

Abstract
An efficient method for the synthesis of nucleoside phosphoramidates prodrugs (6a–f) has been developed that employs a simple protection/deprotection sequence of the nucleoside with benzyloxycarbonyl (Cbz). The coupling reaction of Cbz-protected derivatives (5a–f) with phenyl-(ethoxy-L-alaninyl)-phosphorochloridate (7), followed by Cbz group removal by hydrogenolysis provided the phenyl phosphoramidate ProTides (6a–f) in excellent overall yields.
Keywords: ProTide, Nucleoside, Monophosphate prodrug, Benzyloxycarbonyl, Antiviral
1. Introduction
Over the past 40 years, nucleoside analogs have established themselves as important agents for first-line antiviral therapy and cancer therapy. In most cases, the observed therapeutic activity does not result directly from the nucleoside itself, but from the corresponding nucleoside 5′-triphosphate (NuTP). However, this three step intracellular conversion by kinases is often limited by an inefficient initial phosphorylation step. Nucleotides cannot be considered as therapeutic agents due to their polar nature, their inability to cross the cell membrane and their dephosphorylation in extracellular fluids. In order to overcome these problems, considerable efforts have focused on monophosphate prodrugs1 that mask the monophosphate moiety with various chemically or enzymatically cleaved groups once the compound is transported into the cell. This strategy has the potential to reveal potent compounds that were inactive in their nucleoside form because of a lack of phosphorylation. Among all reported prodrugs approach, the class of aryl phosphoramidate nucleoside prodrug (ProTide) developed by McGuigan2 has been proven to enhance, in vitro, the activity of parent nucleosides by increasing the formation rate of NuTP by improved intracellular transport and/or bypassing the rate limiting monophosphorylation step.
For instance, 2′,3′-didehydro-3′-deoxythymidine monophosphate prodrug (d4T-MP-PD, 1a) demonstrated 100-fold more potency against HIV than d4T.3 The phosphoramidate (d4A-MP-PD, 1b) of 2′,3′-didehydro-2′,3′-dideoxyadenosine (d4A) has markedly enhanced anti-HIV activity (ca. 2000-fold) compared to d4A.4 Recently, application of the ProTide approach significantly improved antiviral potency of the carbocyclic adenosine analog L-Cd4A-MP-PD, 1c (9000-fold) against HIV.5 The antiviral potency of both abacavir and carbovir ProTides (1f and 1g) was boosted by 10- and 20-fold, respectively, against hepatitis B virus (HBV).6 In the same manner, ProTide derivative (1h) of BVDU (brivudin) showed enhanced potency compared to parent compound against colon cancer.7 4′-Azidoadenosine phosphoramidate derivative (1e) also exhibited a more potent antiviral activity against HCV replication than the parent compound.8 More interestingly, phosphoramidates (1d),9 (1i–k),10 and (1l)11 have been shown to exhibit significant antiviral activity against hepatitis C virus (HCV) replication, while their parent nucleoside was devoid of activity (Fig. 1).
Fig. 1.

Antiviral phosphoramidate ProTides (1a–l).
In general, aryl phosphoramidate ProTides are synthesized by direct coupling of an unprotected nucleoside with a phosphorochloridate derivative using either N-methyl imidazole (NMI) or t-butyl magnesium halide (Br or Cl) in tetrahydrofuran (THF). However, the synthesis of most ProTides is plagued with low to moderate yields due to poor chemoselectivity in the ProTide forming reaction and/or poor solubility of nucleoside in THF. Acid labile protective groups, such as isopropylidene12 or cyclopentylidene,8,13 have been used in the past to prepare phosphoramidate prodrugs of ribonucleosides. However, the phosphorochloridate/nucleoside coupling yields remain moderate and the relative instability of the phosphoramidate function to acidic conditions lead to low yields for the deprotection step. Now that pharmaceutical companies, such as Inhibitex,12a Gilead,14 Pharmasset,10 and Idenix15 are developing their own HCV nucleoside ProTides, there is need for an efficient high yielding synthetic route to prepare these compounds involving simple purifications steps. Herein, we are reporting an efficient method to prepare phenyl phosphoramidate ProTides by protection of polar functional groups (OH and NH2) with benzyloxycarbonyl chloride (CbzCl) followed by coupling with phosphoramidate 7 and final deprotection under neutral conditions. The benzyloxycarbonyl (Cbz) group was a particularly attractive protective group for its facile introduction on the sugar moiety (and the base moiety in case of a cytidine analog) and also for its clean removal by simple hydrogenation under mild and neutral conditions without affecting the phosphoramidate functional group.
2. Results and discussions
Cytidine (2a) was used as a model system and we synthesized the Cbz-protected cytidine analog (3a) following previously reported synthetic methods.16 The selective silylation of the 5′-hydroxyl group of 2a was conducted with TBSCl and imidazole in pyridine. Additionally, protection of the two remaining hydroxyl groups along with the amino group was preformed with CbzCl to provide a 93% yield after purification of the fully protected cytidine derivative (3a) (Scheme 1). The silyl group of 3a was removed with Et3N–3HF to afford Cbz-derivative 4a in quantitative yield. With the liphophilic cytidine derivative (4a) in hand, we investigated its coupling with phenyl-(ethoxy-L-alaninyl)-phosphorochloridate (7), which was synthesized by the reaction of phenyl-phosphodichloridate with L-alanine ethyl ester hydrochloric salt.7 The reaction of Cbz-cytidine 4a with phosphorochloridate (7), using the previously reported procedure,7 provided Cbz-cytidine phosphoramidate 5a in 97% yield. Finally, the hydrogenolysis of Cbz-protected cytidine ProTide (5a) in the presence of Pd/C in EtOH provided the cytidine ProTide (6a) in 98% yield. With an overall yield of 86% from cytidine (2a), it is evident that our new CBz protection/deprotection approach represents a valuable alternative to the direct coupling of cytidine 2a with phosphorochloridate (7) (10% yield).
Scheme 1.

Synthesis of the cytidine ProTide 6a.
In order to generalize our strategy, this efficient synthetic route was applied to other natural nucleosides including adenosine (A), guanosine (G), and uridine (U) as summarized in Table 1. The reaction of A and U with TBSCl in pyridine gave 5′-O-silylated A and U in high yield. Due to its low solubility in pyridine, the silylation of G was conducted in DMSO. The reaction of silylated nucleosides (U, A, G) with CbzCl in the presence of DMAP in CH2Cl2 afforded their corresponding Cbz-protected compounds in good yields. In accordance with previously reported results,16a,16b formation of N6-Cbz-protected A and O6-/N2-Cbz-protected G derivatives was never observed, even though an excess of CbzCl was used. The treatment of 5′-O-silylated Cbz-nucleosides (A, G, and U) with Et3N–3HF provided Cbz-protected nucleosides (4b–d) in excellent yields (Table 1). Finally, the coupling reaction of 4b–d with phenoxy phosphorochloridate (7), followed by hydrogenolysis using either Pd/C, H2 (A and G) or catalytic hydrogen transfer (U),16a afforded their corresponding phosphoramidate ProTides (6b–d) in excellent yield.
