

Abstract
Despite increased glucose utilization by hypertrophied myocardium, these hearts exhibit a slower rate of glucose uptake (GU). We hypothesized that, in hypertrophied myocardium, a defect of the insulin-responsive glucose transporter is responsible for impaired GU and metabolism during ischemia, contributing to post-ischemic myocardial dysfunction. In a rabbit model of pressure-overload hypertrophy, GU (31P NMR spectroscopy) and total/phosphorylated insulin-signaling intermediates were assayed: insulin-receptor, insulin-receptor-substrate-1 (IRS-1), phosphatidylinositol-3-kinase (PI3-k), GLUT-4 translocation and contractile function in an isolated heart ischemia/reperfusion model. Total protein was not different between hypertrophied and control hearts. Phosphorylation of IRS-1 and PI3-k activity was significantly lower in hypertrophy during ischemia. GU was impaired pre-ischemia in hypertrophy, remained lower during early reperfusion, and was associated with impaired recovery of contractile function. In conclusion, a defect in IRS-1 phosphorylation and PI3-k activation in hypertrophied hearts restricts insulin-mediated GLUT-4 translocation and ischemia, a known stimulus of GLUT-4 translocation, does not compensate for this defect.
Keywords: Hypertrophy, Ischemia, Glucose transporter, Insulin resistance
Adaptive changes in the development of myocyte hypertrophy include multiplication of sarcomeres, a shift to fetal isoforms of contractile proteins and altered calcium regulation for excitation–contraction coupling. Abnormalities of fatty acid metabolism have also been demonstrated in addition to a switch in substrate preference from predominantly fatty acids to a greater use of glucose for energy production [1,2]. Glucose transport into the cell is rate limiting for glycolysis. Transport across the plasma membrane is passive, and is provided by specific transporter proteins [3], which depend on a concentration gradient. The number of glucose transporter proteins present in the sarcolemma and their affnity for glucose determine the rate of glucose transport across the plasma membrane. We and others have previously shown that the glucose transporter isoform that is predominantly expressed in adult cardiomyocytes is the insulin-sensitive transporter GLUT-4 [4,5]. Under basal conditions, most GLUT-4 protein is localized in intracellular vesicles for storage. Upon maximal insulin stimulation, approximately 40% of the myocardial GLUT-4 molecules are translocated to the plasma membrane, augmenting cellular glucose uptake up to 20-fold [6]. This increase in plasma membrane GLUT-4 molecules appears to be the major mechanism by which insulin acutely stimulates glucose uptake in insulin-sensitive cells [7,8]. After insulin levels drop, the transporters are recycled to the intracellular compartment. The other transporter expressed in the myocardium is GLUT-1 which is more independent of insulin action and is predominantly expressed in fetal and newborn myocardium [4,9]. In the hypertrophied myocardium basal, non-insulin-stimulated, glucose uptake rate is accelerated but insulin-stimulated glucose uptake rate has been shown to be reduced [10–14]. This alteration in glucose uptake rate does not appear to be due to a decrease in GLUT-1 or GLUT-4 protein expression levels [11,14] but is potentially due to an alteration in the insulin-mediated translocation of GLUT-4.
Ischemia has also been shown to induce translocation of insulin-responsive glucose transporter molecules to the plasma membrane and is felt to be a protective mechanism to facilitate glucose metabolism during a period of limited oxygen availability [15]. The effects of insulin on myocardial glucose uptake and glycolysis are not easily distinguished from the effects of ischemia on glucose metabolism. Impaired translocation of glucose transporter to the plasma membrane in response to either insulin or ischemia therefore, likely, has important consequences for the ability of the myocardium to withstand an episode of ischemia. Stimulation of glycolysis under anaerobic conditions is in part mediated by activation of other protein kinases such as the AMP-activated protein kinases [16]. It is also possible that the effects of insulin and ischemia on glucose transport are independent but additive in vivo, as has been shown for insulin and hypoxia in skeletal muscle [17]. Thus, increasing glucose transporter translocation to the plasma membrane may well increase the rate of glycolysis and afford greater protection to the ischemic heart.
Based on our previous observation that glucose uptake is impaired in severely hypertrophied myocardium, we hypothesized that insulin-mediated GLUT-4 translocation in hypertrophied hearts is impaired due to an intrinsic defect in the insulin signaling cascade. This defect in insulin-mediated glucose uptake may result in decreased tolerance of hypertrophied myocardium to ischemia and impaired recovery of post-ischemic contractile function.
