

Abstract
Bone morphogenetic proteins (BMPs) regulate cell differentiation, proliferation, and apoptosis through a canonical SMAD signaling cascade. Absence of BMP signaling causes the formation of intestinal juvenile polyps in the colon cancer-prone syndrome familial juvenile polyposis. As sporadic colon cancers appear to have intact BMP signaling, we evaluated if K-RAS, driving a mitogenic pathway frequently activated in colon cancer, negatively affects BMP growth suppression. We treated non-tumorigenic but activated RAS/ERK FET cells with BMP2, and in combination with pharmacological or genetic inhibition of RAS/ERK, examined BMP-SMAD signaling, transcriptional activity, and cell growth, and also assessed p21WAF1 mRNA, transcriptional activation, and protein levels. BMP2 increased nuclear phospho-SMAD1 2-fold, which increased another 2–3 fold when RAS/ERK was inhibited. BMP2 increased BMP-specific SMAD transcriptional activity 2-fold over control and decreased cell growth, but inhibition of RAS/ERK further enhanced BMP-specific transcriptional activity by an additional 1.5–2 fold and enhanced growth suppression by 20%. BMP-induced growth suppression is mediated in part by p21WAF1, not by transcriptional upregulation but by improved p21 protein stability, which is inhibited by RAS/ERK. In colon cancer cells, BMP-SMAD signaling and growth suppression is facilitated by p21WAF1 but modulated by oncogenic K-RAS to reduce the growth suppression directed by this pathway.
Keywords: Bone morphogenetic protein RAS, ERK, TGFβ, Colorectal cancer
1. Introduction
Bone morphogenetic protein (BMP), a ligand belonging to the TGFβ family, regulates cell growth, apoptosis and differentiation in mesenchymal cells and tissues, but its influence in epithelial-derived cells is not understood. In the autosomal dominantly-transmitted hamartomatous polyposis syndrome, juvenile polyposis (JP), caused by mutations in BMPRIA [1] or its downstream effector SMAD4 [2], affected patients have up to a 12-fold increased lifetime risk for colorectal cancer [3]. Congruent with this, Haramis et. al. found that inhibition of BMP signaling by conditional knockout of BMPRIA results in the formation of numerous ectopic intestinal crypts which mimic the histopathology of JP, and which included the frequent occurrence of intraepithelial neoplasia [4]. These investigations suggest a role for intact BMP-SMAD signaling in mediating growth suppression, and repression of de novo crypt formation and neoplastic growth. In addition, we have shown that key BMP signaling molecules are present in human colorectal cancers, and that BMP induces growth suppression through the BMPRIA receptor [5]. Thus, BMP appears to suppress normal and transformed colon epithelial cells growth, and may be important to prevent colonic neoplastic transformation.
A common finding in colonic neoplasms is activation of RAS and its downstream effectors [6,7]. RAS proteins are proto-oncogenes involved in a variety of signal transduction pathways that normally regulate cellular proliferation, differentiation and death. Activation of RAS (i.e. through EGFR stimulation) or oncogenic RAS (via mutation and constitutive activation) exerts its effects on multiple downstream effector molecules, one of which is RAF, a serine–threonine kinase that activates MEK1 and 2 kinases, which in turn activate Erk kinases 1 and 2 [8–10]. Once phosphorylated, ERK 1 and 2 are free to phosphorylate other cytoplasmic targets as well as translocate to the nucleus and stimulate the activity of an assortment of transcription factors. Indeed, K-RAS develops constitutively activating mutations at codon 12 or 13 in greater than 50% of colorectal adenomas and cancer, and is considered an early event during the progression of colorectal cancer [11–13].
RAS and its downstream effectors might influence BMP signaling and growth effects. BMPs bind to serine–threonine kinases type I receptors (BMPRIA) and type II receptors (BMPRII) [14] that induce phosphorylation of intracellular SMADs 1, 5, or 8 at their carboxy terminus, at serine 463/465 [15]. The activated SMADs then bind to SMAD4, translocate to the nucleus, and in conjunction with specific DNA factors, activate or repress gene transcription [16]. SMAD1 can also be phosphorylated at its mid-protein linker region by activated Erk kinase (downstream of RAS), slowing or inhibiting nuclear accumulation of BMP-activated SMAD1 in mouse mammary cells [17,18].
Because we have previously shown that BMP signaling appears to be intact in primary colon cancers [5], we evaluated if oncogenic K-RAS, driving a mitogenic pathway frequently activated in colorectal cancer, negatively affects BMP signaling and growth suppression. Negative regulation by activated K-RAS would be a means to inhibit BMPs growth suppression in the absence of mutation of BMP signaling components. Here we utilized FET colorectal cancer cells, considered an early model of colorectal cancer due to alterations in Wnt signaling, RAS, and p53 [19], but are non-tumorigenic in nude mice, to examine any influence that RAS has on BMP signaling. We found that FET cells are growth suppressed in response to BMP2 treatment and that oncogenic K-RAS/ERK activation lessens the response to BMP2 in these cells via decreased p21WAF1 protein stabilization, a novel mechanism by which BMP exerts its growth suppressive effects.
