

Abstract
Purpose
The pharmacodynamic properties of MGCD0103, an isotype-selective inhibitor of histone deacetylase (HDAC), were evaluated in preclinical models and patients with a novel whole cell HDAC enzyme assay.
Experimental Design
Boc-Lys(ε-Ac)-AMC, an HDAC substrate with fluorescent read-out, was found to be cell-permeable and was used to monitor MGCD0103-mediated HDAC inhibition in cultured cancer cells in vitro, in peripheral white blood cells ex vivo, in mice in vivo and in human patients.
Results
MGCD0103 inhibited HDAC activity in several human cancer cell lines in vitro and in human peripheral white blood cells ex vivo in a dose-dependent manner. Unlike SAHA, the HDAC inhibitory activity of MGCD0103 was time-dependent and sustained for at least 24 h following drug removal in peripheral white blood cells ex vivo. Inhibitory activity of MGCD0103 was sustained for at least 8 h in vivo in mice, and 48 h in patients with solid tumors. HDAC inhibitory activity of MGCD0103 in peripheral white blood cells correlated with induction of histone acetylation in blood and in implanted tumors in mice. In cancer patients, sustained pharmacodynamic (PD) effect of MGCD0103 was visualized only by dose-dependent enzyme inhibition in peripheral white cells but not by histone acetylation analysis.
Conclusions
This study demonstrates that MGCD0103 has sustained PD effects that can be monitored both in vitro and in vivo with a cell-based HDAC enzyme assay.
Keywords: Histone deacetylase, MGCD0103, pharmacodynamics, cancer clinical trial
INTRODUCTION
Recent evidence shows that cancer is associated with identifiable epigenetic changes and dysfunctional transcriptional regulatory mechanisms thus providing a novel therapeutic strategy (1-3). Both acetylation and deacetylation of histones play a fundamental role in the remodeling of chromatin and epigenetic regulation of gene expression. Transcriptionally active genes are associated with hyperacetylated chromatin, while transcriptionally silent genes are associated with hypoacetylated chromatin. Histone deacetylases (HDACs) remove the acetyl groups from the acetylated lysines in histones and function as transcriptional co-repressors (4-6). Histone deacetylases are grouped in four classes on the basis of the structure of their accessory domains. Class I, II and IV HDACs are Zn2+-dependent enzymes, while Class III enzymes are defined by their dependency on NAD+ and are referred to as sirtuins. Class I HDACs include HDAC 1, 2, 3 and 8; Class II HDACs include HDAC 4, 5, 6, 7, 9, and 10. HDAC 11 belongs to Class IV, but shares features of both Class I and Class II enzymes.
Dysregulation of HDACs and aberrant chromatin acetylation and deacetylation have been implicated in the pathogenesis of cancer. Consequently, inhibition of HDAC activity has been explored as a therapeutic strategy in cancer (4-8). The anticancer activity of HDAC inhibitors is thought to occur through a decrease in transcriptional repression, resulting in inhibition of proliferation, induction of apoptosis, and/or terminal cell differentiation (9, 10). In preclinical studies, several structurally diverse HDAC inhibitors have been found to have potent antitumor activities and tumor specificity. Among these inhibitors are the hydroxamic acids SAHA/vorinostat (Zolinza®) (11-13), NVP-LAQ824 (14), LBH589 (15), and PXD101/belinostat (16, 17); benzamides MS-275 (18-20), and MGCD0103 (21-23); and natural products such as depsipeptide/romidepsin (24). SAHA (Zolinza®) was the first HDAC inhibitor approved and is indicated for treatment of advanced refractory cutaneous T-cell lymphoma (12).
Unlike SAHA, MGCD0103 is a nonhydroxamate isotype-selective HDAC inhibitor which targets HDAC isotypes 1, 2, 3 and 11 (25). Preclinical studies demonstrated that MGCD0103 has significant in vivo antitumor activity with low toxicity. Induction of histone acetylation in tumors by MGCD0103 had been observed to correlate with antitumor activity in mouse models with human tumor xenografts (23).
As efforts in the development of HDAC inhibitors for therapeutic treatment progress, the need for assays to determine the pharmacodynamic (PD) effects of HDAC inhibitors is emerging. In past clinical trials of SAHA and MS-275, PD effects in patients were monitored by analyzing induction of histone acetylation in peripheral white blood cells by immunoblotting, ELISA or FACS (11, 18), or in tumor tissues by immunocytochemistry (18, 20). However, for many of these assays, the sensitivity or limitation of clinical materials has created constraints. In addition, current enzyme assays relying on purified enzymes or lysates do not necessarily reflect in situ activity, since they are likely to disrupt normal protein-protein interactions. Our aim was to develop an HDAC assay using a cell permeable small molecule substrate in intact cells from human patients. Here we present our findings that Boc-Lys(ε-Ac)-AMC, a substrate previously used in vitro for HDAC enzyme assay (26), is cell permeable and is suitable to monitor the inhibitory activity of MGCD0103 in several preclinical models, in vitro and in vivo. This assay was then applied to monitoring PD effects of MGCD0103 in a clinical Phase I study on patients with advanced solid tumors.
MATERIALS AND METHODS
Materials
MGCD0103, N-(2-amino-phenyl)-4-[(4-(pyridin-3-yl)-pyrimidin-2-ylamino)-methyl]-benzamide dihydrobromide, was designed and synthesized at MethylGene (21). The free base form of MGCD0103 was used for all in vitro assays and mouse in vivo analyses. For clinical trials, MGCD0103 was used as the dihydrobromide salt. Compound A (Cpd A) is an inactive analog of MGCD0103 missing an amino group, and was also designed and synthesized at MethylGene. All other comparator HDAC inhibitors used were synthesized at MethylGene. Boc-Lys(ε-Ac)-AMC was purchased from Bachem (Torrance, CA). Fluor-de-Lys Developer was purchased from Biomol (Plymouth Meeting, PA).
Cell culture
Cancer cell lines HCT116 (human colon cancer), Jurkat (human acute T cell leukemia), T24 (human bladder cancer), MCF-7 (human breast adenocarcinoma), PANC-1 (human pancreas carcinoma), HeLa (human cervix carcinoma), A549 (human human non-small cell lung cancer), 293T (human embryonic kidney), DU145 (human prostate cancer), and SW48 (human colorectal adenocarcinoma) were purchased from ATCC (Manassas, VA). HMEC (human mammary epithelial cells) were purchased from BioWhittaker (Walkersville, MD). All cell lines were cultured following vendor instructions.