Materials and methods
Left ventricular hypertrophy model
Pressure-overload hypertrophy was induced by banding of the descending aorta in 10-day-old New Zealand White rabbits as previously described in detail [11]. Development of left ventricular (LV) hypertrophy was determined by measuring LV mass to LV volume ratio by weekly transthoracic echocardiography. We have previously shown that progression of LV hypertrophy reaches a plateau by 4–5 weeks of age in this model, indicative of the compensated phase of hypertrophy [18]. In the present study, severely hypertrophied hearts were studied from banded rabbits that were 5–6 weeks of age and compared to sham operated age-matched controls.
Determination of insulin-signaling pathway intermediates
Following intravenous administration of a mixture of heparin (500 U/kg), ketamine (100 mg/kg), and xylazine (2.5 mg/kg), hypertrophied hearts (n = 6/group) and non-hypertrophied control hearts from age-matched littermates (n = 6/group) were rapidly removed and mounted on an isolated heart perfusion set-up (Langendorff). Hearts were perfused with modified Krebs–Henseleit (KH) solution containing glucose (11 mmol/L) and insulin (10 IU/L), and LV tissue was obtained following 30 min of perfusion, and in a separate set of hearts, after 30 min of normothermic ischemia. The left ventricles were frozen in liquid nitrogen and tissue was further processed by immunoprecipitation as we have previously described in more detail [12]. Immunoprecipitates were then used for gel electrophoresis with 10% sodium dodecyl sulfate (SDS)–(PAGE) gels [12]. After transfer to nitrocellulose membranes, the membranes were incubated with primary antibodies directed against insulin-receptor-β subunit, insulin-receptor substrate-1, phosphatidylinsitol-3 kinase, and tyrosine phosphorylation products (Santa Cruz Biotechnology, Santa Cruz, CA) at a dilution of 1:1000 followed by incubation with horse-radish peroxidase-conjugated secondary antibody (Jackson Immuno Research Labs, West Grove, PA) at a dilution of 1:10,000. The bound antibody was detected by the enhanced chemiluminescence method according to the manufacturer’s instruction (Amersham Life Science, Arlington Heights, IL). After exposure on films, quantitative protein analysis was performed by laser densitometry.
Phosphatidylinositol-3-kinase (PI3-kinase) activity was measured by thin-layer chromatography (TLC) as we have previously described, in ventricular tissue from hypertrophied and age-matched littermates obtained from hearts following perfusion only and also at end-ischemia as described above [12]. TLC plates were developed in CHCl3:CH3OH:H2O:NH4OH (60:47:11.3:2; vol/vol), dried, and visualized by autoradiography. After exposure on films, quantitative analysis was undertaken by laser densitometry.
Determination of activation of GLUT-4 by immunohistochemical staining
Translocation of GLUT-4 from intracellular storage pool to the plasma membrane prior to and during ischemia was determined by immunohistochemical staining of LV cross-sections obtained from hypertrophied and control hearts under baseline conditions and from a separate set of hearts following 30 min of ischemia. Cross-sections of the heart were fixed in 4% paraformaldehyde (PFA) in PBS (pH 7.4), paraffin-embedded, and sectioned. The paraffin-embedded tissue sections were de-paraffinized and rehydrated in a descending alcohol series. The slides were then stained with GLUT-4 primary antibody (R&D Systems, Minneapolis, MN) in the presence of 5% blocking serum in PBS to block unspecific binding sites. Secondary immunoreagent conjugated to the red-fluorescent Alexa-594 (Molecular Probes, Eugene, OR) was used with 590/622 excitation/emission wavelengths. After the staining procedure, sections were covered with glass slides using an anti-fading mounting medium (Dako Corporation, Carpinteria, CA). Slides were visualized using an Axiovert 35 Microscope with a xenon light source. The fluorescent objective of a Nikon 63× objective (NA = 63×/0.75) magnification and excitation/emission filters for the chosen fluorescent probe were used.
Glucose uptake rate by 31P NMR spectroscopy
Glucose uptake rate, defined as glucose transport and phosphorylation over time, was determined by measuring the rate of accumulation of the glucose tracer analogue 2-deoxyglucose. We have shown previously that in this model, 2-deoxyglucose-6-phosphate is irreversibly trapped in the tissue and is neither subjected to further metabolism nor to dephosphorylation. Glucose uptake was determined by 31P nuclear magnetic resonance (NMR) in isolated perfused non-hypertrophied, age-matched control hearts (n = 5) and hypertrophied hearts (n = 5) prior to ischemia and 15 min into reperfusion. NMR spectra were acquired in an 8.45 T vertical bore Bruker spectrometer (Bruker Instruments, Billerica, MA) [11,12]. Spectral peak areas were quantified by integration after baseline correction with software provided by Bruker (Bruker Instruments, Billerica MA). Normalization was carried out by standardizing the 2-deoxyglucose-6-phosphate signal integral to an external standard (500 μmol/L dimethylene phosphonic acid), contained in a balloon adjacent to the heart.