2. Materials and methods
2.1. Cell culture and treatments
FET cells were maintained in Dulbecco’s Modified Eagle Medium:Nutrient Mix F-12 (D-MEM/F-12) (1X), liquid, 1:1, with L-glutamine and HEPES buffer (Invitrogen Corporation, Carlsbad, CA) containing 10% fetal bovine serum and penicillin G/streptomycin (Invitrogen Corporation, Carlsbad, CA). In some assays, cells were pre-treated with PD98059 (an ERK inhibitor; Calbiochem, San Diego, CA) at a concentration of 5 μM for 30 min prior to 100 ng/ml of BMP2 treatment (R and D Systems, Minneapolis, MN).
2.2. Transfections
Dominant negative K-RAS (DN KRAS; a generous gift from Dr. Rik Derynck, University of California, San Francisco), which inhibits the function of activated RAS, and mock vectors were transiently delivered by Transfectin (Promega, Madison, WI) at a ratio of 3:1 of vector to transfection reagent in OPTI-MEM reduced serum free media (GIBCO Carlsbad, CA). After 2–3 h, IMDM with FBS and penicillin G/streptomycin was added to the transfected cells. Two hours post-transfection, complete media was added, and later the cells were used in the experiments.
2.3. Total cell lysis, and immunoblotting
Cells were lysed using total lysis buffer (12 mM Tris–HCl pH 8.3, 100 mM NaCl, 1% SDS, 1% DCA, 1% Triton X-100, 2 mM EDTA, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 50 μM DTT, and 2 mM PMSF). The protein was denatured at 100 °C for 5 min and then loaded onto a 15% polyacrylamide gel. After electrophoresis, the proteins were transferred onto a nylon membrane, blocked for 1 h with 5% milk, and probed overnight with primary antibody at 4 °C. Blotting was done with antibodies to p21WAF1 at a 1:200 dilution (OP64 Calbiochem, San Diego, CA), and PTEN at a 1:200 dilution (SC7974, Santa Cruz Biotechnology, Santa Cruz, CA). The following day, several PBS-Tween 0.1% washes were performed along with appropriate secondary antibody incubation. Blotted proteins were detected with horseradish peroxidase-linked secondary antibodies (Sigma, St. Louis, MO) followed by ECL detection (Amersham, Little Chalfont, UK).
2.4. Total RNA extraction and semi-quantitative reverse transcriptase-polymerase chain reaction
Total RNA extraction was performed using Trizol reagent (Invitrogen Corporation, Carlsbad, CA). Cells grown on 6-well plates were lysed with trizol (1 mL/well) and were combined with chloroform and mixed. Supernatants were then precipitated with isopropanol, and the RNA pellets were washed with 75% ethanol and air-dried, and resuspended in water. Two micrograms of total RNA was converted into cDNA by reverse transcriptase and amplified for BMP2 and BMP7 ligand transcripts (SuperScript II, Invitrogen Corporation). Briefly, following inactivation at 65 °C for 10 min, 1 μL of the reaction mixture was incubated in buffer containing 0.2 mM concentrations of dATP, dCTP, dGTP, dTTP, 0.2 μM concentrations each of oligonucleotide primers, 3 mM MgCl2 and a 10X buffer consisting of 200 mM Tris–HCl (pH 8.0), 500 mM KCl, and 1 U Taq polymerase. The following primers were designed to amplify BMP2 and BMP7: BMP2, forward 5′-CCCAGCGTGAAAAGAGAGAC-3′ and reverse 5′GAGACCGCAGTCCGTCTAAG-3′; BMP7, forward 5′-TCGTGGAACAT-GACAAGGAA-3′ and reverse 5′-CTGATCCGGAACGTCTCATT-3′. Primers for p21/Waf1 were as follows: forward 5′CAGGGGACAGCAGAG-GAAGA-3′ and reverse 5′-TTAGGGCTTCCTCTTGGAGAA-3′. Primers for PTEN are forward 5′-GGACGAACTGGTGTAATGATATG-3′ and reverse 5′-TCTACTGTTTTTGTGAAGTACAGC-3′. GAPDH served as a loading control (forward 5′-ACCACAGTCCATGCCATCAC-3′ and reverse 5′-TCCAC-CACCCTGTTGCTGTA-3′). PCR was performed as follows: denaturation at 95 °C for 3 min and optimized amplification for up to 35 cycles of 94 °C for 30 s, 55 °C for 30 s, and 74 °C for 4 min for BMP7 and GAPDH; for BMP2: denaturation at 95 °C for 3 min and optimized amplification for up to 40 cycles of 94 °C for 30 s, 57 °C for 30 s, and 74 °C for 4 min; for PTEN: denaturation at 95 °C for 3 min; and for p21: denaturation at 95 °C for 3 min and 40 cycles of 94 °C for 30 s, 55 °C for 30 s, and 74 °C for 4 min.
