

Abstract
Coupling of cerebral blood flow (CBF) and cerebral metabolic rate for oxygen (CMRO2) in physiologically activated brain states remains the subject of debates. Recently it was suggested that CBF is tightly coupled to oxidative metabolism in a nonlinear fashion. As part of this hypothesis, mathematical models of oxygen delivery to the brain have been described in which disproportionately large increases in CBF are necessary to sustain even small increases in CMRO2 during activation. We have explored the coupling of CBF and oxygen delivery by using two complementary methods. First, a more complex mathematical model was tested that differs from those recently described in that no assumptions were made regarding tissue oxygen level. Second, [15O] water CBF positron emission tomography (PET) studies in nine healthy subjects were conducted during states of visual activation and hypoxia to examine the relationship of CBF and oxygen delivery. In contrast to previous reports, our model showed adequate tissue levels of oxygen could be maintained without the need for increased CBF or oxygen delivery. Similarly, the PET studies demonstrated that the regional increase in CBF during visual activation was not affected by hypoxia. These findings strongly indicate that the increase in CBF associated with physiological activation is regulated by factors other than local requirements in oxygen.
It was long assumed that changes in cerebral blood flow (CBF) and in the cerebral metabolic rate of oxygen (CMRO2) are tightly coupled in both resting and active brain states. This assumption resulted from the premises that the brain needs oxygen, that CBF is a main homeostatic factor for oxygen supply regulation, and that oxygen availability should be adjusted to meet tissue needs (1). It is known that the brain needs an abundant supply of oxygen and that, at rest, 80–92% of its ATP comes from oxidative metabolism of glucose. Early studies by Kety and Schmidt (2) and Cohen et al. (3) demonstrated that resting CBF does change with hypoxia and hyperoxia, thereby suggesting that CBF regulates oxygen delivery, although it was noted that blood oxygen levels but not tissue oxygen levels likely triggered these CBF changes (1). Therefore, if the cerebral oxygen supply was closely regulated to match tissue demands, then functional activation, which implies the need for additional ATP and oxygen, should cause a coupled increase in both CBF and CMRO2. However, two positron emission tomography (PET) studies conducted by Fox et al. (4, 5) revealed that in humans large, stimulus-induced increases in CBF (≈30% and 50%) were accompanied by only small increases in CMRO2 (≈5%). Others using PET and functional MRI confirmed these findings (6–10). The data indicated that, during short-term functional activation, CBF and CMRO2 are not directly coupled.
Recently, several reports using theoretical models suggested that the apparent uncoupling of CMRO2 and CBF might actually be a tight nonlinear coupling. Mathematical models of oxygen delivery to the brain have been described in which disproportionately large increases in CBF are necessary to sustain even small increases in CMRO2 during activation (11–13).
Buxton and Frank (11) solved the mathematics of a simple capillary model (14) to show that, for any increase in CMRO2, CBF must increase exponentially. For a CMRO2 increase of ≈5%, the calculated CBF increase by this model was similar to that seen by Fox and Raichle (4). Hyder et al. (12) noted that the Buxton and Frank model does not fit all of the available data on CBF and CMRO2 relationships. They augmented the model by allowing changes in the effective diffusivity of the capillary bed. This assumption permitted their model to explain a wide range of data, including studies where ratios of CMRO2 to CBF were closer to 1:1 (7, 15). Using a somewhat different mathematical strategy, Vafaee and Gjedde (13) also allowed oxygen diffusivity to be altered, giving different relationships between CBF and CMRO2. Unfortunately, the addition of variable oxygen diffusivity in the latter two models greatly decreases their predictive power. The authors did not define the conditions in which the changes in oxygen diffusivity are altered.
It is important to note that all of these models assume negligible levels of oxygen in brain tissue. This assumption allows for a much easier solution of the oxygen transport equations and also results in specific relationships between model parameters. Indeed, all three models concluded that CMRO2 is directly proportional to capillary oxygen tension. It is well recognized that CMRO2 can be related to CBF and oxygen extraction as CMRO2 = OEF⋅CBF⋅Ca, where OEF is the oxygen extraction fraction and Ca is the arterial oxygen content. These two relationships allow the mathematical solution of CBF as a function of CMRO2 or vice-versa (11). Essentially, the assumption that oxygen can only move out of the capillary (no back diffusion) at a rate proportionally to capillary oxygen will force CMRO2 to be mathematically coupled to oxygen delivery.
One test of these models would be to create hypoxia during functionally induced increases in neuronal activity. This proposal follows from the fact that all of the models have as a central feature the direct proportionality of CMRO2 to capillary oxygen levels. If arterial oxygen content actually decreases because of hypoxia, maintaining the same CMRO2 level would depend on an increase in CBF to support the total (or average) capillary oxygen content (or oxygen partial pressure). Because of the nonlinear relationship between CMRO2 and oxygen delivery predicted by these models, the addition of hypoxia to functional activation would require even greater CBF changes than that expected in normoxia conditions. Using PET, we tested this hypothesis in healthy subjects.