Table 1.
Synthesis of nucleoside aryloxy phosphoramidate prodrugs 6a–6f
| Entry | Yields
|
|||
|---|---|---|---|---|
| Protection (three steps) | Coupling reaction | Hydrogenolysis (Method A/B)a | Overall yield (%) | |
| 1 | 92% 4a |
96% 5a |
97% (A) 6a |
86(10)b |
| 2 | 96% 4b |
98% 5b |
98% (B) 6b |
92 |
| 3 | 90% 4c |
92% 5c |
94% (A) 6c |
79 |
| 4 | 85% 4d |
90% 5d |
95% (A) 6d |
73 |
| 5 | 96% 4e |
96% 5e |
96% (B) 6e |
87(19.6)c |
| 6 | 95% 4f |
93%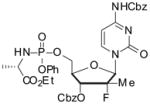 5f |
95% (A) 6f |
86(17)b |
Method A: Pd/C, H2 (1 atm), EtOH; Method B: Pd/C, 1,4-cyclohexadiene, EtOH.
Yield for the coupling of unprotected nucleoside with 7.
Yield for the coupling of unprotected nucleoside with 7, see Ref. 18.
The Cbz approach was also applied to the preparation of both 2′-deoxy-2′-α-fluoro-2′-β-C-methyl uridine and 2′-deoxy-2′-α-fluoro-2′-β-C-methyl-cytidine ProTide 6e and 6f; two anti HCV-agents17 previously prepared by direct coupling of the nucleosides with phosphorochloridate 7 (17% and 20% yields, respectively).18 The key Cbz protected intermediates (4e–f), prepared from their parent nucleosides using the procedure described above (Scheme 1), were reacted with the phenyl-(ethoxy-L-alaninyl)-phosphorochloridate (7) to give Cbz-protected ProTides (5e–f) in excellent yield. Subsequently, they were converted to their corresponding ProTides (6e–f) by hydrogenolysis in quantitative yield (Table 1).
3. Conclusion
In summary, an efficient synthetic procedure for the preparation of aryl phosphoramidate ProTides was successfully developed by utilizing a Cbz protection/deprotection sequence. This method appears to be general and can be applied to ribo or 2′-deoxyribo nucleosides bearing various bases (A, G, C, U). The coupling reaction of a variety of Cbz-protected nucleosides (4a–f) with phenyl-(ethoxy-L-alaninyl)-phosphorochloridate (7) afforded the Cbz-protected ProTides (5a–f) in excellent yields (~90%). The removal of the Cbz group from 5a–f was successfully conducted under hydrogenolysis with Pd/C, hydrogen (1 atm) or catalytic hydrogen transfer to provide the aryl phosphoramidate ProTides (6a–f) in near quantative overall yields. To our knowledge, this approach is one of the most efficient and potentially most general sequences to prepare phosphoramidate nucleoside prodrugs. Its development could significantly impact the multikilo production of clinically relevant compounds, such as 6e and 6f.
4. Experimental section
4.1. General
Nuclear magnetic resonance (NMR) spectra (1H, 13C, 19F, and 31P) were recorded on a Varian Unity Plus 400 MHz fourier transform spectrometer at rt, with tetramethylsilane (TMS) as an internal standard. Chemical shifts (δ) are reported in parts per million (ppm), and signals are quoted as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br (broad), dd (doublet of doublets), or ddd (doublet of doublets of doublets). The phosphoramidates are an approximate 50:50 mixture of diastereomers and the 13C NMR data is reported as observed, that is, some carbon signals overlap. High-resolution mass spectra (HRMS) were recorded on a Micromass Autospec high-resolution mass spectrometer with electrospray ionization (ESI). Thin-layer chromatography (TLC) was performed on 0.25 mm silica gel. Purifications were carried out on silica gel column chromatography (60 Å, 63–200 μm, or 40–75 μm).
4.1.1. 5′-O-tert-Butyldimethylsilyl-N4-2′,3′-O-tris-benzylox-ycarbonylcytidine (3a)
To a solution of cytidine (3.60 g, 14.3 mmol) in 30 mL of anhydrous pyridine was added imidazole (1.25 g, 18.40 mmol) and tert-butyldimethylsilyl chloride (TBDMSCl, 2.40 g, 15.7 mmol) at 0 °C under a N2 atmosphere. The reaction mixture was stirred at rt for 12 h and then treated with methanol (8.0 mL). After stirring for 60 min, the solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography (hexane/EtOAc=10:1 to 1:4 v/v) to give (5.12 g, 12.5 mmol) in 97% yield. Subsequently, to a solution of protected cytidine (2.68 g, 7.50 mmol) and DMAP (5.50 g, 45.0 mmol) in 50 mL of anhydrous CH2Cl2 was added benzyl chloroformate (CbzCl, 4.76 mL, 33.74 mmol) at 0 °C under N2 atmosphere. After stirring for 72 h at rt, the reaction mixture was diluted with CH2Cl2 (200 mL) and then washed with cold 1.0 M HCl aqueous solution (50 mL) then water (100 mL). The solution was dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The residue was purified on silica gel column chromatography (hexane/EtOAc=10:1 to 1:1 v/v) to give 3a (5.47 g, 7.20 mmol) in 93% yield. 1H NMR (400 MHz, CDCl3) δ 8.32 (d, J=8.0 Hz, 1H), 7.58 (br, 1H), 7.42–7.30 (m, 15H), 7.21 (d, J=8.0 Hz, 1H), 6.27 (d, J=3.2 Hz, 1H), 5.35 (t, J=4.0 Hz, 1H), 5.28 (t, J=5.6 Hz, 1H), 5.23 (s, 2H), 5.13 (d, J=2.4 Hz, 2H), 5.10 (d, J=7.6 Hz, 2H), 4.33 (d, J=5.6 Hz, 1H), 4.06 (d, J=10.8 Hz, 1H), 3.79 (d, J=10.8 Hz, 1H), 0.93 (s, 9H), 0.12 (s, 3H), 0.11 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 162.8, 154.7, 153.9, 153.8, 152.5, 144.0,137.2,135.1,134.7, 134.7,130.7,129.2,128.9,128.8,128.7,128.7, 128.6, 128.5, 128.4, 128.3, 117.2, 95.2, 87.8, 81.9, 77.5, 72.7, 70.4, 70.4, 67.9, 61.5, 25.9, 18.4, −5.5, −5.6; MS-ESI+ m/z 760 (M+H+).