Peripheral White Blood Cell Isolation
Whole blood was collected in sodium heparin tubes (BD Biosciences, Mississauga, ON, Canada) and shipped at room temperature within 24 h. Buffy coat cells were separated from erythrocytes by centrifugation at 1850 rpm for 5 min, and further purified with EL lysis buffer (Qiagen, Mississauga, ON, Canada) following the manufactorer’s instructions.
Whole Cell HDAC Enzyme Assay
Boc-Lys(ε-Ac)-AMC (Bachem, Torrance CA) has been previously described as a small molecule substrate for HDAC enzymes in vitro (27). In the present study, it was used with intact cells as sources of HDAC enzymes. Cells were seeded in 50 μl in 96-well culture plates (Corning Inc., Lowell, MA) at a density which had previously been found to be in the linear range of the assay (for HCT116 cells, 1 × 105 cells/well were seeded; for DU145, A549, 293T, T24, Jurkat, MCF-7, PANC-1, and HeLa cells, 5 × 104 cells/well; and for isolated buffy coat cells, 3-8 × 105 cells/well). The reaction was initiated by adding 1 μl of 15 mM Boc-Lys(ε-Ac)-AMC (Bachem, Torrance CA) (stock solution in DMSO), to reach a final concentration of 0.3 mM. Cells were incubated with the substrate for 1 h at 37 °C with 5% CO2. The reaction was then stopped by adding 50 μl of buffer (25 mM Tris-Cl pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2) containing 1 μM TSA (Trichostatin A, BioMol, Plymouth Meeting PA), 1:60 diluted Fluor-de-Lys-Developer™ (BioMol, Plymouth Meeting PA), and 1% NP40 (Sigma-Aldrich Canada, Oakville, ON, Canada). TSA in the stop mixture prevented any further deacetylation, while Fluor-de-Lys-Developer™ released a measurable fluorescent moiety from the deacetylated product, and NP40 lysed the cells to allow intracellular deacetylated product to be accessible to the developer. The reaction was allowed to develop for ≥ 15 min at 37°C, and the fluorescent signal was detected by fluorometer (GeminiXS, Molecular Devices, Sunnyvale CA) at excitation 360 nm, emission 470 nm, and cutoff 435 nm. A standard curve of Boc-Lys-AMC (Bachem, Torrance CA) allowed the conversion of fluorescent signal into picomoles (pmoles) of deacetylated product.
Detection of Histone Acetylation
Isolated peripheral white blood cells or frozen tumor pieces were lysed in the presence of 5 mM sodium butyrate and histones were acid-extracted as described in (28). Protein concentrations were determined using Bradford protein assay reagent (BioRad, Hercules CA), and 10 μg from each sample were resolved by electrophoresis on a 4%-20% SDS polyacrylamide gel. Proteins were transferred to a PVDF membrane, probed with anti-H3Ac antibody (Upstate, 06-599, 1:3000 dilution) and either anti-pan-histones (Chemicon MAB052, 1:2000 dilution) or anti-H4 antibody (Upstate, 07-108, 1:2000 dilution), then revealed with horseradish peroxidase-conjugated anti-rabbit antibody (Sigma, Oakville ON, Canada, 1:5000 dilution) and ECL+ chemiluminescent detection reagents (Amersham Biosciences, Piscataway NJ). The signal was scanned and quantified on Storm860 imaging system (Amersham Biosciences).
In Vivo Studies
Female CD-1 normal mice (Charles River Laboratories, Wilmington, MA) or 8-10 weeks old CD-1 nude mice (Charles River Laboratories, Wilmington, MA) bearing either human colon SW48 tumors or HCT116 tumors were used. MGCD0103 or its inactive analog (Compound A) with an amino group deletion were dissolved in PBS acidified with 0.1 N HCl and animals were dosed once by oral gavage (200 μL) using either vehicle or compounds. At the end of each experiment blood and tumors were collected from animals. For HDAC activity, the blood was stored overnight before processing, to mimic shipment conditions of clinical samples. To evaluate histone acetylation by immunoblot analysis, tumors and blood were harvested and immediately flash-frozen.
PD analyses in cancer patients with advanced solid tumors
An open-label, non-randomized, dose-escalation, multi-center Phase I study was performed in patients with advanced solid malignancies. The trial was conducted at the Kimmel Cancer Center at Johns Hopkins (Baltimore, MD) and Princess Margaret Hospital (Toronto, ON, Canada). The study was approved by the local institutional ethics committee of each institution, and patients provided informed consent for their participation. MGCD0103 was administered in 3-week cycles consisting of an oral dose ranging from 12.5 mg/m2/day to 56 mg/m2/day given 3 times weekly for 2 weeks followed by a 7-day rest period. This cycle was repeated every three weeks. PD samples were collected prior to onset of treatment, as well as at 4 h, 24 h, at day 3 (D3, 48 h after the first dose), and at day 8 (D8, 72 h after the third dose) for evaluation of total HDAC enzyme activity in peripheral white blood cells.
Statistical analysis
In vivo xenograft data were subjected to ANOVA analysis followed by Dunnett’s test comparing each treated group to the vehicle control, with a level of significance of 0.01. The data on inhibition of HDAC activity in clinical samples were subjected to ANOVA analysis followed by a Student-Newman-Keuls procedure for pair-wise comparisons with a level of significance of 0.05. In addition, a linear trend test, which evaluates whether the response is dose-dependent, was applied. To circumvent the influence of an outlier in the D3 time-point data set, the data were also ranked and analysed with ranked linear trend test and ranked ANOVA, followed by ranked Student-Newman-Keuls procedure.
RESULTS
Boc-Lys(ε-Ac)-AMC is a cell-permeable HDAC substrate
In the absence of cells (Fig 1A, bar 1), the substrate Boc-Lys(ε-Ac)-AMC was not converted to product and did not generate fluorescence. Likewise, when no substrate was added, the cells alone generated very low background fluorescence (Fig 1A, bar 5). Measurements of cell-associated and cell-free product showed that Boc-Lys(ε-Ac)-AMC is cell permeable (Fig 1A, bar 2), and that the deacetylated product Boc-Lys-AMC also diffuses out of the cells (Fig 1A, bars 3 and 4). There was no extracellular HDAC activity, since conditioned medium, previously incubated with cells (Fig 1A, bar 6), did not generate more fluorescence than background (Fig 1A, bar 1).
