

Abstract
Although signalling through the type I insulin-like growth factor receptor (IGF-IR) maintains the survival of haematopoietic cells, a specific role of IGF-IR in haematological neoplasms remains largely unknown. Chronic myeloid leukaemia (CML) is the most common subtype of chronic myeloproliferative diseases. Typically, CML evolves as a chronic phase (CP) disease that progresses into accelerated (AP) and blast phase (BP) stages. In this study, we show that IGF-IR is universally expressed in four CML cell lines. IGF-IR was expressed in only 30% and 25% of CP and AP patients, respectively, but its frequency of expression increased to 73% of BP patients. Increased expression levels of IGF-IR with CML progression was supported by quantitative real-time PCR that demonstrated significantly higher levels of IGF-IR mRNA in BP patients. Inhibition of IGF-IR decreased the viability and proliferation of CML cell lines and abrogated their growth in soft agar. Importantly, inhibition of IGF-IR decreased the viability of cells resistant to imatinib mesylate including BaF3 cells transfected with p210 BCR-ABL mutants, CML cell lines and primary neoplastic cells from patients. The negative effects of inhibition of IGF-IR were attributable to apoptosis and cell cycle arrest due to alterations of downstream target proteins. Our findings suggest that IGF-IR could represent a potential molecular target particularly for advanced stage or imatinib-resistant cases.
Keywords: IGF-IR, chronic myeloid leukaemia, BCR-ABL, picropodophyllin, imatinib mesylate
Introduction
The type I insulin-like growth factor receptor (IGF-IR) is a tetrameric transmembrane receptor tyrosine kinase that is composed of two α and two β subunits linked by disulphide bonds [1]. Signalling through IGF-IR contributes to the establishment and progression of human malignancies. In vitro and in vivo experimental approaches have supported the ability of IGF-IR to promote cellular transformation and survival [2, 3]. In addition, IGF-IR plays important roles in regulating cell differentiation, cell shape and migration and metastatic dissemination [4–6]. The oncogenic potential of IGF-IR has been repeatedly documented in solid tumours including cancers of the prostate, breast, colon, ovary, lung, nervous system and skin [7–11]. Although it has been previously demonstrated that IGF-IR is expressed in haematopoietic cells and that signalling through IGF-IR promotes the proliferation and the survival of these cells, few studies have explored the role of IGF-IR in haematological malignancies and most of these studies focused on plasma cell myeloma [12–15].
Chronic myeloid leukaemia (CML) is the most common subtype of chronic myeloproliferative diseases [16]. It typically evolves through three clinicopathological stages: chronic, accelerated and blast phases (CP, AP and BP, respectively). CML is characterized by the t(9; 22)(q34; q11.2) that leads to the expression of the chimeric protein BCR-ABL, which aberrantly functions as a constitutively active tyrosine kinase [17–19]. Currently, targeted inhibition of BCR-ABL by imatinib mesylate is considered first-line therapy in CML [20–22]. Although imatinib is effective in a majority of CML patients in CP, some of these patients develop resistance most frequently through BCR-ABL mutations [23]. Furthermore, CML patients demonstrate significant resistance to imatinib during the more aggressive BP stage of their disease [24, 25]. In the present study, we explored a role of IGF-IR in CML. We tested the expression of IGF-IR in four CML cell lines and in bone marrow and peripheral blood samples from CML patients at different stages of the disease. We used selective and specific antagonism of IGF-IR to investigate its biological contribution to CML. Our findings suggest that targeting IGF-IR could represent a legitimate approach to treat CML patients, particularly during their advanced stage disease and when they develop resistance to imatinib.