4.1.2. N4-2′,3′-O-Tris-benzyloxycarbonylcytidine (4a)
To a solution of 3a (4.50 g, 5.92 mmol) in 20 mL of anhydrous THF was added triethylamine trihydrofluoride (Et3N–3HF, 6.0 mL, 36.8 mmol) at 0 °C under N2 atmosphere. The solution was stirred for 12 h at rt and all volatiles were removed using a rotary evaporator. The residue was dissolved in EtOAc (100 mL) and washed with cold saturated NaHCO3 solution (30 mL×2), and brine (30 mL). The resulting solution was dried over Na2SO4 for 3 h and filtered. The filtrate was adsorbed on silica gel and purified by silica gel column chromatography (CH2Cl2/MeOH=40:1 to 20:1 v/v) to give 4a (3.71 g, 5.74 mmol) in 97% yield. 1H NMR (400 MHz, CDCl3) δ 7.87 (d, J=7.6 Hz, 1H), 7.85 (br, 1H), 7.40–7.30 (m, 15H), 7.27 (d, J=7.6 Hz, 1H), 5.80–5.75 (m, 2H), 5.51 (t, J=4.8 Hz,1H), 5.21 (s, 2H), 5.14–5.07 (m, 4H), 4.33 (m, 1H), 3.99 (d, J=12.0 Hz, 1H), 3.83–3.79 (m, 1H), 3.60 (br, 1H); 13C NMR (100 MHz, CDCl3) δ 174.3, 163.1, 155.2, 154.3, 153.9, 152.3, 146.8, 135.0, 134.8, 128.9, 128.9, 128.8, 128.8, 128.7, 128.6, 95.9, 92.3, 83.8, 75.9, 73.9, 70.6, 70.6, 68.3, 61.5; MS-ESI+ m/z 646 (M+H+).
4.1.3. 5′-O-[Phenyl-(ethoxy-L-alaninyl)]phosphoryl-N4-2′,3′-O- tris-benzyloxycarbonylcytidine (5a)
To a solution of 4a (0.50 g, 0.78 mmol) and 7 (0.46 g, 1.56 mmol) in 10 mL of anhydrous THF was added 1-methylimidazole (0.15 mL, 2.0 mmol) over 10 min at −78 °C under argon atmosphere. After stirring for 2 h at −78 °C, the reaction was maintained for 12 h at rt. The solvent was removed under reduced pressure and the residue was dissolved in CH2Cl2 (20 mL), washed with cold 0.5 M HCl solution (5 mL×2), cold water (10 mL), and brine (5 mL). The organic layer was dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatography (CH2Cl2/MeOH=20:1 to 10:1 v/v) to give 5a (0.68 g, 0.76 mmol), as 1:1 diastereomeric (RP/SP) mixture, in 97% yield. 1H NMR (400 MHz, CDCl3) δ 8.05 (br, 1H), 7.92–7.34 (m, 1H), 7.38–7.09 (m, 21H), 6.12–6.08 (m, 1H), 5.38–5.30 (m, 1H), 5.21 (br, 2H), 5.15–5.06 (m, 4H), 5.06–5.01 (m, 1H), 4.49–4.30 (m, 1H), 4.20–4.08 (m, 3H), 4.07–3.96 (m, 2H), 3.66–3.51 (m, 1H), 1.43–1.33 (m, 3H), 1.28 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 173.4, 173.5, 162.9, 154.8, 154.0, 153.9, 152.3, 150.6, 144.4, 135.2, 134.7, 130.1, 130.0, 129.8, 128.8, 128.8, 128.7, 128.6, 128.5, 128.5, 125.3, 125.0, 120.3, 120.2, 95.7 89.1, 80.1, 76.7, 72.6, 70.9, 68.1, 64.9, 61.8, 50.5, 21.1,14.2; 31P NMR (162 MHz, CDCl3) δ 3.14, 3.00; MS-ESI+ m/z 901 (M+H+); HRMS-ESI+: m/z calcd for C44H45N4O15NaP (M+Na+) 923.2530, found 923.2525.
4.1.4. 5′-O-[Phenyl-(ethoxy-L-alaninyl)]phosphorylcytidine (6a)
To a solution of 5a (0.37 g, 0.41 mmol) in 10 mL of EtOH was added Pd/C (0.02 g, 10% Pd on activated carbon) at rt. The mixture was stirred for 2 h under an atmosphere of H2 (1 atm) and then treated with Celite (1.0 g) and stirred for 30 min. The suspension was filtered and washed with MeOH (10 mL×3). The collected solution was concentrated under reduced pressure and the residue was purified on silica gel column chromatography (MeOH/EtOAc=1:10 v/v) to give 6a (0.20 g, 0.40 mmol), as 1:1. diastereomeric (RP/SP) mixture, in 98% yield. 1H NMR (400 MHz, CD3OD) δ 7.75–7.67 (m, 1H), 7.36–7.31 (m, 2H), 7.24–7.13 (m, 3H), 5.88–5.79 (m, 3H), 4.46–4.26 (m, 2H), 4.18–4.06 (m, 3H), 4.06–4.02 (m, 1H), 3.94–3.85 (m, 1H), 1.33–1.28 (m, 3H), 1.24–1.14 (m, 3H); 13C NMR (100 MHz, CD3OD) δ 175.1, 167.7, 158.6, 142.5, 131.0,130.2,126.4,124.0,121.6,121.5, 96.5, 92.1, 83.6, 76.1, 70.8, 67.2, 62.5, 51.8, 20.6, 14.6; 31P (162 MHz, CD3OD) δ 4.89, 4.74; MS-ESI+ m/z 499 (M+H+); HRMS-ESI+: m/z calcd for C20H27N4O9NaP (M+Na+) 521.1403, found 521.1408.
4.1.5. 5′-O-tert-Butyldimethylsilyl-2′,3′-O-bis-benzyloxycarbonyluridine (3b)
Compound 3b was prepared using the same procedure as 3a: yield 96%; 1H NMR (400 MHz, CDCl3) δ 9.25 (s, 1H), 7.83 (d, J=8.0 Hz, 1H), 7.34–7.31 (m, 10H), 6.28 (d, J=4.8 Hz, 1H), 5.72 (d, J=8.0 Hz, 1H), 5.30–5.26 (m, 3H), 5.16–5.09 (m, 4H), 4.29 (q, J=1.6 Hz, 1H), 3.95 (dd, J=2.0, 11.6 Hz, 1H), 3.82 (dd, J=2.0, 11.6 Hz, 1H), 0.94 (s, 9H), 0.13 (s, 3H), 0.12 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 163.2, 154.2, 154.0, 150.4, 139.6, 134.7, 134.7, 128.9, 128.8, 128.7, 128.5, 103.2, 85.7, 83.0, 74.3, 70.6, 70.5, 62.8, 60.6, 26.0, 18.5, 14.35, −5.4; MS-ESI+ m/z 627 (M+H+).
