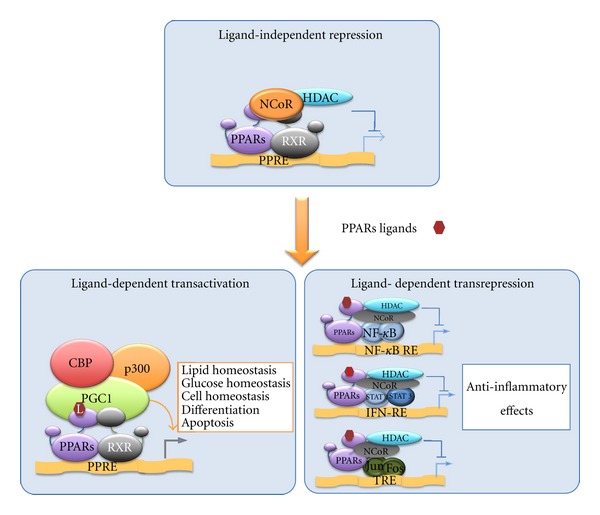
PPARs-mediated mechanisms of transcriptional regulation. In the absence of ligands, PPARs bind the promoters of their target genes and repress transcription by recruiting the corepressor complex. In the presence of ligands, PPARs can induce either ligand-dependent transactivation or transrepression. Transactivation involves PPARs heterodimerization with the retinoid X receptors (RXRs) followed by recognition of specific PPAR response elements (PPREs) and interaction with coactivators. Transrepression involves interference with other signal transduction pathways, including NFκB, STAT, and AP1. NFκB-RE: NFκB response element; IFN-RE = “interferon-stimulated gene factor” responsive element; TRE = O-tetradecanoylphorbol 13-acetate-responsive element.
2. Gastrointestinal Cancers
The inner lining of the digestive tract is composed by high-proliferating cells located at the basis of the epithelium and by differentiated cells undergoing continuous replacement. The rapid cell turnover required for the maintenance of the mucosa homeostasis and the response to an adverse environment, such as toxins and carcinogens present in digested foods, makes the digestive tract a common site of cancer development in humans. In particular, esophagus, stomach, and colon are at high risk of developing cancer: indeed esophageal (EC), gastric (GC), and colorectal cancers (CRC) represent very common malignancies and account for approximately 30% of cancer-related deaths worldwide [21].
Esophageal cancer (EC) is the sixth most common cause of cancer-related death worldwide. Barrett's esophagus is the premalignant condition that appears to predispose to the adenocarcinoma trough a metaplasia-dysplasia-carcinoma sequence. The molecular mechanisms underlying the events leading to the conversion of the normal squamous epithelium to a metaplastic columnar epithelium are poorly understood. However, chronic activation of NFκB, together with the increase of COX-2 and gastrin expression due to gastroesophageal reflux could be responsible for chronic inflammation-related cancer promotion [22, 23].
Gastric cancer (GC) remains the second leading cause of cancer-related death worldwide. More than 90% of these tumors are adenocarcinomas originating from the glandular epithelium of the gastric mucosa [21]. Also in this case, inflammation plays a pivotal role in tumor development. In particular, H. pylori infection is the major causative agent of chronic gastritis and gastrointestinal metaplasia characterized by infiltration of inflammatory cells, enhanced expression of chemokines, NFκB activation, COX-2 overexpression, and upregulation of Wnt signaling pathway leading to aberrant cell proliferation, excessive angiogenesis, and inhibition of apoptosis [24].
Colorectal cancer (CRC) is the third most common cancer in men and the second in women [21, 25]. In the vast majority of cases, CRC occurs sporadically and only in 5–10% is due to inherited mutations [25]. The risk of CRC development increases significantly in people with inflammatory bowel diseases (IBDs), such as ulcerative colitis (UC) and Crohn's disease (CD). Chronic inflammation processes induce development of colitis-associated cancers (CAC) generally initiated by mutations in TP53 or by COX2 overexpression and followed by APC inactivation at later stages [26].
Although the genetic and epigenetic alterations responsible for the different gastrointestinal (GI) cancers are still unknown, a pivotal role of inflammation in their pathogenesis is emerged. In particular, COX2 overexpression contributes to this process inhibiting apoptosis and promoting angiogenesis and invasiveness of tumor cells. Concordantly, epidemiologic studies have demonstrated that the long-term and regular use of nonsteroidal anti-inflammatory drugs (NSAIDs, COX2 inhibitors) reduces the mortality from digestive tract malignancies [24, 25].
In search of new strategies for the treatment of GI cancers, PPARs have attracted increasing attention mainly because of their anti-inflammatory effects, accompanied by prodifferentiation and proapoptotic functions [23–26]. PPARs are heterogeneously expressed along in GI tract and their role in the pathophysiology of these neoplasms is beginning to emerge (Figure 2) [27–29].
Figure 2.
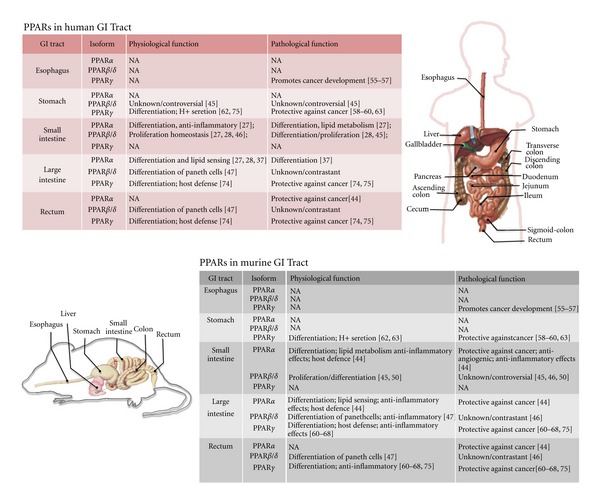
Physiological and pathological functions of the three PPAR isoforms in the human and murine GI tract.