Figure 1. Validation of Whole Cell HDAC Assay.

A: 1. No Cell Control: All reagents were included in the wells except for cells; 2. Total Activity: 293T cells (5 × 104 cells per well) were incubated with the substrate, then they were lysed and the product was converted with Fluor-de-Lys Developer ® (BioMol) into a measurable fluorescent compound; 3. Cell Fraction: 293T cells were incubated with the substrate, but before lysis, the supernatant was replaced with fresh medium; 4. Supernatant: 293T cells were incubated with the substrate, as in 2., but before stopping the reaction, the supernatant was collected, cleared from potentially floating cells by centrifuging, and transferred to empty wells; 5. No Substrate Control: all the components of the reaction were added except for Boc-Lys(ε-Ac)-AMC; 6. Conditioned Medium: 293T cells were incubated in medium without substrate, then the medium was centrifuged to remove any cells, and Boc-Lys(ε-Ac)-AMC was added to the clear, conditioned medium. B: Assay Linearity as a Function of Time. 293T cells were incubated with Boc-Lys(ε-Ac)-AMC for various amounts of time before the reaction was stopped. Error bars represent standard error. C: Assay Linearity as a Function of Cell Number. Cell lines (HCT116 colon carcinoma, A549 lung carcinoma, DU145 prostate carcinoma, and HMEC human mammary epithelial cells) were seeded in 96-well plates at the indicated cell densities, and incubated with Boc-Lys(ε-Ac)-AMC for 1 h before the reaction was stopped. D: Determination of Km in cultured cell lines and peripheral white blood cells (WBC). HDAC activity was measured with various amounts of Boc-Lys(ε-Ac)-AMC for a determined amount of time. The velocity V (pmoles/min) was plotted as a function of substrate concentration S (μM) in a double reciprocal graph.
Characteristics and kinetics of whole cell HDAC enzyme assay in human cancer cell lines in vitro
The assay was time-dependent and linear over 2 h in 293T cells (Figure 1B). It was also cell-number dependent and linear up to 5 × 104 cells/well (for 1 h of reaction time) for several cell lines examined (HCT116, A549, DU145, and HMEC) (Fig 1C). In order to calculate Vmax and Km for several cell lines, HDAC activity was measured as a function of time and substrate concentration (Fig 1D). Vmax values (expressed in pmoles of converted product per minute) were 9.47 ± 0.37 for A549, 14.06 ± 0.70 for DU145, 28.56 ± 1.19 for HCT116, and 6.16 ± 0.38 for peripheral white blood cells (WBC). Km values (μM) were 130 ± 16 for A549, 140 ± 21 for DU145, 255 ± 26 for HCT116, and 122 ± 23 for WBC. Based on these results, the substrate concentration for subsequent assays was set at 300 μM.
Inhibitory activity of MGCD0103 and other HDAC inhibitors in human cancer cell lines in vitro
The whole cell HDAC activity assay was used to evaluate the potency of HDAC inhibitors. Unlike hydroxamate-based compounds such as SAHA or NVP-LAQ-824, MGCD0103 and MS-275 demonstrate increasing potency over time. Their IC50s were time-dependent, shifting with time over a 100-fold range, and only reaching full potency after 16 h of incubation with cells at 37°C (Fig 2). Consequently, the drug exposure time was set at 16-18 h before adding Boc-Lys(ε-Ac)-AMC in further activity assays.
Figure 2. Time dependency of benzamide-based HDAC inhibitors.

HDAC activity was assayed in HCT116 with doses of MGCD0103, MS-275, SAHA, or NVP-LAQ-824 in serial dilutions ranging from 0.006 μM to 60 μM. IC50, defined as the concentration (μM) of compound inhibiting 50% of initial HDAC activity, was determined and plotted as a function of time of pre-treatment (hours).
Alamar Blue ® (BioSource, Invitrogen, Burlington, ON, Canada), an indicator of mitochondrial metabolism, did not reveal any cell cytotoxicity within the exposure time of the assay (data not shown). In addition, other cytotoxic, non-HDAC inhibitors such as paclitaxel did not inhibit HDAC whole-cell activity in this assay (data not shown).
To ensure that the observed decrease in substrate conversion upon treatment with HDAC inhibitors was not due to HDAC down-regulation or depletion, we detected the levels of several major HDAC isotypes by immunoblot analysis (Supplementary Fig S1) and found that, after 16 h of pre-treatment with either MGCD0103 or NVP-LAQ-824 (at a concentration about ten times their respective IC50s), the levels of HDAC1, 2, 3, 6, and 8 were unaffected. HDAC7 was down-regulated by both MGCD0103 and NVP-LAQ-824, as previously reported for other HDAC inhibitors (29). This HDAC7 down-regulation, however, was not correlated with HDAC inhibition measured by the whole cell assay (Supplementary Fig S2). For example, at 7 h of pre-treatment, 5 μM of MGCD0103 (about ten times the IC50) could inhibit HDAC activity without down-regulating HDAC7. Moreover, NVP-LAQ-824 could very efficiently inhibit activity with no time-dependency, even at time points where HDAC7 levels were not yet affected (≤ 3 h pre-treatment). Therefore the measured decrease in substrate deacetylation upon treatment with HDAC inhibitors is probably not much influenced by downregulation of HDAC gene expression. Rather, it is believed to mostly reflect direct inhibition of HDAC activity.
The inhibitory activities of MGCD0103, MS-275, and SAHA were compared using a panel of cell lines (Table 1). Although the measured IC50 varied between cell lines, MGCD0103 was always more potent than the comparator molecules in all cases examined. MGCD0103 was more than 7-fold more potent than SAHA in PANC-1 pancreatic cancer cells, and more than 6-fold in HCT116 colon cancer. Cpd A showed no inhibition in any cell line up to 50 μM.
Table 1.