A second test of these models would be to determine whether the assumption that oxygen content in the brain tissue is negligible was critical to the conclusion that CBF and CMRO2 are tightly coupled. Thereto, we examined human brain oxygen transport by using a more general model of oxygen delivery and diffusion (16, 17). This approach, used by Hudetz (18) to examine oxygen delivery in rat brain, makes no assumptions regarding oxygen content or whether oxygen diffusion out of the capillary is bidirectional. Using values appropriate for human brain cortex, the more general but complex model can be used to test what degree of CBF is necessary to compensate increase in CMRO2 and to maintain adequate oxygen tissue levels. Furthermore, the model can also be used to simulate conditions of normoxia and hypoxia.
Materials and Methods
Mathematical Model of Tissue Oxygen Delivery.
Based directly on previous studies (16–18), a model was implemented that yields the partial pressure of oxygen (pO2) of the capillary along its axis and the oxygen tension in the tissue at points radially outward from the capillary. This model assumes that the capillary supplies oxygen to a cylindrical volume of tissue surrounding the vessel. Oxygen freely diffuses from the capillary and within tissue along radial tissue pO2 gradients; diffusion in the axial direction is considered minimal and ignored (19). Oxygen consumption is assumed to be spatially uniform compared with the scale of the model. These assumptions result in the linear decrease of oxygen content of blood as it travels down the capillary (see Table 1 for definition of variables):
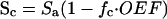 |
1 |
At the end of the capillary, the oxygen content has been reduced by the total amount extracted, or the OEF. As oxygen content decreases, the capillary pO2, presumed to be equal to the pO2 at the capillary-tissue interface, pOi, can be calculated by using hemoglobin's oxygen disassociation curve via the Hill equation:
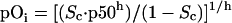 |
2 |
Once the intracapillary oxygen tension is calculated as a function of position down the capillary, the oxygen tension in the tissue radially distributed around the capillary can then be expressed as:
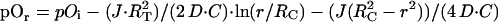 |
3 |
This explicit solution of tissue oxygen tension has been described previously in great detail (16, 19, 20) and results from the solution of Fick's second law of diffusion and standard boundary conditions that there is no net diffusion of oxygen out of the cylinder (r = RT) or across the middle of the cylinder (r = 0; ref. 16). Eq. 3 requires values for several physiologic parameters. These parameters and their values for human gray matter are listed in Table 1. Note that the resting CMRO2 is estimated for gray matter assuming a 4:1 ratio of gray-to-white matter metabolism. A second variable of importance is the assumed distance between capillaries. Intercapillary distances have been measured for a variety of tissues and species. To best illustrate the relationship, we have chosen a value of 64 microns for intercapillary distance, which represents the upper limit of what has been measured on human gray matter.
Table 1.
Variables and human parameter values used in the modeling
| Abbreviation | Parameter | Value |
|---|---|---|
| Sc | Fraction of oxygenated hemoglobin in the capillary | |
| fc | Fractional distance down capillary | |
| pOi | Partial pressure of O2 in the capillary | |
| r | Radial distance from the center of the capillary | |
| pOr | Partial pressure of O2 in tissue along the capillary radius | |
| Sa | Fraction of oxygenated hemoglobin in the artery* | 0.975 |
| OEF | Extracted O2 divided by total available O2† | 0.40 |
| p50 | Partial pressure of O2 for half occupied hemoglobin‡ | 26 mmHg |
| h | Hill constant‡ | 2.8 |
| J | O2 consumption§ | 8.20 × 10−4 cm3 O2/cm3/s |
| RT | Tissue radius¶ | 32 μ** |
| D | O2 diffusivity‖ | 1.8 × 10−5 cm2/sec |
| C | O2 solubility‖ | 2.6 × 10−5 cm3O2/cm3/mmHg |
| RC | Capillary radius¶ | 3 μm** |
Sa = 0.975 yields an arterial pO2 of 96.2 mmHg with the Hill constant and p50 listed below.
Common textbook values, e.g. Voet and Voet (23).
Global CMRO2 of 2.93 ml/min/100 g (24) was multiplied by 8/5 to convert to gray matter CMRO2 and 1.05/6000 to convert units.
Hunziker et al. (25) measured seven groups of human cerebral cortexes (total n = 38) and found intercapillary distances and SD that varied from 54.90–60.52 μm and 1.81–5.73 μm, respectively. We chose one-half of the higher intercapillary distance for the tissue radius.
Clark et al. (26).
For use in Eq. 5, the radii must be expressed as cm to be compatible with other parameters.