3. PPARα
PPARα is mainly expressed in the mucosa of the small and large intestine where “senses” the total flux of dietary fatty acids delivered [27, 28]. In these contexts, PPARα regulates genes involved in lipid metabolism, inflammation, cell cycle progression, and angiogenesis [30–34]. Given its role in these latter processes, PPARα has been suggested to contribute to tumor formation and/or progression. To date, no data are available on its involvement in GC and EC, whereas its role in CRC has been investigated both in vitro and in vivo. PPARα is correlated with a reduced expression of MYC-related genes, such as cyclin D1, caspase3, NFκB, STAT1, and EGFR. PPARα activation inhibits capillary tube formation in vitro and angiogenesis in vivo through a mechanism that involves deconstruction of the cytoskeleton, reduction of bFGF-induced Akt activity and COX-2 expression. PPARα also reduces the neovascularization, modulating the expression of VEGF, FGFs, members of the arachidonic acid P450 mono-oxygenases, thrombospondin, and endostatin [31–35]. In vitro PPARα induces apoptosis through modulation of Bcl-2 and Bad proteins [35, 36]. PPARα ligands, in addition, downregulate oncogenes and upregulate antiproliferative genes, supporting a tumor suppressor role [37]. In CRC cell lines, PPARα is modulated by the activation of the MAPK pathways; specifically, phosphorylation of specific amino acid residues located at the PPARα N-terminus region by JNK and p38 MAPK enhances its ligand-dependent transcriptional activity. This, in turn, promotes apoptosis, differentiation, and anti-inflammatory effects mainly through inhibition of iNOS, COX-2, and TNF-α. On the contrary, activation of ERK-MAPK signaling pathway reduces PPARα activity [38, 39]. Growing evidence obtained in animal models suggests that PPARα has anti-inflammatory effects in vivo but the precise and direct role it plays in intestinal inflammation is not fully elucidated. The data indicate that PPARα has anti-inflammatory effects in a mouse model of chemically induced colitis; PPARα-deficient mice exhibit enhanced inflammation; exposure to PPARα ligands controls colonic inflammation through inhibition of proinflammatory cytokines. Collectively, the evidence supports that PPARα activation leads to mitigation of IBD progression [40–42]. Unfortunately, the precise and correct assessment of PPARα function in CRC is made even more complicated by species-specific differences. The data obtained from mice models indicate that PPARα ligands play a potential role in suppressing polyp formation in Apc-deficient mice, an animal model corresponding to human familial adenomatous polyposis [43]. A significant reduction in PPARα expression is detected in human CRC specimens and UC patients' mucosa, suggesting PPARα as a therapeutic target to prevent adenoma formation also in IBD-induced cancer formation [36, 41, 44]. Thus, in CRC PPARα seems to act as a tumor suppressor with antiangiogenic, anti-inflammatory, and, ultimately, antitumor activities.
4. PPARβ/δ
To date, no studies have demonstrated a role for PPARβ/δ in the esophageal epithelium. In gastric epithelium, it is highly expressed but whether it has any role in tumorigenesis is still poorly understood [27, 28]. In vitro studies report an inverse relationship between PPARβ/δ and NFκB, IL-1β, COX2, and the Wnt-β-catenin/TCF-4 pathways, suggesting a possible protective role in cancer development by virtue of its anti-inflammatory effects [45].
PPARβ/δ is also involved in the homeostatic regulation of proliferation/differentiation and modulation of the inflammatory response in cells of the small and large intestine [27, 28, 46]. Its physiologic role, however, is still unknown as well as it is controversial its function in inflammation and CRC development. In mouse models, PPARβ/δ activation by selective ligands in small and large intestine induces terminal differentiation of epithelial and Paneth cells that play an important role in immunity and host defense [27, 28, 46–48]. Emerging evidence suggests also that PPARβ/δ can suppress inflammatory bowel diseases through a PPARβ/δ-dependent and ligand-independent downregulation of inflammatory signaling [47, 48]. These effects may be due, in part, to interference with NFκB signaling or, alternatively, to inhibition of Paneth cell differentiation that, in turn, could contribute to exacerbate experimentally induced colitis in PPARβ/δ-null mice [47, 48]. In contrast, administration of a highly specific PPARβ/δ ligand does not ameliorate inflammation [49]. The role that PPARβ/δ serves in the interplay between inflammation and cancer and in colon carcinogenesis remains debatable. In fact, in vivo and in vitro experiments have provided conflicting results suggesting that PPARβ/δ ligand activation can either potentiate or attenuate the process [50]. Its expression and/or activity is increased after loss of APC or activation of K-RAS expression [51, 52]. PPARβ/δ has also been shown to be a target of APC/β-catenin/T-cell factor- (TCF-) 4-pathway and, in turn, to modulate further downstream targets, such as c-myc and cyclin D1 [53].
5. PPARγ
PPARγ is the best-studied isoform in the GI cancer context. Its role in esophageal cancer development is debated: its activation in vitro reduces cell growth and induces apoptosis, implying that PPARγ ligands could have a potential use as chemotherapeutic agents in the treatment of patients affected by dysplastic Barrett's esophagus [54]. In contrast, xenografted mice treated with PPARγ agonists show an increased tumor growth. This discrepancy has been ascribed to in vivo effects of “tumor interactions,” differences in PPARγ activation magnitude and PPARγ-independent effects of thiazolidinediones. Recently, it has been reported that PPARγ expression increases in less differentiated human Barrett's adenocarcinoma, supporting a role for PPAR in inhibiting the development of these tumors [54–57].
As far as GC, PPARγ agonists reduce the proliferation of human cells lines in vitro although the effects appear to be dependent upon cell differentiation [58–60]. In contrast, PPARγ silencing in GC cell lines reduces cell viability, suggesting that PPARγ overexpression may induce tumorigenesis [61]. PPARγ agonists induce gastric acid secretion via serum and glucocorticoid inducible kinase (SGK1) upregulation [62]. Although this stimulation should favor the formation of gastric ulcers, PPARγ agonists have been reported to foster ulcer healing, suggesting that the potentially “dangerous” effect on gastric acid secretion is overridden by the simultaneous protective effects [62]. The critical importance of PPARγ in gastric carcinogenesis in vivo has recently been provided: PPARγ heterozygous-deficient mice show an increased susceptibility to carcinogen-induced GC and shorter survival rate than PPARγ wild-type bearing mice, implying a tumor suppressor function. In this animal model, thiazolidinediones act as chemopreventive agents in a PPARγ-dependent manner [63].