Isolated heart perfusion and contractile function measurements
A separate group of animals were administered ketamine (100 mg/kg, IV) and xylazine (2.5 mg/kg, IV) for euthanasia and heparin (500 U/kg IV). The hearts were rapidly excised, the aorta was cannulated and perfused in the Langendorff mode as we have previously described [12]. After a 30-min stabilization period, hypertrophied hearts (n = 6) and non-hypertrophied, age-matched control hearts (n = 6) were arrested with a 2-min infusion of 30 ml normothermic cardioplegia (KH +22.5 mmol/L KCl) and maintained at 37 °C ischemia for 30 min, followed by 30 min of reperfusion. Pressure measurements were obtained with a latex fluid filled balloon, connected to a catheter tip pressure transducer (Millar Instruments, Houston, TX). LV developed pressure (systolic—end-diastolic pressure) at balloon volumes adjusted to produce an end-diastolic pressure in the range of 5–10 mmHg, heart rate and coronary flow were measured pre-ischemia and at the end of the 30-min reperfusion period.
Statistical analysis
Data were analyzed using SPSS software package (version 11.0, SPSS, Chicago, IL) and are reported as means ± standard error of the mean (SEM). ANOVA was used for comparison among and between groups, followed by Bonferroni’s post hoc analysis where appropriate. A value of p ≤ 0.05 was considered statistically significant.
Animal care
All animals received humane care in compliance with the “Principles of Laboratory Animal Care” formulated by the National Society for Medical Research and the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH Publication No. 86-23, revised 1996). The protocol was reviewed and approved by the Institutional Animal Care and Use Committee at Children’s Hospital Boston and Beth-Israel-Deaconess Hospital.
Results
Characteristics of animals
Left ventricular to body weight ratio measured 1.83 ± 0.12 g/kg in non-hypertrophied, age-matched, controls, and 3.39 ± 0.32 g/kg in aortic banded animals (p < 0.0001). None of the aortic banded animals had clinical evidence of heart failure.
Activation of insulin-signaling pathway intermediates
As indicated in Fig. 1, insulin-receptor β-subunit total was not different between control (pre-versus post-ischemia: 108 ± 10 versus 102 ± 15) and hypertrophy (107 ± 17 versus 103 ± 9) and level of phosphorylation was also not different pre-versus post-ischemia (control: 107 ± 4 versus 118 ± 9; hypertrophy: 109 ± 9 versus 100 ± 18). There was no difference in total protein levels of IRS-1 in controls (pre/post-ischemia: 189 ± 4/201 ± 20) versus in hypertrophy (pre/post-ischemia: 185 ± 11/187 ± 15). However, comparing IRS-1 phosphorylation levels, pre-ischemic levels were not different between controls and hypertrophied hearts (114 ± 10 versus 118 ± 16). Ischemia resulted in increased phosphorylation of IRS-1 in control hearts (142 ± 17), but in hypertrophied hearts, phosphorylated IRS-1 levels remained at pre-ischemic values (110 ± 16; p < 0.05 versus control) (see Fig. 2). Phosphatidylinositol-3-kinase (PI3-kinase) activity was significantly lower in hypertrophied hearts pre-ischemia (84 ± 5 in hypertrophy versus 98 ± 4 in controls; p < 0.05) and at end-ischemia compared to controls (91 ± 5 in hypertrophy versus 117 ± 8 in controls; p < 0.01) (see Fig. 3).
Fig. 1.

Insulin-receptor β-subunit is shown as total protein content (A) and the amount phosphorylated (B). As indicated in the representative Western-blot and the summary of densitometry data, there was no difference between control and hypertrophied hearts. Data are expressed as means ± SEM. n = 6/group; C, control, perfusion only; C+I, control, end-ischemia; H, hypertrophy, perfusion only; and H+I, hypertrophy, end-ischemia.
Fig. 2.

Insulin receptor substrate-1 is shown as total protein content (A) and the amount phosphorylated (B). There was no difference between control and hypertrophied hearts in total IRS-1 protein content. With regard to phosphorylation of IRS-1, ischemia resulted in a significant increase in phosphorylation (e.g., activation) of IRS-1 in control hearts but remained at baseline levels in hypertrophied hearts. Data are expressed as means ± SEM. C, control, perfusion only; C+I, control, end-ischemia; H, hypertrophy, perfusion only; and H+I, hypertrophy, end-ischemia. n = 6/group; *p < 0.05 versus C.
Fig. 3.

Representative blot of PI3-kinase activity in anti-IRS-1 immunoprecipitates from left ventricular muscle extracts of hypertrophied hearts compared to hearts from age-matched control animals. The radioactivity at the origin is caused by [32P]ATP residues and other 32P-labeled materials. PIP indicates the position of migration of PI3-P standard. PI3-kinase activity was determined by TLC and a representative blot is shown. Ischemia resulted in a significant increase in PI3-kinase activity in control hearts compared to hypertrophied hearts (n = 6/group; *p < 0.05 versus C).