4.1.6. 2′,3′-O-Bis-benzyloxycarbonyluridine (4b)
Compound 4b was prepared using the same procedure as 4a: yield 99%; 1H NMR (400 MHz, CDCl3) δ 9.69 (s, 1H), 7.64 (d, J=8.0 Hz,1H), 7.35–7.30 (m, 10H), 5.96 (d, J=6.0 Hz, 1H), 5.74 (d, J=8.0 Hz, 1H), 5.54 (dd, J=5.2, 6.0 Hz, 1H), 5.45 (dd, J=3.6, 5.2 Hz, 1H), 5.12–5.05 (m, 4H), 4.25 (d, J=2.4 Hz, 1H), 3.89 (d, J=12.0 Hz, 1H), 3.80 (m, 1H), 3.69 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 163.8, 154.3, 154.0, 150.6, 141.7, 134.7, 134.6, 128.8, 128.8, 128.7, 128.5, 103.3, 88.6, 83.3, 75.8, 74.4, 70.7, 70.5, 61.7, 39.2; MS-ESI+ m/z 513 (M+H+).
4.1.7. 5′-O-[Phenyl-(ethoxy-L-alaninyl)]phosphoryl-2′,3′-O-bis-benzyl-oxycarbonyluridine (5b)
Compound 5b was prepared using the same procedure as 5a: yield 98% (1:1 diastereomeric (RP/SP) mixture). 1H NMR (400 MHz, CDCl3) δ 10.11–10.00 (m, 1H), 7.89 (d, J=8.0 Hz, 0.5H), 7.46 (d, J=8.8 Hz, 0.5H), 7.31–7.22 (m, 15H), 6.13–6.10 (m, 1H), 5.75–5.69 (m, 0.5H), 5.56–5.48 (m, 0.5H), 5.46–5.41 (m, 1H), 5.30 (t, J=5.6 Hz, 0.5H), 5.20 (t, J=5.6 Hz, 0.5H), 5.14–5.04 (m, 4H), 4.48–4.32 (m, 2H), 4.23–4.08 (m, 3H), 4.08–3.96 (m, 1H), 3.85–3.76 (m, 1H), 1.34–1.33 (m, 3H), 1.26–1.18 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 173.6, 173.4, 163.4, 153.9, 150.4, 139.8, 134.5, 129.9, 129.8, 129.6, 129.6, 128.7, 128.6, 128.6, 128.4, 128.4, 128.3, 103.4, 86.9, 86.3, 83.3, 80.2, 76.0, 75.5, 74.6, 73.1, 70.4, 70.2, 65.3, 61.6, 61.4, 60.4, 53.6, 50.2, 20.8, 14.0; 31P (162 MHz, CDCl3) δ 4.13, 4.07; MS-ESI+ m/z 768 (M+H+); HRMS-ESI+: m/z calcd for C36H38N3O14NaP (M+Na+) 790.2001, found 790.1997.
4.1.8. 5′-O-[Phenyl-(ethoxy-L-alaninyl)]phosphoryluridine (6b)
To a solution of 5b (0.50 g, 0.65 mmol) in 10 mL of EtOH was added 1,4-cyclohexadiene (1.30 mL) and Pd/C (0.03 g, 10% Pd on activated carbon) at rt. After stirring for 3 h at rt, the reaction mixture was treated with Celite (2.0 g) and stirred for 30 min. The suspension was filtered and washed with MeOH (15 mL×3). The collected solution was concentrated under reduced pressure and the residue was purified by silica gel column chromatography (MeOH/EtOAc=1:10 v/v) to give 6b (0.32 g, 0.64 mmol), as a 1:1 diastereomeric (RP/SP) mixture, in 98% yield. 1H NMR (400 MHz, CD3OD) δ 7.70 (d, J=8.0 Hz, 0.5H), 7.63 (d, J=8.0 Hz, 0.5H), 7.39–7.33 (m, 2H), 7.27–7.18 (m, 3H), 5.91 (d, J=8.8 Hz, 0.5H), 5.89 (d, J=8.8 Hz, 0.5H), 5.69 (d, J=8.0 Hz, 0.5H), 5.61 (d, J=8.0 Hz, 0.5H), 4.44–4.29 (m, 2H), 4.19–4.09 (m, 5H), 3.98–3.91 (m, 1H), 3.87–3.76 (m, 1H), 1.36–1.30 (m, 3H), 1.25–1.21 (m, 3H); 13C NMR (100 MHz, CD3OD) δ 175.2, 166.2, 152.2, 142.4, 131.0, 130.8, 126.4, 126.2, 121.5, 103.2, 90.8, 84.0, 75.3, 71.1, 67.4, 62.5, 62.4, 51.7, 20.6, 14.6; 1P (162 MHz, CD3OD) δ 4.94, 4.76; MS-ESI+ m/z 500 (M+H+); HRMS-ESI+: m/z calcd for C20H26N3O10NaP (M+Na+) 522.1253, found 522.1256.
4.1.9. 5′-O-tert-Butyldimethylsilyl-2′,3′-O-bis-benzylox-ycarbonyladenosine (3c)
Compound 3c was prepared using the same procedure as 3a: yield 92% (two steps); 1H NMR (400 MHz, CDCl3) δ 8.33 (s, 1H), 8.15 (s, 1H), 7.37–7.29 (m, 10H), 6.33 (d, J=6.4 Hz, 1H), 5.81 (dd, J=4.8, 6.0 Hz, 1H), 5.66 (br, 2H), 5.52 (dd, J=4.8, 6.0 Hz, 1H), 5.17–5.05 (m, 4H), 4.37 (q, J=2.8 Hz, 1H), 3.95 (dd, J=2.8, 11.6 Hz, 1H), 3.80 (dd, J=2.8, 11.6 Hz, 1H), 0.94 (s, 9H), 0.13 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 155.6, 154.4, 153.9, 153.6, 150.3, 142.8, 138.9, 134.8, 134.7, 128.9, 128.9, 128.9, 128.8, 128.6, 120.0, 85.0, 83.3, 77.0, 74.7, 70.6, 70.5, 62.9, 26.1, −5.2, −5.3; MS-ESI+ m/z 650 (M+H+).
