

Abstract
In this multicenter evaluation, the VERSANT HCV RNA 3.0 Assay (bDNA) (Bayer Diagnostics, Tarrytown, N.Y.) was shown to have excellent reproducibility, linearity, and analytical sensitivity across specimen collection matrices (serum, EDTA, ACD-A), and hepatitis C virus (HCV) genotypes 1 to 6. The VERSANT HCV bDNA Assay has a reportable range of 615 to 7,690,000 (7.69 × 106) IU/ml. The total coefficient of variation (CV) ranged from 32.4% at 615 IU/ml to 17% at 6.8 × 106 IU/ml. The assay was linear across the reportable range. Analytical specificity of 98.8% was determined by testing 999 specimens from volunteer blood donors. Evaluation of HCV genotypes using RNA transcripts of representative clones of 1a, 1b, 2a, 2b, 2c, 3a, 4a, 5a, and 6a and patient specimens showed that the largest difference between genotype 1, upon which the assay is standardized, and non-1 genotypes was within 1.5-fold. Testing of potentially interfering endogenous substances and exogenous substances and conditions found no interference in HCV-positive or HCV-negative specimens except for unconjugated bilirubin at concentrations of ≥20 mg/dl and protein at concentrations of ≥9 g/dl. Biological variability was estimated from 29 clinically stable individuals not on HCV therapy who were tested weekly over an 8-week period. The combined estimate of total (biologic plus assay) variability was 0.15 log10 standard deviation (CV, 36.1%), a fold change of 2.6. Thus, the observed fold change between any two consecutive HCV RNA measures is expected to be less than 2.6-fold (equivalent to 0.41 log10 IU/ml) 95% of the time in clinically stable individuals.
Hepatitis C virus (HCV) infection is a major health care problem, with an estimated 4 million individuals infected in the United States (1) and 170 million infected worldwide (26). Approximately 70% of those infected will develop chronic HCV infection, which is a leading cause of chronic liver disease (13). Interferon monotherapy was the first antiviral regimen approved for treatment of chronic HCV infection. Sustained response rates were low (8 to 12%) (3), but newer antiviral treatments (interferon plus ribavirin, peginterferon plus ribavirin) have improved the response rates, particularly for patients infected with non-1 genotypes.
Quantitative HCV RNA results have been used prior to initiating interferon-based anti-HCV treatment to assess the likelihood of sustained virological response, defined as a negative result in a qualitative HCV RNA assay 6 months after the end of treatment. HCV RNA levels or changes from baseline have also been used early in treatment to attempt to accurately predict response to treatment (6, 10, 14, 15, 18, 20, 27).
The performance characteristics of an assay, i.e., its accuracy, reproducibility, and predictive values, determine the interpretation and utility of the assay's results (12, 18). The VERSANT HCV bDNA Assay, utilizing a signal amplification technique, is a fundamentally simple hybridization-based procedure involving unique nucleic acid probes directed to highly conserved sequences within the 5′ untranslated and core regions of the HCV genome (7). Advances in branched-DNA (bDNA) technology have been incorporated into version 3.0 of the VERSANT HCV bDNA Assay, which has greater specificity, greater sensitivity, and a broader reportable range than version 2.0 of the assay. The enhancements include the use of novel nucleotides (iso-cytidine and iso-guanidine) and a preamplifier (4). The changes incorporated into version 3.0 of the assay result in a theoretical signal amplification approximately fivefold greater than that of version 2.0.
In this study, the performance characteristics of the assay, including the conversion factor between copies per milliliter and international units per milliliter, detection cutoff and analytical specificity, reproducibility, linearity, HCV genotype detection, effects of potentially interfering endogenous and exogenous substances and conditions, effects of specimen collection and handling, and biological variability, were assessed by using multiple operators, sites, and kit lots.
MATERIALS AND METHODS
Testing by the VERSANT HCV bDNA Assay was performed at seven centers: Memorial Blood Center, Minneapolis, Minn.; Johns Hopkins Medical Institutions, Baltimore, Md.; Center for Blood Research, Sacramento, Calif.; Ochsner Foundation Hospital, New Orleans, La.; Bayer Reference Testing Laboratory, Berkeley, Calif.; University of California—San Francisco, San Francisco, Calif.; and Bayer Diagnostics NAD, East Walpole, Mass. All clinical specimens used in this study were collected with Institutional Review Board approval and informed consent. Clinical specimens were centrifuged and frozen at −60 to −80°C within 4 h of collection unless otherwise stated.
Assay procedure.
After HCV genomic RNA is released from the virions, the RNA is captured onto a microwell by a set of specific, synthetic oligonucleotide capture probes during an overnight incubation. A set of target probes is also added. The capture probes and the target probes bind to the 5′ untranslated and core regions of the HCV genome. A preamplifier probe that hybridizes to the target probes is then added. The amplifier probe subsequently hybridizes to the preamplifier, forming a bDNA complex. Multiple copies of an alkaline phosphatase-labeled probe are then hybridized to this immobilized complex. Detection is achieved by incubating the alkaline phosphatase-bound complex with a chemiluminescent substrate. Light emission is directly related to the amount of HCV RNA present in each sample, and results are recorded as relative light units (RLUs) by the Bayer System 340 bDNA Analyzer. Total hands-on time for processing one 96-well plate is less than 50 min (8).
Analytical specificity.
Evaluation of analytical specificity was performed to validate the detection cutoff (DC) at 615 IU/ml (3,200 HCV RNA copies/ml) previously established during preclinical testing. The DC is the value on the quantitation scale where the assay has a specificity of ≥95% with 95% confidence (i.e., the lower 95% confidence limit is ≥95%). Testing was performed using 999 HCV-seronegative blood donor specimens collected at three geographically distinct locations in the United States.
Reproducibility, linearity, and 95% detection limit (95% DL).