Translocation of GLUT-4 in response to ischemia
The effects of ischemia on translocation of GLUT-4 in hypertrophied and age-matched controls are shown in Fig. 4. Fluorescently labeled GLUT-4 protein is uniformly distributed during normal perfusion with glucose and insulin (10 U/L) containing perfusate in control hearts as well as hypertrophied hearts. During ischemia however, there is a marked increase in fluorescence in the cell membrane indicating GLUT-4 translocation in control hearts but this effect is markedly blunted in hypertrophied hearts.
Fig. 4.

Representative immunohistochemical cross-sections of the LV stained with GLUT-4 showed that pre-ischemia GLUT-4 protein was evenly distributed in hypertrophied and control hearts and during ischemia, in control hearts, GLUT-4 translocated to membrane to a higher degree than in hypertrophied hearts.
Glucose uptake
Figs. 5A and B show the rate of 2-deoxyglucose-6-phosphate (2-DG-6-P) accumulation over a period of 15 min in the aortic banded groups and control animals prior to ischemia and following a 30-min period of normothermic, no-flow ischemia. Overall, 2-DG-6-P accumulation in the presence of 10 U/L insulin was significantly lower in hypertrophied hearts (2.46 ± 0.18 μmol/g dry weight) compared to normal age-matched control hearts (4.21 ± 0.26 μmol/g dry weight; p < 0.05) with a slower rate of rise and lower total accumulation after 15 min. During early reperfusion, glucose uptake rate recovered faster in control hearts (6.24 ± 0.66 μmol/g dry weight) compared to hypertrophied hearts (3.79 ± 0.15 μmol/g dry weight; p < 0.05).
Fig. 5.

(A,B) Glucose uptake rate pre-ischemia and during the first 15 min post-ischemia in controls (A) and hypertrophy (A) are shown. Glucose uptake rate under maximal insulin stimulation was significantly lower in hypertrophied hearts compared to normal, age-matched, control hearts and during early reperfusion, glucose uptake rate recovered faster in control hearts compared to hypertrophied hearts.
Isolated heart perfusion and contractile function measurements
There was no difference in pre-ischemic coronary flow, heart rate or developed pressure measurements between hypertrophied and control hearts (data not shown). Post-ischemic recovery of developed pressure was worse however, in hypertrophied hearts (pre-versus post-ischemia: 94 ± 3 mmHg versus 70 ± 5 mmHg; p < 0.001) compared to control hearts (pre-versus post-ischemia: 99 ± 2 mmHg versus 91 ± 1 mmHg; p < 0.001 versus hypertrophy post-ischemia) (see Fig. 6).
Fig. 6.
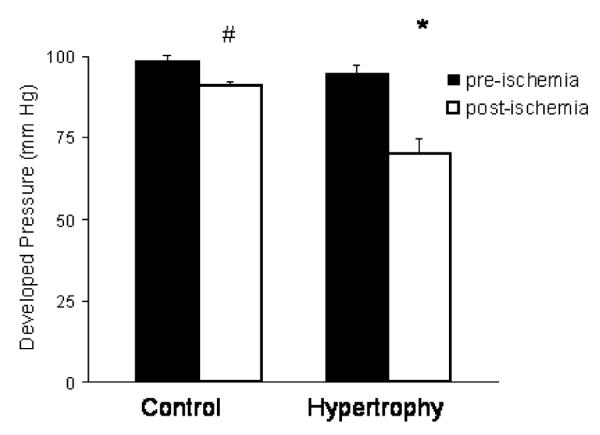
Developed pressure measured prior to a 30-min period of normothermic ischemia and 30 min after reperfusion in hypertrophied hearts and age-matched non-hypertrophied control hearts. n = 6/group, *p < 0.001 versus pre-ischemia hypertrophied hearts. #p < 0.001versus post-ischemia hypertrophied hearts.