2.5. Luciferase assays
Transient transfection of colon cancer cells with the BRE-Luc plasmid, which contains a consensus promoter element specific to BMP-SMAD activation (a gift from Dr. Peter ten Dijke, Netherlands Cancer Institute, Amsterdam) was done to assess the effects of BMP2 on BMP-specific transactivation. The pWWP-luc plasmid, containing the promoter of p21WAF1, was transfected to assess the effects of BMP2 on p21WAF1 transactivation, and the PTEN-luc plasmid, containing the wild type protoer of PTEN, was used to assess the effects of BMP2 on PTEN transactivation. Reporter vectors (0.75 μg/ml) and the pRL-TK vector (20 ng/well) are transiently delivered by Transfectin (Promega, Madison, WI) in 12-well plates with a ratio of 3:1 of vector to transfection reagent in OPTI-MEM reduced serum free media (GIBCO Carlsbad, CA). Two hours post-transfection, 1 ml of complete media was added per well and twelve to 16 h post-transfection, cells were treated with 50 ng/ml of BMP2 or BMP7. Luciferase activity was measured by a dual-luciferase kit (Promega, Madison, WI) 20 to 24 h after the treatment; normalization was performed using the Renilla luciferase activity expressed by the co-transfected pRL-TK vector.
2.6. Deconvolution digital immunofluorescent microscopy
Cells were plated in four-chambered slides (Nunc Inc. Naperville, IL) at 1000 cells/chamber. Once cells became 80% confluent on the coverslip, (approximately 48 h after plating), the cells were exposed to pharmacological inhibitor or media alone for 30 min followed by the addition of 100 ng/ml BMP2. After 1 h of exposure, the slides were placed at 4 °C and washed 3 times with phosphate buffered saline and then fixed in 3.7% formaldehyde in the same buffer at room temperature for 30 min and permeabilized with 0.3% Triton X-100. The slides were then blocked with 5% bovine serum albumin in PBS for 1 h. The anti-phosphoSMAD1 primary antibody (Chemicon, Temecula, CA) was diluted 1:100 in 5% bovine serum albumin in phosphate buffered saline and added for 2 h at room temperature. Biotin SP-conjugated Affinity Pure Donkey anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA) was then added at a 1:250 dilution for 2 h in 5% bovine serum albumin in phosphate buffered saline and incubated at room temperature. Hoescht 33342 dye (Molecular Probes, Eugene, OR) was also added to the secondary antibody solution at a concentration of 1:10,000. Slides were then stained with Streptavidin Alexa 488 (Molecular Probes, Eugene, OR) at 1:1000 in 5% bovine serum albumin in phosphate buffered saline and incubated at room temperature for 2 h. Coverslips were then mounted on the glass slides using Gelvatol (Air Products and Chemicals, Inc., Allentown, PA). Images were captured with a DeltaVision Restoration microscope system (Applied Precision Inc., Issaquah, WA) using a Photometrics Sony Coolsnap HQ charged-coupled device (CCD) camera system attached to an inverted, wide-field fluorescent microscope (Nikon TE-200). Optical sections were acquired using a 60× Nikon (NA 1.4) oil immersion objective in 0.2 μm steps in the z-axis. Images were saved, processed, and analyzed on Silicon Graphics Workstations (O2, Octane) using the DeltaVision software package Softworx (Version 2.50). Images are normalized to the autofluorescence of unstained cells and cells stained with only the secondary antibody and Streptavidin Alexa 488 probe. Two independent observers counted the number of fluorescent dots in the nucleus and the cytoplasm to quantify the data.
2.7. MTT assay
The effect of BMP2 treatment on cell growth was assessed by using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) based metabolic assay. Cells were seeded in 48-well plates at a density of 10 to 20,000 cells/well in 0.4 ml of culture medium supplemented with BMP2 [100 ng/ml]. Metabolic activity, corresponding to growth, was assayed after 4 and 6 days of incubation at fixed intervals for MTT-dependent absorbency. For this, cells were stained 3 h with MTT dye, the reaction product released by lysis with SDS, and absorbency detected at 570λ using Beckman-Coulter DU640B spectrophotometer (Beck-man-Coulter, Fullerton, CA).
2.8. Cell growth assay
Cells were seeded at a density of 10,000 cells per well and 24 h later were treated with 100 ng/ml BMP2 in the presence of FBS. After 4 and 6 days, cells were lysed in 0.5 ml of 0.05% trypsin and counted using a hemacytometer.
2.9. Statistical analysis
Statistical significance was determined using either the student’s t-test or two-factor without replication ANOVA. Probability values less than 0.05 were considered to be significant.
