

Abstract
Background&Aim
Signal transducer and activator of transcription 3 (STAT3), a key mediator of anti-inflammatory cytokine signaling, is essential for heme oxygenase-1 (HO-1)-induced cytoprotection. The phosphoinositide 3-kinase (PI3K)/phosphatase and tensin homolog delete on chromosome 10 (PTEN) pathways regulate diverse innate immune responses. This study was designed to investigate the role of STAT3 in the regulation of PI3K/PTEN cascade after HO-1 induction in a mouse model of innate immune-dominated liver ischemia/reperfusion injury (IRI).
Methods&Results
Mice subjected to Ad-HO-1 transfer were resistant to liver IRI, and this cytoprotective effect correlated with increased intrahepatic PI3K/Akt and diminished PTEN expression. In contrast, mice undergoing adjunctive Ad-HO-1 treatment and STAT3 knockdown (siRNA) remained susceptible to IR-mediated local inflammatory response and hepatocellular damage. Consistent with decreased cell apoptosis and inhibited TLR4 expression after PI3K/Akt activation, treatment with specific PI3k inhibitor increased local inflammation and recreated liver IRI despite Ad-HO-1 gene transfer. Parallel in vitro studies with bone marrow derived-macrophages have confirmed that HO-1 - STAT3 axis-induced PI3K/Akt negatively regulated PTEN expression in TLR4-dependent fashion.
Conclusion
These findings underscore the role of HO-1 induced STAT3 in modulating PI3K/PTEN in liver IRI cascade. Activating PI3K/Akt provides negative feedback mechanism for TLR4-driven inflammation. Identifying molecular pathways of STAT3 modulation in the innate immune system provides the rationale for novel therapeutic approaches for the management of liver inflammation and IRI in transplant patients.
Introduction
Liver ischemia and reperfusion injury (IRI), an innate immunity-dominated local inflammation, occurs in clinical settings involving interruption of the blood supply, such as surgical interventions, trauma or transplantation. In the latter, liver IRI represents one of the most critical problems, as it often leads to primary graft non-function, predisposes to chronic rejection, and contributes to shortage of organs available for transplantation [1].
IR insult to the liver is a multifaceted process that combines elements of “warm” and “cold” injury. The warm organ damage, occurring in-situ in low flow states, is dominated by Kupffer cell-derived cytotoxic molecule-mediated hepatocellular injury. Cold IRI experienced during ex-vivo preservation, is dominated by local inflammation mediated by neutrophils, damage to liver sinusoidal endothelial cells (SEC), and disruption of the microcirculation. These seemingly distinct processes share overlapping effects upon liver non-parenchyma (Kupffer cell/lymphocyte) and parenchyma (hepatocyte) cell functions, both of which lead to the organ failure [2-4]. Activated Kupffer cells release superoxide radicals, TNF-α and IL-1, which promote NF-κB activation, and recruitment of activated CD4+ T cells into the liver [5-7].
Heme oxygenase-1 (HO-1) exerts potent pro-survival functions against oxidative stress and organ damage [8]. Our group has pioneered the concept of HO-1-mediated cytoprotection against organ IRI in transplant recipients [1,9,10]. Moreover, HO-1 induction ameliorates macrophage infiltration in vivo [9], whereas mice receiving ex-vivo genetically modified macrophages overexpressing HO-1 are resistant to IRI [11]. In contrast, HO-1 deficiency increases the susceptibility to oxidative stress, enhances endothelial damage [12], and promotes allograft rejection [13].
We have shown that TLR4 signaling provides the triggering mechanism in the innate immune activation during liver IRI via downstream IFN regulatory factor (IRF) 3 [14]. The inhibition of HO-1 restored hepatic IRI [15], whereas Ad-HO-1 transfer rescued TLR4-deficient mice from hyperoxia-induced lung apoptosis by upregulating Bcl-2 and phosphorylated-Akt [16]. The protective role of HO-1 might depend on STAT3 activation [17], consistent with the ability of STAT3 to regulate immune homeostasis and influence cell differentiation [18]. STAT3 disruption leads to overactivated host innate immune responses [19]. Moreover, evidence suggests phosphatase and tensin homolog delete on chromosome 10 (PTEN), counter-regulated phosphoinositide 3-kinase (PI3K) activity in cell survival and growth [20-22]. Macrophage PTEN deficiency enhanced PI3K activity and decreased TLR-mediated cytokine expression [23]. Furthermore, PTEN inhibition increased ischemic myocardium survival [24] and reduced ischemic brain injury [25]. These functions associate with PI3K/Akt signaling [26], crucial for migration of neutrophils/monocytes to the inflammation site [27]. Pharmacologic inhibition of PI3K diminished NF-κB and inflammatory gene expression [28-30]. Although these protective effects relate to PI3K/PTEN signaling, little is known about the role of STAT3 in the regulation of this very process.
This study was designed to analyze the role of STAT3 in TLR4-driven innate immune response in the context of HO-1 cytoprotection. Ad-HO-1-induced STAT3 prevented liver IRI and inhibited TLR4-mediated inflammation. In parallel, STAT3 phosphorylation triggered PI3K/Akt signaling and prevented PTEN activation induced by IR. In contrast, STAT3 silencing increased TLR4/NF-κB expression and pro-inflammatory gene programs, inevitably leading to IR-hepatocellular damage. Hence, by orchestrating PI3K/PTEN signaling, the HO-1-STAT3 axis is essential in the mechanism of TLR4-driven inflammation in liver IRI immune cascade.
Materials and Methods
Animals
Male C57BL/6 wild-type (WT) mice at 6-8 weeks of age were used (The Jackson Laboratory, Bar Harbor, ME). Animals, housed in UCLA animal facility under specific pathogen-free conditions, received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” (NIH publication 86-23 revised 1985).
Preparation of siRNA
The siRNA against STAT3 was designed using the siRNA selection program [31]. The sense and antisense strands of murine STAT3 siRNA were 5′-CCUCCAGGACGACUUUGAU-3′ (sense) and 5′-AUCAAAGUCGUCCUGGAGG-3′ (antisense). The non-silencing (NS) siRNA were 5′-UUCUCCGAACGUGUCACGU-3′ (sense) and 5′-ACGUGACACGUUCGGAGAA-3′ (antisense). All siRNAs were synthesized in 2′-deprotected, duplexed, desalted and purified siRNA form (Qiagen Inc., Chatsworth, CA).