Discussion
Several studies have suggested that a rapid recovery of glycolysis is important for the recovery of heart function following ischemia [19,20]. Also, alterations in energy metabolism have been implicated as contributing to worse recovery of contractile function in the hypertrophied myocardium following ischemia [21,22]. During ischemia, hypertrophied hearts exhibit accelerated loss of high-energy nucleotides, greater accumulation of tissue lactate, earlier onset of ischemic contracture, and accelerated accumulation of cytosolic calcium during early reperfusion [23,24]. During ischemia, anaerobic glycolysis is the primary source for ATP production since lack of oxygen and accumulation of NADH in the mitochondria rapidly inhibit oxidative phosphorylation. Although the mechanisms responsible for the greater impairment of post-ischemic mechanical function in hypertrophied hearts remain to be fully elucidated, we speculate that regulation of glucose transport across the plasma-membrane plays a significant role. The slower glucose uptake rate in the hypertrophied hearts may in part explain the accelerated loss of high-energy phosphates during ischemia and worse recovery during reperfusion [25]. As we have previously shown, even though energy metabolism, and therefore ATP production, recovered in the hypertrophied hearts, postischemic mechanical function remained impaired. Recovery of oxidative metabolism is also crucial during the reperfusion period [26]. In normal hearts, fatty acid oxidation is the predominant source for energy production in general and also during reperfusion [27,28]. In hypertrophied myocardium, fatty acid oxidation is impaired [1,29] and glucose metabolism takes over a more important role in energy metabolism. Therefore, the translocation of GLUT-4 into the sarcolemmal membrane during ischemia also has potential implications for rates of glucose metabolism during early reperfusion since GLUT-4 protein continues to reside at higher levels in the sarcolemma.
Glucose utilization in the myocardium is controlled by many factors including cardiac work, ischemia, hypoxia, and insulin [30]. The rate of glucose uptake is largely determined by the number and activity of GLUT-4 in the sarcolemmal membrane. It has been shown that both hypoxia (e.g., ischemia) and insulin cause translocation of myocardial glucose transporters from an intracellular site to the sarcolemmal membrane [31]. Insulin binds to a specific cell surface receptor which spans across the cell membrane. Insulin receptor binding induces autophosphorylation and activation of the protein tyrosine kinase domain, located in the β-subunit. The main target for the activated kinase is insulin-receptor substrate-1 (IRS-1) protein which after phosphorylation associates with the enzyme PI3-kinase [32]. PI3-kinase activation subsequently leads to translocation of GLUT-4. Under basal conditions, GLUT-4 protein is stored in intracellular vesicles, and upon stimulation, is translocated to the plasma membrane, increasing the membrane capacity for glucose transport. Myocardial insulin insensitivity, as seen in non-insulin-dependent diabetes mellitus, has been shown to be associated with impaired glucose uptake during ischemia [33]. In hypertrophied myocardium, relative insensitivity to insulin has been described, which results in a slower rate of glucose entry into the myocytes and a decline in insulin-mediated glucose uptake and non-oxidative glucose metabolism [11–13,34]. However, the total expression of GLUT-4 is not the limiting factor since protein expression is not different in hypertrophied heart compared to the normal, age-matched controls [10,11]. In this study, we have found that failure of insulin-mediated recruitment of GLUT-4 to the plasma membrane is responsible for impaired glucose transport in hypertrophied myocardium. Besides insulin, stimuli such as exercise, hypoxia, hormones, and ischemia have also been shown to cause translocation of GLUT-4 molecules in fat and skeletal muscle [16,31,35–38]. Diabetic myocardium and potentially other states of insulin-resistance appear to have an impaired ability to increase glucose uptake in response to either insulin or ischemia [39–42]. In addition, intracellular acidosis inhibits the insulin-mediated signaling pathway and an intracellular pH of 6.75 seems to be the threshold [32]. As we have previously reported hypertrophied hearts develop lower pH levels than non-hypertrophied control hearts. An intracellular pH of 6.66 was found in our model of pressure-overload hypertrophy at the end of a 30-min ischemic period compared to a pH of 6.88 in control hearts [26].
There is some controversy as to whether or not insulin and ischemia have an additive effect on GLUT-4 translocation and subsequent increase in glucose uptake. Brosius et al. [43] did not find that ischemia in the presence of insulin resulted in additional GLUT-4 translocation. However, when hyperinsulinemia was present, Russell et al. [44] reported an increase in GLUT-4 as well as GLUT-1 translocation rate. If indeed alternate pathways regulate ischemia-induced GLUT-4 regulation as effectively as insulin-mediated GLUT-4 activation, then we could show in this study that in hypertrophied hearts not only insulin-regulated glucose uptake is impaired but ischemia as additional stimulus for GLUT-4 translocation did not ameliorate this effect. Our results suggest that there is a lack of glucose transporter translocation to the sarcolemmal membrane in hypertrophied hearts during ischemia. The defect in activation of GLUT-4 translocation appears to be in the early steps of tyrosine kinase phosphorylation of insulin-receptor substrate-1 and inhibition of the insulin-signaling pathway could be due to lower pH occurring in hypertrophied hearts during ischemia. Ultimately, impaired glucose uptake during ischemia and also during early reperfusion in hypertrophied hearts is associated with impaired recovery of contractile function in hypertrophied hearts.