Tissue pO2 levels are the smallest in the tissue farthest from incoming blood (the outer edge of the cylinder at the venous end of the capillary), or the “lethal corner.” For the capillary to successfully supply oxygen to all surrounding brain tissue, cellular pO2 levels must not drop below 1 mmHg (1 mmHg = 133 Pa; ref. 27). When cellular pO2 is above 1 mmHg, ADP is the limiting factor in metabolism, and oxygen consumption is maintained. For any given configuration of the model the pO2 level in the “lethal corner” indicates whether all brain tissue is successfully oxygenated.
The model equations were used to calculate the spatial distribution of oxygen levels in the tissue in six conditions. We simulated a baseline state, neuronal activation, hypoxia, and neuronal activation + hypoxia. For the neuronal activation, we allowed both 5 and 15% increases in CMRO2, which brackets the range of reported changes in CMRO2 during visual activation studies (4, 7). Hypoxia was simulated by dropping arterial pO2 to 45 mmHg, similar to that achieved in our human PET experiments. To examine the assumption that CBF and CMRO2 changes are not directly coupled, CBF was not altered during these simulations. Thus, these simulations tested whether CBF increases are necessary to provide sufficient oxygen to the brain tissue. The levels of pO2 in the “lethal corner” were examined for each condition. In another set of simulations, the pO2 of arterial blood was lowered until the pO2 of the “lethal corner” fell below 1 mmHg and pO2 was recorded as the lowest level of hypoxia allowable without requiring increased CBF.
CBF and Oxygen Delivery Assessment in Human Subjects.
Human subjects.
Nine healthy right-handed subjects, four females and five males, aged 20 to 26 years old, were recruited from the Washington University community. The Human Studies Committee and the Radioactive Drug Research Committee of our institution approved the protocol of this study. Written informed consent was obtained from every subject.
PET imaging.
Eight quantitative CBF scans, in two blocks of four, were performed on each of nine normal subjects by using PET. The first block was obtained while the subject breathed room air (baseline) through a mouthpiece. The second block was obtained during breathing of a reduced oxygen air mixture with fraction of inspired oxygen of ≈12% to create mild hypoxia. This level of hypoxia is easily tolerated while at rest and is similar to standing on a summit of ≈14,000 feet (4267 m; e.g., Pike's Peak in Colorado). Studies were done by using a Siemens (Iselin, NJ)/CTI ECAT EXACT HR 47 tomograph (28). This scanner collects 47 simultaneous slices with 3.125-mm spacing encompassing an axial field of view of 15 cm. Transaxial and axial spatial resolution are ≈4.3 mm full-width half-maximum (FWHM) at slice center. Studies were done in the two-dimensional acquisition mode. All subjects had a left antecubital i.v. catheter and a right radial artery catheter. The arterial catheter (21-gauge) was placed under local anesthesia after confirmation of a negative Allen's test. A 12-lead ECG was attached, and a pulse oximeter was placed on an index finger.
The subject was positioned within the PET scanner (Siemens 961 ECAT HR EXACT) with the entire brain within the axial field-of-view and the cantho-meatal line (line between the closed eyelids and the external opening of the ear) parallel to the imaging planes. A computer-driven monitor was placed ≈61 cm from the subject's eyes. A mouthpiece was placed in the subject's mouth, and the subjects were reminded to keep their lips closed around the mouthpiece at all times. The mouthpiece was connected via a short stem (≈5 cm) to a T-valve. The inflow was connected to a manifold system and a reservoir, which was vented to the room. A nose clip was applied, and a thermoplastic mask was placed over the face and attached to the head holder.
A transmission scan for attenuation correction of all emission data was obtained. Each quantitative scan for CBF was obtained by injecting 50 mCi [15O] water by i.v. bolus, acquiring a 180-s dynamic PET scan (framing: 35 × 2 s, 10 × 5 s, 6 × 10 s) and arterial blood sampling by continuous arterial blood withdrawal over a shielded scintillator to count blood activity. Twelve minutes was allowed between tracer injections for [15O] decay (half-life of 2.02 min). Between the two blocks of scans, the air being breathed by the subject was switched from room air to a mixture with 1 part compressed air (20% O2) and 4 parts of a 90% nitrogen/10% oxygen mixture. Arterial blood gas measurements were performed approximately every 15 min. After stabilization of the arterial pO2 and arterial oxygen content, the second block of PET scans were begun. In one subject, the last CBF scan data were discarded because of elevated pO2 on arterial gases, which was determined to be due to leakage of room air in through a faulty valve in the T-valve system.
Each subject was questioned after the initiation of breathing room air through the mouthpiece, after initiation of hypoxia, and at the end of the hypoxia. Using hand signals (yes/no/intermediate), the subject could indicate any anxiety, shortness of breath, lightheadedness, or inability to concentrate. After the study was completed, the subjects were reinterviewed to confirm their responses.
Stimulation paradigm.