IC50 Values of Compounds for HDAC Inhibition in Cancer Cell Lines
| IC50 (μM) | ||||||||
|---|---|---|---|---|---|---|---|---|
| HCT116 | Jurkat | T24 | MCF-7 | PANC-1 | HeLa | A549 | 293T | |
| MGCD0103 | 0.45* ± 0.05 (n=11) | 0.11 ± 0.02 (n=5) | 0.22 ± 0.04 (n=6) | 0.21 ± 0.01 (n=5) | 0.09 ± 0.03 (n=3) | 0.08 ± 0.01 (n=4) | 0.38 ± 0.04 (n=9) | 0.45 ± 0.02 (n=57) |
| MS-275 | 2.50 ± 0.26 (n=5) | 0.26 ± 0.01 (n=5) | 0.27 ± 0.04 (n=5) | 0.36 ± 0.01 (n=5) | 0.45 ± 0.04 (n=3) | 0.34 ± 0.03 (n=4) | 0.54 ± 0.16 (n=4) | 2.65 ± 1.06 (n=5) |
| SAHA | 2.98* ± 0.18 (n=4) | 0.46 ± 0.06 (n=5) | 0.43 ± 0.07 (n=5) | 0.63 ± 0.11 (n=5) | 0.69 ± 0.32 (n=3) | 0.39 ± 0.03 (n=4) | 0.53 ± 0.06 (n=4) | 2.59 ± 0.52 (n=5) |
| Cpd A | >50 (n=3) | >50 (n=4) | >50 (n=4) | >50 (n=4) | >50 (n=2) | >50 (n=4) | >50 (n=3) | NA |
Cells were seeded on Day 0 at 1×105 (HCT116), or at 5×104 (all other lines) cells per well in a 96-well culture plate, and assayed for HDAC activity on Day 2 following an 18h treatment with various doses of MGCD0103, MS-275, SAHA, or Cpd A. The numbers represent the concentration (μM) inhibiting 50% of the total HDAC enzyme activity. The error represented is the standard error to the mean. NA: not available.
these values were presented in (1) and are used here for the sake of comparison.
Profile and persistence of MGCD0103 inhibitory activity in human peripheral white blood cells ex vivo
The whole-cell HDAC enzyme activity assay was also shown to behave in a linear fashion as a function of time and cell number in healthy human peripheral white blood cells ex vivo (Figure 3A). MGCD0103 induced significant inhibition of HDAC activity in a dose-dependent manner in human peripheral white blood cells treated ex vivo for 18 h (Figure 3B), and the calculated IC50 of MGCD0103 was 0.35 μM (± 0.08 s.e.m.; n = 16). Inhibition with MGCD0103 reached a plateau at 1.5 μM, which corresponds approximately to a 70% reduction of total HDAC activity. Some enzyme activity remained uninhibited even at very high doses of the compound (up to 50 μM). Non-selective hydroxamate-based inhibitors such as SAHA and NVP-LAQ-824, on the other hand, were able to inhibit the cellular pool of HDAC activity almost completely, with an IC50 of 0.9 μM for SAHA, and 0.05 μM for NVP-LAQ-824.
Figure 3. Whole Cell HDAC Assay in Human Peripheral White Blood Cells (Ex Vivo).
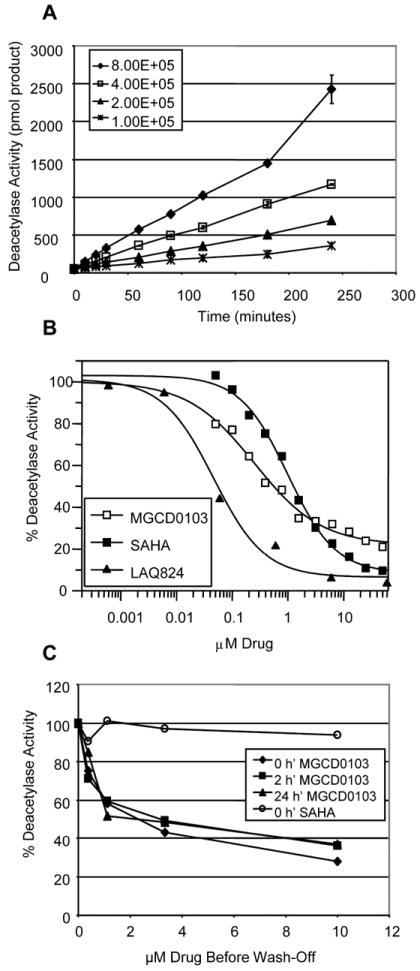
HDAC enzyme activity was monitored with Boc-Lys(ε-Ac)-AMC in human peripheral white blood cells (from buffy coat) isolated from volunteer blood. A: Time dependency. B: Human peripheral white blood cells were treated with a range of doses of either SAHA, MGCD0103, or NVP-LAQ-824 for 16 h, then assayed for HDAC activity with Boc-Lys(ε-Ac)-AMC. C: Human peripheral white blood cells were treated with a range of doses of either SAHA or MGCD0103 for 16 h before compounds were removed from the medium by washing cells with PBS. Cells were then resuspended in fresh medium at time 0 h’ and assayed at indicated time points post-wash for whole cell HDAC activity with Boc-Lys(ε-Ac)-AMC.
The HDAC inhibitory activity of MGCD0103 was long-lasting in human peripheral white blood cells treated ex vivo with MGCD0103, and whole cell HDAC inhibition was sustained even 24 h after removal of the compound (Figure 3C). In contrast, SAHA demonstrated very rapid reversal of HDAC inhibition upon drug removal, as enzyme activity was fully restored as soon as the compound was removed.
In vivo PD effects in tumor and peripheral white blood cells following oral dosing of MGCD0103 in mice
With the aim of using the whole-cell HDAC activity assay as a PD endpoint during clinical trials, it was necessary to validate the use of blood as a surrogate tissue for tumors. Mice (n = 5 per group) were administered MGCD0103 (free base) orally at 90 mg/kg, a dose which has antitumor efficacy in all xenograft models we had previously tested (23). At several time points, blood was collected and stored overnight to mimic shipment conditions of clinical samples. As shown in Fig 4A, the HDAC inhibitory activity was time dependent, with maximal inhibition (40% of initial level) rapidly achieved within the first hour following administration. Inhibition was sustained for approximately 8 h, and HDAC activity returned to baseline levels by 24 h. Inhibition was highly significant, with p<0.01 as determined by post-ANOVA Dunnett’s test, for all time-points up to and including 8 h.
Figure 4. HDAC Activity in Mice In Vivo.