Mouse liver IRI model
We used well-established mouse model of warm hepatic ischemia followed by reperfusion [7,11,14,15,31]. Mice were injected with heparin (100U/kg) and an atraumatic clip was used to interrupt the arterial/portal venous blood supply to the cephalad liver lobes. After 90min the clip was removed; mice were sacrificed at 6h of reperfusion. Ad-HO-1 or Ad-β-gal (2.5×109 pfu i.v.) was injected 24h prior to ischemia. STAT3 siRNA or nonspecific siRNAs (2mg/kg i.v.) was administered 4h prior to ischemia. PI3K inhibitor (LY294002, 0.5mg/kg i.p.) was given 30min prior to ischemia.
Hepatocellular damage assay
Serum glutamic-pyruvic transaminase (sGPT) levels, an indicator of hepatocellular injury, were measured with an autoanalyzer (ANTECH Diagnostics, Los Angeles, CA).
Histology, immunohistochemistry and double-immunofluorescence staining
Livers sections (5-μm) were stained with hematoxylin and eosin (H&E). The severity of IRI was graded using Suzuki's criteria on a scale from 0-4 [32]. Liver macrophages were detected using primary mAb against mouse CD11b (Mac-1, M1/70; BD Biosciences, San Jose, CA) followed by incubating with secondary Ab, biotinylated goat anti-rat IgG (Vector, Burlingame, CA). HO-1 and STAT3 double-positive cells were identified by immunoflurorescence. Goat anti-mouse CD68 (macrophage) and rabbit anti-mouse HO-1 or phos-STAT3 (Cell Signaling Technology, Danvers, MA) mAbs were used. After incubation with secondary goat anti-rabbit FITC-conjugated IgG (Sigma-Aldrich, Inc., St. Louis, MO) and rabbit anti-goat Texas Red-conjugated IgG (Vector), the samples were pre-mounted with VECTASHIELD medium with DAPI (Vector). Positive cells were counted blindly in 10 HPF/section ( ×200).
Myeloperoxidase activity assay
The presence of myeloperoxidase (MPO) was used as an index of hepatic neutrophil accumulation [11]. The change in absorbance was measured spectrophotometrically at 655nm. One unit of MPO activity was defined as the quantity of enzyme degrading 1μmol peroxide/min at 25°C per gram of tissue.
Caspase-3 activity assay
Proteins (30 μg/sample) from BMMs were incubated with 200μM of enzyme-specific colorimetric caspase-3 substrate at 37°C for 2h [11]. Caspase-3 activity was assessed by measuring the absorbance at a wavelength of 405nm. To determine cellular activity, the inhibitor-treated protein extracts and the purified caspase-3 (standard) were used.
Malachite green phosphate assay
Liver protein lysates were immunoprecipitated with anti-PTEN Ab and incubated with protein A/G agarose beads. The PTEN malachite green assay was performed with beads-bound PTEN (Echelon Biosciences Inc., Salt Lake City, UT). The released phosphate was determined relative to a phosphatase standard curve.
TUNEL assay
The Klenow-FragEL DNA Fragmentation Detection Kit (EMD Chemicals, Gibbstown, NJ) was used to detect the DNA fragmentation characteristic of apoptosis in formalin-fixed paraffin-embedded liver sections [11].
Quantitative RT-PCR analysis
Quantitative real-time PCR was performed using the DNA Engine with Chromo 4 Detector (MJ Research, Waltham, MA). In a final reaction volume of 25μl, the following were added: 1 ×SuperMix (Platinum SYBR Green qPCR Kit; Invitrogen), cDNA and 10μM of each primer. Amplification conditions were: 50°C (2min), 95°C (5min), followed by 50 cycles of 95°C (15sec) and 60°C (30sec). Primers used to amplify specific gene fragments have been published [11,14,33]
Cell isolation and cultures
Murine bone marrow macrophages (BMM) were generated, as described [11]. Cells (1 ×106/well) were cultured for 7 days, and then transfected with Ad-HO-1 or Ad-β-gal (at multiplicity of infection [MOI]=10). After 24-48h, cells were added with 100ng/ml of LPS for additional 6h. In some experiments, cells were pretreated for 1h with either 10μM of PI3K inhibitor LY294002 (Calbiochem) or 6.5μl/ml of DMSO.
ELISA assay
Murine BMM culture supernatants were harvested for cytokine analysis. Mouse ELISA kits were used to measure TNF-α/IL-6 levels (eBiosciences).
Western blot analysis
Proteins (30μg/sample) from cell cultures or livers were subjected to 12% SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane (Bio-Rad, Hercules, CA). Polyclonal rabbit anti-mouse HO-1 (StressGen Biotech, Victoria, BC, Canada), TLR4 (Imgenex, San Diego, CA), phos-Stat3, Stat3, phos-Akt, Akt, PTEN, p-IκBα, IκBα, cleaved caspase-3, Bcl-2, Bcl-xl, and β-actin Abs (Cell Signaling Technology) were used. The relative quantities of proteins were determined by densitometer, and expressed in absorbance units (AU).
Statistical analysis
Data are expressed as mean±SD. Statistical comparisons were analyzed by Student's t-test. All differences were considered statistically significant at the p-value of <0.05.
Results
HO-1 - STAT3 signaling ameliorates liver IRI
We analyzed the hepatocellular damage in mouse livers subjected to 90min of warm ischemia followed by 6h reperfusion. The hepatocellular function, measured by sGPT levels (IU/L), improved in WT mice subjected to Ad-HO-1 but not Ad-β-gal (292±108 and 6003±2316, respectively, p<0.05) gene transfer (Fig. 1A). However, disruption of STAT3 signaling by using siRNA in Ad-HO-1-treated recipients, increased sGPT levels, compared to NS siRNA-treated controls (14059±5560 and 524±206, respectively, p<0.05). Liver serum enzyme data correlated with Suzuki's histological grading of IRI (Fig. 1B,C). Unlike Ad-β-gal-treated mice, which showed moderate-severe sinusoidal congestion, cytoplasmic vacuolization, and hepatocellular necrosis (Panel c; score=3.33±0.52), those given Ad-HO-1 had minimal pathological changes (Panel b; score=0.83±0.4, p<0.0001). Livers in animals conditioned with siSTAT3 and Ad-HO-1 revealed significant edema, sinusoidal congestion/cytoplasmic vacuolization, and 30-50% necrosis (Panel d; score=3.5±0.55). In contrast, livers in mice treated with NS siRNA showed mild-moderate edema and no necrosis (Panel e; score=1.2±0.41, p<0.0001). Furthermore, MPO assay (U/gm, Fig. 1D), reflecting neutrophil activity, was decreased in livers subjected to Ad-HO-1 (1.42±0.42) or NS siRNA (1.54±0.18), compared with Ad-β-gal (4.49±0.38, p<0.05) or siSTAT3 (5.69±0.31, p<0.05).