3. Results
3.1. BMP-SMAD signaling is intact in FET colon cancer cells and is growth suppressive
Non-tumorigenic FET colon cancer cells have been described to contain intact TGFβ signaling [20], but the BMP pathway status is not known. We assessed the pathway by (a) immunofluorescence of phospho-SMAD1 after BMP2 treatment, (b) BMP-SMAD specific transactivation and (c) cell growth. BMP2 treatment induced a 2.5 fold increase in phospho-SMAD1 immunofluorescence into the nucleus when compared to controls (Fig. 1A and B). Additionally, BMP2 treatment induced a three fold increase in BMP-SMAD specific transcriptional activity in FET cells as measured by a BRE-Luc luciferase reporter assay (Fig. 2). Furthermore, BMP2 treatment caused a 20–25% reduction in cell growth by cell counting (Fig. 3) and MTT assay (not shown) over untreated cells. These data indicate that BMP-SMAD signaling and transactivation is intact in FET cells, and that BMP2 is growth suppressive in these cells.
Fig. 1.

BMP2 induces nuclear phosphoSMAD1, which is improved with RAS/ERK inhibition. (A) Selected images from a deconvolution digital immunofluorescent microscope showing immunofluorescence of FET cells using phosphoSMAD1 antibody (green) with nuclei stained with DAPI (blue), with or without RAS/ERK inhibition. Inset in left upper panel is the negative control without antibody. Twelve or more images were acquired for each experiment. (B) Summary graph with relative quantification of nuclear phosphoSMAD1 immunofluorescence data. Average of 3 quantifiable images per experiment is represented in the data. Cells were treated with 100 ng/mL of BMP2 where indicated and imaged 0–2 h after treatment. Dosage of PD98059 was 5 μM. *P≤0.05.
Fig. 2.
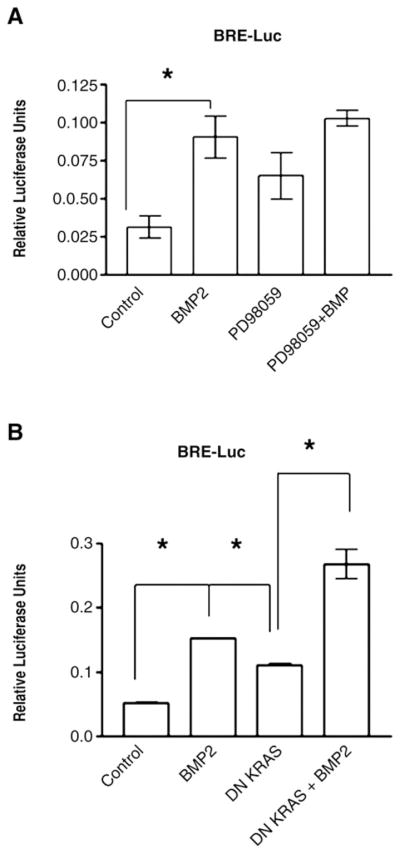
BMP-specific transactivation is improved with RAS/ERK inhibition. The BRE-Luc construct was used with and without pharmacologic inhibition of RAS/ERK with (A) PD98059 (5 μM) or after transfection with (B) DN KRAS. Cells were treated with 100 ng/mL of BMP2 where indicated and assayed 16–20 h after treatment. Data are from 5 independent experiments. *P≤0.05.
Fig. 3.

Inhibition of RAS/ERK improves BMP2-induced growth suppression. Cells were treated with 100 ng/mL of BMP2 with or without PD98059 (5 μM) where indicated every 2 days, and cells were counted. Results are expressed as percent change in growth with BMP2 treatment compared to untreated controls. With DN KRAS transfection, the slowing of cellular growth manifested early and lasted up to 6 days despite the transient transfection (see Supplemental Fig. 1). pcDNA3.0, the vector for DN KRAS, was used as an additional control. Day 6 data are shown. Data are from 3 independent experiments. *P≤0.05.
3.2. BMP-SMAD signaling and growth suppression is modulated by K-RAS/ERK
It has been demonstrated in osteoblasts that BMP2 can stimulate RAS/MAPK activity [21]. We assessed if BMP2 treatment can stimulate phospho-ERK activity in FET cells. BMP2 treatment activated ERK (in conjunction with increased nuclear phospho-SMAD1), by 10 min after BMP2 treatment that persisted through 120 min. The phospho-ERK activation is in addition to the constitutive levels stimulated by oncogenic RAS in these cells (Fig. 4), as FET colon cancer cells contain a mutated K-RAS gene (codon 12 mutation: GGT>GCT) [6], making K-RAS constitutively active. BMP2 activation of phospho-ERK may act as a “brake” on BMP-induced growth suppression by regulating or limiting this response in the correct setting. Treatment of cells with the ERK inhibitor PD98059 abolished both constitutive phospho-ERK activation and BMP2-induced phospho-ERK activation in these cells (Fig. 4).