An eight-member panel with target concentrations between 480 and 7,690,000 IU/ml (2,500 to 40,000,000 HCV RNA copies/ml) was prepared for this study. Six of the eight members contained virus-inactivated, β-propiolactone-treated, HCV RNA-positive patient serum specimens (genotype 1) and had target concentrations of 7.69 × 105, 7.69 × 104, 7.69 × 103, 1.9 × 103, 9.6 × 102, and 4.8 × 102 IU/ml, respectively. The remaining two members were prepared by using recombinant single-stranded bacteriophage DNA with target concentrations of 7.69 × 106 and 7.69 × 104 IU/ml, respectively. Bacteriophage DNA was used to ensure a source of testing material for the upper end of the assay's reportable range. Both types of panel members were prepared by serial dilution of the panel member with the highest concentration into pooled HCV-negative recalcified plasma. The actual concentrations were lower than the target concentrations and are shown in Table 1 as the “value assignment” values (corresponding to expected concentrations in Fig. 1). Value assignments were established prior to the clinical trial in multiple runs using multiple operators in which a reference standard curve (directly linked to the National Institute of Standards and Technology phosphate standard) (5) was used in place of the kit standards. The quantitation values produced by the reference standard curve for each of the diluted panel members were multiplied by the gravimetrically determined dilution factors used to prepare them. The mean of these values was used to assign a value to the highest (undiluted) panel members (QC1 for recombinant bacteriophage DNA-containing members and QC2 for β-propiolactone-treated HCV-containing members). The dilution factors were then used to determine the expected values for the diluted panel members with lower concentrations. During the clinical trial, two operators at each of three sites performed the testing. Using three distinct kit lots, each operator performed two runs per day for two days, with triplicates of each panel member per run, resulting in 216 determinations per panel member.
TABLE 1.
Value assignment, within-run CV, between run CV, total CV, and total log SD for the HCV RNA reproducibility panel members
| Panel member | n | Value assignment (IU/ml)a | CV (%)
|
Total log SD | ||
|---|---|---|---|---|---|---|
| Within- run | Between- run | Total | ||||
| UQLb | NAc | 7.69 × 106 | 9.2 | 2.4 | 17.1 | 0.07 |
| QC1d | 216 | 6.8 × 106 | 13.9 | 2.7 | 17.0 | 0.07 |
| QC2 | 216 | 6.1 × 105 | 9.1 | 8.6 | 17.0 | 0.07 |
| QC3d | 216 | 6.8 × 104 | 10.2 | 7.5 | 15.0 | 0.06 |
| QC4 | 216 | 6.1 × 104 | 9.3 | 7.9 | 16.4 | 0.07 |
| QC5 | 216 | 6.1 × 103 | 10.9 | 11.5 | 18.8 | 0.08 |
| QC6 | 216 | 1.5 × 103 | 18.9 | 12.6 | 24.2 | 0.10 |
| QC7 | 216 | 7.6 × 102 | 24.2 | 9.4 | 28.2 | 0.12 |
| DCb | NA | 6.15 × 102 | 26.9 | 13.4 | 32.4 | 0.14 |
| QC8 | 216 | 4.8 × 102 | 33.1 | 22.8 | 42.2 | 0.18 |
Panel members ranged from 35,000,000 to 2,500 copies/ml (6.8 × 106 to 4.8 × 102 IU/ml). The reportable range of the assay is 7.69 × 106 to 615 IU/ml (40,000,000 to 3,200 copies/ml). The DC of 615 IU/ml (the value that provided 95% specificity with 95% confidence) and the UQL of 7.69 × 106 IU/ml were established in preclinical testing and validated in the clinical trial.
Estimates at the DC and UQL were obtained by interpolation and extrapolation, respectively.
NA, not applicable.
Panel members QC1 and QC3 were prepared by using recombinant single-stranded bacteriophage DNA.
FIG. 1.

Linearity of the VERSANT HCV bDNA Assay using the reproducibility panel (QC panel) and a pooled patient serum specimen. The reproducibility panel members (⧫) ranged from 6.8 × 106 to 3.8 × 102 IU/ml (35,500,000 to 2,000 copies/ml). The dilutions of the pooled serum specimen (□) ranged from 5.1 × 106 to 480 IU/ml (2.6 × 107 to 2,500 copies/ml). Note that the lowest member of each panel was below the DC of 615 IU/ml (3,200 copies/ml). The DC is the value that results in ≥95% specificity with ≥95% confidence.
Reproducibility, linearity, and the 95% DL (the lowest concentration of HCV RNA that produces a quantitative result in 95% of replicate specimens) were also evaluated for serially diluted, untreated, pooled patient specimens collected in Vacutainer serum separator tubes (SST), K2 EDTA tubes, or acid citrate dextrose (ACD) solution A tubes (all from Becton Dickinson, Franklin Lakes, N.J.). Each specimen collection matrix was tested at six concentrations: 3.8 × 106, 3.8 × 105, 3.8 × 103, 1.9 × 103, 9.6 × 102, and 4.8 × 102 IU/ml. Three operators performed 18 runs, 6 runs per kit lot, with quadruplicate testing of each panel member per run, for 72 determinations per panel member per anticoagulant matrix.
Conversion factor between copies per milliliter and international units per milliliter.
Seven different kit lots of the VERSANT HCV bDNA Assay were used to determine the conversion factor between copies per milliliter, the unit used for the VERSANT HCV bDNA Assay, and international units per milliliter, the unit used for the World Health Organization (WHO) International Standard for HCV RNA, NIBSC code 96/790, genotype 1 (22). The WHO standard was reconstituted according to the manufacturer's instructions. A six-member panel was created by preparing serial dilutions of the WHO standard, targeting a range of 2.5 × 104 to 780 IU/ml. Three operators performed a total of nine runs per kit lot. Each panel member was tested in quadruplicate in each run, for 252 determinations per panel member. Because the WHO standard is based on consensus results and lot-to-lot reproducibility has not yet been demonstrated (22, 23), the assay results are presently reported in both copies per milliliter and international units per milliliter by the assay software.