Figure 1.
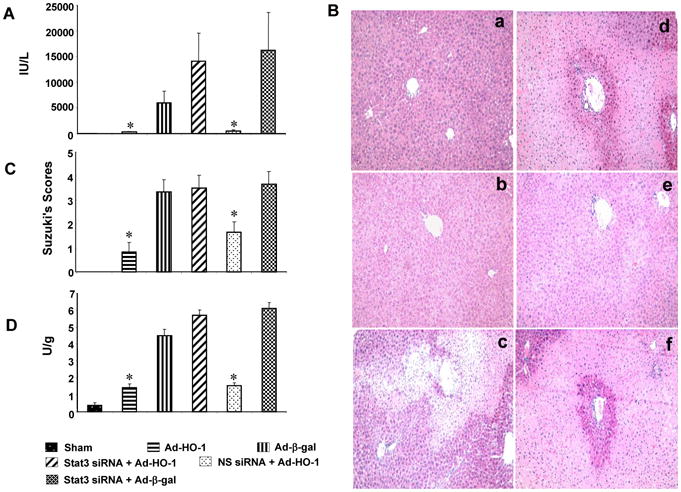
HO-1-STAT3 signaling reduces severity of liver IRI. Mice were subjected to 90min of partial liver warm ischemia, followed by 6h reperfusion.
(A). Hepatocellular function evaluated by sGPT (IU/L). Mean±SD (n=4-6 samples/group). *p<0.05.
(B/C). The severity of liver IRI by Suzuki's histological grading: (a) Sham; (b) Ad-HO-1 (0.83±0.4); (c) Ad-β-gal (3.33±0.52); (d) siSTAT3+Ad-HO-1 (3.5±0.55); (e) nonspecific siRNA+Ad-HO-1 (1.2±0.41); (f) siSTAT3+Ad-β-gal (3.6±0.51). Representative of 4-6 mice/group; original magnification ×200.
(D). Neutrophil accumulation analyzed by MPO activity (U/gm). Mean±SD (n=4-6 samples/group). *p< 0.05.
HO-1 - STAT3 signaling promotes PI3K/Akt and reduces IR induced-apoptosis
We found that by 6h of reperfusion after 90min of ischemia, the liver expression of phos-Akt, Bcl-2, and Bcl-xL was strongly up-regulated after Ad-HO-1, but not Ad-β-gal transfer (Fig. 2A,B). The expression of cleaved caspase-3 was inhibited in Ad-HO-1 but not Ad-β-gal group. However, knockdown of STAT3 in Ad-HO-1-transfected livers downregulated phos-Akt, Bcl-2, and Bcl-xL, yet upregulated cleaved caspase-3, compared with NS siRNA-controls. These findings were confirmed by increased caspase-3 activity in siSTAT3- but not NS siRNA-treated mice (Fig. 2C: 4.12±0.34 and 1.17+0.18, p<0.001). Ad-HO-1 but not Ad-β-gal decreased caspase-3 activity (0.95±0.22 and 3.96±0.35, respectively; p<0.001).
Figure 2.
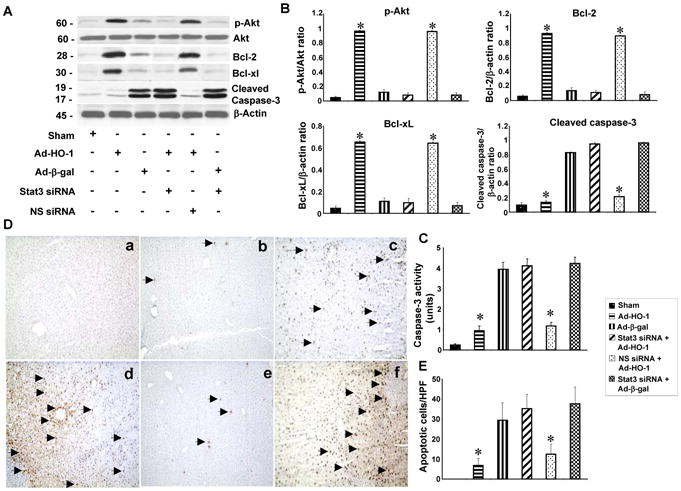
HO-1-STAT3 axis activates hepatic PI3K/Akt signaling and reduces IR-apoptosis.
(A/B) Western-assisted analysis of phos-Akt, Bcl-2, Bcl-xl and cleaved caspase-3. Representative of three experiments.
(C). Caspase-3 activity. Mean±SD; n=4-6 samples/group. *p<0.001.
(D/E). Liver apoptosis by TUNEL staining. (a) Sham; (b) Ad-HO-1; (c) Ad-β-gal; (d) siSTAT3+Ad-HO-1; (e) nonspecific siRNA+Ad-HO-1; (f) siSTAT3+Ad-β-gal. Apoptotic cells are marked (arrow). Results scored semi-quantitatively by averaging the number of apoptotic cells (mean±SD) per field at 200×magnification (representative of 4-6 mice/group).
We analyzed liver apoptosis by TUNEL staining (Fig. 2D,E). Ad-HO-1 decreased the frequency of apoptotic TUNEL+ cells in ischemic livers (Panel b: 6.8±3.6), compared with Ad-β-gal (Panel c: 29.5±8.8, p<0.0005). In contrast, livers treated with siSTAT3 showed increased frequency of TUNEL+ cells (Panel d: 35.3±7.2), compared with NS siRNA controls (Panel e: 12.5±5, p<0.001).