Materials and methods
Antibodies
Antibodies obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA) included Bcl-2 (catalogue number: sc-7382), cyclin B1 (sc-7393), cyclin E (sc-198), Cdc2 (sc-52316), pCdc2 (Thr14/Tyr15; sc-12340-R) and p16 (sc-56330); from Cell Signaling Technology (Danvers, MA, USA) were pIGF-IR (Tyr1131; 3021), pBCR-ABL (p-c-Abl; Tyr412; 2865), Akt (9272) and pAkt (Ser473; 587F11); from Zymed Laboratories (South San Francisco, CA, USA) were IGF-IRβ (39–6700) and Bcl-XL (18–0217); from Calbiochem (Gibbstown, NJ, USA) was BCR-ABL (c-Abl; OP19); from R&D Systems (Minneapolis, MN, USA) was STAT5 (MAB2174); from GeneTex Incorporation (San Antonio, TX, USA) was pSTAT5 (Tyr694; GTX52364) and from Sigma (St. Louis, MO, USA) was β-Actin (A-2228).
Cell lines and treatments
Four CML cell lines – K562, KBM-5, MEG01 and BV173 – were used. The P6 (BALB/c3T3 mouse fibroblasts overexpressing human IGF-IR) and R− (mouse fibroblast 3T3-like cells with a targeted ablation of Igf1r gene) cell lines were a generous gift from Dr. R. Baserga (Philadelphia, PA, USA) and were used as positive and negative controls for the expression of IGF-IR, respectively [26]. BaF3 cells expressing wild-type (WT) p210 BCR-ABL, BCR-ABL mutants or empty vector were kindly provided by Dr. C. Sawyers (New York, NY, USA) [27]. The normal human skin fibroblast cell line AG01523 (Coriell Institute for Medical Research, Camden, NJ, USA) was used as a negative control for the treatment by the cyclolignan picropodophyllin (PPP; Clontech, Mountain View, CA, USA) [14, 28]. Cell lines were maintained in RPMI 1640 (CML cell lines and BaF3 cells permanently transfected with WT p210 BCR-ABL, BCR-ABLE255K or BCR-ABLT315I), DMEM (P6 and R− cell lines) or EEMEM (AG01523 cells) medium supplemented with 10% FBS (15% FBS for AG01523) (Sigma), glutamine (2 mM), penicillin (100 U/ml) and streptomycin (100 μg/ml) at 37°C in humidified air with 5% CO2. RPMI 1640 was additionally supplemented with recombinant murine IL-3 (1 ng/ml; PeproTech, Rocky Hill, NJ, USA) and used to culture BaF3 cells transfected with empty vector. Selective targeting of IGF-IR was achieved by PPP after being dissolved in ethanol to a concentration of 0.5 mM (final concentration of ethanol was less than 0.4% by volume). Imatinib was dissolved in sterile water to a concentration of 1 mM. We also tested the effects of inhibition of IGF-IR by PPP in primary neoplastic cells from five CML (two in CP and three in BP) patients with resistance to imatinib. Approval of the institutional ethical research committee was secured prior to the initiation of the studies that included patient material. In addition, in some experiments, specific down-regulation of IGF-IR was achieved by transient transfection with SMARTpool-designed siRNA (mixture of four different constructs; Dharmacon, Lafayette, CO, USA). The siCONTROL non-targeting siRNA was used as a negative control (Dharmacon). Transfection of the cells by siRNA was performed with the Nucleofector solution as recommended by the manufacturer (Amaxa Biosystems, Gathersburg, PA, USA) (V-solution for K562, R-solution for BV173 and C-solution for KBM-5; T-016 program for K562 and BV173 and X-005 program for KBM-5).
Cell viability, proliferation, apoptosis, cell cycle and colony formation in soft agar assays
Cell viability was evaluated by exclusion of staining by trypan blue dye. Apoptosis was examined by using flow cytometry (FACscan, BD Pharmingen, San Jose, CA, USA) after staining the cells with annexin V-FITC and propidium iodide (PI) (BD Pharmingen). Analysis of the cell cycle was performed with a commercially available kit (CycleTEST PLUS DNA Reagent Kit, BD Pharmingen), whereby PI-stained nuclei were analysed by flow cytometry. In addition, morphological changes consistent with apoptosis and cell cycle arrest were assessed after staining cytospin-prepared slides with Giemsa. Cell proliferation analysis was performed with BrdU staining in an ELISA-based kit (Roche, Mannheim, Germany). Evaluation of colony formation in soft agar was performed with cell transformation detection kit (Chemicon, Temecula, CA, USA). Colonies were stained and photographed with FluorChem 8800 imaging system (Alpha Innotech, San Leandro, CA, USA).