Each block of four PET scans consisted of a visual fixation (“control” state) scan, two successive visual activation tasks, and a final repeat visual fixation scan (ABBA design). In the visual fixation scan, the subject was instructed to fixate his/her gaze on a crosshair displayed in white on a black background on the monitor. For the visual activation task, subjects were asked to fixate their gaze on the same crosshair in the center of the monitor, but they were instructed to press a button with their right forefinger whenever the crosshair dimmed. The dimmings were programmed to occur at pseudorandom intervals averaging once per 5 s during the 180 s of scanning. Surrounding the crosshair, there was a black/white annular reversing (8 Hz) checkerboard similar to that used in Fox and Raichle (4).
Image processing.
The first step in the quantitation of CBF was to sum a set of 20 two-second frames from each dynamic PET scan to create 40-s “autoradiographic” scans (29). The first frame used to create the “autoradiographic” scan was determined by inspecting the whole-brain time-activity curve and selecting the frame 6 s after the tracer bolus arrived at the brain. The arterial blood activity curve was deconvolved for dispersion within the tubing to the scintillator and shifted in time to match the whole-brain time-activity curve. Each PET scan resulted in a quantitative image of CBF (30).
The eight autoradiographic CBF images for each subject were corrected for intrasession head motion (31). Using a standard threshold based on the summed image, the global, or whole brain, CBF value was calculated for each image. All data were spatially transformed to a standard atlas format (32) for creation of images summing the responses across all subjects (33). Images of the average CBF increase (with global normalization) from visual activation across all subjects were made in standard atlas coordinates.
Regional oxygen delivery was calculated as the product of regional CBF and measured arterial O2 content.
Volume of interest analysis.
A unique region-of-interest centered over the area of visual cortex activation was determined for each subject. First, all eight CBF images were normalized by their global averages. For each subject, all four visual fixation scans were summed and subtracted from all four visual activation scans. This individual summed subtraction image was then searched for the spherical region (13 mm diameter) of highest CBF change (34). This region was then applied to each of the quantitative CBF scans for calculation of the measured regional CBF.
The mean and standard deviation (SD) of regional CBF and global CBF for each of the four states (normoxia/fixation; normoxia/visual activation; hypoxia/fixation; and hypoxia/visual activation) were calculated and analyzed by using t tests (two-tailed, significance set at P < 0.05).
Results
Estimated Capillary and Tissue Oxygen Levels.
Fig. 1 demonstrates the spatial distribution of oxygen, as pO2, in capillary and brain tissue as estimated by the mathematical model for the baseline, or control state, condition. The graph shows the large gradient of oxygen down the capillary, as well as the oxygen concentration in the tissue moving radially out from the capillary. The “lethal corner” is seen to be well above critical level in this control state. In Fig. 2, the consequences of hypoxia, neuronal activation and hypoxia plus neuronal activation to the “lethal corner” are displayed. These simulations demonstrate that, even without increasing blood flow, the tissue can maintain oxygen tissue levels adequate for oxidative metabolism under all of these conditions. The level of arterial pO2 that can be reached before blood flow would have to increase because of lack of tissue oxygen was determined to be 30.9 mmHg.
Figure 1.

Simulation results for pO2 distribution at baseline levels of CBF and CMRO2 (see Table 1). Oxygen tension decreases proceeding along the capillary and moving out radially from the capillary. In this model, the site of lowest oxygen tension, or “lethal corner,” is found in the brain tissue farthest from the capillary and at the level of the capillary exit. The figure shows that more than adequate oxygen levels exist for the entire brain volume.
Figure 2.

Brain tissue oxygen levels in “lethal corner,” using model stimulation of hypoxia and neuronal activation. Hypoxia was superimposed in all three conditions, assuming an arterial pO2 of 45 mmHg. In all simulations, CBF was held fixed at baseline values to determine whether tissue pO2 would decrease below viable levels. The data show that, for all conditions, the brain maintains adequate levels of oxygen without need for raising CBF.
Human Blood Flow and Oxygen Delivery.
Table 2 demonstrates the effects of hypoxia on arterial blood. Breathing air with reduced O2 resulted in a mean fall of 55.7% in arterial pO2. Arterial O2 content during the mild hypoxia was reduced by 14.5% from baseline levels. Arterial pCO2 and pH were changed minimally during hypoxia. pH fell 0.021 units (P < 0.05) and pCO2 fell by 2.02 mmHg (P > 0.05). Measurements of pH, pCO2, pO2, and O2 content were highly stable within the room air and hypoxia blocks, however.
Table 2.
Summary of arterial blood gas for nine normal volunteers
| pO2 (mmHg) | O2 content, ml/dl | pCO2, mmHg | pH | |
|---|---|---|---|---|
| Room air (mean ± SD) | 100.7 ± 7.9 | 15.7 ± 1.5 | 36.5 ± 4.9 | 7.38 ± 0.02 |
| Hypoxia (mean ± SD) | 45.0 ± 4.2 | 13.4 ± 1.2 | 34.5 ± 5.0 | 7.40 ± 0.02 |
| Change with hypoxia | −55.7%** | −14.5%** | −5.5% | 0.3%* |
, P < 0.05; **, P < 0.0001.