Acknowledgments
This work was supported by National Heart, Lung, and Blood Institute Grants HL-075430 (to I. Friehs), HL-063095 (to P.J. del Nido) and HL 063609 (Beth-Israel-Deaconess NMR Research Center).
References
- [1].Allard MF, Schoenekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am. J. Physiol. 1994;267:H742–H750. doi: 10.1152/ajpheart.1994.267.2.H742. [DOI] [PubMed] [Google Scholar]
- [2].Kagaya Y, Kanno Y, Takeyama D, Ishide N, Maruyama Y, Takahashi T, Ido T, Takishima T. Effects of long-term pressure overload on regional myocardial glucose and free fatty acid uptake in rats. A quantitative autoradiographic study. Circulation. 1990;81:1353–1361. doi: 10.1161/01.cir.81.4.1353. [DOI] [PubMed] [Google Scholar]
- [3].Pessin JE, Bell GI. Mammalian facilitative glucose transporter family: structure and molecular regulation. Annu. Rev. Physiol. 1992;54:911–930. doi: 10.1146/annurev.ph.54.030192.004403. [DOI] [PubMed] [Google Scholar]
- [4].Friehs I, Cao-Danh H, Stamm C, Cowan DB, McGowan FX, del Nido PJ. Postnatal increase in insulin-sensitive glucose transporter expression is associated with improved recovery of postischemic myocardial function. J. Thorac. Cardiovasc. Surg. 2003;126:263–271. doi: 10.1016/s0022-5223(03)00034-5. [DOI] [PubMed] [Google Scholar]
- [5].Santalucia T, Camps M, Castello A, Munoz P, Nuel A, Testar X, Palacin M, Zorzano A. Developmental regulation of Glut-1 (erythroid/Hep G2) and Glut-4 (muscle/fat) glucose transporter expression in rat heart, skeletal muscle and brown adipose tissue. Endocrinology. 1992;130:837–846. doi: 10.1210/endo.130.2.1370797. [DOI] [PubMed] [Google Scholar]
- [6].Slot JW, Geuze HJ, Gigengack S, James DE, Lienhard GE. Translocation of the glucose transporter GLUT-4 in cardiac myocytes of the rat. Proc. Natl. Acad. Sci. USA. 1991;88:7815–7819. doi: 10.1073/pnas.88.17.7815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cushman SW, Wardzala LJ. Potential mechanisms of insulin action on glucose transport in the isolated rat adipose cell: apparent translocation of intracellular transport systems to the plasma membrane. J. Biol. Chem. 1980;255:4758–4762. [PubMed] [Google Scholar]
- [8].Suzuku K, Kono T. Evidence that insulin causes translocation of glucose transport activity to the plasma membrane from an intracellular storage site. Proc. Natl. Acad. Sci. USA. 1980;77:2542–2545. doi: 10.1073/pnas.77.5.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gould GW, Holman GD. The glucose transporter family: structure, function and tissue-specific expression. Biochem. J. 1993;295:329–341. doi: 10.1042/bj2950329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Friehs I, del Nido PJ. Increased susceptibility of hypertrophied hearts to ischemic injury. Ann. Thorac. Surg. 2003;75:S678–S684. doi: 10.1016/s0003-4975(02)04692-1. [DOI] [PubMed] [Google Scholar]
- [11].Friehs I, Moran AM, Stamm C, Colan SD, Takeuchi K, Cao-Danh H, Rader CM, McGowan FX, del Nido PJ. Impaired glucose transporter activity in pressure-overload hypertrophy is an early indicator of progression to failure. Circulation. 1999;100(Suppl. 19):II187–II193. doi: 10.1161/01.cir.100.suppl_2.ii-187. [DOI] [PubMed] [Google Scholar]
- [12].Friehs I, Stamm C, Cao-Danh H, McGowan FX, Jr., del Nido PJ. Insulin-like growth factor-1 improves postischemic recovery in hypertrophied hearts. Ann. Thorac. Surg. 2001;72:1650–1656. doi: 10.1016/s0003-4975(01)03098-3. [DOI] [PubMed] [Google Scholar]
- [13].Paternostro G, Clarke K, Heath J, Seymour AM, Radda GK. Decreased GLUT-4 mRNA content and insulin-sensitive deoxyglucose uptake show insulin resistance in the hypertensive rat heart. Cardiovasc. Res. 1995;30:205–211. [PubMed] [Google Scholar]