4.1.10. 2′,3′-Bis-benzyloxycarbonyladenosine (4c)
Compound 4c was prepared using the same procedure as 4a: yield 98%; 1H NMR (400 MHz, CDCl3) δ 8.25 (s,1H), 7.71 (s,1H), 7.36–7.27 (m,10H), 7.00 (d, J=10.8 Hz, 2H), 6.85 (br, 2H), 6.10 (dd, J=4.8, 7.6 Hz, 1H), 6.03 (d, J=7.6 Hz, 1H), 5.70 (d, J=4.8 Hz, 1H), 5.16–5.01 (m, 4H), 4.43 (s, 1H), 3.97 (d, J=12.4 Hz, 1H), 3.83 (t, J=12.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 156.4, 154.2, 153.6, 152.7, 148.3, 140.2, 134.7, 134.5, 128.8, 128.7, 128.7, 128.6, 128.5, 87.7, 85.8, 76.1, 75.6, 70.6, 70.3, 62.6, 60.4, 21.1, 14.2; MS-ESI+ m/z 536 (M+H+).
4.1.11. 5′-O-[Phenyl-(ethoxy-L-alaninyl)]phosphoryl-2′,3′-O-bis-benzyl-oxycarbonyladenosine (5c)
Compound 5c was prepared using the same procedure as 5a: yield 92% (1:1 diastereomeric (RP/SP) mixture); 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 0.5H), 8.26 (s, 0.5H), 7.99 (s, 0.5H), 7.93 (s, 0.5H), 7.34–7.19 (m, 15H), 6.25 (br, 2H), 6.19–6.17 (m, 1H), 6.02–5.93 (m, 1H), 5.72 (m, 1H), 5.13–5.03 (m, 7H), 4.54–4.37 (m, 2H), 4.14–3.96 (m, 2H),1.35–1.25 (m, 3H),1.18 (m, 3H); δ −158.03; 13C NMR (100 MHz, CDCl3) δ 173.7, 173.6, 155.8, 154.1, 153.8, 153.3, 150.7, 149.8, 139.6, 134.6, 129.9, 128.9, 128.8, 128.7, 128.6, 125.1, 120.4, 120.3, 120.2, 85.9, 80.7, 75.8, 73.6, 70.6, 65.3, 50.4, 47.9, 21.0, 14.2; 31P (162 MHz, CDCl3) δ 3.23, 3.16; MS-ESI+ m/z 791 (M+H+); HRMS-ESI+: m/z calcd for C37H40N6O12P (M+H+) 791.2435, found 791.2436.
4.1.12. 5′-O-[Phenyl-(ethoxy-L-alaninyl)]phosphoryladenosine (6c)
Compound 6c was prepared using the same procedure as 6a: yield 94% (1:1 diastereomeric (RP/SP) mixture); 1H NMR (400 MHz, CD3OD) δ 8.25–8.16 (m, 2H), 7.31–7.28 (m, 2H), 7.20–7.11 (m, 3H), 6.02 (t, J=4.8 Hz, 1H), 4.66–4.61 (m, 1H), 4.45–4.30 (m, 3H), 4.29–4.23 (m, 1H), 4.09–4.01 (m, 2H), 3.91–3.08 (m, 1H), 1.27–1.20 (m, 3H), 1.18–1.13 (m, 3H); 13C NMR (100 MHz, CD3OD) δ 175.1, 157.4, 154.1, 152.2, 150.8, 141.1, 130.9, 130.1, 126.3, 121.5, 121.5, 120.6, 90.0, 84.5, 75.5, 71.7, 67.4, 62.5, 51.7, 20.5, 14.6; 31P (162 MHz, CD3OD) δ 4.93, 4.78; MS-ESI+ m/z 523 (M+H+); HRMS-ESI+: m/z calcd for C21H27N6O8NaP (M+Na+) 545.1517, found 545.1515.
4.1.13. 5′-O-tert-Butyldimethylsilyl-2′,3′-O-bis-benzyloxycarbonyl-guanosine (3d)
To a solution of guanosine (2.0 g, 7.06 mmol) in 20 mL of anhydrous DMSO was added DMAP (0.09 g, 0.71 mmol), Et3N (1.07 g, 10.6 mmol), and TBSCl (1.17 g, 7.76 mmol) at 0 °C under N2 atmosphere. After stirring for 24 h at rt, 20 mL of anhydrous CH2Cl2, CbzCl (4.82 g, 28.24 mmol), and DMAP (4.32 g, 35.30 mmol) was added to the solution at 0 °C under N2 atmosphere. After stirring for 48 h at rt, the reaction mixture was concentrated under reduced pressure and the residue was dissolved in CH2Cl2 (100 mL) and washed with cold 0.5 M HCl solution (30 mL×2), cold water (30 mL), and brine (30 mL). The organic layer was dried over Na2SO4, and filtered. The filtrate was concentrated and purified by silica gel column chromatography (CH2Cl2/MeOH=30:1 to 10:1 v/v) to give 3d (4.32 g, 6.49 mmol) in 92% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.74 (br, 1H), 7.85 (s, 1H), 7.40–7.29 (m, 10H), 6.57 (br, 2H), 5.99 (d, J=7.2 Hz, 1H), 5.67 (dd, J=5.2, 7.2 Hz, 1H), 5.40 (dd, J=2.8, 5.2 Hz, 1H), 5.18–5.10 (m, 4H), 4.30 (q, J=3.6 Hz, 1H), 3.87 (d, J=3.6 Hz, 2H), 0.89 (s, 9H), 0.09 (3H), 0.08 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 173.7, 156.6, 154.1, 153.5, 153.1, 151.3, 135.0, 134.8, 134.3, 128.7, 128.6, 128.5, 128.4, 128.2, 116.4, 82.9, 82.4, 76.1, 74.6, 69.9, 69.7, 62.8, 47.3, 33.9, 25.8, 25.5, 18.0, −5.5, −5.6; MS-ESI+ m/z 666 (M+H+); HRMS-ESI+: m/z calcd for C32H40N5O9Si (M+H+) 666.2601, found 666.2603.
4.1.14. 2′,3′-O-Bis-benzyloxycarbonylguanosine (4d)
Compound 4d was prepared using the same procedure as 4a: yield 92%; 1H NMR (400 MHz, DMSO-d6) δ 10.72 (br, 1H), 7.99 (s, 1H), 7.38–7.29 (m, 10H), 6.55 (br, 2H), 5.98 (d, J=7.6 Hz, 1H), 5.72 (dd, J=5.2, 7.6 Hz, 1H), 5.47 (t, J=5.2 Hz,1H), 5.42 (dd, J=2.0, 5.2 Hz,1H), 5.17–5.06 (m, 5H), 4.26 (q, J=2.0 Hz,1H), 3.68 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 186.5, 175.4, 173.7, 156.6, 154.0, 153.6, 153.1, 151.2, 135.2, 135.0, 134.9, 128.6, 128.6, 128. 5, 128.5, 128.4, 128.2, 116.5, 83.1, 83.1, 76.1, 75.1, 69.8, 69.6, 60.9, 47.3; MS-ESI+ m/z 552 (M+H+); HRMS-ESI+: m/z calcd for C26H26N5O9 (M+H+) 552.2601, found 552.2603.