Several studies have addressed the role of PPARG in CRC development. PPARG mRNA is detected in the normal human mucosa of the caecum and colon, as well as in adenocarcinomas and CRC-derived cell lines. Although PPARγ function in colon carcinogenesis has been controversial for long time, more recent evidence supports a role as tumor suppressor [64–68]. PPARγ agonists induce cell cycle arrest, differentiation, and apoptosis. In particular, p16, p21 and p27, as well as the tumor suppressor gene, PTEN are upregulated while β-catenin, COX-2, VEGF, Bcl-2, and NFκB target genes are downregulated. Finally, PPARγ reduces the epithelial mesenchymal transition (EMT), a well-known process that allows cancer cells to acquire invasive ability, a prerequisite for metastasis formation. Consistent with the evidence in vitro, mouse models have shown that PPARγ ligands reduce the growth of tumors originated from subcutaneously injected human CRC cells and the number of aberrant crypt foci (ACF) in a chemically induced model of IBD [60, 69].
Loss-of-function mutations in PPARG have been reported in 8% of human CRCs, implying a protective effect [70]. Although these mutations have been classified as “very rare events” [70, 71], increasing evidence suggests that PPARγ activity is attenuated during the transition from adenoma to adenocarcinoma, likely explaining why PPARγ agonists are effective in blocking the early stages of tumorigenesis (i.e., ACF formation is inhibited while little or no effect is detected in advanced tumor stages) [71–74]. PPARγ-reduced activity may, at least in part, involve its phosphorylation by the mitogen-activated kinase ERK1/2, and its ligand-independent SUMOylation, two posttranslational modifications that negatively modulate PPARγ activity [72, 73]. In addition to loss-of-function mutations and inactivating posttranslational modifications, low PPARG expression has been found in 35% of sporadic CRCs due to epigenetic events such as DNA methylation and repressive histone modifications [75–77]. Interestingly, the epigenetic repression appears to be associated with a more aggressive course, EMT activation, and patients' worse prognosis, further supporting the notion that PPARγ is an independent prognostic factor in CRC [75, 78]. Reduced PPARγ levels have been detected in patients affected by IBDs, such as UC and CD, suggesting that impaired PPARG expression precedes and is not secondary to the inflammatory process and likely contributes to the pathogenesis of IBDs [79–81]. Concordantly, PPARG genetic variations have recently been correlated with a different risk of IBD incidence [81, 82]. Low PPARγ levels have also been found in peripheral mononuclear cells of IBD patients in the absence of specific PPARG mutations. Epigenetic events or abnormal signaling pathways carried out by natural ligands or microorganisms of the colon microenvironment might account for the impaired PPARG expression in UC and CD patients [82].
6. PPARs and ncRNA
PPARs deregulation during tumorigenesis of the GI tract has been attributed to gene mutations, altered levels of expression and, more recently, epigenetic modifications. These latter events, however, have been identified as “critical” only for PPARG expression while no evidence has been provided for their involvement in PPARα and PPARβ/δ regulation [69, 76, 77]. A novel mechanism of gene regulation is emerging that involves noncoding RNAs (ncRNAs). They are recognized as important regulators of physiological and pathological processes playing critical roles in DNA structure, RNA production, protein translation, and protein functions [83]. The term ncRNA includes both micro-RNAs (miRNA) and long noncoding RNAs (lncRNA). MicroRNAs are small noncoding RNAs that inhibit protein translation or induce degradation of their target mRNAs upon binding to cognate recognition sites usually located in the 3′ untranslated region [84]. Over one third of protein-coding genes is potentially regulated by miRNAs thus affecting important biological functions among which tumorigenesis [84, 85]. A handful of miRNAs have been identified to promote or inhibit tumor initiation, progression, and metastasis, influencing oncogenes or tumour suppressor genes or acting as oncogenes or tumor suppressors themselves [86, 87]. Although the role of ncRNAs in the regulation of PPARs expression or activity is beyond the scope of this review, we would like to mention the miRNAs directly involved in PPARs regulation. miR-21 and miR-10b downregulate PPARα in liver, while miR-506 targets this receptor in human CRC cell lines [88–91]. PPARγ is negatively regulated by miR-27 and miR-130 family members in preadipocytes, hampering adipocyte differentiation [92–97]. In addition, miR-27 reduces PPARγ expression in LPS-stimulated macrophages, inhibiting its anti-inflammatory activity [92]. More recently, miR-27 has been implicated in downregulation of PPARγ in cardiomyocytes and also in neuroblastoma and breast cancer [95, 97]. miR-122 targets PPARβ/δ in liver [98]. Lastly, PPAR activity may also be repressed via miR-dependent targeting of PPARs-coregulators [99]. All together these observations indicate that miRNAs may exert coordinating and redundancy-limiting actions on the gene-expression networks controlled by PPARs [99].
In addition to miRNAs, also long noncoding RNAs (lcnRNAs) regulate nuclear receptors and thus, potentially, PPARs expression and activity. LncRNAs are, in general, defined as non-protein coding transcripts longer than 200 nucleotides that might directly affect gene expression through the interaction with transcriptional activators/repressors inducing or repressing gene transcription [83]. Several studies suggest a critical role of lncRNAs also in the epigenetic-dependent gene regulation by orienting chromatin-modifying factors/complexes to specific locations in the genome and in the nucleus [83, 100, 101]. The direct involvement of lncRNA in PPARs expression has not been described so far; the ncRNA SRA (steroid receptor RNA activator) has recently been reported to associate with PPARγ and modulate transcription of PPAR-target genes [102]. A more recent work provides evidence of a new mechanism of nuclear receptor activity regulation: the ncRNA Gas5 acts as a decoy RNA inhibiting the activity of the glucocorticoid receptor on its target genes [103].