HCV genotype studies.
Two genotype studies were performed. The first study, which evaluated the quantitation of non-1a genotypes relative to that of genotype 1a (upon which the assay standards and controls are based), used RNA transcripts from the 5′ untranslated regions of representative clones of HCV genotypes 1a, 1b, 2a, 2b, 2c, 3a, 4a, 5a, and 6a. Transcripts were synthesized, purified, and characterized according to size and integrity by agarose gel electrophoresis (5). HCV RNA concentrations were determined by using phosphate analysis and confirmed by A260 and hyperchromicity (5). Each transcript was diluted to target concentrations of 3.8 × 105 and 1.3 × 103 IU/ml. Two operators performed two runs per day, each using three kit lots. Both concentrations of each genotype were tested in triplicate in each run, for 36 determinations per genotype.
A second study was performed to evaluate and compare assay precision, linearity, and analytical sensitivity in patient specimens for each of HCV genotypes 1 to 6. Genotypes were determined by using either nucleic acid sequencing or the INNOLiPA HCV II Assay (Bayer Diagnostics, Berkeley, Calif.). This study used serial dilutions of at least three patient specimens for each of the genotypes represented in the transcript study, with one exception: only one patient specimen was found for genotype 5. Target concentrations of the dilution series were 3.8 × 105, 1.9 × 103, 9.6 × 102, and 4.8 × 102 IU/ml. Six operators performed 36 runs—12 runs with each of three kit lots—for 24 determinations per panel member and 72 determinations per genotype concentration, except for genotype 5, for which 24 determinations per genotype concentration were made.
Potentially interfering endogenous substances.
The following endogenous substances were evaluated: hemoglobin (to 500 mg/dl; Sigma, St. Louis, Mo.); conjugated bilirubin (to 40 mg/dl; Promega Corp., Madison, Wis.) and unconjugated bilirubin (40 mg/dl; ICN Pharmaceuticals, Costa Mesa, Calif.); triglycerides (Intralipid, to 3,000 mg/dl; Fresenius Kabi Clayton, Clayton, N.C.); protein (human serum albumin, to 11 g/dl; Sigma); and elevated liver enzymes (serum glutamic-pyruvic transaminase [SGPT, or ALT] at levels ≥2 times the upper normal limit [UNL] of 50 U/liter in females or 70 U/liter in males [mean, 14.6 times the UNL; median, 6.2 times the UNL], serum glutamic-oxaloacetic transaminase [SGOT, or AST] at levels higher than the UNL of 47 U/liter [mean, 20.7 times the UNL; median, 5.6 times the UNL], and serum gamma-glutamyl transpeptidase [SGGT, or GGT] at levels higher than the UNL of 80 U/liter [mean, 8.0 times the UNL; median, 4.4 times the UNL]). Specimens for all substances except liver enzymes were prepared by spiking aliquots of HCV-negative serum with an HCV-positive serum to a target concentration of 1.3 × 103 IU/ml and then spiking in the endogenous substances. All spike volumes were ≤5% of the total specimen volume. Thirty-five to 45 HCV-negative serum specimens, depending on the number of concentrations tested, and 25 HCV-positive serum specimens were assayed per substance.
Another study was performed to evaluate the ability of the Lysis Working Reagent (LWR) to disrupt the interaction of cryoprecipitates and HCV RNA. LWR is a kit component used in the first step of the assay to solubilize and denature proteins, thus lysing the HCV virions and releasing the RNA. Because cryoprecipitates include a complex of protein and HCV RNA, inability of LWR to disrupt this complex would result in a reduced HCV RNA signal for a specimen. To test the effect of LWR on cryoprecipitates, serum specimens were collected from 40 HCV-positive patients. Cryoprecipitates were generated by placing one aliquot of each specimen on ice for 1 h. Specimens contained cryoprecipitates if, after chilling on ice and centrifugation at 16,000 × g for 5 min, there was a >30% reduction in HCV RNA quantification compared to that for the matched, unchilled, uncentrifuged aliquot. Specimens that contained cryoprecipitates were divided into two aliquots, both of which were placed on ice. LWR was added to the first aliquot, which was vortexed and tested after 1 h. This aliquot served as the control. LWR was also added to the second aliquot, which was centrifuged after 1 h, and the supernatant was tested. If the viral quantification for the control condition (total specimen) was equivalent to that for the test condition (supernatant), the conclusion was drawn that the addition of LWR to the specimen (HCV-LWR) had disrupted the interaction of cryoprecipitates and HCV RNA, i.e., cryoprecipitates did not cause a loss of signal in the assay.
Potentially interfering exogenous substances and conditions.