The reversing checkerboard stimulus induced large increases in regional blood flow to visual cortex, including Brodman areas 17 and 18 (Fig. 3), in both normoxia and hypoxia conditions (Table 3). Regional CBF in the visual cortex was quantitated with and without normalizing by whole brain CBF. During normoxia, activation resulted in a normalized visual cortex CBF increase of 35.6% (P < 0.0005). Without normalization, the absolute regional CBF change was 32.3% (P < 0.0005). During hypoxia, activation resulted in a normalized visual cortex CBF increase of 31.6%, which was not significantly different from that measured during normoxia. Without normalization, the absolute regional mean CBF change from activation during hypoxia was the same as during normoxia (32.2%). The data show that the increase in CBF from visual activation was not affected by the presence of hypoxia.
Figure 3.

Blood flow images displayed as a midsagittal slice after averaging subject data in a standard atlas coordinate system. Image data were derived from quantitative PET blood flow scans that were normalized for variations in global blood flow. Each of nine subjects underwent scanning in two blocks: one block during breathing room air and a second block during hypoxia induced by reducing the inspired O2 fraction. In each block, PET studies were done during two control tasks and two visual activation tasks. (A) Average blood flow image from the control scans. (B) Mean subtraction image (visual activation minus control task) while breathing room air. (C) Average subtraction (visual activation minus visual fixation) image during hypoxia scaled to the same maximum as B. The black circle illustrates the relative size and position of the spherical region-of-interest used for quantitation of the regional visual cortex CBF. Note similarity in magnitude and distribution of the increased blood flow during visual activation. No augmentation of the blood flow response is seen despite the presence of reduced arterial oxygen content.
Table 3.
Mean ± SD values for global and regional CBF
| CBF, ml/min/100 g | Control | Hypoxia | Activation | Activation + hypoxia |
|---|---|---|---|---|
| Global | 56.65 ± 10.87* | 61.59 ± 10.00* | 54.81 ± 8.68 | 61.48 ± 11.20 |
| Visual cortex | 68.50 ± 17.64** | 71.45 ± 16.93 | 89.00 ± 17.13** | 94.04 ± 22.72 |
, P = 0.038; **, P < 0.0005.
Independent of neuronal activation, hypoxia alone resulted in small increases in CBF during the control task. Global CBF in the control scans demonstrated a small but statistically significant rise (8.7%; P = 0.038) during hypoxia, compared with normoxia. However, visual activation was not associated with significant changes in global CBF in either normoxia or hypoxia states. Hypoxia was not associated with any significant regional changes in visual cortex CBF in the resting state.
As shown in Fig. 4, when activation was compared with the baseline state, there was a rise of 3.2 O2 ml/min/100 g in oxygen delivery. However, when activation + hypoxia was compared with the baseline state, the rise was only 1.9 O2 ml/min/100 g in oxygen delivery, a 39% decrease compared with activation alone (P < 0.05). These data show that the addition of hypoxia does not induce compensating regulation of blood flow. More importantly, the regulation of the blood flow increase during activation does not appear to tightly maintain oxygen delivery to the tissue, even during times of increased metabolic rate.
Figure 4.

Effect of visual activation and hypoxia on visual cortex oxygen delivery. The regional oxygen delivery was calculated from the product of the subject's arterial oxygen content and PET-measured regional CBF. The data shown represent the mean and standard deviation of nine subjects (with each subject value representing the mean of two measurements in each of the four states). Oxygen delivery during activation + hypoxia was significantly less than oxygen delivery during activation alone (P < 0.05), demonstrating the lack of compensatory increase in CBF.
There was no change in mental status or cognitive performance. There were no subjective signs of hypoxia. All subjects denied shortness of breath, anxiety, drowsiness, or lightheadedness. There was no significant change in either detection of dimming events (averaging 83.5% at baseline and 86.9% during hypoxia) or in the reaction times (averaging 511 ms at baseline and 501 ms during hypoxia). Thus, there was no evidence for hypoxia-induced loss of attention.
Discussion
Previous assumptions that behaviorally induced increases in local blood flow would be reflected in similar local increases in the oxygen metabolism (1) have been contradicted by brain imaging studies with PET (4, 5) and functional MRI (8). Fox and his colleagues (4, 5) demonstrated that, in normal, awake adult humans, stimulation of the visual or somatosensory cortex results in dramatic increases in blood flow but minimal increases in oxygen consumption. Increases in glucose utilization occur in parallel with blood flow (5, 35), an observation fully anticipated by the work of others (36, 37). These results suggested that the additional metabolic requirements associated with short-term increase of neuronal activity might be supplied largely through glycolysis alone.