Although the relationship between PPARs and ncRNAs in cancer is only at the beginning to emerge, it is conceivable that miRNAs may regulate PPARs expression influencing the development of GI cancers at different levels.
7. Conclusions and Future Directions
Dietary, environmental, and genetic factors contribute to the etiology, pathogenesis and risk with of gastrointestinal cancers. The link between PPARs and environmental factors in the development of GI tumors is strong, reciprocal but still poorly understood at molecular level. Inflammation plays a crucial role in the development of premalignant lesions of the esophagus, stomach, and colon rectum that, up to now, has only indirectly been proved through expression/correlation studies. PPARs impact diverse aspects of cancer development such as signaling pathways, metabolic interactions, cell cycle, and inflammation.
Here, we have overviewed the most recent evidence of the literature supporting the hypothesis that the events underlying chronic inflammatory conditions and their evolution towards GI tumors could be at least in part orchestrated by the pro- and antitumor effects mediated by PPARs. Their expression and activity in tumor cells are modulated by genetic and epigenetic alterations; miRNAs are emerging as a new pathogenetic player. Intriguingly, dietary and life styles as well as environmental factors may influence PPARs function and impact cancer predisposition through epigenetic mechanisms. Hence, understanding how the individual genetic background and environmental factors contribute to PPARs deregulation and hence to the establishment of an inflammatory status or a tumor condition is mandatory. The studies reported here suggest, in addition, a rationale for novel strategies in cancer treatment whereby PPARs ligands might directly interfere with tumor growth and promote anticancer activity. More direct data and deeper evidence are still awaited to appraise the benefits that these agonists may provide in the prevention and treatment of GI tract inflammations and tumors. Clinical trials suggest that PPARs ligands may not be so effective as a single agent in advanced tumors but they could be effective in combination with a classical chemotherapy and additional anticancer agents such as epigenetic drugs, recently introduced into the therapeutic armamentarium. Another promising translational outcome of these studies is the possibility to identify PPAR alterations in premalignant lesions so that they can be used as prognostic biomarkers. In conclusion, elucidation of these pathways could provide biomarkers or new therapeutic targets with broad implications for cancer prevention, risk prediction, and prognosis.
Authors' Contribution
A. Fucci and T. Colangelo contributed equally to the work.
References
- 1.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocrine Reviews. 1999;20(5):649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 2.Kersten S, Wahli W. Peroxisome proliferator activated receptor agonists. EXS. 2000;89:141–151. doi: 10.1007/978-3-0348-8393-1_9. [DOI] [PubMed] [Google Scholar]
- 3.Michalik L, Desvergne B, Wahli W. Peroxisome-proliferator-activated receptors and cancers: complex stories. Nature Reviews Cancer. 2004;4(1):61–70. doi: 10.1038/nrc1254. [DOI] [PubMed] [Google Scholar]
- 4.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nature Medicine. 2004;10(4):355–361. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 5.Nahlé Z. PPAR trilogy from metabolism to cancer. Current Opinion in Clinical Nutrition and Metabolic Care. 2004;7(4):397–402. doi: 10.1097/01.mco.0000134360.30911.bb. [DOI] [PubMed] [Google Scholar]
- 6.Michalik L, Auwerx J, Berger JP, et al. International union of pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacological Reviews. 2006;58(4):726–741. doi: 10.1124/pr.58.4.5. [DOI] [PubMed] [Google Scholar]
- 7.Michalik L, Wahli W. Involvement of PPAR nuclear receptors in tissue injury and wound repair. The Journal of Clinical Investigation. 2006;116(3):598–606. doi: 10.1172/JCI27958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fajas L, Fruchart JC, Auwerx J. PPARγ3 mRNA: a distinct PPARγ mRNA subtype transcribed from an independent promoter. FEBS Letters. 1998;438(1-2):55–60. doi: 10.1016/s0014-5793(98)01273-3. [DOI] [PubMed] [Google Scholar]
- 9.Al-Shali K, Cao H, Knoers N, Hermus AR, Tack CJ, Hegele RA. A single-base mutation in the peroxisome proliferator-activated receptor γ4 promoter associated with altered in vitro expression and partial lipodystrophy. The Journal of Clinical Endocrinology & Metabolism. 2004;89(11):5655–5660. doi: 10.1210/jc.2004-0280. [DOI] [PubMed] [Google Scholar]
- 10.Zhu Y, Alvares K, Huang Q, Rao MS, Reddy JK. Cloning of a new member of the peroxisome proliferator-activated receptor gene family from mouse liver. Journal of Biological Chemistry. 1993;268(36):26817–26820. [PubMed] [Google Scholar]
- 11.Meirhaeghe A, Amouyel P. Impact of genetic variation of PPARγ in humans. Molecular Genetics and Metabolism. 2004;83(1-2):93–102. doi: 10.1016/j.ymgme.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 12.Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPARγ2: tissue-specific regulator of an adipocyte enhancer. Genes and Development. 1994;8(10):1224–1234. doi: 10.1101/gad.8.10.1224. [DOI] [PubMed] [Google Scholar]