Sixteen microbial agents known to cause human infection (Escherichia coli, Pseudomonas aeruginosa, Klebsiella pneumoniae, Haemophilus influenzae, Enterobacter cloacae, Pseudomonas fluorescens, Staphylococcus aureus, Serratia marcescens, Streptococcus pneumoniae, Staphylococcus epidermidis, group B streptococcus, Candida albicans, cytomegalovirus [CMV], human immunodeficiency virus type 1 [HIV-1], hepatitis A virus [HAV], and hepatitis B virus [HBV]) and 21 therapeutic drugs (prednisone, ganciclovir, indinavir, lamivudine [3TC], pegylated interferon alfa-2b [PEG-INTRON], cyclosporine, acyclovir, zidovudine (AZT), ritonavir, ribavirin, alpha interferon [Intron A], tacrolimus, didanosine (ddI), nelfinavir, stavudine (d4T), mycophenolate mofetil, rapamycin, azathioprine, saquinavir, amantadine, and trimethoprim-sulfamethoxazole) were tested. The microbial agents were selected as being either the most common bacterial agents, the most common coinfectious agents in HCV-infected individuals (HBV and HIV), or other viral causes of liver disease (HAV, HBV, and CMV). Therapeutic drugs were selected based on treatment of HCV infection and HIV-HCV coinfection. Microbial agents and drugs were spiked into 25 HCV-positive specimens and 45 HCV-negative specimens, as described above, and tested. Bacteria and fungi were spiked to a final concentration of approximately 103 CFU/ml. HIV-1 and HBV were spiked to a final concentration of 106 RNA or DNA copies/ml, respectively. HAV was spiked to a final concentration of 106 50% tissue culture infective doses/ml, and CMV was spiked to a final concentration of 103.5 50% tissue culture infective doses/ml. With the exception of HIV-1, which was obtained from the Aaron Diamond Cancer Center, New York, N.Y., and HBV, obtained from BloodSource, Sacramento, Calif., all microbial agents were purchased from American Type Culture Collection, Manassas, Va. Therapeutic drugs, obtained from Sigma or from the drug manufacturer, were tested at 5 times the peak serum or plasma concentration (Cmax), except for lamivudine, which, because of solubility, was tested at one-half the reported Cmax.
A second study evaluated assay specificity by testing 10 clinical specimens each from HCV-seronegative individuals infected with one of the viruses HAV, HBV, and HIV-1 or with one of the following six diseases: anti-nuclear antigen, rheumatoid factor, primary biliary cirrhosis, autoimmune hepatitis, alcoholic cirrhosis, and nonalcoholic steatohepatitis. The results from these 90 specimens were compared to those from 100 HCV-negative volunteer blood donor specimens.
A third study evaluated the effects of as many as four freeze-thaw cycles on viral quantitation. In this study, specimens were collected from HCV-positive and HCV-negative individuals into Vacutainer SST, plastic K2 EDTA tubes, and ACD solution A tubes, centrifuged, divided into four aliquots, and frozen at −60 to −80°C. A freeze-thaw cycle was performed by thawing an aliquot on ice for 2 h and refreezing at −60 to −80°C for at least 12 h before the commencement of a subsequent freeze-thaw cycle. Specimens meeting these specifications were collected and tested from 25 HCV-positive and 40 HCV-negative individuals.
Specimen collection and handling conditions.
The following Becton Dickinson Vacutainer collection devices were evaluated: SST (the reference condition), plastic K2 EDTA tubes, ACD solution A tubes, and K2 plasma preparation tubes (PPT). Multiple tubes of each collection device were used to collect matched specimens from 25 HCV-positive and 45 HCV-negative individuals. All tubes were centrifuged, and the serum or plasma from one tube was immediately placed at −60 to −80°C. The matched serum or plasma specimens (separate tubes for SST and PPT; aliquots for plastic K2 EDTA and ACD solution A) were held at 2 to 8°C for 8, 24, or 48 h before freezing.
Within-subject variability.
Specimens were collected weekly for 8 weeks from 29 clinically stable, chronically HCV infected individuals who were not receiving anti-HCV treatment. The inclusion criteria for this study were as follows: individuals had to be at least 18 years of age, be repeatedly reactive by a Food and Drug Administration-licensed HCV enzyme-linked immunosorbent assay, have taken no antiviral medication and had no clinical events within 3 months of the start of the study, and have detectable HCV RNA in serum or plasma. Individuals were excluded from analysis if they missed more than two of the eight visits, had any significant clinical events, had changes in medication, or initiated antiviral therapy during the 8-week study.
Statistical analyses.
HCV RNA quantitations were log-transformed prior to analysis. Variance components in the reproducibility study were estimated using restricted maximum likelihood methods. In the genotype transcript study, relative recovery was defined as the ratio of the non-1a geometric mean quantitation to the 1a geomean quantitation adjusted for the respective gravimetrically determined concentration. Assay performance using HCV-positive specimens under a test condition, such as interfering substances, was evaluated by comparing the mean log quantitations for the test and reference conditions. A test condition was considered equivalent to the reference condition if the 90% confidence interval (90% CI) of the log difference was within 0.16 log10 IU/ml. A mean quantitation difference of 0.16 log10 IU/ml would result in a decrease of as much as 5% in the percent concordance between assay measurements from the test and reference conditions. Percent concordance is defined as the percentage of specimens tested in both the reference and test conditions for which the results are not statistically different. Because results expressed in international units per milliliter are not reported for specimens containing <615 IU/ml, assay performance using HCV-negative specimens under a test condition was evaluated by comparing the mean log RLUs of the signals for the test and reference conditions. A test condition was considered equivalent to the reference condition if the 95% upper confidence limit of the difference between the test and reference mean log RLUs was less than 0.08 log10 IU/ml. An increase of <0.08 log10 IU/ml in the mean log RLU would result in an assay specificity of at least 95%. In determining within-subject variability, the fold change between two successive measurements from a clinically stable individual that was unlikely to have occurred by chance was calculated by using the following formula: fold change = 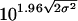 .
.
Excluded specimens.
Seven specimen results were excluded from analysis because of insufficient specimen volume, a missing specimen, or operator error. One series of HCV-positive unconjugated-bilirubin specimens (at 10, 20, and 40 mg/dl) was excluded because the reference condition specimen was lost. One HCV-positive conjugated-bilirubin specimen was excluded because the tube was mislabeled. Two EDTA specimens and one serum specimen in the specimen-collection-and-handling study were excluded because of insufficient specimen volume or failure to add specimen to the well. If the operators knew at the time that a specimen result would be invalid because of a procedural error, testing of that specimen was repeated, if possible, and the repeat results were used in the analyses.
RESULTS
Analytical specificity.