Interpretations of these blood flow-metabolism relationships during changes in functional brain activity are presently controversial. Several schools of thought have emerged. One hypothesis that addressed the role of glycolysis in brain functional activation was substantially described by P. Magistretti and colleagues based on their work with cultured astrocytes (38, 39). This theory suggests that increases in neuronal activity stimulated by the excitatory amino acid transmitter glutamate result in relatively large increases in glycolytic metabolism in astrocytes. The energy supplied through glycolysis in the astrocyte is used to metabolize glutamate to glutamine before being recycled to neurons. The increase in glycolytic processing in astrocytes leads to a transient overproduction of lactate. The lactate then leaves the astrocyte and is taken up and oxidatively metabolized by neurons. Additional support for this hypothesis came from in vivo observations that increases in neuronal activity are associated with glycogenolysis in astrocytes (40) and astrocytes are uniquely equipped enzymatically for using this convenient source of readily available energy (38, 40).
A popular alternative hypothesis is based on optical imaging of physiologically stimulated visual cortex of anesthetized cats by Malonek and Grinvald (41). Using wavelengths of light sensitive to deoxyhemoglobin and oxyhemoglobin, they noted an almost immediate increase in deoxyhemoglobin concentration followed, after a brief interval, by an increase in oxyhemoglobin. This latter increase, while centered at the same location as the change in deoxyhemoglobin, is greater in magnitude and extended over a much larger area of the cortex than did the changes in deoxyhemoglobin (41). They interpret these results to mean that increases in neuronal activity are associated with highly localized increases in oxygen consumption that stimulate a vascular response, delayed by several seconds, that is large in relation to both the magnitude of the increase in oxygen consumption and the area of cerebral cortex that is actually active.
A theoretical rationale for the large vascular response came from work by Buxton and Frank (11). In their modeling of brain oxygen delivery, they showed that, in an idealized capillary tissue cylinder in the brain, an increase in blood flow in excess of the increased oxygen metabolic demands of the tissue is required to maintain proper oxygenation of the tissue. This conclusion originates from the limited diffusivity and poor solubility of oxygen in brain tissue. In this theory, blood flow remains coupled to oxidative metabolism but in a nonlinear fashion. Large increases appear to overcome the diffusion and solubility limitations of oxygen in brain tissue to maintain adequate tissue oxygenation.
In the models proposed by Buxton and Frank (11) and others (12, 13), any level of hypoxia results in more than compensatory increase in CBF to maintain CMRO2, a net increase in oxygen delivery and reduction in oxygen extraction. Neuronal activation during hypoxia would then raise CBF even further. It follows from the equation of the Crone model (11, 14) that the percentage change in CBF from activation would be larger when the baseline oxygen extraction is lower. Thus, hypoxia should augment CBF changes. Our data demonstrated that hypoxia has no effect on the magnitude of CBF change during activation. For both normalized CBF and absolute CBF change, hypoxia made no impact. Hypoxia alone resulted in a net decrease of oxygen delivery because there was insufficient compensatory CBF increase.
The recent models assume that the human brain has unidirectional flux of oxygen out of the capillary. This assumption appears justified by the finding that there is minimal oxygen in brain tissue compared with blood, using transit time measures (42). However, because of the very high oxygen content of blood (from hemoglobin), the actual level of human brain oxygen is difficult to estimate with these techniques. We have implemented a model that does not make any assumption regarding brain oxygen levels; however, the mathematical solution is considerably more complex. We show with a simulation of human gray matter that brain oxygen is most likely present to a significant degree. The presence of tissue oxygen, which is diffusing back into the capillary, implies that, if usage increases or supply decreases, tissue oxygen levels can be maintained in the viable range without the need for increased CBF.
It is noted that no current tissue model of oxygen delivery and transport can completely reflect the known complexity of the capillary bed. For instance, whereas our model assumes a Krogh cylinder, which is consistent with concurrent flow, Reneau et al. (16) argue that its results are not significantly different from a countercurrent geometry, such as the frustum of a right circular cone where the arterial and venous tissue radii differ. As long as the tissue volume remains constant, basic results remain the same. Whereas the location of the least supplied tissue, or “lethal corner,” is a function of capillary arrangement, its oxygen tension is not significantly higher for countercurrent flow patterns. Furthermore, the model prediction that human gray matter can tolerate decreases in pO2 to 30 mmHg before neuronal viability is threatened fits well with the literature. Experimental measurements of CBF during controlled hypoxia in human subjects show that a large increase in CBF begins as pO2 is reduced to 30–35 mmHg (43).
It would be easy to assume that, because blood flow and glucose utilization change in parallel during neuronal activation and both increase more than oxygen utilization, the increase in blood flow serves to deliver required amount of glucose. Data from Powers and his colleagues (44) suggest otherwise. They noted no change in the magnitude of the normalized regional blood flow response to physiological stimulation of the human brain during stepped hypoglycemia. They concluded that the increase in blood flow associated with physiological brain activation is not regulated by a mechanism that matches local cerebral glucose supply to local cerebral glucose demand (44). We would now extend that suggestion to include oxygen as well.