4.1.15. 5′-O-[Phenyl-(ethoxy-L-alaninyl)]phosphoryl-2′,3′-O-bis-benzyl-oxycarbonylguanosine (5d)
Compound 5d was prepared using the same procedure as 5a: yield 90% (1:1 diastereomeric (RP/SP) mixture); 1H NMR (400 MHz, CD3OD) δ 7.73 (s, 1H), 7.31–7.13 (m, 15H), 7.10 (s, 1H), 7.00 (s, 1H), 6.07 (t, J=6.0 Hz, 1H), 5.89 (t, J=6.0 Hz, 0.5H), 5.81 (t, J=6.0 Hz, 0.5H), 5.69–5.64 (m, 1H), 5.16–5.01 (m, 5H), 4.45–4.32 (m, 3H), 4.09–4.04 (m, 2H), 3.94–3.85 (m, 1H), 1.29–1.23 (m, 3H), 1.16 (t, J=5.8 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ 175.2, 175.0, 159.4, 155.6, 155.3, 153.0, 152.2, 138.9, 138.4, 136.6, 131.0, 130.9, 130.0, 129.8, 129.8, 129.6, 129.6, 129.5, 128.1, 126.4, 122.3, 121.6, 87.2, 82.0, 77.2, 75.2, 71.6, 66.8, 62.5, 51.7, 34.1, 20.5,14.6; 31P (162 MHz, CD3OD) δ 4.98, 4.84; MS-ESI+ m/z 807 (M+H+); HRMS-ESI+: m/z calcd for C37H40N6O13P (M+H+) 807.2399, found 807.2399.
4.1.16. 5′-O-[Phenyl-(ethoxy-L-alaninyl)]phosphorylguanosine (6d)
Compound 6d was prepared using the same procedure as 6a: yield 95% (1:1 diastereomeric (RP/SP) mixture); 1H NMR (400 MHz, DMSO-d6) δ 10.73 (br, 1H), 7.84 (s, 1H), 7.38–7.14 (m, 5H), 6.57 (br, 2H), 6.11–6.02 (m, 1H), 5.76–5.71 (m, 1H), 5.56 (m, 1H), 5.37–5.33 (m, 1H), 4.43–4.39 (m, 1H), 4.24–3.98 (m, 6H), 3.80 (m, 1H), 1.23–1.18 (m, 3H), 1.15–1.10 (m, 3H); 13C NMR (100 MHz, DMSO-d6) δ 173.3, 173.2, 156.8, 153.8, 151.5, 150.7, 135.3, 130.0, 129.6, 124.6, 120.2, 116.7, 86.3, 82.5, 73.2, 70.2, 66.0, 60.6, 49.8, 19.7, 14.0; 31P (162 MHz, DMSO-d6) δ 4.66, 4.65; MS-ESI+ m/z 539 (M+H+); HRMS-ESI+: m/z calcd for C21H27N6O9NaP (M+Na+) 561.1468, found 561.1469.
4.1.17. 5′-O-tert-Butyldimethylsilyl-3′-O-benzyloxycarbonyl-2′-fluoro-2′-methyluridine (3e)
Compound 3e was prepared using the same procedure as 3a: yield 98% (two steps); 1H NMR (400 MHz, CDCl3) δ 9.90 (br, 1H), 8.05 (d, J=8.0 Hz, 1H), 7.40–7.35 (m, 5H), 6.27 (d, J=17.6 Hz, 1H), 5.72 (dd, J=1.6, 8.0 Hz, 1H), 5.20 (dd, J=12.0, 25.6 Hz, 2H), 5.16 (dd, J=9.6, 22.0 Hz, 1H), 4.26 (dd, J=1.2, 9.6 Hz, 1H), 4.10 (dd, J=1.2, 12.0 Hz, 1H), 3.71 (dd, J=1.2, 12.0 Hz, 1H), 1.42 (d, J=22.4 Hz, 3H), 0.91 (s, 9H), 0.07 (s, 3H), 0.06 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 163.5, 154.4, 150.7, 139.5, 134.6, 129.0, 128.8, 128.7, 103.1, 100.7, 98.9, 89.0 (d, J=39.3 Hz), 79.6, 77.4, 73.7 (d, J=15.4 Hz), 70.9, 60.0, 25.9, 18.5, 17.1 (d, J=24.7 Hz), −5.5, −5.7; 19F (376 MHz, CDCl3) δ −159.70, 159.74; MS-ESI+ m/z 509 (M+H+); HRMS-ESI+: m/z calcd for C24H34FN2O7Si (M+H+) 509.2126, found 509.2127.
4.1.18. 3′-Benzyloxycarbonyl-2′-fluoro-2′-methyluridine (4e)
Compound 4e was prepared using the same procedure as 4a: yield 98%; 1H NMR (400 MHz, CD3OD) δ 8.06 (d, J=8.0 Hz, 1H), 7.40–7.31 (m, 5H), 6.13 (d, J=18.4 Hz, 1H), 5.73 (d, J=8.0 Hz, 1H), 5.20 (s, 2H), 5.18 (dd, J=8.8, 20.8 Hz, 1H), 4.17 (dd, J=1.2, 8.8 Hz, 1H), 3.95 (dd, J=2.0, 13.2 Hz, 1H), 3.71 (dd, J=2.8, 13.2 Hz, 1H), 1.36 (d, J=22.8 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ 165.8, 156.0, 152.3, 142.0, 136.7, 129.8, 129.8, 129.5, 103.4, 102.0, 100.2, 91.1 (d, J=38.2 Hz), 81.4, 75.9 (d, J=15.5 Hz), 71.6, 60.1, 17.8 (d, J=25.5 Hz); 19F (376 MHz, CD3OD) δ −159.56; MS-ESI+ m/z 395 (M+H+); HRMS-ESI+: m/z calcd for C18H19FN2O7Na(M+Na+) 417.1069, found 417.1069.