- 13.Diradourian C, Girard J, Pégorier JP. Phosphorylation of PPARs: from molecular characterization to physiological relevance. Biochimie. 2005;87(1):33–38. doi: 10.1016/j.biochi.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 14.Renaud JP, Moras D. Structural studies on nuclear receptors. Cellular and Molecular Life Sciences. 2000;57(12):1748–1769. doi: 10.1007/PL00000656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83(6):841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 16.Berger J, Moller DE. The mechanisms of action of PPARs. Annual Review of Medicine. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 17.Ricote M, Glass CK. PPARs and molecular mechanisms of transrepression. Biochimica et Biophysica Acta. 2007;1771(8):926–935. doi: 10.1016/j.bbalip.2007.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yessoufou A, Wahli W. Multifaceted roles of peroxisome proliferator-activated receptors (PPARs) at the cellular and whole organism levels. Swiss Medical Weekly. 2010;140:p. w13071. doi: 10.4414/smw.2010.13071. [DOI] [PubMed] [Google Scholar]
- 19.Torchia J, Glass C, Rosenfeld MG. Co-activators and co-repressors in the integration of transcriptional responses. Current Opinion in Cell Biology. 1998;10(3):373–383. doi: 10.1016/s0955-0674(98)80014-8. [DOI] [PubMed] [Google Scholar]
- 20.Li M, Pascual G, Glass CK. Peroxisome proliferator-activated receptor γ-dependent repression of the inducible nitric oxide synthase gene. Molecular and Cellular Biology. 2000;20(13):4699–4707. doi: 10.1128/mcb.20.13.4699-4707.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas RM, Sobin LH. Gastrointestinal cancer. Cancer. 1995;75(1):154–170. doi: 10.1002/1097-0142(19950101)75:1+<154::aid-cncr2820751305>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 22.Al-Taie OH, Graf T, Illert B, et al. Differential effects of PPARγ activation by the oral antidiabetic agent pioglitazone in Barrett’s carcinoma in vitro and in vivo. Journal of Gastroenterology. 2009;44(9):919–929. doi: 10.1007/s00535-009-0086-y. [DOI] [PubMed] [Google Scholar]
- 23.Konturek PC, Nikiforuk A, Kania J, Raithel M, Hahn EG, Mühldorfer SM. Activation of NFκB represents the central event in the neoplastic progression associated with Barrett’s esophagus: a possible link to the inflammation and overexpression of COX-2, PPARγ and growth factors. Digestive Diseases and Sciences. 2004;49(7-8):1075–1083. doi: 10.1023/b:ddas.0000037790.11724.70. [DOI] [PubMed] [Google Scholar]
- 24.Hafner C, Reichle A, Vogt T. New indications for established drugs: combined tumor-stroma-targeted cancer therapy with PPARγ agonists, COX-2 inhibitors, mTOR antagonists and metronomic chemotherapy. Current Cancer Drug Targets. 2005;5(6):393–419. doi: 10.2174/1568009054863591. [DOI] [PubMed] [Google Scholar]
- 25.Schoen RE, Pinsky PF, Weissfeld JL, et al. Colorectal-cancer incidence and mortality with screening flexible sigmoidoscopy. The New England Journal of Medicine. 2012;366(25):2345–2357. doi: 10.1056/NEJMoa1114635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartnett L, Egan LJ. Inflammation, DNA methylation and colitis-associated cancer. Carcinogenesis. 2012;33(4):723–731. doi: 10.1093/carcin/bgs006. [DOI] [PubMed] [Google Scholar]
- 27.Huin C, Corriveau L, Bianchi A, et al. Differential expression of peroxisome proliferator-activated receptors (PPARs) in the developing human fetal digestive tract. The Journal of Histochemistry & Cytochemistry. 2000;48(5):603–611. doi: 10.1177/002215540004800504. [DOI] [PubMed] [Google Scholar]
- 28.Braissant O, Wahli W. Differential expression of peroxisome proliferator-activated receptor- α, -β 1, and -γ during rat embryonic development. Endocrinology. 1998;139(6):2748–2754. doi: 10.1210/endo.139.6.6049. [DOI] [PubMed] [Google Scholar]
- 29.Kliewer SA, Forman BM, Blumberg B, et al. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(15):7355–7359. doi: 10.1073/pnas.91.15.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cuzzocrea S, di Paola R, Mazzon E, et al. Role of endogenous and exogenous ligands for the peroxisome proliferators activated receptors alpha (PPAR-α) in the development of inflammatory bowel disease in mice. Laboratory Investigation. 2004;84(12):1643–1654. doi: 10.1038/labinvest.3700185. [DOI] [PubMed] [Google Scholar]
- 31.Griffioen M, Steegenga WT, Ouwerkerk IJM, Peltenburg LTC, Jochemsen AG, Schrier PI. Repression of the minimal HLA-B promoter by c-myc and p53 occurs through independent mechanisms. Molecular Immunology. 1998;35(13):829–835. doi: 10.1016/s0161-5890(98)00074-1. [DOI] [PubMed] [Google Scholar]
- 32.Watson JD, Oster SK, Shago M, Khosravi F, Penn LZ. Identifying genes regulated in a Myc-dependent manner. Journal of Biological Chemistry. 2002;277(40):36921–36930. doi: 10.1074/jbc.M201493200. [DOI] [PubMed] [Google Scholar]
- 33.Varet J, Vincent L, Mirshahi P, et al. Fenofibrate inhibits angiogenesis in vitro and in vivo. Cellular and Molecular Life Sciences. 2003;60(4):810–819. doi: 10.1007/s00018-003-2322-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kasai T, Miyauchi K, Yokoyama T, Aihara K, Daida H. Efficacy of peroxisome proliferative activated receptor (PPAR)-α ligands, fenofibrate, on intimal hyperplasia and constrictive remodeling after coronary angioplasty in porcine models. Atherosclerosis. 2006;188(2):274–280. doi: 10.1016/j.atherosclerosis.2005.10.047. [DOI] [PubMed] [Google Scholar]