The analytical specificity of the assay was 98.8% (987 of 999), with a lower 95% confidence limit of 98.1%. The specificity across the three kit lots ranged from 98.2 to 99.2%. Specimens with quantitations above the DC of 615 IU/ml had values that ranged from 680 to 1,300 IU/ml.
Reproducibility, linearity, and 95% DL.
Table 1 shows the value assignment, total log standard deviation (SD), and total coefficient of variation (CV) for each panel member. Components of variance analysis showed that the majority of the variability was attributable to within-run (range, 9.1% for QC2 to 26.9% at the DC) and between-run (range, 2.7% for QC1 to 13.4% at the DC) components, with less variability due to day, lot, operator, and site. Because total variability and linearity were acceptable at the DC (total CV, 32.4%) and at 7.69 × 106 IU/ml (total CV, 17.1%), these values defined the lower and upper limits of the reportable range of the assay.
Figure 1 presents the linearity data from the quality control (QC) panel and the patient serum panel. The data show that the assay is linear over the reportable range of 615 to 7.69 × 106 IU/ml. The largest log10 differences between the expected and observed values were 0.05 in the QC panel and −0.04 in the patient serum panel. Linearity studies using patient specimens collected in K2 EDTA and ACD solution A tubes provided results that were equivalent to those for the QC panel and patient serum panel, with the greatest log10 difference between expected and observed values being −0.05 IU/ml.
The 95% DL is the concentration at which repeated testing of the same specimen yields a quantitative value (i.e., ≥DC) 95% of the time. Panel member QC6, with 1,500 IU/ml, was detected 100% of the time, and panel member QC7, with 760 IU/ml, was detected 72.7% of the time. By use of interpolation, the estimated 95% DL was 990 IU/ml, with an upper 95% CI of 1,200 IU/ml.
Conversion factor between copies per milliliter and international units per milliliter.
The conversion factor for the VERSANT HCV bDNA Assay was as follows: 1 IU/ml = 5.2 copies/ml. Two studies were performed; the first study used four kit lots for an initial determination of the conversion factor. The mean factor was 5.23, rounded to 5.2. This factor was subsequently confirmed by using the three clinical kit lots, with a mean of 5.27. The CV for the seven conversion factor determinations was 8.3%.
HCV genotype studies.
Figure 2 shows the recovery of the non-1a genotypes relative to that of genotype 1a for the high (3.8 × 105 IU/ml) and low (1.3 × 103 IU/ml) concentrations of HCV RNA transcripts. All non-1a genotype transcripts had relative recoveries that were within 1.5-fold of the recovery of the genotype 1a transcript.
FIG. 2.

Relative recovery of non-1a HCV genotype transcripts compared to that of the genotype 1a transcript. Transcripts were tested at two concentrations. The target concentration of the high panel members was 3.8 × 105 IU/ml (solid bars). The target concentration of the low panel members was 1.3 × 103 IU/ml (open bars).
Results from the serially diluted patient specimens of genotypes 1 through 6 demonstrated equivalent reproducibility, linearity, and 95% DLs for all genotypes. Total CVs were less than 36% for all genotype panel members with mean quantitations above the DC. In the linearity assessment, all log differences between expected and observed values for panel members above the DC were within ±0.07 log10 IU/ml for all genotypes. The 95% DLs were estimated by interpolating between the 5th percentiles of quantitation distributions of the two concentration levels whose sensitivity bracketed 95%. For all non-1 genotypes, mean 95% DLs were within 1.4-fold of the mean 95% DL for genotype 1. The fold changes in the 95% DL for specimens containing genotypes 2 to 6 relative to genotype 1 were 0.8, 1.3, 0.7, 0.7, and 0.8, respectively. These results were similar to the relative recovery results seen in the genotype transcript study.
Potentially interfering substances and conditions.
None of the endogenous substances (including cryoprecipitates) or exogenous substances (disease pathogens, therapeutic drugs) or conditions (as many as four freeze-thaw cycles) tested interfered with assay performance with HCV-positive or HCV-negative specimens, except for unconjugated bilirubin at ≥20 mg/dl (a 42% decrease in geomean quantitation, or a 0.23 log decrease) and protein at ≥9 g/dl (a 32% decrease in geomean quantitation, or a 0.17 log decrease) in HCV-positive specimens.
In the cryoprecipitate study, 20 of the 40 chilled specimens contained cryoprecipitates. By using these 20 specimens, the ratio of viral quantifications between the uncentrifuged specimens treated with LWR and the supernatant of the specimen treated with LWR was found to be 1.00 (90% CI, 0.97 to 1.02), indicating that LWR disrupted the cryoprotein-HCV RNA complex.
In the study evaluating possible interference from other disease pathogens by using HCV-negative clinical specimens from individuals with other viral and nonviral liver diseases, there was no significant difference in the geomean RLU between the “other-disease” specimens and healthy blood donor specimens (geomean RLUs,1.56 and 1.57, respectively).
Specimen collection and handling.
Vacutainer SST, K2 EDTA tubes, K2 PPT, and ACD solution A tubes gave equivalent performance and are acceptable for use in the assay. In HCV-positive specimens, the ratio of the geomean quantitations for the anticoagulants compared to serum ranged from 0.96 to 1.16. Testing of matched specimens centrifuged within 2 to 4 h of collection and held at 2 to 8°C for 8, 24, and 48 h before freezing at −60 to −80°C showed that serum and plasma can be held at 2 to 8°C for as long as 48 h prior to freezing. Evaluation of precision and analytical sensitivity showed that all the specimen collection matrices tested generated equivalent performance characteristics in the assay. The P values for equivalence of precision across serum, EDTA, and ACD collection matrices were all greater than 0.05, ranging from 0.19 to 0.75, indicating that there were no statistically significant differences in precision at any concentration. The 95% DLs for serum, EDTA, and ACD were 1,200, 980, and 1,100 IU/ml, respectively, and were within 1.2-fold of each other.
Within-subject variability.