PCR studies
RT-PCR was used for the detection of IGF-IR mRNA in CML cell lines. Briefly, total RNA was isolated using RNeasy Mini Kit (Qiagen, Valencia, CA, USA). Reverse transcription was performed with the Qiagen One-step RT-PCR kit (Qiagen). The primers for IGF-IRα were: forward, 5′-GTAGCTTGCCGCCACTACTACT-3′ and reverse, 5′-GGAGCATCTGAGCAGAAGTAACAGA-3′ (product size 307 bp). Amplification was performed at 94°C for 30 sec., 58°C for 30 sec. and 72°C for 30 sec. for 35 cycles and a final elongation at 72°C for 10 min. in a thermal cycler (PTC100, MJ Research, Watertown, MA, USA). β-Actin (RDP-38, R&D Systems) was used as an internal control. The PCR products were detected by ethidium bromide staining on a 1% agarose gel and visualized by FluorChem 8800 imaging system (Alpha Innotech).
In addition, quantitative real-time PCR was used to measure the relative expression of IGF-IR mRNA in primary peripheral blood neoplastic cells from CML patients. Cells were separated using RBC lysis buffer (eBioscience, San Diego, CA, USA). Total RNA was isolated and cDNA was synthesized using iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions. PCR was performed in a MicroAmp Optical 96-well reaction plate (Applied Biosystems, Foster City, CA, USA) using 1.0 μl of cDNA template, 12.5 μl of 2× TaqMan Master Mix (Applied Biosystems), 1.25 μl of 20× Assay-on-Demand gene expression product (assay ID Hs00181385_m1 for IGF-IR target gene, product number 4319413E for human 18s rRNA as endogenous control, Applied Biosystems). The RT reaction was performed in triplicate wells for 2 min. at 50°C, followed by hot-start PCR (10 min. at 95°C) and 40 cycles of 15 sec. of denaturation at 95°C and 1 min. annealing/extension at 60°C using an ABI 7500 sequence detection system (Applied Biosystems). The relative quantification of IGF-IR gene expression was calculated according to the following formula: 2−ΔCT (ΔCT= CT of IGF-IR - CT of 18s rRNA), where CT is the cycle threshold.
Immunoprecipitation and Western blotting
Cell lysates were obtained using standard techniques and lysis buffer that was composed of 25 mM HEPES (pH 7.7), 400 mM NaCl, 1.5 mM MgCl2, 2 mM ethylenediaminetetraacetic acid, 0.5% Triton X-100, 0.1 mM PMSF, 3 mM DTT, phosphatase inhibitor (20 mM β-GP, 1 mM Na3VO4; Roche) and protease inhibitor cocktails (10 μg/ml leupeptine, 2 μg/ml pepstatin, 50 μg/ml antipain, 1× benzamidine, 2 μg/ml aprotinin, 20 μg/ml chymostatin; Roche). For immunoprecipitation, lysates were incubated with primary antibody overnight at 4°C. Agarose beads conjugated with A/G were added and incubated for 2 hrs at 4°C. The immunocomplexes were spun, washed three times with cold PBS and once with lysis buffer and subjected to SDS-PAGE. For Western blotting, proteins (50–80 μg) were electrophoresed on 6–12% SDS-PAGE. The proteins were transferred to nitrocellulose membranes and probed with specific primary antibodies and then with the appropriate horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology; and GE Healthcare, Cardiff, UK). Proteins were detected using a chemiluminescence-based kit (Amersham Life Sciences, Arlington Heights, IL, USA).