A. CD-1 mice (n=5/group) were given oral MGCD0103 free base (90mg/kg). At indicated time points, blood was collected, peripheral white blood cells were isolated from individual mice and assayed for HDAC activity using 4 × 105 cells per well. The bars represent averages for each group of mice, and the error bars represent the standard error. The significance indicated (p<0.01) was determined by a Dunnett’s test following ANOVA analysis. B and C. CD-1 nude mice (n=3/group) with SW48 colon cancer xenografts (about 1000 mm3) were given a single oral administration of MGCD0103 free base (90 mg/kg), then were sacrificed after specified amount of time. Both blood and tumors were collected and peripheral white blood cells were isolated from blood. Histones were acid-extracted from individual animals and pooled for immunoblot analysis (10 μg per lane) using antibodies against acetylated H3 histones and total H4 histones (B). Signals were quantified by densitometry and normalized by dividing the H3Ac signal with total H4, in order to correct for uneven loading (C). D. CD-1 nude mice (n=5/group) with HCT116 colon cancer xenografts were treated with a single oral dose of MGCD0103 free base (90 mg/kg). At specified times, mice were sacrificed and tumors were collected. Histones were acid-extracted and analyzed by immunoblot (10 μg per lane) using antibodies against acetylated H3 histones and pan-histones. Signals were quantified and normalized by dividing the H3Ac signal with the signal for pan-histones.
The observed in vivo HDAC inhibition was associated with an increase in histone acetylation in SW48 tumors implanted in nude mice. As shown in Fig 4B and 4C, MGCD0103 induced histone H3 hyperacetylation in SW48 tumors as early as 4 h following acute administration of MGCD0103 free base (90mg/kg). Acetylation increased over time, with a 4.3-fold peak at 16 h. After 48 h, the levels of H3 acetylation in tumors returned to baseline values. In peripheral white blood cells of the same mice, H3 acetylation levels, normalized with total H4 histone levels, paralleled those of SW48 tumors, although to a lesser degree with a 2.6-fold peak after 16 h, with values returning to baseline levels within 48 h (Fig 4B, 4C). H3 hyperacetylation in both tumor cells and peripheral white blood cells correlated with inhibition of HDAC activity (Fig 4A). In this model, H3 acetylation in peripheral white blood cells appears to be a valid surrogate for MGCD0103 activity in the tumor.
H3 acetylation was also examined in HCT116 tumors implanted in nude mice (Fig 4D). In this model, MGCD0103 induced a very modest, but highly significant (p<0.01 with post-ANOVA Dunnett’s test) H3 hyperacetylation. The effect reached a plateau 8 h post-treatment and lasted for at least 48 h.
Baseline whole cell HDAC activity levels in human peripheral white blood cells from volunteers
We determined the natural variability in HDAC activity levels in peripheral white blood cells from eight healthy volunteers at two different time points, two months apart. Fig 5A shows that the baseline HDAC activity levels in white blood cells, as determined by this whole cell HDAC activity assay, are constant across individuals (with a coefficient of variation of 6.6% for the first time-point and 8.2% for the second) and across time points (with coefficients of variation ranging from 1.0% to 11.3%). Moreover, we tested various shipment-mimicking conditions with blood from healthy donors. Blood was collected in heparin-coated regular or BD-Vacutainer-CPT tubes, stored at 4°C or ambient temperature for 0 h, 24 h, or 48 h after collection prior to isolation of peripheral white blood cells. No significant difference in baseline HDAC activity was observed among these conditions (data not shown).
Figure 5. HDAC Activity in White Cells from Healthy Volunteers and Cancer Patients In Vivo.
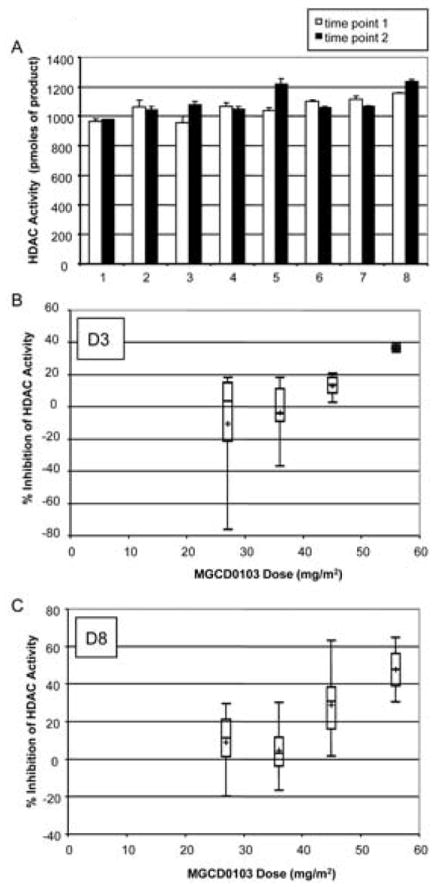
A. Basal HDAC activity levels were measured in peripheral white blood cells (8 × 105 cells per well) isolated from eight different volunteers, at two time-points two months apart, using the whole cell HDAC activity assay. B-C. Phase I trial subjects with solid tumors were treated three times a week with MGCD0103 at 27 mg/m2, 36 mg/m2, 45 mg/m2, or 56 mg/m2. At specified time points, HDAC activity was evaluated in peripheral white blood cells (8 × 105 cells per well) from all evaluable subjects. The results were expressed relative to the 0 h time-point (prior to onset of treatment) and graphed as boxplots. The boxes are delimited by the lower and upper quartiles, while the middle bar represents the median. The whiskers reach to the smallest and the largest data points of each set. The cross represents the average. B: Day 3 (D3) time-point, collected 48 h after onset of treatment and prior to the second dose; at 27 mg/m2, n=6; at 36 mg/m2, n=6; at 45 mg/m2, n=9; at 56 mg/m2, n=2. C: Day 8 (D8) time-point, collected 72 h after the third dosing; at 27 mg/m2, n=6; at 36 mg/m2, n=4; at 45 mg/m2, n=11; at 56 mg/m2, n=2).
PD effects of MGCD0103 in peripheral white blood cells from patients with solid tumors in Phase I trial
The whole cell HDAC enzyme assay was used to monitor the inhibitory activity of MGCD0103 in peripheral white blood cells from subjects with solid tumors in a Phase I clinical trial. The overall clinical outcome of MGCD0103 in this trial and PD effects of MGCD0103 at 24 h were reported in a separate publication (30). A closer examination of the dose- and time-dependent PD effects at later time-points is presented here.