The purpose of this study was to estimate biological variability in HCV RNA levels and to combine this estimate with the assay variability estimate so as to determine the maximum fold change between successive measurements that could occur by chance. This estimated value is important if a change in measurement is being used to assess treatment efficacy. The study subjects consisted of 16 men and 13 women, aged 33 to 76. Twenty-three subjects were Caucasian, four were African-American, and two were Hispanic. Twenty-one percent of the subjects had a diagnosis of cirrhosis. Baseline HCV RNA levels ranged from 1.4 × 104 to 7.5 × 106 IU/ml. As shown in Fig. 3, when ratios between successive quantitations were plotted against the mean of the two quantitations, the data indicated that variation in ratios was independent of viral load; that is, there is no indication that patients with a lower viral load will demonstrate greater biological variability between measurements than patients with a high viral load. There was no statistical difference in variability between specimens from males and specimens from females. The estimate of biological variability was 0.11 log10 SD (CV, 26.6%), or a 2.1-fold change between successive measurements. The combined estimate of assay variability, using combined data from all members of the reproducibility panel (see Table 1), was 0.10 log10 SD (CV, 23.5%), or a 1.9-fold change. The combined estimate of total (biological plus assay) variability was 0.15 log10 SD (CV, 36.1%), a fold change of 2.6. Thus, the observed fold change between any two consecutive HCV RNA measures is expected to be less than 2.6-fold (equivalent to 0.41 log10 IU/ml) 95% of the time in clinically stable individuals.
FIG. 3.

Scatter plot of serial quantitation ratios versus geomean quantitations (in international units per milliliter). Specimens were collected weekly for 8 weeks from individuals with chronic HCV infection and stable disease. Both members of a serial quantitation pair had to be within the reportable range of the VERSANT HCV bDNA Assay to be included in the plot.
DISCUSSION
Numerous quantitative HCV assays are available for use as an aid in the management of HCV-infected patients. However, results expressed as copies of HCV RNA per milliliter are not comparable between assays. This lack of comparability poses a significant problem for patient management, because a treatment decision threshold in copies per milliliter from one assay will not necessarily be the same for another. Consequently, standardization of assay results to international units per milliliter and use of international units per milliliter for patient care decisions are necessary (17, 19). In a comparative study, Sherman et al. (24) found significantly different international-units-per-milliliter results by the VERSANT HCV bDNA Assay versus the Cobas Amplicor HCV Monitor, version 2.0, for 205 specimens from HCV-infected patients and HCV-HIV-coinfected patients. For this reason, they recommend using the same assay to test longitudinally collected patient specimens. This recommendation was also made in the 2002 National Institutes of Health Consensus Development Conference Statement on the management of hepatitis C (17). The reasons for discordant measurements of viral load in international units between different assays remain unknown and could be related to the performance of a given assay with a given patient specimen and the role of interfering substances in serum in the biochemical reaction (J.-M. Pawlotsky et al., unpublished data).
The analytical specificity of 98.8% for the VERSANT HCV bDNA Assay is similar to those reported in other studies of this assay. Trimoulet et al. (25) reported a specificity of 98.2% with 383 HCV-negative specimens. Two other studies (2, 21) used a lower DC of 480 IU/ml (2,500 copies/ml) and, as would be expected, reported somewhat lower specificities (96.8 and 98%, respectively).
In some assays, the 95% DL, which provides a quantitative result in 95% of replicate determinations, is the lowest value reported by the assay. In the VERSANT HCV bDNA Assay, results are reported below the 95% DL, down to the DC. The results between the 95% DL and DC were shown in the trial to be specific (≥98.8%), precise (≤32.4%), and linear (±0.1 log unit) and can provide useful information to the clinician. As with other quantitative HCV assays, a result below the DC of 615 IU/ml cannot be used to imply eradication of virus or to detect active viral replication in anti-HCV-positive individuals. For either of these purposes, specimens with quantitative results below the DC must be retested by using one of the more sensitive qualitative assays, with 95% DLs of 6 to 50 IU/ml.
A critical factor for maintaining high specificity is the production of a homogeneous specimen by efficient mixing prior to addition of the specimen to the microwell, and by paying particular attention to specimen vials with limited air space that may impede proper vortexing. A homogeneous specimen provides the necessary serum- or plasma-derived protein matrix required as a blocking agent during the hybridization steps. Lack of sufficient plasma or serum protein limits the blocking process and can lead to high backgrounds and false-positive results.
The linearity, reproducibility, and 95% DL results obtained in this trial were also similar to previously published results (2, 21, 24, 25). Three of these studies (2, 21, 25) reported slopes for linearity close to 1.0 but intercepts that varied from −0.177 to 0.468. The intercept numbers reported in the present trial, which were closer to zero, may be due to corrections made in quantitations based on volumetric measurements of all dilutions.
Other studies that have evaluated the reproducibility of the VERSANT HCV bDNA Assay, using considerably fewer replicates, have shown low CVs for within-run and between-run results, similar to those reported here (2, 21, 24, 25). The uncomplicated specimen preparation step, in which 50 μl of undiluted plasma or serum is added directly to the capture well containing lysis buffer, contributes significantly to the low CVs of the VERSANT HCV bDNA Assay. Assays using complex specimen-processing procedures, including manual nucleic acid extraction and purification, are prone to higher CVs (9).
This is the first study to present comparative results for the different genotypes by using HCV transcripts, which were assigned HCV RNA concentrations by an independent method, phosphate analysis. Previously published studies have compared the quantitations of patient specimens containing different HCV genotypes only for the VERSANT HCV bDNA Assay versus other assay methods (2, 11, 21). This study also evaluated reproducibility, linearity, and 95% DL across genotypes by using patient specimens. Results obtained in the transcript study, which showed that the non-1a genotypes all had quantitations within 1.5-fold of that of genotype 1a, were corroborated in the study using patient specimens, where the 95% DL determinations of non-1 genotypes were within 1.4-fold of that of genotype 1. Linearity and reproducibility were equivalent across the genotypes tested. The bDNA technology is well adapted to accurate measurement of HCV RNA in the presence of the minor target sequence variations that account for the different HCV genotypes (7). Multiple target and capture probes containing single nucleic acid substitutions within the conserved 5′ untranslated and core regions of HCV increase the probability that the different genotypes will be detected with similar hybridization efficiencies.