- 35.Pozzi A, Capdevila JH. PPARα ligands as antitumorigenic and antiangiogenic agents. PPAR Research. 2008;2008:8 pages. doi: 10.1155/2008/906542.906542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinasso G, Oraldi M, Trombetta A, et al. Involvement of PPARs in cell proliferation and apoptosis in human colon cancer specimens and in normal and cancer cell lines. PPAR Research. 2007;2007:9 pages. doi: 10.1155/2007/93416.93416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gizard F, Amant C, Barbier O, et al. PPARα inhibits vascular smooth muscle cell proliferation underlying intimal hyperplasia by inducing the tumor suppressor p16INK4a. The Journal of Clinical Investigation. 2005;115(11):3228–3238. doi: 10.1172/JCI22756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barger PM, Browning AC, Garner AN, Kelly DP. p38 mitogen-activated protein kinase activates peroxisome proliferator-activated receptor α: a potential role in the cardiac metabolic stress response. Journal of Biological Chemistry. 2001;276(48):44495–44501. doi: 10.1074/jbc.M105945200. [DOI] [PubMed] [Google Scholar]
- 39.Burns KA, Vanden Heuvel JP. Modulation of PPAR activity via phosphorylation. Biochimica et Biophysica Acta. 2007;1771(8):952–960. doi: 10.1016/j.bbalip.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Riccardi L, Mazzon E, Bruscoli S, et al. Peroxisome proliferator-activated receptor-α modulates the anti-inflammatory effect of glucocorticoids in a model of inflammatory bowel disease in mice. Shock. 2009;31(3):308–316. doi: 10.1097/SHK.0b013e31818339e7. [DOI] [PubMed] [Google Scholar]
- 41.Moraes LA, Piqueras L, Bishop-Bailey D. Peroxisome proliferator-activated receptors and inflammation. Pharmacology and Therapeutics. 2006;110(3):371–385. doi: 10.1016/j.pharmthera.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 42.Pyper SR, Viswakarma N, Yu S, Reddy JK. PPARα: energy combustion, hypolipidemia, inflammation and cancer. Nuclear receptor signaling. 2010;8, article e002 doi: 10.1621/nrs.08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mutoh M, Niho N, Wakabayashi K. Concomitant suppression of hyperlipidemia and intestinal polyp formation by increasing lipoprotein lipase activity in Apc-deficient mice. Biological chemistry. 2006;387(4):381–385. doi: 10.1515/BC.2006.051. [DOI] [PubMed] [Google Scholar]
- 44.Matthiessen MW, Pedersen G, Albrektsen T, Adamsen S, Fleckner J, Brynskov J. Peroxisome proliferator-activated receptor expression and activation in normal human colonic epithelial cells and tubular adenomas. Scandinavian Journal of Gastroenterology. 2005;40(2):198–205. doi: 10.1080/00365520410009573. [DOI] [PubMed] [Google Scholar]
- 45.Wu CW, Yu J, Sung JJY. Peroxisome proliferator-activated receptor δ and gastric cancer. Oncology Reports. 2009;22(3):451–457. [PubMed] [Google Scholar]
- 46.Peters JM, Hollingshead HE, Gonzalez FJ. Role of peroxisome-proliferator-activated receptor β/δ (PPARβ/δ) in gastrointestinal tract function and disease. Clinical Science. 2008;115(3-4):107–127. doi: 10.1042/CS20080022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Varnat F, Heggeler BB, Grisel P, et al. PPARβ/δ regulates paneth cell differentiation via controlling the hedgehog signaling pathway. Gastroenterology. 2006;131(2):538–553. doi: 10.1053/j.gastro.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 48.Escher P, Braissant O, Basu-Modak S, Michalik L, Wahli W, Desvergne B. Rat PPARs: quantitative analysis in adult rat tissues and regulation in fasting and refeeding. Endocrinology. 2001;142(10):4195–4202. doi: 10.1210/endo.142.10.8458. [DOI] [PubMed] [Google Scholar]
- 49.Hollingshead HE, Morimura K, Adachi M, et al. PPARβ/δ protects against experimental colitis through a ligand-independent mechanism. Digestive Diseases and Sciences. 2007;52(11):2912–2919. doi: 10.1007/s10620-006-9644-9. [DOI] [PubMed] [Google Scholar]
- 50.Takayama O, Yamamoto H, Damdinsuren B, et al. Expression of PPARδ in multistage carcinogenesis of the colorectum: implications of malignant cancer morphology. British Journal of Cancer. 2006;95(7):889–895. doi: 10.1038/sj.bjc.6603343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He TC, Chan TA, Vogelstein B, Kinzler KW. PPARδ is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell. 1999;99(3):335–345. doi: 10.1016/s0092-8674(00)81664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shao J, Sheng H, DuBois RN. Peroxisome proliferator-activated receptors modulate K-Ras-mediated transformation of intestinal epithelial cells. Cancer Research. 2002;62(11):3282–3288. [PubMed] [Google Scholar]
- 53.He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281(5382):1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 54.Wang W, Wang R, Zhang Z, Li D, Yu Y. Enhanced PPAR-γ expression may correlate with the development of Barrett’s esophagus and esophageal adenocarcinoma. Oncology Research. 2011;19(3-4):141–147. doi: 10.3727/096504011x12935427587849. [DOI] [PubMed] [Google Scholar]
- 55.Naito Y, Takagi T, Yoshikawa T. Gastrointestinal cytoprotection by PPARγ ligands. PPAR Research. 2010;2010:8 pages. doi: 10.1155/2010/108632.108632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tselepis C, Perry I, Dawson C, et al. Tumour necrosis factor-α in Barrett’s oesophagus: a potential novel mechanism of action. Oncogene. 2002;21(39):6071–6081. doi: 10.1038/sj.onc.1205731. [DOI] [PubMed] [Google Scholar]
- 57.Abdel-Latif MMM, O’Riordan J, Windle HJ, et al. NF-κB activation in esophageal adenocarcinoma: relationship to Barrett’s metaplasia, survival, and response to neoadjuvant chemoradiotherapy. Annals of Surgery. 2004;239(4):491–500. doi: 10.1097/01.sla.0000118751.95179.c6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sato H, Ishihara S, Kawashima K, et al. Expression of peroxisome proliferator-activated receptor (PPAR)γ in gastric cancer and inhibitory effects of PPARγ agonists. British Journal of Cancer. 2000;83(10):1394–1400. doi: 10.1054/bjoc.2000.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kitamura S, Miyazaki Y, Hiraoka S, et al. PPARγ inhibits the expression of c-MET in human gastric cancer cells through the suppression of Ets. Biochemical and Biophysical Research Communications. 1999;265(2):453–456. doi: 10.1006/bbrc.1999.1715. [DOI] [PubMed] [Google Scholar]