HO-1 - STAT3 signaling reduces TLR4/NF-κB-mediated IR-inflammation
We next investigated the role of STAT3 in the regulation of PTEN, TLR4 and NF-κB in vivo. Ad-HO-1 treatment enhanced HO-1 expression and STAT3 phosphorylation (Fig. 3A and 3B) in ischemic livers, compared with Ad-β-gal. However, STAT3 knockdown applied after Ad-HO-1 treatment diminished HO-1 and phos-STAT3, whereas NS siRNA plus Ad-HO-1 did maintain high HO-1 and STAT3 phosphorylation levels. Indeed, Ad-HO-1 decreased PTEN, TLR4 and phos-IκBα expression, as compared with Ad-β-gal. Conversely, PTEN, TLR4 and phos-IκBα increased sharply after disruption of STAT3 in Ad-HO-1- but not NS siRNA-treated group.
Figure 3.
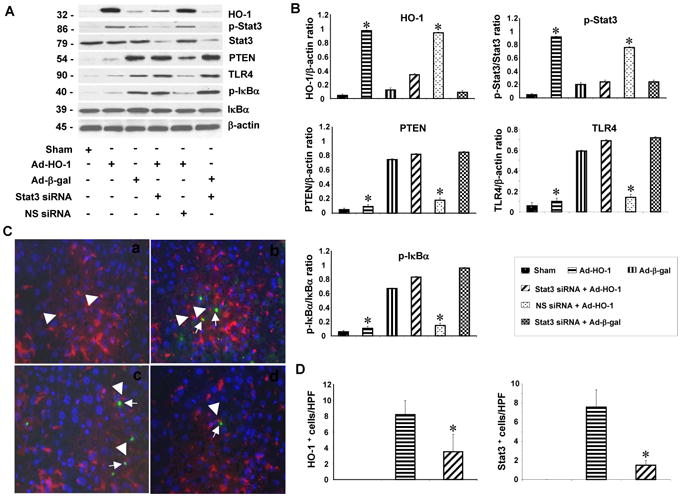
HO-1-STAT3 signaling reduces IR-induced TLR4 and NF-κB expression.
(A/B) Western-assisted detection of HO-1, phos-STAT3, PTEN, TLR4, and phos-IκBα. Representative of three experiments.
(C/D). Immunofluorescence staining of HO-1 and phos-STAT3 in ischemic livers. (a) Sham; (b) HO-1 expression in Ad-HO-1 treated livers; (c) Phos-STAT3 expression in Ad-HO-1 treated livers; (d) Phos-STAT3 expression in siSTAT3 treated livers. Note; positive staining in macrophages (head arrow), detected with CD68 mAb. Green: HO-1 and p-STAT3; Red: macrophage marker; Blue: DAPI nuclear stain. Results scored semi-quantitatively by averaging number of positively-stained cells (mean±SD)/field at 200×magnification. Representative of 4-6 mice/group.
We used immunofluorescence staining to identify HO-1 or phos-STAT3 double-positive cells. As shown in Figure 3C, resident hepatic CD68 macrophages stained positive for HO-1 and phos-STAT3. Although Ad-HO-1 gene transfer increased the number of double-positive HO-1 and phos-STAT3 cells, STAT3 knockdown decreased the frequency of HO-1+ and STAT3+ cells (Fig. 3D), suggesting macrophages are the major targets for Ad-HO-1 and siSTAT3 treatment. We observed marginal HO-1 and STAT3 expression in SEC after Ad-HO-1 transfer.
We used immunohistochemistry to study whether STAT3 may affect macrophage function. CD11b+ cells were decreased in the ischemic liver lobes after Ad-HO-1 but not Ad-β-gal treatment (Fig. 4A; Panel b: 12.5±6.2 and Panel c: 35.2±8.7, p<0.001). Disruption of STAT3 in Ad-HO-1-livers increased CD11b+ infiltration (Panel d: 41.2±7.7) compared to NS siRNA-treated group (Panel e: 20.2±7.9, p<0.001). Consistent with immunostaining data, our qRT-PCR results (Fig. 4C) showed mRNA levels coding for TNF-α, IL-6, MCP-1 and IP-10 to be consistently reduced in Ad-HO-1 but not Ad-β-gal-treated livers. However, the expression of proinflammatory cytokines increased sharply after STAT3 silencing despite concomitant Ad-HO-1. These results were supported by PTEN mRNA expression and PTEN phosphate release assay, when STAT3 knockdown increased PTEN mRNA expression and activity (Fig. 4D, E), indicating enhanced macrophage function after blocking STAT3 activation in the ischemic livers.
Figure 4.
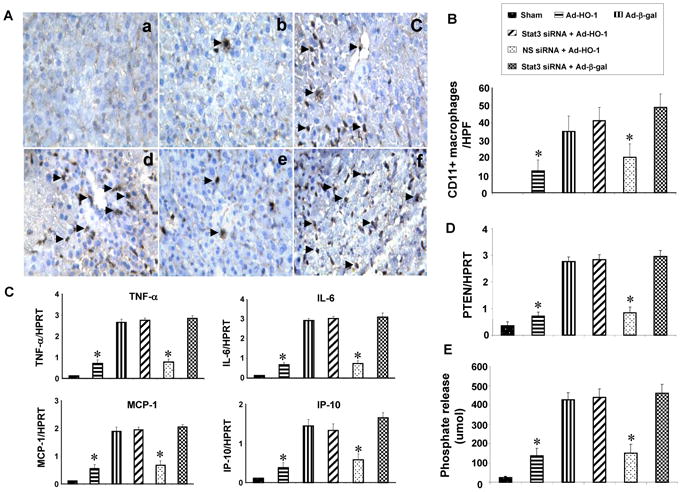
HO-1-STAT3 signaling regulates macrophage functions.
(A/B). Immunohistochemical staining of CD11b+ cells. (a) Sham; (b) Ad-HO-1; (c) Ad-β-gal; (d) siSTAT3+Ad-HO-1; (e) nonspecific siRNA+Ad-HO-1; (f) siSTAT3+Ad-β-gal. Results were scored semi-quantitatively by averaging the number of positively-stained cells (mean±SD)/field at 200×magnification. Representative of 4-6 mice/group.
(C). Quantitative RT-PCR-assisted cytokine/chemokine gene expression. Mean±SD (n=3-4 samples/group). *p<0.005.
(D). Quantitative RT-PCR-assisted detection of PTEN gene expression. Mean±SD (n=3-4 samples/group). *p<0.001.
(E). PTEN activity. Mean±SD; n=4-6 samples/group. *p<0.001.