Tyrosine kinase activity assay
IGF-IR or BCR-ABL tyrosine kinase activity in CML cell lines was measured using a commercially available kit (Takara Bio, Otsu, Shiga, Japan). Cell lysates were prepared and the specific antibody was used for immunoprecipitation. Agarose beads conjugated with protein A/G were added. Before kinase reactions were initiated with ATP-2Na in immobilized wells and blocked by blocking solution, PPP was added at the indicated concentrations in tyrosine kinase buffer at room temperature for 30 min. Horseradish peroxidase-conjugated antiphosphotyrosine (PY20) antibody was added to each well and developed by addition of HRP substrate solution (TMBZ). The reaction was stopped with 1N H2SO4 and absorbance was measured at 450 nm in a microplate reader (Bio-Tek Instruments, Winooski, VT, USA). The ATP-free reaction was used as a negative control. In some experiments, endogenous IGF-IR tyrosine kinase activity was calculated using the equation of the PTK activity standard curve, normalized to the protein amount in each well, and expressed as units per microgram protein.
Measurement of pIGF-IR levels by an ELISA-based assay
pIGF-IR level in CML cell lines was measured using the human pIGF-IR DuoSet IC ELISA kit as described by the manufacturer (DYC1770–2; R&D Systems). Total proteins of 125 μg were added onto each well. Absorbance was measured at 450 nm in a microplate spectrophotometer (Bio-Tek Instruments) and the reference wavelength was 540 nm.
Patients, tissue microarray and immunohistochemical staining
CML patients and the tissue microarray were previously described [29, 30]. Immunohistochemical staining was performed on sections from the tissue microarray as well as on formalin-fixed and paraffin-embedded sections from cellblocks prepared from cell lines using standard techniques as previously described [29–31]. Antibody dilution was 1:30 for IGF-IR. Photomicrographs were obtained using a Nikon Microphot FXA microscope (Nikon Instruments, Melville, NY, USA) and an Olympus DP70 camera (Olympus America, Melville, NY, USA).
Statistical analysis
Statistical analysis was performed using Student’s t-test for unpaired data or two-way ANOVA and Bonferroni post hoc test where appropriate in Prism 5 software (version 5.0b, GraphPad Software, La Jolla, CA, USA). P < 0.05 was considered to be statistically significant.
Results
Expression of IGF-IR in CML cell lines and primary human tumours
Immunohistochemical studies demonstrated strong expression of IGF-IR protein in four CML cell lines including K562, KBM-5, MEG01 and BV173 (Fig. 1A). Western blotting and RT-PCR confirmed these results and showed the expression of IGF-IR protein and mRNA in CML cell lines, respectively (Fig. 1B and C). The K562 cell line demonstrated higher expression levels of IGF-IR protein and mRNA compared with other CML cell lines. The mouse fibroblast cell lines R− and P6 were used as negative and positive controls for the expression of IGF-IR, respectively. It is important to note that the overexpression of IGF-IR in P6 cells is not physiologic. Although all of the CML cell lines expressed IGF-IR, ELISA showed that pIGF-IR levels were higher in the K562 and BV173 cell lines compared with KBM-5 and MEG01 (Fig. 1D). In addition, tyrosine kinase activity assay showed the KBM-5 and MEG01 cell lines to possess significantly lower tyrosine kinase activity (Fig. 1E).
Fig 1.