In conclusion, intact brain tissue has excess oxygen delivery compared with utilization. At rest, this excess oxygen diffuses back to the capillary, but, in neuronal activation, it may be used for oxidation without necessary increase of CBF. In healthy human subjects, CBF increase in activated state does not change with addition of hypoxia, which implies that CBF must be regulated by factors other than just local requirements in oxygen.
Acknowledgments
We would like to thank Lori Groh and Lenis Lich for skilled technical assistance and express appreciation for the support of the National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke and NIH/National Heart, Lung, and Blood Institute.
Abbreviations
- CBF
cerebral blood flow
- CMRO2
cerebral metabolic rate for oxygen
- PET
positron emission tomography
- pO2
partial pressure of oxygen
References
- 1.Siesjo B K. Brain Energy Metabolism. New York: Wiley; 1978. [Google Scholar]
- 2.Kety S S, Schmidt C F. J Clin Invest. 1948;27:484–491. doi: 10.1172/JCI101995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen P J, Alexander S C, Smith F C, Reivich M, Wollman H. J Appl Physiol. 1967;23:183–189. doi: 10.1152/jappl.1967.23.2.183. [DOI] [PubMed] [Google Scholar]
- 4.Fox P T, Raichle M E. Proc Natl Acad Sci USA. 1986;83:1140–1144. doi: 10.1073/pnas.83.4.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fox P T, Raichle M E, Mintun M A, Dence C. Science. 1988;241:462–464. doi: 10.1126/science.3260686. [DOI] [PubMed] [Google Scholar]
- 6.Kuwabara H, Ohta S, Brust P, Meyer E, Gjedde A. Prog Brain Res. 1992;91:209–215. doi: 10.1016/s0079-6123(08)62337-7. [DOI] [PubMed] [Google Scholar]
- 7.Seitz R J, Roland P E. Acta Neurol Scand. 1992;86:60–67. doi: 10.1111/j.1600-0404.1992.tb08055.x. [DOI] [PubMed] [Google Scholar]
- 8.Kim S G, Ugurbil K. Magn Reson Med. 1997;38:59–65. doi: 10.1002/mrm.1910380110. [DOI] [PubMed] [Google Scholar]
- 9.Davis T L, Kwong K K, Weisskoff R M, Rosen B R. Proc Natl Acad Sci USA. 1998;95:1834–1839. doi: 10.1073/pnas.95.4.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujita H, Kuwabara H, Reutens D C, Gjedde A. J Cereb Blood Flow Metab. 1999;19:266–271. doi: 10.1097/00004647-199903000-00004. [DOI] [PubMed] [Google Scholar]
- 11.Buxton R B, Frank L R. J Cereb Blood Flow Metab. 1997;17:64–72. doi: 10.1097/00004647-199701000-00009. [DOI] [PubMed] [Google Scholar]
- 12.Hyder F, Shulman R G, Rothman D L. J Appl Physiol. 1998;85:554–564. doi: 10.1152/jappl.1998.85.2.554. [DOI] [PubMed] [Google Scholar]
- 13.Vafaee M S, Gjedde A. J Cereb Blood Flow Metab. 2000;20:747–754. doi: 10.1097/00004647-200004000-00012. [DOI] [PubMed] [Google Scholar]
- 14.Crone C. Acta Physiol Scand. 1963;58:292–305. doi: 10.1111/j.1748-1716.1963.tb02652.x. [DOI] [PubMed] [Google Scholar]
- 15.Roland P E, Eriksson L, Atone-Elander S, Widen L. J Neurosci. 1987;7:2373–2389. [PMC free article] [PubMed] [Google Scholar]
- 16.Reneau D D, Bruley D F, Knisely M H. J Assoc Adv Med Instrum. 1970;4:211–223. [Google Scholar]
- 17.Reneau D D, Knisely M H. Chem Eng Prog. 1971;67:18–27. [Google Scholar]
- 18.Hudetz A G. Brain Res. 1999;817:75–83. doi: 10.1016/s0006-8993(98)01200-1. [DOI] [PubMed] [Google Scholar]
- 19.Reneau D D, Bruley D F, Knisely M H. AIChE J. 1969;15:916–925. [Google Scholar]
- 20.Hudetz A G, Halsey J H, Horton C R, Conger K A, Reneau D D. Stroke. 1982;13:693–700. doi: 10.1161/01.str.13.5.693. [DOI] [PubMed] [Google Scholar]
- 21.Powers W J, Grubb R L, Baker R P, Mintun M A, Raichle M E. J Neurosurg. 1985;62:539–546. doi: 10.3171/jns.1985.62.4.0539. [DOI] [PubMed] [Google Scholar]