4.1.19. 5′-O-[Phenyl-(ethoxy-L-alaninyl)]phosphoryl-3′-O-benzyloxy-carbonyl-2′-fluoro-2′-methyluridine (5e)
Compound 5e was prepared using the same procedure as 5a: yield 96% (1:1 diastereomeric (RP/SP) mixture); 1H NMR (400 MHz, CDCl3) δ 9.42–9.28 (m, 1H), 7.49–7.42 (m, 1H), 7.39–7.28 (m, 6H), 7.24–7.13 (m, 4H), 6.20 (br, 1H), 5.71–5.50 (m, 1H), 5.23–5.14 (m, 2H), 5.12–5.03 (m, 1H), 4.61–4.55 (m, 1H), 4.38–4.33 (m, 2H), 4.27–4.21 (m, 1H), 4.19–4.11 (m, 2H), 4.08–3.96 (m, 1H), 1.43–1.30 (m, 6H), 1.28–1.20 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 174.2, 162.9, 154.4, 150.3, 139.3, 134.7, 130.1, 129.8, 129.1, 129.1, 128.9, 128.9, 128.7, 128.6, 125.4, 125.0, 120.3, 120.2, 103.4, 100.1, 98.3, 77.7, 74.7, 71.1, 61.9, 50.5, 21.2, 17.6, 14.3; 19F (376 MHz, CDCl3) δ −159.56; 31P (162 MHz, CDCl3) δ 5.87, 5.90; MS-ESI+ m/z 650 (M+H+); HRMS-ESI+: m/z calcd for C29H34FN3O11P (M+H+) 650.1918, found 650.1923.
4.1.20. 5′-O-[Phenyl-(ethoxy-L-alaninyl)]phosphoryl-2′-fluoro-2′-methyluridine (6e)
Compound 6e was prepared using the same procedure as 6b: yield 96% (1:1 diastereomeric (RP/SP) mixture); 1H NMR (400 MHz, CDCl3) δ 10.05 (br, 1H), 7.50–7.14 (m, 6H), 6.17 (d, J=18.4 Hz, 1H), 5.70–5.54 (m, 1H), 4.59–4.31 (m, 3H), 4.30–4.22 (m, 1H), 4.20–4.11 (m, 2H), 4.07–3.96 (m, 1H), 3.94–3.78 (m, 1H), 3.46 (m, 1H), 1.41–1.28 (m, 6H), 1.27–1.22 (m, 3H); 19F (376 MHz, CDCl3) δ −162.41; 31P (162 MHz, CDCl3) δ 4.01, 3.58; MS-ESI+ m/z 516 (M+H+).
4.1.21. 5′-O-tert-Butyldimethylsilyl-N4-3′-O-bis-benzyloxycarbonyl-2′-fluoro-2′-methylcytidine (3f)
Compound 3f was prepared using the same procedure as 3a: yield 93% (two steps); 1H NMR (400 MHz, CDCl3) δ 8.44 (d, J=7.2 Hz, 1H), 7.40–7.26(m, 11H) 7.20 (br, 1H), 6.39 (d, J=17.2 Hz, 1H), 5.24–5.12 (m, 5H), 4.28 (dd, J=1.2, 9.2 Hz, 1H), 4.12 (dd, J=1.6, 12.0 Hz, 1H), 3.72 (dd, J=1.6, 12.0 Hz, 1H), 1.37 (d, J=24.4 Hz, 3H), 0.93 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 174.3, 162.7, 154.4, 144.1, 137.3, 135.1, 134.6, 130.5, 129.4, 129.0, 128.9, 128.8, 128.7, 128.6, 128.4, 127.6, 127.1, 117.3, 101.0, 99.1, 89.8 (d, J=3.9 Hz), 79.5, 73.6 (d, J=1.6 Hz), 70.8, 68.1, 65.3, 60.0, 26.0, 18.6, 17.0 (d, J=1.6 Hz), −5.5, −5.6; MS-ESI+ m/z 642 (M+H+); HRMS-ESI+: m/z calcd for C32H41FN3O8Si (M+H+) 642.2665, found 642.2642.
4.1.22. N4-3′-O-Bis-benzyloxycarbonyl-2′-fluoro-2′-methylcytidine (4f)
Compound 4f was prepared using the same procedure as 4a: yield 99%; 1H NMR (400 MHz, CDCl3) δ 8.45 (br, 1H), 8.16 (s, 1H), 7.45–7.32(m, 10H) 7.05 (s, 1H), 6.33 (d, J=7.6 Hz, 1H), 5.41 (s, 1H), 5.20–5.15 (m, 4H), 4.26 (d, J=9.2 Hz, 1H), 4.12 (d, J=12.8 Hz, 1H), 3.79 (d, J=12.0 Hz, 1H), 1.38 (d, J=22.4 Hz, 1H), 1.381 (d, J=22.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 162.9, 154.9, 148.7, 144.8, 137.3, 135.1, 134.4, 130.6, 129.3, 129.0, 128.9, 128.8, 128.8, 128.6, 128.5, 117.3, 101.0, 95.9, 80.0, 74.2 (d, J=1.6 Hz), 70.8, 70.1, 68.1, 65.3, 59.2, 17.0 (d, J=2.6 Hz); 19F (376 MHz, CDCl3) δ −159.89; MS-ESI+ m/z 528 (M+H+); HRMS-ESI+: m/z calcd for C26H27FN3O8 (M+H+) 528.1776, found 528.1777.
4.1.23. 5′-O-[Phenyl-(ethoxy-L-alaninyl)]phosphoryl-N4-3′-O-bis-benzyloxycarbonyl-2′-fluoro-2′-methylcytidine (5f)
Compound 5f was prepared using the same procedure as 5a: yield 93% (1:1 diastereomeric (RP/SP) mixture); 1H NMR (400 MHz, CDCl3) δ 8.14 (s, 1H), 7.86 (m, 1H), 7.46–7.06(m, 16H) 6.35 (br, 1H), 5.24–5.15 (m, 4H), 5.14–5.05 (m, 1H), 4.62–4.55 (m, 1H), 4.42–4.33 (m, 2H), 4.29–4.22 (m, 1H), 4.20–4.12 (m, 2H), 4.09–3.93 (m, 1H), 1.41–1.35 (m, 3H), 1.34–1.31 (m, 3H), 1.26–1.20 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 173.7, 162.8, 154.9, 154.3, 152.3, 150.7, 143.8, 137.3, 135.1, 134.4, 130.8, 130.1, 130.0, 129.4, 129.1, 128.9, 128.6, 128.6, 128.5, 125.4, 120.2, 120.1, 117.3, 100.4, 95.8, 77.5, 74.7, 71.0, 70.0, 68.2, 65.5, 64.9, 61.8, 50.5, 21.1, 17.4, 14.3; 19F (376 MHz, CDCl3) δ −158.94; MS-ESI+ m/z 783 (M+H+); HRMS-ESI+: m/z calcd for C37H41FN4O12P (M+H+) 783.2447, found 783.2451.