- [14].Allard MF, Wambolt RB, Longnus SL, Grist M, Lydell CP, Parsons HL, Rodrigues B, Hall JL, Stanley WC, Bondy GP. Hypertrophied rat hearts are less responsive to the metabolic and functional effects of insulin. Am. J. Physiol. Endocrinol. Metab. 2000;279:E487–E493. doi: 10.1152/ajpendo.2000.279.3.E487. [DOI] [PubMed] [Google Scholar]
- [15].Sun DW, Nguyen N, DeGrado TR, Schwaiger M, Grosius FC. Ischemia induces translocation of the insulin-responsive glucose transporter Glut-4 to the plasma membrane of cardiac myocytes. Circulation. 1994;89:793–798. doi: 10.1161/01.cir.89.2.793. [DOI] [PubMed] [Google Scholar]
- [16].Russel RB, Bergeron R, Shulman GI, Young LH. Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMP: by AICAR. Am. J. Physiol. 1999;277:H643–H649. doi: 10.1152/ajpheart.1999.277.2.H643. [DOI] [PubMed] [Google Scholar]
- [17].Cartee GD, Douen AG, Ramlal T, Klip A, Holloszy JO. Stimulation of glucose transport in skeletal muscle by hypoxia. J. Appl. Physiol. 1991;70:1593–1600. doi: 10.1152/jappl.1991.70.4.1593. [DOI] [PubMed] [Google Scholar]
- [18].Moran AM, Friehs I, Takeuchi K, Stamm C, Hammer PE, McGowan FX, del Nido PJ, Colan SD. Non-invasive serial evaluation of myocardial mechanics in pressure overload hypertrophy of rabbit myocardium. Herz. 2003;28:52–62. doi: 10.1007/s00059-003-2392-0. [DOI] [PubMed] [Google Scholar]
- [19].Jeremy RW, Korestsune Y, Marban E, Becker LC. Relation between glycolysis and calcium homeostasis in postischemic myocardium. Circ. Res. 1992;70:1180–1190. doi: 10.1161/01.res.70.6.1180. [DOI] [PubMed] [Google Scholar]
- [20].Mallet RT, Hartman DA, Bunger R. Glucose requirement for postischemic recovery of perfused working heart. Eur. J. Biochem. 1990;188:481–493. doi: 10.1111/j.1432-1033.1990.tb15426.x. [DOI] [PubMed] [Google Scholar]
- [21].Anderson PG, Allard MF, Thomas GD, Bishop SP, Digerness SB. Increased ischemic injury but decreased hypoxic injury in hypertrophied rat hearts. Circ. Res. 1990;67:948–959. doi: 10.1161/01.res.67.4.948. [DOI] [PubMed] [Google Scholar]
- [22].Cunningham MJ, Apstein CS, Weinberg EO, Vogel WM, Lorell BH. Influence of glucose and insulin on the exaggerated diastolic and systolic dysfunction of hypertrophied rat hearts during hypoxia. Cunningham Circ. Res. 1990;66:406–415. doi: 10.1161/01.res.66.2.406. [DOI] [PubMed] [Google Scholar]
- [23].Allard MF, Flint JD, English JC, Henning SL, Salamanca MC, Kamimura CT, English DR. Calcium overload during reperfusion is accelerated in isolated hypertrophied rat hearts. J. Mol. Cell. Cardiol. 1994;26:1551–1563. doi: 10.1006/jmcc.1994.1175. [DOI] [PubMed] [Google Scholar]
- [24].Sink JD, Pellom GL, Currie WD, Hill RC, Olsen CO, Jones RN, Wechsler AS. Response of hypertrophied myocardium to ischemia: correlation with biochemical and physiological parameters. J. Thorac. Cardiovasc. Surg. 1981;81:865–872. [PubMed] [Google Scholar]
- [25].Gaasch WH, Zile MR, Hoshino PK, Weinberg EO, Rodes DR, Apstein CS. Tolerance of the hypertrophic heart to ischemia. Studies in compensated and failing dog hearts with pressure overload hypertrophy. Circulation. 1990;81:1644–1653. doi: 10.1161/01.cir.81.5.1644. [DOI] [PubMed] [Google Scholar]
- [26].Stamm C, Friehs I, Cowan DB, Moran AM, Cao-Danh H, Duebener LF, del Nido PJ, McGowan FX. Inhibition of tumor necrosis factor-a improves postischemic recovery of hypertrophied hearts. Circulation. 2001;104(Suppl. I):I-350–I-355. doi: 10.1161/hc37t1.094851. [DOI] [PubMed] [Google Scholar]
- [27].Lopaschuk GD, Wambolt RB, Barr RL. An imbalance between glycolysis and glucose oxidation is a possible explanation for the detrimental effects of high levels of fatty acids during aerobic reperfusion of ischemic hearts. J. Pharmacol. Exp. Ther. 1993;264:135–144. [PubMed] [Google Scholar]