PI3K/Akt inhibition recreates liver IRI
To address the functional role of PI3K/Akt signaling in liver cytoprotection, we used adjunctive treatment with PI3K inhibitor (LY294002) and Ad-HO-1. Increased sGPT levels were consistently found in mice treated with PI3K inhibitor with or without Ad-HO-1 (Fig. 5A, 12,286±3585 and 15930±3500, respectively), compared with Ad-HO-1 alone (362±100, p<0.05). Unlike Ad-HO-1 treated recipients, which showed minimal liver damage (Fig. 5B,C, Panel c; Suzuki score=0.83±0.41, p<0.0001), those given PI3K inhibitor revealed significant edema, sinusoidal congestion, cytoplasmic vacuolization, and hepatocellular necrosis (30-50%; Panel b; score=3.67±0.52). Livers in animals conditioned with PI3K inhibitor after Ad-HO-1 showed moderate-severe hepatocellular changes (Panel d; score=3.33±0.51). As shown in Fig. 5D, MPO levels (U/gm) were elevated in PI3K inhibitor-treated mice (5.89±0.49), compared with DMSO controls (3.45±0.38, p<0.05). In contrast, Ad-HO-1 treated livers showed decreased MPO activity (1.49±0.24), compared with PI3K inhibitor plus Ad-HO-1 (5.2±0.46, p<0.05).
Figure 5.
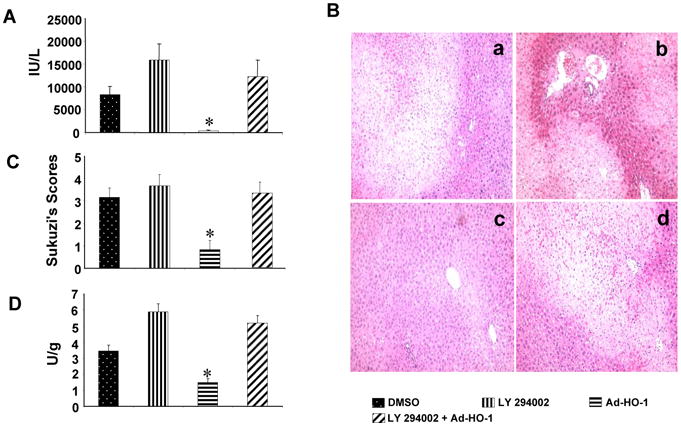
PI3K blockade recreates liver IRI in mice undergoing Ad-HO-1 gene transfer.
(A) The sGPT levels (IU/L). Mean±SD (n=4-6 samples/group). *p<0.05.
(B/C) Severity of liver IRI by Suzuki's histological scores. (a) DMSO control (3.16±0.41); (b) PI3K inhibitor LY294002 (3.67±0.52); (c) Ad-HO-1 (0.83±0.41); (d) LY294002+Ad-HO-1 (3.33±0.51). Representative of 4-6 mice/group; original magnification×200.
(D) Liver neutrophil accumulation (MPO activity; U/gm). Mean±SD; n=4-6 samples/group. *p< 0.05.
STAT3-induced PI3K/Akt negatively regulates PTEN/TLR4 expression in vitro
Our in vivo data suggest that STAT3 triggers PI3K/Akt signaling to promote HO-1-mediated cytoprotection. We then used a well-defined cell culture system to test a hypothesis that STAT3-induced PI3K/Akt may indeed regulate TLR4 in vitro. Thus, LPS-stimulated BMM were first pretreated with PI3K inhibitor (LY294002), and then transfected with Ad-HO-1. Indeed, Ad-HO-1 alone not only increased STAT3 and Akt phosphorylation (Fig. 6A,B), but also inhibited PTEN along with TLR4/IκB expression. However, inhibition of PI3K abolished Akt expression in BMMs, in association with increased PTEN mRNA (Fig. 6C) and protein expression, enhanced TLR4/IκB activation, regardless of the concomitant Ad-HO-1. Consistent with the latter, TNF-α and IL-6 production markedly increased in LPS-stimulated BMMs after treatment with PI3K inhibitor despite adjunctive Ad-HO-1 (Fig. 6D). These results document that STAT3-induced PI3K/Akt modulates TLR4-driven inflammatory response mediated by PTEN, confirming the key role of PI3K/PTEN signaling in the regulation of innate immune response.
Figure 6.
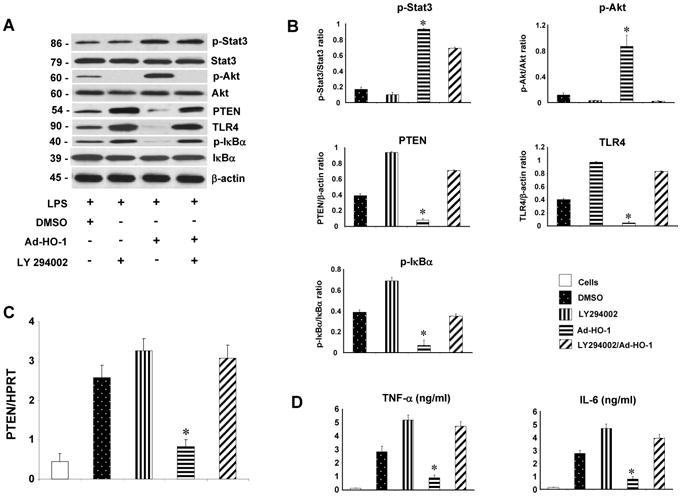
STAT3-induced PI3K/Akt signaling negatively regulates PTEN/TLR4 pathways in LPS-stimulated BMM in vitro.
(A/B) Western-assisted expression of phos-STAT3, Akt, PTEN, TLR4 and IκBα in LPS- stimulated BMMs after treatment with PI3K inhibitor (LY294002) and Ad-HO-1. Representative of three experiments.
(C) Quantitative RT-PCR-assisted detection of PTEN gene expression. Mean±SD (n=3-4 samples/group). *p<0.001.
(D) ELISA-based TNF- α/IL-6 levels in cell culture supernatants. Mean±SD (n=3-4 samples/group). *p<0.0005.