- 22.Carpenter D A, Grubb R L, Tempel L W, Powers W E. J Cereb Blood Flow Metab. 1991;11:837–844. doi: 10.1038/jcbfm.1991.143. [DOI] [PubMed] [Google Scholar]
- 23.Voet D, Voet J G. Biochemistry. 2nd Ed. New York: Wiley; 1995. pp. 216–223. [Google Scholar]
- 24.Mintun M A, Raichle M E, Martin W R W, Herscovitch P. J Nucl Med. 1984;25:177–187. [PubMed] [Google Scholar]
- 25.Hunziker O, Al S A, Schulz U. J Gerontol. 1979;34:345–350. doi: 10.1093/geronj/34.3.345. [DOI] [PubMed] [Google Scholar]
- 26.Clark D K, Erdmann W, Halsey J H, Strong E. Adv Exp Med Biol. 1977;94:697–704. doi: 10.1007/978-1-4684-8890-6_96. [DOI] [PubMed] [Google Scholar]
- 27.Guyton A C, Hall J E. Textbook of Medical Physiology. 9th Ed. Philadelphia: Saunders; 1996. pp. 513–523. [Google Scholar]
- 28.Wienhard H, Dahlbom M, Eriksson L, Michel C, Bruckbauer T, Pietrzyk U, Heiss W-D. J Comput Assist Tomogr. 1994;18:110–118. [PubMed] [Google Scholar]
- 29.Raichle M E, Martin W R, Herscovitch P, Mintun M A, Markham J. J Nucl Med. 1983;24:790–798. [PubMed] [Google Scholar]
- 30.Herscovitch P, Mintun M A, Raichle M E. J Nucl Med. 1985;26:416–417. [PubMed] [Google Scholar]
- 31.Snyder A Z. In: Quantification of Brain Function Using PET. Myers R, Cunningham V, Bailey D, Jones T, editors. San Diego: Academic; 1996. [Google Scholar]
- 32.Talairach J, Tournoux P. Co-Planar Stereotaxic Atlas of the Human Brain. New York: Thieme; 1988. [Google Scholar]
- 33.Ojemann J G, Buckner R L, Akbudak E, Snyder A Z, Ollinger J M, McKinstry R C, Rosen B R, Petersen S E, Raichle M E, Conturo T E. Hum Brain Mapp. 1998;6:203–215. doi: 10.1002/(SICI)1097-0193(1998)6:4<203::AID-HBM2>3.0.CO;2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mintun M A, Fox P T, Raichle M E. J Cereb Blood Flow Metab. 1989;9:96–103. doi: 10.1038/jcbfm.1989.13. [DOI] [PubMed] [Google Scholar]
- 35.Blomqvist G, Seitz R J, Sjogren I, Halldin C, Stone-Elander S, Widen L, Solin O, Haaparanta M. Acta Physiol Scand. 1994;151:29–43. doi: 10.1111/j.1748-1716.1994.tb09718.x. [DOI] [PubMed] [Google Scholar]
- 36.Sokoloff L, Reivich M, Kennedy C, Des Rosiers M H, Patlak C S, Pettigrew K D, Sakurada O, Shinohara M. J Neurochem. 1977;28:897–916. doi: 10.1111/j.1471-4159.1977.tb10649.x. [DOI] [PubMed] [Google Scholar]
- 37.Yarowsky P, Kadekaro M, Sokoloff L. Proc Natl Acad Sci USA. 1983;80:4179–4183. doi: 10.1073/pnas.80.13.4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bittar P G, Charnay Y, Pellerin L, Bouras C, Magistretti P. J Cereb Blood Flow Metab. 1996;16:1079–1089. doi: 10.1097/00004647-199611000-00001. [DOI] [PubMed] [Google Scholar]
- 39.Tsacopoulos M, Magistretti P J. J Neurosci. 1996;16:877–885. doi: 10.1523/JNEUROSCI.16-03-00877.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harley C A, Bielajew C H. J Comp Neurol. 1992;322:377–389. doi: 10.1002/cne.903220307. [DOI] [PubMed] [Google Scholar]
- 41.Malonek D, Grinvald A. Science. 1996;272:551–554. doi: 10.1126/science.272.5261.551. [DOI] [PubMed] [Google Scholar]
- 42.Kassissia I G, Goresky C A, Rose C P, Schwab A J, Simard A, Huet P-M, Bach G G. Circ Res. 1995;77:1201–1211. doi: 10.1161/01.res.77.6.1201. [DOI] [PubMed] [Google Scholar]
- 43.Shimojyo S, Scheinberg P, Kogure K, Reinmuth O M. Neurology. 1968;18:127–133. doi: 10.1212/wnl.18.2.127. [DOI] [PubMed] [Google Scholar]
- 44.Powers W J, Hirsch I B, Cryer P E. Am J Physiol. 1996;270:H554–H559. doi: 10.1152/ajpheart.1996.270.2.H554. [DOI] [PubMed] [Google Scholar]