In order to determine the dynamic range of the HDAC enzyme assay, we measured the inhibitory activity of MGCD0103 in 4 groups of subjects treated orally at doses of 27 mg/m2, 36 mg/m2, 45 mg/m2, or 56 mg/m2 MGCD0103. As shown in Fig 5B and 5C, MGCD0103-mediated enzyme inhibition was dose-dependent and sustained for at least 48 h after the first administration (D3, Fig 5B) and up to 72 h after the third administration (D8, Fig 5C). At the highest dose (56 mg/m2), HDAC inhibition was significantly higher than at 27 mg/m2 and 36 mg/m2 in both D3 and D8 time-points, as determined by the Student-Newman-Keuls procedure following ANOVA analysis, with a significance level of 0.05. However, one of the samples from the D3 data set treated at 27 mg/m2 met all the criteria of an outlier, explaining why the average in this group was so different from the median (Fig 5B). It was therefore justifiable to perform a non parametric ranked analysis. When the ranked Student-Newman-Keuls test was applied to the D3 data set, it appeared that HDAC inhibition at 56 mg/m2 was also significantly higher than at 45 mg/m2 (p<0.05). The ranked analysis did not change conclusions for the D8 time-point. Moreover, whether the data were analyzed raw or ranked, the linear trend test showed that the dose-dependency was highly significant (p<0.01) at both time-points.
DISCUSSION
The whole cell HDAC enzyme assay allows the assessment of HDAC activity in a context in which interactions of HDAC enzymes with associated proteins remain intact. This is important as the complexes involving HDACs differ in various cell lines and tissues. Indeed, the differences in IC50s reported in Table 1 reflect this disparity. In contrast, in vitro measurements rely on the purification of specific HDAC isotypes, which may create artificial conditions and alter normal protein-protein interactions. Therefore, assessing HDAC activity in a native, whole cell context may be more physiologically relevant.
The cell-based activity assay was also informative regarding inhibitor selectivity. Only a portion of total cellular HDAC activity could be abrogated by MGCD0103, while SAHA and NVP-LAQ-824 almost completely inactivated the cellular enzyme pool at high concentrations. Obviously, the cellular HDAC activity measured in this assay is defined by the isotypes which can use the substrate. Boc-Lys(ε-Ac)-AMC is a pan-HDAC substrate, although Class I enzymes use it with a greater turnover rate than Class II or Class III isotypes (Lu A.-H and Rahil J, unpublished data). Class III isotypes (Sirtuins) are not inhibited by NVP-LAQ-824, SAHA (31), or TSA, and yet those inhibitors almost completely abrogate the measured activity, so it is unlikely that the deacetylase activity measured in this assay comes from Sirtuins. Therefore the isotypes contributing to the total measured activity are Class I and Class II enzymes, with a preference for Class I isotypes. Unlike NVP-LAQ-824 or SAHA, MGCD0103 specifically inhibits only a subset of the measured HDAC isotypes. The uninhibited enzymes account for approximately 35% of the total measured cellular HDAC activity at 1.5 μM in peripheral white blood cells. These results are consistent with the finding that MGCD0103 in vitro primarily inhibits only human HDACs 1 and 2, and less potently HDACs 3 and 11 (25, 32).(1)
Interestingly, the IC50s of MGCD0103 and other benzamide-based inhibitors, such as MS-275, shift with time over a 100-fold range. Closer examination of their biochemical properties revealed that benzamide-based inhibitors have the characteristics of a slow-tight inhibitor, which will be described elsewhere (A.-H. Lu, J. Rahil et al., unpublished data). Consequently, one might argue that the inevitable delay between blood collection and time of assay, due to shipment, might amplify the apparent MGCD0103 inhibitory activity. However, the shift in IC50 is dramatic only in the first 8 h of exposure in cultured cell lines, and, therefore, clinical samples at later time-points are not likely to be meaningfully affected by this factor. Otherwise, we found no significant differences in the basal level of HDAC activity between samples from healthy donors, whether they were fresh or stored for 24 or 48 h, or taken at several time points. Thus, the delay due to shipment does not appear to affect the measurement of HDAC activity per se, and changes in HDAC activity levels during clinical trials are believed to be accounted for by the treatment, rather than by natural fluctuations.
As a corollary of the slow-tight kinetic properties of MGCD0103, the off-binding rate is very slow and can account for the prolonged inhibitory effect of MGCD0103 observed in ex vivo treated cells after the compound had been removed (Fig 3C). One implication of these results is that patients may receive fewer doses of MGCD0103 while the PD effects are maintained. Another implication is that detection of HDAC inhibition in isolated peripheral white blood cells from patients would not be compromised by unavoidable washing steps during the isolation protocol. The clinical PD data are, therefore, assumed to be relevant to the actual inhibitory activity of MGCD0103.
The utility of blood as a surrogate tissue for solid tumors was validated in nude mice bearing implanted tumors (Fig 4). At a dose previously demonstrated to have efficacy against implanted tumors (90 mg/kg) (23), we observed reduction in peripheral white blood cell HDAC activity in a time-dependent manner, concomitant with induction of histone acetylation both in peripheral white blood cells and in implanted tumors. These data indicate that MGCD0103 was biologically active as expected. Moreover, in mice, t1/2 for MGCD0103 was found to be about 0.7 h (J. Wang et al., unpublished data), while HDAC inhibition was sustained for at least 8 h and histone acetylation peaked at 16 h in SW48 tumors and lasted for at least 48 h in HCT116 tumors. Therefore, in mice, the PD effect of MGCD0103, as monitored by HDAC inhibition and histone acetylation, outlasted detectable plasma exposures.
PK parameters for MGCD0103 in plasma from treated patients have been analyzed (32-34). Correlation with HDAC inhibition has been observed and was described in detail elsewhere (30). Briefly, in patients receiving 27 mg/m2 dose, Cmax was 71.6 ng/ml (± 50.4 SD), which translates to 0.18 μM (± 0.12 SD). In patients receiving 56 mg/m2 dose, Cmax was 172.0 ng/ml (± 28.3 SD), which corresponds to 0.43 μM (± 0.07 SD). T1/2 was about 10 h and Tmax was around 1 h, regardless of the dose. The magnitude of HDAC inhibition was, not surprisingly, rather limited in the 27 mg/m2 dose group since the Cmax was well below the IC50 measured in ex vivo treated peripheral white blood cells. In the 56 mg/m2 dose group, however, Cmax was above the IC50 as determined in ex vivo assay (0.35 μM). In this dose group, a significant reduction of HDAC enzyme inhibition was seen 24 h (30) and 48 h after the first administration, as well as 72 h after the third. The whole-cell HDAC enzyme assay appears to be applicable to determine the PD effect of MGCD0103, at least in peripheral white blood cells from patients with solid tumors. In leukemia patients, whose peripheral white blood cells contain leukemia blasts, we need to consider that apoptotic cells get rapidly cleared from the blood (35). If MGCD0103-mediated HDAC inhibition increased apoptosis of cancer cells, as has been suggested elsewhere,(1) the detected HDAC inhibition in peripheral white blood cells from leukemia patients would likely be underestimated.