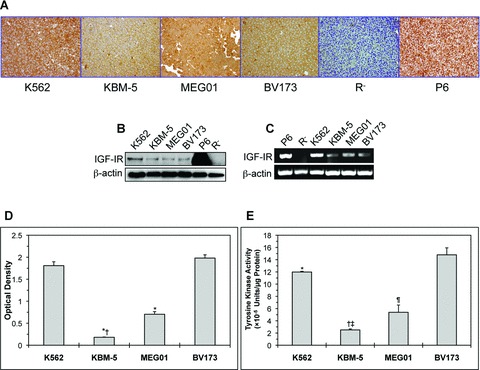
Expression of IGF-IR in CML cell lines. (A) Immunohistochemical staining shows strong and universal expression of IGF-IR in four CML cell lines including K562, KBM-5, MEG01 and BV173. (B) Western blotting confirms the expression of IGF-IR protein in all of the CML cell lines. The K562 cell line demonstrates a higher level of IGF-IR protein compared with the other three cell lines. β-Actin confirms equal loading. (C) RT-PCR shows the expression of IGF-IR mRNA in CML cell lines. Similar to IGF-IR protein levels, K562 cells appear to express the highest level of IGF-IR mRNA. β-Actin supports equal loading. In all of these experiments, R− and P6 cells served as negative and positive controls for the expression of IGF-IR, respectively. (D) The KBM-5 and MEG01 cell lines demonstrate lower levels of pIGF-IR in comparison with K562 and BV173. The results represent the means ± S.D. of three experiments. *: P < 0.0001 compared with K562 and BV173 and †: P < 0.0001 compared with MEG01. (E) The KBM-5 and MEG01 cell lines possess the lowest levels of IGF-IR tyrosine kinase activity. Depicted results represent the means ± S.D. of three experiments. *: P < 0.05 compared with BV173, †: P < 0.0001 compared with K562 and BV173, ‡: P < 0.05 compared with MEG01 and ¶: P < 0.001 compared with K562 and BV173.
The expression of IGF-IR was also studied in a tissue microarray that included primary bone marrow specimens from CML patients at different stages of the disease (Fig. 2A). Only 3 of 10 (30%) and 2 of 8 (25%) of CP and AP CML patients expressed IGF-IR, respectively. Nonetheless, the frequency of IGF-IR expression increased to 11 of 15 (73%) of CML patients during the advanced BP. Increased expression levels of IGF-IR with the progression of CML was further supported by quantitative real-time PCR that demonstrated significantly higher levels of IGF-IR mRNA in the majority of BP patients compared with CP and AP patients (Fig. 2B).
Fig 2.
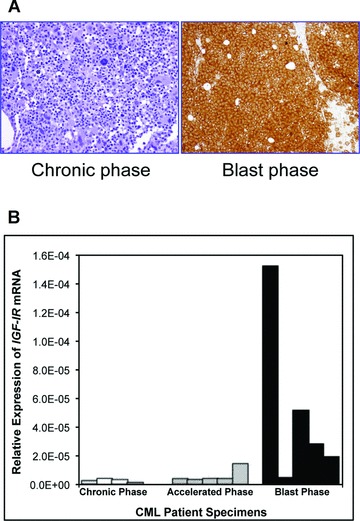
Expression of IGF-IR in primary human CML specimens. (A) A representative bone marrow specimen from a patient with CML in BP demonstrates strong expression of IGF-IR protein compared with bone marrow specimen from a CML patient in CP that is negative for IGF-IR. (B) The markedly increased expression of IGF-IR in the majority of BP CML patients (four out of five patients) compared with patients in CP (four patients) or AP (five patients) is also demonstrated by real-time quantitative PCR analysis of primary neoplastic cells.