Discussion
This study documents the essential role of HO-1-STAT3 axis in orchestrating PI3K/PTEN signaling in the mechanism of TLR4-driven innate immune inflammation in liver IRI cascade. First, Ad-HO-1 cytoprotection was accompanied by STAT3-dependent increased hepatic PI3K/Akt and diminished PTEN expression. Second, consistent with decreased cell apoptosis and depressed TLR4 signaling after PI3K/Akt activation, pre-treatment with PI3k inhibitor has augmented local inflammation and recreated liver IRI despite Ad-HO-1 transfer. Third, HO-1-STAT3 pathway-induced PI3K/Akt negatively regulated PTEN expression. Our results also show that TLR4-mediated in vivo activation of IκB/NF-κB signaling via PI3K/Akt pathway. Thus, inhibition of PI3K/Akt promoted IκB phosphorylation and enhanced TLR4-driven inflammation in the liver. PTEN is known to antagonize PI3K/Akt by decreasing translocation of Akt to the cellular membrane [34]. In the current study, increased PTEN expression diminished phos-Akt, increased proinflammatory cytokine program and cell apoptosis. Our findings are in agreement with PI3K/Akt pathway serving as an endogenous negative-feedback or compensatory in vivo mechanism that can negatively regulate TLR4 to limit the hepatic inflammatory events [35]. By using double-immunofluorescence staining, we have identified macrophages as the major targets for Ad-HO-1 and siSTAT3 treatment. These results are consistent with our previous findings that macrophage HO-1 overexpression is important for cytoprotection in liver IRI [36].
We have first shown that STAT3 activation after Ad-HO-1 treatment improved the hepatocellular function in a mouse model of segmental liver warm IRI. Indeed, STAT3 knockdown in Ad-HO-1-treated mice increased sGPT levels, implicating STAT3 as an essential mediator in HO-1 cytoprotection, consistent with STAT3 mediated HO-1 transcription by increasing STAT3 DNA binding activity to HO-1 promoter [37]. A number of factors may contribute to protective effects of STAT3 activation. First, macrophages are critical in the initial uptake of pathogens, such as PAMPs and DAMPs, to further trigger inflammatory responses by activating TLR4 and NF-κB. Indeed, STAT3 knockdown enhanced inflammation by augmenting TLR4/NF-κB expression in Ad-HO-1-treated livers. Second, neutrophils contribute to local inflammation, and during liver IRI they enter hepatic parenchyma and become activated by TNF-α/IL-1, which in turn may lead to NF-κB activation and downstream gene transcription. In addition, neutrophil adherence to ICAM-1 and binding of leukocyte adhesion molecules to cellular and matrix ligands can trigger endothelial cell signaling and SEC apoptosis [3]. We have shown that prevention of neutrophil activation is essential for halting innate inflammation in hepatic IRI [38]. Our present results show that STAT3 activation in Ad-HO-1-treated mice decreased neutrophil activation, whereas STAT3 disruption reversed the process. This data was strengthened by histopathology evaluation, as STAT3 knockdown in Ad-HO-1 treated mice resulted in severe liver damage, compared with minimal pathological changes in mice conditioned with Ad-HO-1 with or without control siRNA. Hence, STAT3 disruption leads to neutrophil overactivation, which further enhance hepatic innate immune cascade. These findings need to be confirmed in STAT3-deficient mice when such become commercially available.
PTEN may function as a negative feedback repressor of PI3K/Akt activation [39]. Regulation of PI3K/Akt activity by cell-specific PTEN deficiency markedly reduced TNF-α release from primary macrophages [40]. However, the role of STAT3 in modulation of PI3K/PTEN signaling is poorly understood. Our data demonstrate that Ad-HO-1-induced STAT3 decreased PTEN mRNA levels and its protein expression, suggesting Ad-HO-1 prevents PTEN mRNA transcription induced by IR in a STAT3-dependent manner. Indeed, IR-induced PTEN expression was significantly decreased after activation of STAT3 by Ad-HO-1, whereas disruption of STAT3 resulted in the suppression of phos-Akt but activation of PTEN. These findings were in agreement with our in vitro experiments in which inhibition of PI3K/Akt by LY294002 resulted in PTEN upregulation, enhanced TLR4/NF-κB activity, and increased production of proinflammatory cytokines. Consistent with the ability of PI3K/Akt to modulate PTEN transcription/protein expression during inflammatory response, in vivo disruption of PI3K/Akt abolished local cytoprotection and recreated TLR4-driven IRI, despite Ad-HO-1 transfer. This implicates the critical role of STAT3-induced PI3K/Akt in the regulation of PTEN/TLR4 pathway against hepatic IRI. Indeed, TLR4 signaling is not only critical for triggering local inflammation [14] but can also dictate the severity of IR-induced liver damage [41]. Since Akt acts as an anti-apoptotic signaling molecule, it can inhibit caspase-mediated cell death through phosphorylation of Bcl-2/Bcl-xL-associated death promoter (BAD), releasing Bcl-2 family members, and direct phosphorylation of caspase protease [42]. Our results support the regulatory role of PI3K/Akt by downregulating caspase-3 activity, and upregulating anti-apoptotic Bcl-2/Bcl-xL, which in turn decrease apoptotic liver cell death. Consistently, STAT3 knockdown inactivated Akt and enhanced PTEN, evidenced by increased frequency of apoptotic cells in Ad-HO-1 treated livers.
Figure 7 depicts putative molecular mechanisms by which HO-1-STAT3 axis may regulate innate immunity in hepatic IRI. HO-1-induced STAT3 activates PI3K/Akt signaling and depresses PTEN activity. This process is STAT3-dependent. PI3K/Akt further promotes antiapoptotic Bcl-2/Bcl-xL expression, which in turn reduces hepatocellular apoptosis. In addition, PI3K/Akt inhibits downstream NF-κB activation, resulting in the suppression of hepatic inflammatory gene programs. PI3k/Akt may also provide negative regulatory feedback for TLR4 pathway to inhibit IκB phosphorylation and NF-κB translocation.
Figure 7.

HO-1-STAT3 axis in the regulation of innate inflammation signaling pathways in liver IRI.
In conclusion, HO-1-STAT3 axis regulates innate immune responses in liver IRI. The PI3K/PTEN signaling is crucial in, and PI3K/Akt provides the negative feedback mechanism for hepatic TLR4-driven inflammation. By identifying molecular pathways in STAT3 modulation in the innate immune system, our studies provide the rationale for novel therapeutic approaches to manage hepatic inflammation and IRI in liver transplant recipients.