4.1.24. 5′-O-[Phenyl-(ethoxy-L-alaninyl)]phosphoryl-2′-fluoro-2′-methylcytidine (6f)
Compound 6f was prepared using the same procedure as 6a: yield 95% (1:1 diastereomeric (RP/SP) mixture); 1H NMR (400 MHz, CD3OD) δ 7.66–7.50 (m, 1.0 H), 7.40–7.29 (m, 2H), 7.26–7.16 (m,3H), 6.22 (br,1H), 5.86–5.78 (m,1H), 4.60–4.47 (m,1H), 4.45–4.35 (m, 1H), 4.33–4.22 (m, 1H), 4.17–4.06 (m, 3H), 3.97–3.77 (m, 2H), 1.38–1.18 (m, 9H); 19F (376 MHz, CD3OD) δ −162.81; 31P (162 MHz, CD3OD) δ 4.94, 4.80; MS-ESI+ m/z 515 (M+H+).
Acknowledgments
This work was supported in part by NIH grant 1RO1-AI071846, 5R37-AI-025899, 5R37-AI-041980, 5P30-AI-50409 (CFAR), and by the Department of Veterans Affairs. Dr. Schinazi is the founder and a major shareholder of RFS Pharma, LLC. Emory received no funding from RFS Pharma, LLC to perform this work and vice versa.
References and notes
- 1.For reviews on phosphates and phosphonates prodrugs: Bobeck DR, Coats SJ, Schinazi RF. Antiviral Ther. 2010;15:935–950. doi: 10.3851/IMP1667.Erion MD, Hecker SJ. J Med Chem. 2008;51:2328–2345. doi: 10.1021/jm701260b.Schultz C. Bioorg Med Chem. 2003;11:885–898. doi: 10.1016/s0968-0896(02)00552-7.
- 2.For a recent review on the ProTide approach see: Mehellou Y, Balzarini J, McGuigan C. ChemMedChem. 2009;4:1779–1791. doi: 10.1002/cmdc.200900289.
- 3.Siddiqui AQ, Ballatore C, McGuigan C, De Clercq E, Balzarini J. J Med Chem. 1999;42:393–399. doi: 10.1021/jm9803931. [DOI] [PubMed] [Google Scholar]
- 4.McGuigan C, Wedgwood OM, De Clercq E, Balzarini J. Bioorg Med Chem Lett. 1996;6:2359–2362. [Google Scholar]
- 5.McGuigan C, Hassan-Abdallah A, Srinivasan S, Wang Y, Siddiqui A, Daluge SM, Gudmundsson KS, Zhou H, McLean EW, Peckham JP, Burnette TC, Marr H, Hazen R, Condreay LD, Johnson L, Balzarini J. J Med Chem. 2006;49:7215–7226. doi: 10.1021/jm060776w. [DOI] [PubMed] [Google Scholar]
- 6.McGuigan C, Harris SA, Daluge SM, Gudmundsson KS, McLean EW, Burnette TC, Marr H, Hazen R, Condready LD, Johnson L, De Clercq E, Balzarini J. J Med Chem. 2005;48:3504–3515. doi: 10.1021/jm0491400. [DOI] [PubMed] [Google Scholar]
- 7.(a) McGuigan C, Thiery JC, Daverio F, Jiang WG, Davies G, Mason M. Bioorg Med Chem. 2005;13:3219–3227. doi: 10.1016/j.bmc.2005.02.041. [DOI] [PubMed] [Google Scholar]; (b) Congiatu C, Brancale A, Mason MD, Jiang WG, McGuigan C. J Med Chem. 2006;49:452–455. doi: 10.1021/jm0509896. [DOI] [PubMed] [Google Scholar]
- 8.Perrone P, Daverio F, Valente R, Rajyaguru S, Martin JA, Leveque V, Pogam SL, Najera I, Klumpp K, Smith DB, McGuigan C. J Med Chem. 2007;50:5463–5470. doi: 10.1021/jm070362i. [DOI] [PubMed] [Google Scholar]
- 9.Perrone P, Luoni GM, Kelleher MR, Daverio F, Angell A, Mulready S, Congiatu C, Rajyaguru S, Martin JA, Leveque V, Pogam SL, Najera I, Klumpp K, Smith DB, McGuigan C. J Med Chem. 2007;50:1840–1849. doi: 10.1021/jm0613370. [DOI] [PubMed] [Google Scholar]
- 10.Sofia MJ, Bao D, Chang W, Du J, Nagarathnam D, Rachakondfa S, Reddy PG, Ross BS, Wang P, Zhang HR, Bansal S, Espiritu C, Keilman M, Lam AM, Steuer HMM, Niu C, Otto MJ, Furman PA. J Med Chem. 2010;53:7202–7218. doi: 10.1021/jm100863x. [DOI] [PubMed] [Google Scholar]
- 11.Rondla R, Coats SJ, McBrayer TR, Grier J, Johns M, Tharnish PM, Whitaker T, Zhou L, Schinazi RF. Antiviral Chem Chemother. 2009;20:99–106. doi: 10.3851/IMP1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) McGuigan C, Gilles A, Madela K, Aljarah M, Holl S, Jones S, Vernachio J, Hutchins J, Ames B, Bryant KD, Gorovits E, Ganguly B, Hunley D, Hall A, Kolykhalov A, Liu Y, Muhammad J, Raja N, Walters R, Wang J, Chamberlain S, Henson G. J Med Chem. 2010;53:4949–4957. doi: 10.1021/jm1003792. [DOI] [PubMed] [Google Scholar]; (b) McGuigan C, Perrone P, Madela K, Neyts J. Bioorg Med Chem Lett. 2009;19:4316–4320. doi: 10.1016/j.bmcl.2009.05.122. [DOI] [PubMed] [Google Scholar]
- 13.Derudas M, Brancale A, Naesens L, Neyts J, Balzarini J, McGuigan C. Bioorg Med Chem. 2010;18:2748–2755. doi: 10.1016/j.bmc.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Birkus G, Kutty N, He GX, Mulato A, Lee W, McDermott M, Cihlar T. Antiviral Res. 2007;74:A57. [Google Scholar]
- 15.Zhou XJ, Pietropaolo K, Chen J, Khan S, Sullivan-Bolyai J, Mayers D. Antimicrob Agents Chemother. 2011;55:76–81. doi: 10.1128/AAC.01101-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Johnson DC, II, Widlanski TS. Org Lett. 2004;6:4643–4646. doi: 10.1021/ol048426w. [DOI] [PubMed] [Google Scholar]; (b) Watkins BE, Kiely JS, Rapoport H. J Am Chem Soc. 1982;104:5702–5708. [Google Scholar]; (c) Wandzik I, Bieg T, Kadela M. Nucleosides, Nucleotides Nucleic Acids. 2008;27:1250–1256. doi: 10.1080/15257770802458303. [DOI] [PubMed] [Google Scholar]
- 17.Sofia MJ, Du J, Wang P, Nagarathnam D. WO2008121634. 2008
- 18.Our attempts to apply this method to ProTides with a 2′-β-C-methyl sugar have been unsuccessful due to formation of a 2′,3′-carbonate during the CbzCl reaction.