Selective or specific antagonism of IGF-IR induces negative biological effects in CML cell lines
PPP, a selective inhibitor of IGF-IR, decreased the viability of CML cell lines. This effect was both concentration and time dependent and was more pronounced in the cell lines KBM-5 and MEG01 when compared with K562 and BV173 (Fig. 3A). At 48 hrs, PPP IC50 was 0.3 μM and 0.9 μM for KBM-5 and MEG01 cell lines, respectively. In spite of being less effective in K562 and BV173 cells, PPP at a concentration of 2.0 μM induced 36% and 37% decrease in the cell viability of these two cell lines at 48 hrs, respectively (Fig. 3A). The human skin fibroblast cells AG01523 were used as a negative control for the effect of the treatment with PPP [14]. Also, PPP induced concentration- and time-dependent increase in apoptotic cell death in CML cell lines. Apoptosis was demonstrated by flow cytometric analysis of annexin V binding (Fig. 3B). Cells were considered apoptotic when stained for annexin V only or when stained simultaneously for annexin V and PI. Similar to the changes in cell viability, inhibition of IGF-IR by PPP was associated with more apoptotic death in the two cell lines KBM-5 and MEG01. Furthermore, PPP induced G2/M-phase cell cycle arrest in the four cell liens included in the study (Fig. 3C). Apoptosis and cell cycle arrest were also morphologically documented after staining of the cells with Giemsa (Fig. 3D). Morphological changes consistent with apoptosis included nuclear fragmentation and condensation and cell shrinkage. The occurrence of the G2/M-phase was demonstrated in the form of significantly increased atypical mitotic figures. In addition, Fig. 3E and F demonstrate that antagonism of IGF-IR by PPP induced significant concentration-dependent decrease in the cell proliferation and cell growth in soft agar of the CML cell lines K562, KBM-5 and MEG01. The BV173 cell line showed less response to PPP in these two experiments. The number of colonies corresponding to each of the cell lines is shown in the left panel of Fig. 3F, and representative culture plates from K562 and KBM-5 cell lines are depicted in the right panel.
Fig 3.
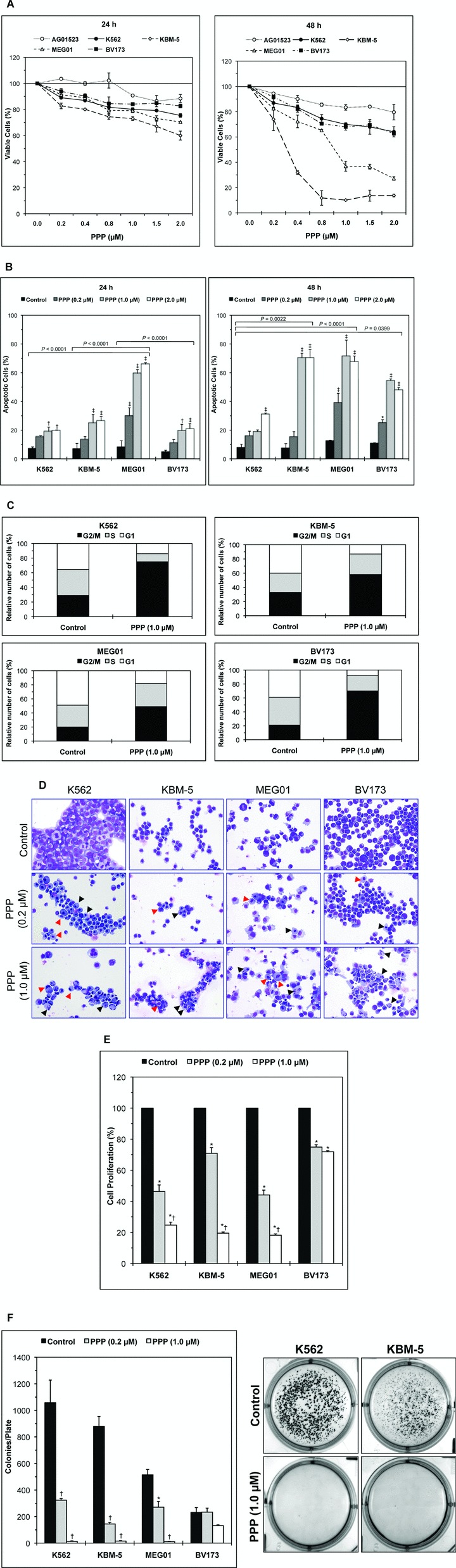
Selective inhibition of IGF-IR induces negative biological effects in CML cell lines. (A) The selective inhibitor of IGF-IR PPP induces concentration- and time-dependent decrease in the viability of CML cell lines. Results shown are the means ± S.D. of three consistent experiments (24 hrs in the left panel and 48 hrs in the right panel). The effect of PPP is more pronounced in the KBM-5 and MEG01 cell lines. At 24 hrs, the decrease in cell viability was statistically significant between the four cell lines and the control cells AG01523 (P < 0.0001). At 48 hrs, the decrease in the cell viability remained statistically significant between the four cell lines and the control cells AG01523, albeit more significant in KBM-5 and MEG01 cells (P= 0.017 for K562 and BV173 and P < 0.0001 for KBM-5 and MEG01). In addition, the difference in PPP-induced decrease in cell viability between both K562 and BV173 cell lines versus KBM-5 and MEG01 