Fig. 4.

BMP2 induces ERK activation above constitutively active levels, and RAS/ERK inhibition blocks both stimuli for ERK activation. Cells were treated with 100 ng/mL of BMP2 with or without PD98059 (5 μM) where indicated for up to 2 h, followed by Western blotting of extracted proteins. GAPDH was used as a loading control. The lower panel shows the relative density of phospho-ERK (pERK) with respect to GAPDH. Data are representative of 3 independent experiments.
Because BMP-SMAD signaling is intact in FET cells, which has also been described in primary colon cancers [5], we examined if the RAS/ERK mitogenic pathway, often constitutively activated in colon cancers and further induced by BMP, could negatively affect BMP-SMAD signaling and thus negate its growth suppressive effects. Use of the ERK inhibitor PD98059 nearly abolishes constitutive ERK activation in FET cells with as little as 5 μM PD98059 (Supplemental Fig. 1). Transfection of FET cells with DN RAS also completely abolished constitutive ERK activation through 48 h after transfection (Supplemental Fig. 1). Both pharmacological inhibition of ERK (with the inhibitor PD98059) and transfected DN-KRAS resulted in a 2–3-fold increase in phospho-SMAD1 nuclear fluorescence over control, suggesting improved autocrine or paracrine BMP-SMAD signaling (Fig. 1A and B; see Fig. 5D for endogenous BMP transcripts). With exogenous BMP2 treatment, nuclear phospho-SMAD1 immunofluorescence was increased another 2–3 fold over PD98059 or DN KRAS alone, indicating improved nuclear translocation with RAS/ERK inhibition (Fig. 1A and B). Inhibition of RAS/ERK by PD98059 or DN KRAS also improved BMP-SMAD transcriptional activity, both endogenously (with BMP signaling presumably via autocrine stimulation) and with exogenous BMP2 treatment. As shown in Fig. 2A and B, PD98059 or DN KRAS increased BMP-SMAD specific transcriptional activity 2 fold over controls, and the addition of exogenous BMP2 further increased this up to another 2 fold. Inhibition of RAS/ERK also improved BMP2-induced growth suppression, with an additional 25–40% growth suppression by cell counts (Fig. 3) and MTT assay (not shown) over BMP2 treatment alone. These data demonstrate that activated RAS/ERK slows BMP-SMAD induced phospho-SMAD1 nuclear translocation, BMP-SMAD specific gene transactivation, and inhibits some of the growth suppression that is induced by BMP signaling.
Fig. 5.

No alteration of expression of PTEN and BMP ligands with BMP2 treatment. (A) semi-quantitative RT-PCR of PTEN, with relative expression with GAPDH in the bar graph; (B) PTEN-Luc luciferase assay; (C) Western blot of PTEN, with relative expression with tubulin in the bar graph; (D) semi-quantitative RT-PCR of BMP2 and BMP7. Cells were treated with 100 ng/mL of BMP2 where indicated and assayed 0–48 h for the RT-PCR experiments, and up to 96 h for Western blots after treatment. Data are representative from 2 independent experiments. GAPDH (RT-PCR) and tubulin (Western) were used as loading controls.
3.3. BMP-SMAD signaling stabilizes p21WAF1 protein as one mechanism for growth suppression, and is inhibited by activated RAS/ERK
To understand the mechanism of growth suppression by BMP2 in FET cells, we examined two suppressor gene products: PTEN and p21WAF1. BMP2 has been reported to increase PTEN protein levels through reduced degradation in breast cancer cells [22], and BMP2 can upregulate p21WAF1 in breast, prostate, and gastric cancers [23–29]. BMP2 treatment did not alter PTEN mRNA levels as assessed by semi-quantitative RT-PCR, nor PTEN promoter activity as assessed by PTEN-luc transcription (Fig. 5A and B). Additionally, the steady state level of PTEN protein was not altered by BMP2 treatment (Fig. 5C). We also demonstrate that FET cells produce both endogenous BMP2 and BMP7 and there is no evidence of BMP2 induced negative feedback regulation of these ligands (Fig. 5D). These data demonstrate that in FET colon cancer cells that BMP2 does not affect PTEN, BMP2, or BMP7 expression.