Interestingly, HDAC enzyme inhibition was sustained in MGCD0103-treated cancer patients for up to 48-72 h (Fig 5B-C) even though plasma concentrations of the drug were decreased to below the limits of detection at these time-points. This finding is further supported by a similar observation in both the ex vivo assay with peripheral white blood cells from human volunteers and in vivo in mice. This suggests that a less frequent dosing schedule may be desired to reach the best therapeutic index. Indeed, the observed half-life of MGCD0103, combined with the sustained PD, allow for dosing schedules of 2-3 times per week (32, 36).
Histone acetylation was measured in samples from MGCD0103-treated solid-tumor patients (30). A modest but significant induction of histone acetylation (approximately 1.8 fold), correlating with HDAC inhibition, was observed transiently 24 h after the first administration in patients who received the highest dose of MGCD0103 (30). However, no significant histone acetylation was found in peripheral white blood cells from subjects treated with MGCD0103 at the other dose levels or time points (unpublished data). This lack of sensitivity for the histone acetylation assay in human patients may be due to the constraints of the clinical trial set-up and the fact that the samples were evaluated after overnight shipment rather than fresh. In contrast, the whole cell HDAC activity assay revealed inhibitory activity at all time points and the dynamic range was wider than for the acetylation assay under the current shipment conditions.
In conclusion, the data presented here confirm MGCD0103’s HDAC-inhibitory activity and ability to induce histone acetylation in mice, and demonstrate that the MGCD0103-mediated HDAC inhibition can be monitored by a novel whole cell HDAC enzyme assay in patients. This enzyme assay is more sensitive than histone acetylation assays, provides rapid results while using few peripheral white blood cells from patients. Thus, it could prove to be a useful PD assay in future clinical studies of MGCD0103 and other HDAC inhibitors.
Acknowledgments
We appreciate assistance and discussions from our MethylGene colleagues Theresa Yan, Jianhong Liu, Marielle Fournel, Dr. A. Robert MacLeod, Hélène Ste-Croix, Laura Pearce, Tracy-Ann Patterson, Dr. Christiane Maroun, Dr. Jubrail Rahil, Dr. James Wang, Dr. Lori Martell, and Eugene Kovtun. We also thank our colleagues from Pharmion Corporation and Taiho Pharmaceuticals Co. (Japan), especially Drs. Eric Laille for discussion of PK parameters of MGCD0103, Richard McNally for helping with the statistical analysis, and Carla Heise for discussion of experimental results. We are grateful to the clinical teams at Sidney Kimmel Cancer Center at John Hopkins University, and at Princess Margaret Hospital, University of Toronto, MethylGene Inc. as well as all patients and their families, for their participation in the clinical trial.
This work was financially supported by MethylGene Inc., Taiho Pharmaceuticals (Japan) and Pharmion Corporation.
Abbreviations
- HDAC
histone deacetylase
- PD
pharmacodynamics
- PK
pharmacokinetics
- SD
standard deviation
- TSA
Trichostatin A
Footnotes
Fournel M, Bonfils C, Hou Y, Yan PT, Trachy-Bourget M-C, Kalita A, Liu J, Lu A-H, Zhou NZ, Robert M-F, Gillespie J, Wang JJ, Ste-Croix H, Rahil J, Lefebvre S, Moradei O, Delorme D, MacLeod AR, Besterman JM, and Li Z. MGCD0103, a Novel Isotype-Selective Histone Deacetylase Inhibitor, Has Broad-Spectrum Antitumor Activity In Vitro and In Vivo. Submitted for publication.
References
- 1.Gore SD, Baylin S, Sugar E, et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res. 2006;66:6361–9. doi: 10.1158/0008-5472.CAN-06-0080. [DOI] [PubMed] [Google Scholar]
- 2.Rasheed WK, Johnstone RW, Prince HM. Histone deacetylase inhibitors in cancer therapy. Expert Opin Investig Drugs. 2007;16:659–78. doi: 10.1517/13543784.16.5.659. [DOI] [PubMed] [Google Scholar]
- 3.Santini V, Gozzini A, Ferrari G. Histone deacetylase inhibitors: molecular and biological activity as a premise to clinical application. Curr Drug Metab. 2007;8:383–93. doi: 10.2174/138920007780655397. [DOI] [PubMed] [Google Scholar]
- 4.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 5.Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1:287–99. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 6.Acharya MR, Sparreboom A, Venitz J, Figg WD. Rational development of histone deacetylase inhibitors as anticancer agents: a review. Mol Pharmacol. 2005;68:917–32. doi: 10.1124/mol.105.014167. [DOI] [PubMed] [Google Scholar]
- 7.Monneret C. Histone deacetylase inhibitors for epigenetic therapy of cancer. Anticancer Drugs. 2007;18:363–70. doi: 10.1097/CAD.0b013e328012a5db. [DOI] [PubMed] [Google Scholar]
- 8.Garcia-Manero G, Issa JP. Histone deacetylase inhibitors: a review of their clinical status as antineoplastic agents. Cancer Invest. 2005;23:635–42. doi: 10.1080/07357900500283119. [DOI] [PubMed] [Google Scholar]
- 9.Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 10.Gallinari P, Di Marco S, Jones P, Pallaoro M, Steinkuhler C. HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res. 2007;17:195–211. doi: 10.1038/sj.cr.7310149. [DOI] [PubMed] [Google Scholar]
- 11.Kelly WK, O’Connor OA, Krug LM, et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005;23:3923–31. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marks PA. Discovery and development of SAHA as an anticancer agent. Oncogene. 2007;26:1351–6. doi: 10.1038/sj.onc.1210204. [DOI] [PubMed] [Google Scholar]
- 13.Duvic M, Vu J. Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin Investig Drugs. 2007;16:1111–20. doi: 10.1517/13543784.16.7.1111. [DOI] [PubMed] [Google Scholar]