The simplicity of adding undiluted plasma or serum directly to capture wells is preferable, in terms of labor, ergonomic issues, and supplies, to complex specimen extraction. Nevertheless, the crude lysate may contain exogenous and endogenous matter that could potentially interfere with assay results. The analysis of potentially interfering substances, both endogenous and exogenous, revealed that only high concentrations of unconjugated bilirubin (≥20 mg/dl) and protein (≥9 g/dl) reduced quantification. Chronic hepatitis C patients with very high concentrations of unconjugated bilirubin are unlikely candidates for antiviral treatment. Whereas chronic hepatitis patients often have conjugated bilirubinemia, high levels of the unconjugated form usually occur in patients with fulminant hepatitis, decompensated cirrhosis, rare inborn errors of metabolism (e.g., Crigler-Najjar syndrome), or hemolytic diseases. Elevated protein concentrations may theoretically reduce HCV RNA concentrations in only a small fraction of samples. In the NHANES III study (16), protein levels of >9 g/dl were found in 0.1% of the general population and in 2.6% of subjects positive for both anti-HCV antibodies and HCV RNA. Despite the theoretical role of the effect of elevated protein concentrations on HCV RNA measurement, the clinical utility study performed as part of this clinical trial (unpublished data) demonstrated the efficacy of this assay at predicting failure to respond to antiviral therapy in a diverse population.
The current indications for the use of quantitative HCV RNA assays require the availability of accurate, precise, and specific assays with accurate pan-genotypic quantitations. The VERSANT HCV bDNA Assay has a reportable range of 615 to 7.69 × 106 IU/ml, high reproducibility and specificity, and quantification of non-1a genotypes within 1.5-fold of the quantification of genotype 1a. The broad and accurate reportable range facilitates rapid and economical specimen testing by curtailing the number of specimens exceeding the upper quantitation limit (UQL); such specimens require dilution and retesting, increasing the expense and lengthening the duration of the procedure. Fewer than 1% of the specimens from patients with chronic HCV infection that were tested as part of this clinical trial had values greater than the UQL (unpublished data). Finally, the VERSANT HCV bDNA Assay's uncomplicated assay procedures, high throughput, simple hardware operating procedures, and minimal infrastructure requirements deliver a practical assay to the clinical laboratory setting.
Acknowledgments
This study was supported by test kits and funding from Bayer Diagnostics, Tarrytown, N.Y. N. Terrault is a consultant with Bayer Diagnostics, and T. Elbeik is and has been a speaker for Bayer Diagnostics.
REFERENCES
- 1.Alter, M. J., D. Kruszon-Moran, O. V. Nainan, G. M. McQuillan, F. Gao, L. A. Moyer, R. A. Kaslow, and H. S. Margolis. 1999. The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N. Engl. J. Med. 341:556-562. [DOI] [PubMed] [Google Scholar]
- 2.Beld, M., R. Sentjens, S. Rebers, C. Weegink, J. Weel, C. Sol, and R. Boom. 2002. Performance of the new Bayer VERSANT HCV RNA 2.0 Assay for quantitation of hepatitis C virus RNA in plasma and serum: conversion to international units and comparison with the Roche COBAS Amplicor HCV Monitor, version 2.0, Assay. J. Clin. Microbiol. 40:788-793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carithers, R. L., and S. S. Emerson. 1997. Therapy of hepatitis C: meta-analysis of interferon alfa-2b trials. Hepatology 26(Suppl.):83S-88S. [DOI] [PubMed] [Google Scholar]
- 4.Collins, M. L., B. Irvine, D. Tyner, E. Fine, C. Zayati, C.-A. Chang, T. Horn, D. Ahle, J. Detmer, L.-P. Shen, J. Kolberg, S. Bushnell, M. S. Urdea, and D. D. Ho. 1997. A branched DNA signal amplification assay for quantification of nucleic acid targets below 100 molecules/mL. Nucleic Acids Res. 25:2979-2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collins, M. L., C. Zayati, J. J. Detmer, B. Daly, J. A. Kolberg, T.-A. Cha, B. D. Irvine, J. Tucker, and M. S. Urdea. 1995. Preparation and characterization of RNA standards for use in quantitative branched DNA hybridization assays. Anal. Biochem. 226:120-129. [DOI] [PubMed] [Google Scholar]
- 6.Davis, G. L., R. Esteban-Mur, V. Rustgi, J. Hoefs, S. C. Gordon, C. Trepo, M. L. Shiffman, S. Zeuzem, A. Craxi, M.-H. Ling, and J. Albrecht for the International Hepatitis Interventional Therapy Group. 1998. Interferon alfa-2b alone or in combination with ribavirin for the treatment of relapse of chronic hepatitis C. N. Engl. J. Med. 339:1493-1499. [DOI] [PubMed] [Google Scholar]