In contrast, BMP2 treatment caused a doubling of p21WAF1 protein levels in FET cells (Fig. 6A and B). However, utilizing semi-quantitative RT-PCR for p21WAF1 transcripts, there was minimal change in p21WAF1 mRNA levels with BMP2 treatment (which was further verified by qualitative RT-PCR — data not shown), and this was not augmented with inhibition of RAS/ ERK by PD98059 (Fig. 6C). Additionally, using pWWP-luc, a p21-specific transcriptional reporter, we only noticed minimal increases in transcription over control (1.2-fold) that was not augmented with inhibition of RAS/ERK by PD98059 or DN KRAS transfection (Fig. 6D). Because we saw only minimal changes in p21WAF1 mRNA levels or transcriptional activity, we hypothesized the BMP2-induced increased p21WAF1 levels were from reduced degradation of p21WAF1 protein. To test this hypothesis, we stimulated p21WAF1 protein levels with BMP2, and then inhibited new protein synthesis with cyclohexamide. Cells were then restimulated with BMP2 to assess the effect on p21WAF1 protein levels. As shown in Fig. 7A, BMP2-stimulated p21WAF1 protein is rapidly degraded by 4 h, but re-stimulation with BMP2 induced some p21WAF1 protein stabilization that lasted through 12 h post-re-stimulation. Thus, BMP2 induces p21WAF1 stabilization as the principal mechanism to increase p21WAF1 protein levels, more so than transcription of p21WAF1 mRNA.
Fig. 6.

BMP2 increases p21WAF1 protein with no change in p21WAF1 transcription. (A) Western blot of p21WAF1; (B) relative density of p21WAF1 from Western blot in (A); (C) semi-quantitative RT-PCR of p21WAF1 in the presence or absence of RAS/ERK inhibition; (D) pWWP-Luc activity, measuring p21WAF1-specific promoter activity, in the presence or absence of RAS/ERK inhibition. Dosage of PD98059 was 5 μM. Cells were treated with 100 ng/mL of BMP2 where indicated and assayed 0–48 h for the RT-PCR experiments, and up to 96 h for Western blots after treatment. Data are representative from 3 independent experiments. GAPDH (RT-PCR) and tubulin (Western) were used as loading controls.
Fig. 7.

p21WAF1 protein is stabilized with BMP2 treatment, and is further improved with RAS/ERK inhibition. Cells were serum starved for 24 h, followed by BMP2 (100 ng/mL) to stimulate p21WAF1 protein expression. Further protein synthesis was then inhibited with cyclohexamide 12 h after the initial BMP2 stimulation. Cells were then treated as indicated: (A) without and with BMP2 restimulation, and (B) PD98059 (5 μM) alone or with BMP2 restimulation. Cells were assayed 36 h later. Tubulin was used as a loading control. Bar graphs in A and B show relative expression of p21WAF1 to tubulin. Data are representative from 3 independent experiments. *P<0.05.
We next sought if inhibition of RAS/ERK could alter BMP2-induced p21WAF1 protein stabilization, and performed the same experiments in the presence or absence of RAS/ERK inhibition. The ERK inhibitor PD98059 alone did not affect p21WAF1 stabilization; however p21WAF1 protein levels were stabilized through 36 h after PD98059 and re-stimulation with BMP2, indicating less p21WAF1 degradation as compared to BMP2 re-stimulation alone (Fig. 7B). Similar results were observed in FET cells transfected with DN KRAS (not shown). Taken together, RAS/ERK appears to inhibit BMP2-induced p21WAF1 stabilization, which likely modulates the observed growth change in FET cells.
4. Discussion
BMPs, part of the TGFβ family (which includes TGFβ and activin), appear to be regulators to control epithelial cell growth. Among colorectal cancers, key receptors for TGFβ and activin signaling are often mutated, muting SMAD signaling and subsequent growth suppression [30–32]. We have previously shown intact BMP signaling in primary colon cancers and cell lines [5], suggesting that if BMP growth suppression was important for growth control, other mechanisms for inhibiting BMP-induced growth suppression in addition to receptor or SMAD4 mutation seen with the familial JP syndrome may be operative. Here, utilizing the non-tumorigenic but K-RAS-mutated FET colon cells, we demonstrate that (a) BMP signaling and transactivation is intact, (b) BMP2 induces growth suppression, (c) BMP2 stabilizes p21WAF1 protein, with minimal increases in p21WAF1 transcription, and (d) BMP-SMAD signaling is slowed by activated RAS/ERK, which attenuates BMP2’s growth suppressive effects. Thus, RAS/ERK is a negative regulator of BMP signaling, and appears to interfere with BMP2-induced p21WAF1 stabilization to avert growth suppression.
BMP2 treatment has been shown to upregulate p21WAF1 in breast, prostate, and gastric cancers [23–29] and the mechanism has been presumed increased transcription of p21WAF1. In other colon cancer cell lines with more modest BMP2-induced growth suppression, p21WAF1 transcription was not elevated [5]. In FET colon cancer cells, which we show are responsive to BMP2 induced growth suppression, p21WAF1 transcription was also not induced. However, the steady state levels of p21WAF1 protein doubled, and we demonstrated that BMP2-induces stabilization of existing p21WAF1 protein, limiting its degradation and allowing p21WAF1 to exert its function for growth suppression. Stabilization of p21WAF1 protein has also been described after TGFβ treatment in FET cells suggesting that both BMP and TGFβ mediate a portion of their growths’ suppression by this mechanism [33]. BMP2 treatment has also been shown to increase the tumor suppressor PTEN in MCF7 breast cancer cells through protein stabilization [34]. We did not find any change in PTEN protein levels after BMP2 treatment in FET colon cancer cells, suggesting that p21WAF1 is more important as a mediator of growth retardation than PTEN in these cells.