- 14.Kato Y, Salumbides BC, Wang XF, et al. Antitumor effect of the histone deacetylase inhibitor LAQ824 in combination with 13-cis-retinoic acid in human malignant melanoma. Molecular cancer therapeutics. 2007;6:70–81. doi: 10.1158/1535-7163.MCT-06-0125. [DOI] [PubMed] [Google Scholar]
- 15.Giles F, Fischer T, Cortes J, et al. A Phase I Study of Intravenous LBH589, a Novel Cinnamic Hydroxamic Acid Analogue Histone Deacetylase Inhibitor, in Patients with Refractory Hematologic Malignancies. Clin Cancer Res. 2006;12:4628–35. doi: 10.1158/1078-0432.CCR-06-0511. [DOI] [PubMed] [Google Scholar]
- 16.Qian X, LaRochelle WJ, Ara G, et al. Activity of PXD101, a histone deacetylase inhibitor, in preclinical ovarian cancer studies. Molecular cancer therapeutics. 2006;5:2086–95. doi: 10.1158/1535-7163.MCT-06-0111. [DOI] [PubMed] [Google Scholar]
- 17.Plumb JA, Finn PW, Williams RJ, et al. Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Molecular cancer therapeutics. 2003;2:721–8. [PubMed] [Google Scholar]
- 18.Gojo I, Jiemjit A, Trepel JB, et al. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood. 2007;109:2781–90. doi: 10.1182/blood-2006-05-021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hess-Stumpp H, Bracker TU, Henderson D, Politz O. MS-275, a potent orally available inhibitor of histone deacetylases-The development of an anticancer agent. Int J Biochem Cell Biol. 2007 doi: 10.1016/j.biocel.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 20.Ryan QC, Headlee D, Acharya M, et al. Phase I and pharmacokinetic study of MS-275, a histone deacetylase inhibitor, in patients with advanced and refractory solid tumors or lymphoma. J Clin Oncol. 2005;23:3912–22. doi: 10.1200/JCO.2005.02.188. [DOI] [PubMed] [Google Scholar]
- 21.Moradei O, Leit S, Zhou N, et al. Substituted N-(2-aminophenyl)-benzamides, (E)-N-(2-aminophenyl)-acrylamides and their analogues: novel classes of histone deacetylase inhibitors. Bioorg Med Chem Lett. 2006;16:4048–52. doi: 10.1016/j.bmcl.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 22.Bonfils C, Kalita A, Liu J, Besterman JM, Li Z. Development of whole cell HDAC enzyme assay to analyze inhibitory activity of MGCD0103 in vitro and in vivo. Proc Am Assoc Cancer Res; 96th annual meeting of the AACR; 2005. Abs 606. [Google Scholar]
- 23.Li Z, Zhou N, Fournel M, et al. Antitumor activities of MGCD0103, a novel isotype-selective histone deacetylase inhibitor. 16th EORTC-NCI-AACR Symp Mol Targets Cancer Ther. Eur J Cancer Suppl. 2004;2 Abst 83. [Google Scholar]
- 24.Byrd JC, Marcucci G, Parthun MR, et al. A phase 1 and pharmacodynamic study of depsipeptide (FK228) in chronic lymphocytic leukemia and acute myeloid leukemia. Blood. 2005;105:959–67. doi: 10.1182/blood-2004-05-1693. [DOI] [PubMed] [Google Scholar]
- 25.Vaisburg A. Discovery and Development of MGCD0103 - an orally active HDAC inhibitor in Human clinical trials. XIXth International Symposium on Medicinal Chemistry. Int Symp Med Chem. 2006 Abs 055. [Google Scholar]
- 26.Heltweg B, Dequiedt F, Verdin E, Jung M. Nonisotopic substrate for assaying both human zinc and NAD+-dependent histone deacetylases. Analytical biochemistry. 2003;319:42–8. doi: 10.1016/s0003-2697(03)00276-8. [DOI] [PubMed] [Google Scholar]
- 27.Heltweg B, Jung M. A microplate reader-based nonisotopic histone deacetylase activity assay. Analytical biochemistry. 2002;302:175–83. doi: 10.1006/abio.2001.5542. [DOI] [PubMed] [Google Scholar]
- 28.Zweidler A. Resolution of histones by polyacrylamide gel electrophoresis in presence of nonionic detergents. Methods in cell biology. 1978;17:223–33. [PubMed] [Google Scholar]
- 29.Dokmanovic M, Perez G, Xu W, et al. Histone deacetylase inhibitors selectively suppress expression of HDAC7. Molecular cancer therapeutics. 2007;6:2525–34. doi: 10.1158/1535-7163.MCT-07-0251. [DOI] [PubMed] [Google Scholar]
- 30.Siu LL, Pili R, Duran I, et al. A Phase I Study of MGCD0103 Given as a Three-Weekly Oral Dose in Patients with Advanced Solid Tumors. Journal of Clinical Oncology. doi: 10.1200/JCO.2007.14.5730. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–52. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- 32.Siu LL, Carducci M, Patterson T, et al. Phase I study of isotype-selective histone deacetylase (HDAC) inhibitor MGCD0103 given as a three-times weekly oral dose in patients (pts) with advanced solid tumors. 18th EORTC-NCI-AACR Cancer Symposium. Eur J Cancer. 2006;4(12) Abs 300. [Google Scholar]
- 33.Carducci M, Siu LL, Sullivan R, et al. Phase I study of isotype-selective histone deacetylase (HDAC) inhibitor MGCD0103 given as three-times weekly oral dose in patients (pts) with advanced solid tumors. Proc Am Soc Clin Oncol; 2006 ASCO Annual Meeting; 2006. Abs 3007. [Google Scholar]
- 34.Siu LL, Carducci M, Pearce L, et al. Phase I study of isotype-selective histone deacetylase (HDAC) inhibitor MGCD0103 given as three-times weekly oral dose in patients (pts) with advanced solid tumors. AACR-NCI-EORTC Intl Conf Mol Targ Cancer Ther. Clin Cancer Res. 2005;11(23 Suppl) Abs C77. [Google Scholar]
- 35.Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature. 2000;407:784–8. doi: 10.1038/35037722. [DOI] [PubMed] [Google Scholar]
- 36.Garcia-Manero G, Yang AS, Giles F, et al. Phase I/II study of the oral isotype-selective histone deacetylase (HDAC) inhibitor MGCD0103 in combination with azacitidine in patients (pts) with high-risk Myelodysplastic Syndrome (MDS) or Acute Myelogenous Leukemia (AML). 48th American Society of Hematology (ASH) Blood. 2006;108(11) Abs 1954. [Google Scholar]