Supplementary Material
Acknowledgments
NIH Grants DK 062357-07; DK 062357-06S1; DK 083408-01A1 and The Dumont Research Foundation.
Abbreviations
- Ad-HO-1
recombinant adenovirus encoding hemeoxygenase-1
- Ad-β-gal
recombinant adenovirus β-galactosidase reporter gene
- BMMs
bone marrow derived-macrophages
- IRI
ischemia/reperfusion injury
- MPO
myeloperoxidase
- PI3K
phosphoinositide 3-kinase
- PTEN
phosphatase and tensin homolog delete on chromosome 10
- sGPT
serum glutamic pyruvic transaminase
- siRNA
small interfering RNA
- SEC
sinusoidal endothelial cells
- TLR4
Toll-like receptor 4
- TUNEL
terminal deoxyribonucleotidyl transferase (TdT)-mediated dUTP-digoxigenin Nick End Labeling
Footnotes
Financial Disclosure: The authors have no financial arrangements and potential conflicts.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fondevila C, Busuttil RW, Kupiec-Weglinski JW. Hepatic ischemia/reperfusion injury--a fresh look. Exp Mol Pathol. 2003;74(2):86–93. doi: 10.1016/s0014-4800(03)00008-x. [DOI] [PubMed] [Google Scholar]
- 2.Jaeschke H, Bautista AP, Spolarics Z, Spitzer JJ. Superoxide generation by Kupffer cells and priming of neutrophils during reperfusion after hepatic ischemia. Free Radic Res Commun. 1991;15(5):277–284. doi: 10.3109/10715769109105223. [DOI] [PubMed] [Google Scholar]
- 3.Sindram D, Porte RJ, Hoffman MR, Bentley RC, Clavien PA. Platelets induce sinusoidal endothelial cell apoptosis upon reperfusion of the cold ischemic rat liver. Gastroenterology. 2000;118:183–191. doi: 10.1016/s0016-5085(00)70427-6. [DOI] [PubMed] [Google Scholar]
- 4.Samarasinghe DA, Tapner M, Farrell GC. Role of oxidative stress in hypoxia- reoxygenation injury to cultured rat hepatic sinusoidal endothelial cells. Hepatology. 2000;31:160–165. doi: 10.1002/hep.510310124. [DOI] [PubMed] [Google Scholar]
- 5.Colletti LM, Remick DG, Burtch GD, Kunkel SL, Strieter RM, Campbell AD., Jr Role of tumor necrosis factor-alpha in the pathophysiologic alterations after hepatic ischemia/reperfusion injury in the rat. J Clin Invest. 1990;85(6):1936–1943. doi: 10.1172/JCI114656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zwacka RM, Zhang Y, Halldorson J, Schlossberg H, Dudus L, Engelhardt JF. CD4(+) T-lymphocytes mediate ischemia/reperfusion-induced inflammatory responses in mouse liver. J Clin Invest. 1997;100(2):279–289. doi: 10.1172/JCI119533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen XD, Ke B, Zhai Y, Amersi F, Gao F, Anselmo DM, et al. CD154-CD40 T-cell costimulation pathway is required in the mechanism of hepatic ischemia/reperfusion injury, and its blockade facilitates and depends on heme oxygenase-1 mediated cytoprotection. Transplantation. 2002;74:315–319. doi: 10.1097/00007890-200208150-00005. [DOI] [PubMed] [Google Scholar]
- 8.Otterbein LE, Soares MP, Yamashita K, Bach FH. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. 2003;24:449–455. doi: 10.1016/s1471-4906(03)00181-9. [DOI] [PubMed] [Google Scholar]
- 9.Amersi F, Buelow R, Kato H, Ke B, Coito AJ, Shen XD, et al. Upregulation of heme oxygenase-1 protects genetically fat Zucker rat livers from ischemia/reperfusion injury. J Clin Invest. 1999;104:1631–9. doi: 10.1172/JCI7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katori M, Busuttil RW, Kupiec-Weglinski JW. Heme oxygenase 1 system in organ transplantation. Transplantation. 2002;74:905–12. doi: 10.1097/00007890-200210150-00001. [DOI] [PubMed] [Google Scholar]
- 11.Ke B, Shen XD, Gao F, Ji H, Qiao B, Zhai Y, et al. Adoptive transfer of ex vivo HO- 1 modified bone marrow-derived macrophages prevents liver ischemia and reperfusion injury. Mol Ther. 2010;18:1019–1025. doi: 10.1038/mt.2009.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yachie A, Niida Y, Wada T, Igarashi TN, Kaneda H, Toma T, et al. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J Clin Invest. 1999;103:129–135. doi: 10.1172/JCI4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soares MP, Lin Y, Anrather J, Csizmadia E, Takigami K, Sato K, et al. Expression of heme oxygenase-1 can determine cardiac xenograft survival. Nat Med. 1998;4:1073–1077. doi: 10.1038/2063. [DOI] [PubMed] [Google Scholar]
- 14.Zhai Y, Shen XD, O'Connell R, Gao F, Lassman CR, Busuttil RW, et al. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J Immunol. 2004;173:7115–7119. doi: 10.4049/jimmunol.173.12.7115. [DOI] [PubMed] [Google Scholar]
- 15.Shen XD, Ke B, Zhai Y, Gao F, Busuttil RW, Cheng G, et al. Toll-like receptor and heme oxygenase-1 signaling in hepatic ischemia/reperfusion injury. Am J Transplant. 2005;5:1793–1800. doi: 10.1111/j.1600-6143.2005.00932.x. [DOI] [PubMed] [Google Scholar]
- 16.Zhang X, Shan P, Qureshi S, Homer R, Medzhitov R, Noble PW, et al. Cutting edge: TLR4 deficiency confers susceptibility to lethal oxidant lung injury. J Immunol. 2005;175:4834–4838. doi: 10.4049/jimmunol.175.8.4834. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Shan P, Jiang G, Zhang SS, Otterbein LE, Fu XY, et al. Endothelial STAT3 is essential for the protective effects of HO-1 in oxidant-induced lung injury. FASEB J. 2006;20:2156–2158. doi: 10.1096/fj.06-5668fje. [DOI] [PubMed] [Google Scholar]