- 60.Grommes C, Landreth GE, Heneka MT. Antineoplastic effects of peroxisome proliferator-activated receptor γ agonists. Lancet Oncology. 2004;5(7):419–429. doi: 10.1016/S1470-2045(04)01509-8. [DOI] [PubMed] [Google Scholar]
- 61.Liu SH, Shen CC, Yi YC, et al. Honokiol inhibits gastric tumourigenesis by activation of 15-lipoxygenase-1 and consequent inhibition of peroxisome proliferator-activated receptor-γ and COX-2-dependent signals. British Journal of Pharmacology. 2010;160(8):1963–1972. doi: 10.1111/j.1476-5381.2010.00804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rotte A, MacK AF, Bhandaru M, et al. Pioglitazone induced gastric acid secretion. Cellular Physiology and Biochemistry. 2009;24(3-4):193–200. doi: 10.1159/000233245. [DOI] [PubMed] [Google Scholar]
- 63.Lu J, Imamura K, Nomura S, et al. Chemopreventive effect of peroxisome proliferator-activated receptor γ on gastric carcinogenesis in mice. Cancer Research. 2005;65(11):4769–4774. doi: 10.1158/0008-5472.CAN-04-2293. [DOI] [PubMed] [Google Scholar]
- 64.Saez E, Tontonoz P, Nelson MC, et al. Activators of the nuclear receptor PPARγ enhance colon polyp formation. Nature Medicine. 1998;4(9):1058–1061. doi: 10.1038/2042. [DOI] [PubMed] [Google Scholar]
- 65.Sarraf P, Mueller E, Jones D, et al. Differentiation and reversal of malignant changes in colon cancer through PPARγ . Nature Medicine. 1998;4(9):1046–1052. doi: 10.1038/2030. [DOI] [PubMed] [Google Scholar]
- 66.Drori S, Girnun GD, Tou L, et al. Hic-5 regulates an epithelial program mediated by PPARγ . Genes and Development. 2005;19(3):362–375. doi: 10.1101/gad.1240705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Su W, Bush CR, Necela BM, et al. Differential expression, distribution, and function of PPAR-γ in the proximal and distal colon. Physiological Genomics. 2007;30(3):342–353. doi: 10.1152/physiolgenomics.00042.2007. [DOI] [PubMed] [Google Scholar]
- 68.Niho N, Takahashi M, Shoji Y, et al. Dose-dependent suppression of hyperlipidemia and intestinal polyp formation in Min mice by pioglitazone, a PPARγ ligand. Cancer Science. 2003;94(11):960–964. doi: 10.1111/j.1349-7006.2003.tb01385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sabatino L, Fucci A, Pancione M, Colantuoni V. PPARG epigenetic deregulation and Its role in colorectal tumorigenesis. PPAR Research. 2012;2012:12 pages. doi: 10.1155/2012/687492.687492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sarraf P, Mueller E, Smith WM, et al. Loss-of-function mutations in PPARγ associated with human colon cancer. Molecular Cell. 1999;3(6):799–804. doi: 10.1016/s1097-2765(01)80012-5. [DOI] [PubMed] [Google Scholar]
- 71.Ikezoe T, Miller CW, Kawano S, et al. Mutational analysis of the peroxisome proliferator-activated receptor γ in human malignancies. Cancer Research. 2001;61(13):5307–5310. [PubMed] [Google Scholar]
- 72.Burgermeister E, Seger R. MAPK kinases as nucleo-cytoplasmic shuttles for PPARγ . Cell Cycle. 2007;6(13):1539–1548. doi: 10.4161/cc.6.13.4453. [DOI] [PubMed] [Google Scholar]
- 73.Garcia-Bates TM, Lehmann GM, Simpson-Haidaris PJ, Bernstein SH, Sime PJ, Phipps RP. Role of peroxisome proliferator-activated receptor gamma and its ligands in the treatment of hematological malignancies. PPAR Research. 2008;2008:18 pages. doi: 10.1155/2008/834612.834612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thompson EA. PPARγ physiology and pathology in gastrointestinal epithelial cells. Molecules and Cells. 2007;24(2):167–176. [PubMed] [Google Scholar]
- 75.Pancione M, Forte N, Sabatino L, et al. Reduced β-catenin and peroxisome proliferator-activated receptor-γ expression levels are associated with colorectal cancer metastatic progression: correlation with tumor-associated macrophages, cyclooxygenase 2, and patient outcome. Human Pathology. 2009;40(5):714–725. doi: 10.1016/j.humpath.2008.08.019. [DOI] [PubMed] [Google Scholar]
- 76.Pancione M, Sabatino L, Fucci A, et al. Epigenetic silencing of peroxisome proliferator- activated receptor γ is a biomarker for colorectal cancer progression and adverse patients’ outcome. PLoS One. 2010;5(12) doi: 10.1371/journal.pone.0014229.e14229 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 77.Sabatino L, Fucci A, Pancione M, et al. UHRF1 coordinates peroxisome proliferator activated receptor gamma (PPARG) epigenetic silencing and mediates colorectal cancer progression. doi: 10.1038/onc.2012.3. Oncogene. In press. [DOI] [PubMed] [Google Scholar]
- 78.Ogino S, Shima K, Baba Y, et al. Colorectal cancer expression of peroxisome proliferator-activated receptor γ (PPARG, PPARgamma) is associated with good prognosis. Gastroenterology. 2009;136(4):1242–1250. doi: 10.1053/j.gastro.2008.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yamamoto-Furusho JK, Peñaloza-Coronel A, Sánchez-Muñoz F, Barreto-Zuñiga R, Dominguez-Lopez A. Peroxisome proliferator-activated receptor-gamma (PPAR-γ) expression is downregulated in patients with active ulcerative colitis. Inflammatory Bowel Diseases. 2011;17(2):680–681. doi: 10.1002/ibd.21322. [DOI] [PubMed] [Google Scholar]