- [28].McVeigh JJ, Lopaschuk GD. Dichloroacetate stimulation of glucose oxidation improves recovery of ischemic rat hearts. Am. J. Physiol. 1990;259:H1079–H1085. doi: 10.1152/ajpheart.1990.259.4.H1079. [DOI] [PubMed] [Google Scholar]
- [29].El Alaoui-Talibi Z, Landormy S, Loireau A, Moravec J. Fatty acid oxidation and mechanical performance of volume-overloaded rat hearts. Am. J. Physiol. 1992;262:H1068–H1074. doi: 10.1152/ajpheart.1992.262.4.H1068. [DOI] [PubMed] [Google Scholar]
- [30].Depre C, Vanoverschelde JL, Taegtmeyer H. Glucose for the heart. Circulation. 1999;99:578–588. doi: 10.1161/01.cir.99.4.578. [DOI] [PubMed] [Google Scholar]
- [31].Wheeler TJ. Translocation of glucose transporters in response to anoxia in heart. J. Biol. Chem. 1988;263:19447–19454. [PubMed] [Google Scholar]
- [32].Beauloye C, Bertrand L, Krause U, Marsin A-S, Dresselaers T, Vanstapel F, Vanoverschled J-L, Hue L. No-flow ischemia inhibits insulin signaling in heart by decreasing intracellular pH. Circ. Res. 2001;88:513–519. doi: 10.1161/01.res.88.5.513. [DOI] [PubMed] [Google Scholar]
- [33].Stanley WC, Hall JL, Hacker TA, Hernandez LA, Whitesell LF. Decreased myocardial glucose uptake during ischemia in diabetic swine. Metabolism. 1997;46:168–172. doi: 10.1016/s0026-0495(97)90297-3. [DOI] [PubMed] [Google Scholar]
- [34].Paolisso G, Galzerano D, Gambardella A, Varricchio G, Saccomanno F, D’Amore A, Varricchio M, D’Onofrio F. Left ventricular hypertrophy is associated with a stronger impairment of non-oxidative glucose metabolism in hypertensive patients. Eur. J. Clin. Invest. 1995;25:529–533. doi: 10.1111/j.1365-2362.1995.tb01740.x. [DOI] [PubMed] [Google Scholar]
- [35].Kahn BB. Facilitative glucose transporters: regulatory mechanisms and dysregulation in diabetes. J. Clin. Invest. 1992;89:1367–1374. doi: 10.1172/JCI115724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Watanabe T, Smith MM, Robinson RW, Kono T. Insulin action on glucose transport in cardiac muscle. J. Biol. Chem. 1984;259:13117–13122. [PubMed] [Google Scholar]
- [37].Rattigan S, Appleby GJ, Clark MG. Insulin-like action of catecholamines and Ca2+ to stimulate glucose transport and GLUT-4 translocation in perfused rat heart. Biochim. Biophys. Acta. 1988;1094:217–223. doi: 10.1016/0167-4889(91)90012-m. [DOI] [PubMed] [Google Scholar]
- [38].Zaninetti D, Greco-Perotto R, Jeanrenaud B. Heart glucose transport and transporters in rat heart: regulation by insulin, workload and glucose. Diabetologia. 1988;31:108–113. doi: 10.1007/BF00395557. [DOI] [PubMed] [Google Scholar]
- [39].Randle PJ, Newsholme E, Garland PB. Regulation of glucose uptake by muscle. Biochem. J. 1964;93:632–665. doi: 10.1042/bj0930665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Morgan HE, Cadenas E, Regen DM, Park CR. Regulation of glucose uptake in muscle. II. Rate-limiting steps and effects of insulin and anoxia in heart muscle from diabetic rats. J. Biol. Chem. 1961;236:262–268. [PubMed] [Google Scholar]
- [41].Barrett EJ, Schwartz RG, Young LH, Jacob R, Zaret BL. Effect of chronic diabetes on myocardial fuel metabolism and insulin sensitivity. Diabetes. 1988;37:943–948. doi: 10.2337/diab.37.7.943. [DOI] [PubMed] [Google Scholar]
- [42].Saad MJ. Molecular mechanisms of insulin resistance. Braz. J. Med. Biol. Res. 1994;27:941–957. [PubMed] [Google Scholar]
- [43].Brosius FC, Nguyen N, Egert S, Lin Z, Deeb GM, Haas F, Schwaiger M, Sun D. Increased sarcolemmal glucose transporter abundance in myocardial ischemia. Am. J. Cardiol. 1997;30:A77–A84. doi: 10.1016/s0002-9149(97)00460-8. [DOI] [PubMed] [Google Scholar]
- [44].Russell RR, Yin R, Caplan MJ, Hu X, Ren J, Shulman GI, Sinusas AJ, Young LH. Additive effects of hyperinsulinemia and ischemia on myocardial GLUT1 and GLUT4 translocation in vivo. Circulation. 1998;98:2180–2186. doi: 10.1161/01.cir.98.20.2180. [DOI] [PubMed] [Google Scholar]