RAS/ERK activation is normally regulated by ligand stimulation, turning on a mitogenic pathway that causes cellular proliferation [11]. Indeed, BMP2 activates ERK, counterintuitive to its SMAD-mediated and p21WAF1 mechanistic growth suppression. We hypothesize that this ERK activation is a brake to regulate or limit BMP-SMAD signaling and growth suppression. On the other hand, oncogenic RAS, causing constitutive stimulation of this mitogenic pathway, continuously interferes with intact BMP-SMAD signaling, more permanently muting BMP-SMAD induced signaling and growth suppression. We propose this phenomenon is due to the mechanism suggested by Kretzschmar et al. in which RAS/ERK causes phosphorylation of the linker region of SMAD1, slowing nuclear translocation and subsequent transcription [17]. Indeed, in our FET cells, slowing of SMAD translocation by RAS/ERK may mediate all of the effects observed by BMP2-induced SMAD signaling and growth suppression.
In conclusion, we demonstrate that RAS/ERK attenuates BMP2-SMAD signaling and growth suppression in FET cells. The growth suppression is mediated through BMP-induced increases in p21WAF1 protein through enhanced stability of p21WAF1, and not through increased p21WAF1 transcriptional activity. The BMP-induced p21WAF1 stability is also reduced by activated RAS/ERK. We propose RAS/ERK activation, common in colorectal adenomas and cancer, as one mechanism to prohibit the growth suppressive effects of intact BMP signaling.
Supplementary Material
{kind=link}
Acknowledgments
Supported by the U.S. Public Health Service (T32-HL07212 for SEB; CA90231 and DK067287 to JMC, and DK074019 to BHJ) and the VA Research Service. A portion of this work was presented in abstract form at the February 2006 TGFβ in Cancer and Other Diseases meeting of the American Association for Cancer Research in La Jolla, California, and the May 2006 annual meeting of the American Gastroenterological Association in Los Angeles, California. We thank Dr. Peter ten Dijke, Netherlands Cancer Institute, Amsterdam, the Netherlands for the BRE-Luc construct; Dr. Rik Derynck, University of California, San Francisco for the DN-KRAS construct; and Dr. Burt Vogelstein, Johns Hopkins University, Baltimore, Maryland for the pWWP construct. We would also like to thank UCSD Cancer Center Digital Imaging Shared Resource for use of the deconvolution microscope.
Abbreviations
- BMP
bone morphogenetic protein
- TGFβ
transforming growth factor beta
- ERK
extracellular signal-related kinase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- RT-PCR
reverse transcription polymerase chain reaction
- DN
dominant negative
- JP
juvenile polyposis syndrome
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.cellsig.2007.01.017.
References
- 1.Howe JR, Bair JL, Sayed MG, Anderson ME, Mitros FA, Petersen GM, Velculescu VE, Traverso G, Vogelstein B. Nat Genet. 2001;28:184. doi: 10.1038/88919. [DOI] [PubMed] [Google Scholar]
- 2.Howe JR, Roth S, Ringold JC, Summers RW, Jarvinen HJ, Sistonen P, Tomlinson IP, Houlston RS, Bevan S, Mitros FA, Stone EM, Aaltonen LA. Science. 1998;280:1086. doi: 10.1126/science.280.5366.1086. [DOI] [PubMed] [Google Scholar]
- 3.Huang SC, Chen CR, Lavine JE, Taylor SF, Newbury RO, Pham TTT, Ricciardiello L, Carethers JM. Cancer Res. 2000;60:6882. [PubMed] [Google Scholar]
- 4.Haramis AP, Begthel H, van den Born M, van Es J, Jonkheer S, Offerhaus GJ, Clevers H. Science. 2004;303:1684. doi: 10.1126/science.1093587. [DOI] [PubMed] [Google Scholar]
- 5.Beck SEJB, Fiorino A, Gomez J, Del Rosario E, Cabrera BL, Huang SC, Chow YC, Carethers JM. Am J Physiol: Gasterointest Liver Physiol. 2006;291:G135. doi: 10.1152/ajpgi.00482.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kopreski MS, Benko FA, Borys DJ, Khan A, McGarrity TJ, Gocke CD. J Natl Cancer Inst. 2000;92:918. doi: 10.1093/jnci/92.11.918. [DOI] [PubMed] [Google Scholar]