- 80.Zhang Z-F, Yang N, Zhao G, Zhu L, Wang L-X. Association between the Pro12Ala polymorphism of peroxisome proliferator-activated receptor gamma 2 and inflammatory bowel disease: a meta-analysis. PLoS One. 2012;7(1) doi: 10.1371/journal.pone.0030551.e30551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Andersen V, Christensen J, Ernst A, et al. Polymorphisms in NF-κB, PXR, LXR, PPARγ and risk of infammatory bowel disease. World Journal of Gastroenterology. 2011;17(2):197–206. doi: 10.3748/wjg.v17.i2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dubuquoy L, Rousseaux C, Thuru X, et al. PPARγ as a new therapeutic target in inflammatory bowel diseases. Gut. 2006;55(9):1341–1349. doi: 10.1136/gut.2006.093484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Turner AMW, Morris KV. Controlling transcription with noncoding RNAs in mammalian cells. BioTechniques. 2010;48(6):9–16. doi: 10.2144/000113442. [DOI] [PubMed] [Google Scholar]
- 84.Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nature Reviews Molecular Cell Biology. 2008;9(3):219–230. doi: 10.1038/nrm2347. [DOI] [PubMed] [Google Scholar]
- 85.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nature Reviews Genetics. 2008;9(2):102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 86.Garzon R, Calin GA, Croce CM. MicroRNAs in cancer. Annual Review of Medicine. 2009;60:167–179. doi: 10.1146/annurev.med.59.053006.104707. [DOI] [PubMed] [Google Scholar]
- 87.Krutovskikh VA, Herceg Z. Oncogenic microRNAs (OncomiRs) as a new class of cancer biomarkers. BioEssays. 2010;32(10):894–904. doi: 10.1002/bies.201000040. [DOI] [PubMed] [Google Scholar]
- 88.Kida K, Nakajima M, Mohri T, et al. PPARα is regulated by miR-21 and miR-27b in human liver. Pharmaceutical Research. 2011;28(10):2467–2476. doi: 10.1007/s11095-011-0473-y. [DOI] [PubMed] [Google Scholar]
- 89.Zhou J, Wang K-C, Wu W, et al. MicroRNA-21 targets peroxisome proliferators-activated receptor-α in an autoregulatory loop to modulate flow-induced endothelial inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(25):10355–10360. doi: 10.1073/pnas.1107052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zheng L, Lv GC, Sheng J, Yang YD. Effect of miRNA-10b in regulating cellular steatosis level by targeting PPAR-α expression, a novel mechanism for the pathogenesis of NAFLD. Journal of Gastroenterology and Hepatology. 2010;25(1):156–163. doi: 10.1111/j.1440-1746.2009.05949.x. [DOI] [PubMed] [Google Scholar]
- 91.Tong JL, Zhang CP, Nie F, et al. MicroRNA 506 regulates expression of PPAR alpha in hydroxycamptothecin-resistant human colon cancer cells. FEBS Letters. 2011;585(22):3560–3568. doi: 10.1016/j.febslet.2011.10.021. [DOI] [PubMed] [Google Scholar]
- 92.Jennewein C, von Knethen A, Schmid T, Brüne B. MicroRNA-27b contributes to lipopolysaccharide-mediated peroxisome proliferator-activated receptor γ (PPARγ) mRNA destabilization. Journal of Biological Chemistry. 2010;285(16):11846–11853. doi: 10.1074/jbc.M109.066399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Karbiener M, Fischer C, Nowitsch S, et al. microRNA miR-27b impairs human adipocyte differentiation and targets PPARγ . Biochemical and Biophysical Research Communications. 2009;390(2):247–251. doi: 10.1016/j.bbrc.2009.09.098. [DOI] [PubMed] [Google Scholar]
- 94.Kim SY, Kim AY, Lee HW, et al. miR-27a is a negative regulator of adipocyte differentiation via suppressing PPARγ expression. Biochemical and Biophysical Research Communications. 2010;392(3):323–328. doi: 10.1016/j.bbrc.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 95.Lee J-J, Drakaki A, Iliopoulos D, Struhl K. MiR-27b targets PPARγ toinhibit growth, tumor progression and the inflammatory response in neuroblastoma cells. doi: 10.1038/onc.2011.543. Oncogene. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee EK, Lee MJ, Abdelmohsen K, et al. miR-130 suppresses adipogenesis by inhibiting peroxisome proliferator-activated receptor γ expression. Molecular and Cellular Biology. 2011;31(4):626–638. doi: 10.1128/MCB.00894-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang J, Song Y, Zhang Y, et al. Cardiomyocyte overexpression of miR-27b induces cardiac hypertrophy and dysfunction in mice. Cell Research. 2012;22(3):516–527. doi: 10.1038/cr.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gatfield D, Le Martelot G, Vejnar CE, et al. Integration of microRNA miR-122 in hepatic circadian gene expression. Genes and Development. 2009;23(11):1313–1326. doi: 10.1101/gad.1781009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Eendebak RJAH, Lucassen PJ, Fitzsimons CP. Nuclear receptors and microRNAs: Who regulates the regulators in neural stem cells? FEBS Letters. 2011;585(5):717–722. doi: 10.1016/j.febslet.2011.01.039. [DOI] [PubMed] [Google Scholar]
- 100.Wang X, Song X, Glass CK, Rosenfeld MG. The long arm of long noncoding RNAs: roles as sensors regulating gene transcriptional programs. Cold Spring Harbor Perspectives in Biology. 2011;3(1, article a003756) doi: 10.1101/cshperspect.a003756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nie L, Wu H-J, Hsu J-M, et al. Long non-coding RNAs: versatile master regulators of gene expression and crucial players in cancer. American Journal of Translational Research. 2012;4(2):127–150. [PMC free article] [PubMed] [Google Scholar]
- 102.Xu B, Gerin I, Miao H, et al. Multiple roles for the non-coding RNA SRA in regulation of adipogenesis and insulin sensitivity. PLoS One. 2010;5(12) doi: 10.1371/journal.pone.0014199.e14199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kino T, Hurt DE, Ichijo T, Nader N, Chrousos GP. Noncoding RNA Gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Science Signaling. 2010;3(107):p. ra8. doi: 10.1126/scisignal.2000568. [DOI] [PMC free article] [PubMed] [Google Scholar]