- 7.Detmer, J., R. Lagier, J. Flynn, C. Zayati, J. Kolberg, M. Collins, M. Urdea, and R. Sanchez-Pescador. 1996. Accurate quantification of hepatitis C virus (HCV) RNA from all HCV genotypes using branched-DNA technology. J. Clin. Microbiol. 34:901-907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elbeik, T., R. Delassandro, Y.-M. A. Chen, S. V. Soutchkov, R. A. Loftus, and S. Beringer. 2003. Global cost modeling analysis of HIV-1 and HCV viral load assays. Expert Rev. Phamacoecon. Outcomes Res. 3:383-407. [DOI] [PubMed] [Google Scholar]
- 9.Elbeik, T., R. A. Loftus, and S. Beringer. 2002. Health care industries' perspective of viral load assays: the VERSANT HIV-1 RNA 3.0 assay. Expert Rev. Mol. Diagn. 2:275-285. [DOI] [PubMed] [Google Scholar]
- 10.Fried, M. W., M. L. Shiffman, K. R. Reddy, C. Smith, G. Marinos, F. L. Gonçales, D. Häussinger, M. Diago, G. Carosi, D. Dhumeaux, A. Craxi, A. Lin, J. Hoffman, and J. Yu. 2002. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 347:975-982. [DOI] [PubMed] [Google Scholar]
- 11.Germer, J. J., P. J. Heimgartner, D. M. Ilstrup, W. S. Harmsen, G. D. Jenkins, and R. Patel. 2002. Comparative evaluation of the VERSANT HCV RNA 3.0, QUANTIPLEX HCV RNA 2.0, and COBAS AMPLICOR HCV MONITOR version 2.0 assays for quantification of hepatitis C virus RNA in serum. J. Clin. Microbiol. 40:495-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hodinka, R. L. 1998. The clinical utility of viral quantitation using molecular methods. Clin. Diagn. Virol. 10:25-47. [DOI] [PubMed] [Google Scholar]
- 13.Hoofnagle, J. H. 1997. Hepatitis C: the clinical spectrum of disease. Hepatology 26(Suppl. 1):15S-20S. [DOI] [PubMed] [Google Scholar]
- 14.Lee, S. S., E. J. Heathcote, K. R. Reddy, S. Zeuzem, M. W. Fried, T. L. Wright, P. J. Pockros, D. Häussinger, C. I. Smith, A. Lin, and S. C. Pappas. 2002. Prognostic factors and early predictability of sustained viral response with peginterferon alfa-2a (40KD). J. Hepatol. 37:500-506. [DOI] [PubMed] [Google Scholar]
- 15.Manns, M. P., J. G. McHutchison, S. C. Gordon, V. K. Rustgi, M. Shiffman, R. Reindollar, Z. D. Goodman, K. Koury, M.-H. Ling, J. K. Albrecht, and the International Hepatitis Interventional Therapy Group. 2001. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 358:958-965. [DOI] [PubMed] [Google Scholar]
- 16.National Center for Health Statistics. 1996. National Health and Nutrition Examination Survey (NHANES) III, 1988-94. [Online.] http://www.cdc.gov/nohs/about/major/nhanes/datalink.htm#NHANESIII.
- 17.Pawlotsky, J.-M. 2002. Use and interpretation of virological tests for hepatitis C. Hepatology 36(Suppl. 1):S65-S73. [DOI] [PubMed] [Google Scholar]
- 18.Pawlotsky, J.-M., M. Bouvier-Alias, C. Herzode, F. Darthuy, J. Remire, and D. Dhumeaux. 2000. Standardization of hepatitis C virus RNA quantification. Hepatology 32:654-659. [DOI] [PubMed] [Google Scholar]
- 19.Poynard, T., J. McHutchison, Z. Goodman, M.-H. Ling, and J. Albrecht for the ALGOVIRC Project Group. 2000. Is an “à la carte” combination interferon alfa-2b plus ribavirin regimen possible for the first line treatment in patients with chronic hepatitis C? Hepatology 31:211-218. [DOI] [PubMed] [Google Scholar]
- 20.Ross, R. S., S. Viazov, S. Sarr, S. Hoffmann, A. Kramer, and M. Roggendorf. 2002. Quantitation of hepatitis C virus RNA by third generation branched DNA-based signal amplification assay. J. Virol. Methods 101:159-168. [DOI] [PubMed] [Google Scholar]
- 21.Saldanha, J., N. Lelie, A. Heath, and the WHO Collaborative Study Group. 1999. Establishment of the first international standard for nucleic acid amplification technology (NAT) assays for HCV RNA. Vox Sang. 76:149-158. [DOI] [PubMed] [Google Scholar]
- 22.Saldanha, J., A. Heath, N. Lelie, G. Pisani, M. Nübling, M. Yu, and the Collaborative Study Group. 2000. Calibration of HCV working reagents for NAT assays against the HCV international standard. Vox Sang. 78:217-224. [DOI] [PubMed] [Google Scholar]
- 23.Seeff, L. B., and J. H. Hoofnagle. 2003. Appendix: the National Institutes of Health consensus development conference management of hepatitis C 2002. Clin. Liver Dis. 7:261-287. [DOI] [PubMed] [Google Scholar]
- 24.Sherman, K. E., S. D. Rouster, and P. S. Horn. 2002. Comparison of methodologies for quantification of hepatitis C virus (HCV) RNA in patients coinfected with HCV and human immunodeficiency virus. Clin. Infect. Dis. 35:482-487. [DOI] [PubMed] [Google Scholar]
- 25.Trimoulet, P., P. Halfon, E. Pohier, H. Khiri, G. Chêne, and H. Fleury. 2002. Evaluation of the VERSANT HCV RNA 3.0 Assay for quantification of hepatitis C virus RNA in serum. J. Clin. Microbiol. 40:2031-2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.World Health Organization. October 2000, revision date. Fact sheet no. 164: hepatitis C. [Online.] http://who.int/inf-fs/en/fact164.html.
- 27.Zeuzem, S., S. V. Feinman, J. Rasenack, E. J. Heathcote, M.-Y. Lai, E. Gane, J. O'Grady, J. Reichen, M. Diago, A. Lin, J. Hoffman, and M. J. Brunda. 2000. Peginterferon alfa-2a in patients with chronic hepatitis C. N. Engl. J. Med. 343:1666-1672. [DOI] [PubMed] [Google Scholar]