- 7.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. N Engl J Med. 1988;319:525. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 8.Blank JL, Gerwins P, Elliott EM, Sather S, Johnson GL. J Biol Chem. 1996;271:5361. doi: 10.1074/jbc.271.10.5361. [DOI] [PubMed] [Google Scholar]
- 9.Morrison DK, Kaplan DR, Rapp U, Roberts TM. PNAS. 1988;85:8855. doi: 10.1073/pnas.85.23.8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reuter CWM, Catling AD, Jelinek T, Weber MJ. J Biol Chem. 1995;270:7644. doi: 10.1074/jbc.270.13.7644. [DOI] [PubMed] [Google Scholar]
- 11.Carethers JM. Biology of Colorectal Cancer. In: Rustgi A, Crawford J, editors. Gastrointestinal Cancers: Biology and Clinical Management. 1. Saunders; Philadelphia, PA: 2003. p. 407. [Google Scholar]
- 12.Derynck R, Zhang YE. Nature. 2003;425:577. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 13.Rebollo A, Martinez AC. Blood. 1999;94:2971. [PubMed] [Google Scholar]
- 14.Liu F, Ventura F, Doody J, Massague J. Mol Cell Biol. 1995;15:3479. doi: 10.1128/mcb.15.7.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu F, Hata A, Baker JC, Doody J, Carcamo J, Harland RM, Massague J. Nature. 1996;381:620. doi: 10.1038/381620a0. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Musci T, Derynck R. Curr Biol. 1997;7:270. doi: 10.1016/s0960-9822(06)00123-0. [DOI] [PubMed] [Google Scholar]
- 17.Kretzschmar M, Doody J, Massague J. Nature. 1997;389:618. doi: 10.1038/39348. [DOI] [PubMed] [Google Scholar]
- 18.Kretzschmar M, Liu F, Hata A, Doody J, Massague J. Genes Dev. 1997;11:984. doi: 10.1101/gad.11.8.984. [DOI] [PubMed] [Google Scholar]
- 19.Gayet J, Zhou XP, Duval A, Rolland S, Hoang JM, Cottu P, Hamelin R. Oncogene. 2001;20:5025. doi: 10.1038/sj.onc.1204611. [DOI] [PubMed] [Google Scholar]
- 20.Ye SC, Foster JM, Li W, Liang J, Zborowska E, Venkateswarlu S, Gong J, Brattain MG, Willson JKV. Cancer Res. 1999;59:4725. [PubMed] [Google Scholar]
- 21.Lai CF, Cheng SL. J Biol Chem. 2002;277:15514. doi: 10.1074/jbc.M200794200. [DOI] [PubMed] [Google Scholar]
- 22.Waite KA, Eng C. Hum Mol Genet. 2003;12:679. [PubMed] [Google Scholar]
- 23.Brubaker KD, Corey E, Brown LG, Vessella RL. J Cell Biochem. 2004;91:151. doi: 10.1002/jcb.10679. [DOI] [PubMed] [Google Scholar]
- 24.Ghosh-Choudhury N, Ghosh-Choudhury G, Celeste A, Ghosh PM, Moyer M, Abboud SL, Kreisberg J. Biochim Biophys Acta. 2000;1497:186. doi: 10.1016/s0167-4889(00)00060-4. [DOI] [PubMed] [Google Scholar]
- 25.Ghosh-Choudhury N, Woodruff K, Qi W, Celeste A, Abboud SL, Ghosh-Choudhury G. Biochem Biophys Res Commun. 2000;272:705. doi: 10.1006/bbrc.2000.2844. [DOI] [PubMed] [Google Scholar]
- 26.Pouliot F, Blais A, Labrie C. Cancer Res. 2003;63:277. [PubMed] [Google Scholar]
- 27.Wen XZ, Miyake S, Akiyama Y, Yuasa Y. Biochem Biophys Res Commun. 2004;316:100. doi: 10.1016/j.bbrc.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 28.Yue J, Frey RS, Mulder KM. Oncogene. 1999;18:2033. doi: 10.1038/sj.onc.1202521. [DOI] [PubMed] [Google Scholar]
- 29.Yue J, Hartsough MT, Frey RS, Frielle T, Mulder KM. J Cell Physiol. 1999;178:387. doi: 10.1002/(SICI)1097-4652(199903)178:3<387::AID-JCP13>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 30.Grady WM, Markowitz SD. Annu Rev Genomics Human Genet. 2002;3:101. doi: 10.1146/annurev.genom.3.022502.103043. [DOI] [PubMed] [Google Scholar]
- 31.Jung B, Doctolero RT, Tajima A, Nguyen AK, Keku T, Sandler RS, Carethers JM. Gastroenterology. 2004;126:654. doi: 10.1053/j.gastro.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 32.Jung B, Smith EJ, Doctolero RT, Gervaz P, Alonso JC, Miyai K, Keku T, Sandler RS, Carethers JM. Int J Cancer. 2006;118:2509. doi: 10.1002/ijc.21710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gong J, Ammanamanchi S, Ko TC, Brattain MG. Cancer Res. 2003;63:3340. [PubMed] [Google Scholar]
- 34.Waite KA, Eng C. Nat Rev, Genet. 2003;4:763. doi: 10.1038/nrg1178. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.