- 18.Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 19.Welte T, Zhang SS, Wang T, Zhang Z, Hesslein DG, Yin Z, et al. STAT3 deletion during hematopoiesis causes Crohn's disease-like pathogenesis and lethality: a critical role of STAT3 in innate immunity. Proc Natl Acad Sci U S A. 2003;100:1879–1884. doi: 10.1073/pnas.0237137100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 21.Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16:64–67. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- 22.Marsh DJ, Dahia PL, Zheng Z, Liaw D, Parsons R, Gorlin RJ, et al. Germline mutations in PTEN are present in Bannayan-Zonana syndrome. Nat Genet. 1997;164:333–334. doi: 10.1038/ng0897-333. [DOI] [PubMed] [Google Scholar]
- 23.Schabbauer G, Tencati M, Pedersen B, Pawlinski R, Mackman N. PI3K-Akt pathway suppresses coagulation and inflammation in endotoxemic mice. Arterioscler Thromb Vasc Biol. 2004;24:1963–1969. doi: 10.1161/01.ATV.0000143096.15099.ce. [DOI] [PubMed] [Google Scholar]
- 24.Cai Z, Semenza GL. PTEN activity is modulated during ischemia and reperfusion: involvement in the induction and decay of preconditioning. Circ Res. 2005;97:1351–1359. doi: 10.1161/01.RES.0000195656.52760.30. [DOI] [PubMed] [Google Scholar]
- 25.Omori N, Jin G, Li F, Zhang WR, Wang SJ, Hamakawa Y, et al. Enhanced phosphorylation of PTEN in rat brain after transient middle cerebral artery occlusion. Brain Res. 2002;954:317–322. doi: 10.1016/s0006-8993(02)03366-8. [DOI] [PubMed] [Google Scholar]
- 26.Lee JH, Kim KY, Lee YK, Park SY, Kim CD, Lee WS, et al. Cilostazol prevents focal cerebral ischemic injury by enhancing casein kinase 2 phosphorylation and suppression of phosphatase and tensin homolog deleted from chromosome 10 phosphorylation in rats. J Pharmacol Exp Ther. 2004;308:896–903. doi: 10.1124/jpet.103.061853. [DOI] [PubMed] [Google Scholar]
- 27.Curnock AP, Logan MK, Ward SG. Chemokine signalling: pivoting around multiple phosphoinositide 3-kinases. Immunology. 2002;105:125–136. doi: 10.1046/j.1365-2567.2002.01345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arbibe L, Mira JP, Teusch N, Kline L, Guha M, Mackman N, et al. Toll-like receptor 2-mediated NF-kappa B activation requires a Rac1-dependent pathway. Nat Immunol. 2000;1:533–540. doi: 10.1038/82797. [DOI] [PubMed] [Google Scholar]
- 29.Ojaniemi M, Glumoff V, Harju K, Liljeroos M, Vuori K, Hallman M. Phosphatidylinositol 3-kinase is involved in Toll-like receptor 4-mediated cytokine expression in mouse macrophages. Eur J Immunol. 2003;33:597–605. doi: 10.1002/eji.200323376. [DOI] [PubMed] [Google Scholar]
- 30.Rhee SH, Kim H, Moyer MP, Pothoulakis C. Role of MyD88 in phosphatidylinositol 3-kinase activation by flagellin/toll-like receptor 5 engagement in colonic epithelial cells. J Biol Chem. 2006;281:18560–18568. doi: 10.1074/jbc.M513861200. [DOI] [PubMed] [Google Scholar]
- 31.Ke B, Shen XD, Gao F, Qiao B, Ji H, Busuttil RW, et al. Small interfering RNA targeting heme oxygenase-1 (HO-1) reinforces liver apoptosis induced by ischemia-reperfusion injury in mice: HO-1 is necessary for cytoprotection. Hum Gene Ther. 2009;20(10):1133–1142. doi: 10.1089/hum.2009.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki S, Toledo-Pereyra LH, Rodriguez FJ, Cejalvo D. Neutrophil infiltration as an important factor in liver ischemia and reperfusion injury. Modulating effects of FK506 and cyclosporine. Transplantation. 1993;55:1265–1272. doi: 10.1097/00007890-199306000-00011. [DOI] [PubMed] [Google Scholar]
- 33.Meng F, Henson R, Wehbe–Janek H, Ghoshal K, Jacob ST, Patel T, et al. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–658. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuki A, de la Pompa JL, Stambolic V, Elia AJ, Sasaki T, del Barco Barrantes I, et al. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol. 1998;8:1169–1178. doi: 10.1016/s0960-9822(07)00488-5. [DOI] [PubMed] [Google Scholar]
- 35.Fukao T, Koyasu S. PI3K and negative regulation of TLR signaling. Trends Immunol. 2003;24:358–363. doi: 10.1016/s1471-4906(03)00139-x. [DOI] [PubMed] [Google Scholar]
- 36.Kato H, Amersi F, Buelow R, Melinek J, Coito AJ, Ke B, et al. Heme oxygenase-1 overexpression protects rat livers from ischemia/reperfusion injury with extended cold preservation. Am J Transplant. 2001;1:121–128. [PubMed] [Google Scholar]
- 37.Lee PJ, Camhi SL, Chin BY, Alam J, Choi AM. AP-1 and STAT mediate hyperoxia-induced gene transcription of heme oxygenase-1. Am J Physiol Lung Cell Mol Physiol. 2000;279:L175–L182. doi: 10.1152/ajplung.2000.279.1.L175. [DOI] [PubMed] [Google Scholar]
- 38.Uchida Y, Freitas MC, Zhao D, Busuttil RW, Kupiec-Weglinski JW. The inhibition of neutrophil elastase ameliorates mouse liver damage due to ischemia and reperfusion. Liver Transpl. 2009;15:939–947. doi: 10.1002/lt.21770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, et al. Nagative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 40.Luyendyk JP, Schabbauer GA, Tencati M, Holscher T, Pawlinski R, Mackman N. Genetic analysis of the role of the PI3K-Akt pathway in lipopolysaccharide-induced cytokine and tissue factor gene expression in monocytes/macrophages. J Immunol. 2008;180:4218–4226. doi: 10.4049/jimmunol.180.6.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uchida Y, Ke B, Freitas MC, Yagita H, Akiba H, Busuttil RW, et al. T-Cell Immunoglobulin Mucin-3 determines severity of liver ischemia/reperfusion injury in mice in a TLR4-dependent manner. Gastroenterology. 2010;139:2195–206. doi: 10.1053/j.gastro.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gatoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.