was statistically significant (P < 0.0001 at both 24 and 48 hrs). (B) Antagonism of IGF-IR by PPP induces concentration- and time-dependent increase in apoptotic cell death in CML cell lines (results collected at 24 hrs are shown in the left panel and at 48 hrs in the right panel). Results shown represent the means ± S.D. of three consistent experiments. *: P < 0.05, †: P < 0.01, and ‡: P < 0.001 compared with control untreated cells. At 24 hrs, the cell line MEG01 demonstrated significantly more apoptotic cell death compared with the other three cell lines. After 48 hrs, apoptosis was more pronounced in KBM-5 and MEG01. Comparing the collective response of K562 and BV173 to PPP versus KBM-5 and MEG01 cell lines showed that P= 0.0021 at 24 hrs and P= 0.0004 at 48 hrs. (C) Treatment of CML cell lines with PPP induces G2/M-phase cell cycle arrest as demonstrated by flow cytometric analysis after staining with PI. (D) Morphological changes consistent with apoptosis are shown, and include nuclear condensation and fragmentation and cell shrinkage (black arrowheads). Markedly increased atypical mitotic forms are consistent with G2/M-phase cell cycle (red arrowheads). Original magnification is ×500. (E) Antagonism of IGF-IR signalling by PPP induces concentration-dependent decrease in the proliferation of CML cell lines at 24 hrs. At a PPP concentration of 1.0 μm, this effect is less pronounced in the BV173 cell line. The results shown are the means ± S.D. of three experiments. *: P < 0.001 compared with control untreated cells and †: P < 0.001 compared with PPP (0.2 μM). (F) Treatment of CML cell lines with PPP markedly decreases their growth in soft agar. The effect is more prominent in K562, KBM-5 and MEG01. The means ± S.D. of the numbers of the colonies of each cell line from three consistent experiments are shown in the left panel. *: P < 0.01 and †: P <0.001 compared with control untreated cells. Representative cell culture plates from K562 and KBM-5 cells are depicted in the right panel.
Because PPP might have induced some nonspecific effects, we have also initiated experiments using IGF-IR siRNA. IGF-IR siRNA decreased the viability of CML cell lines. The effect of IGF-IR siRNA was more pronounced at 48 hrs (Fig. 4A). Consistently, treatment of these cells with IGF-IR siRNA was associated with significant increase in apoptotic cell death that became more prominent at 72 hrs (Fig. 4B). Similar to the results obtained with PPP, the KBM-5 cell line was generally more sensitive to the specific down-regulation of IGF-IR by siRNA compared with K562 and BV173.
Fig 4.
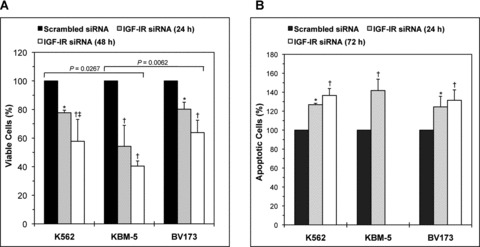
Specific blockade of IGF-IR decreases cell viability and induces apoptosis in CML cell lines. (A) IGF-IR siRNA decreases the viability of K562, KBM-5 and BV173 cell lines. The decrease in cell viability is time-dependent and becomes more pronounced at 48 hrs after transfection of the cells. At 24 hrs, the KBM-5 cell line appears to be more sensitive to the effects of IGF-IR siRNA when compared with K562 and BV173. *: P < 0.05 and †: P < 0.001 versus control untreated cells and ‡: P < 0.05 versus IGF-IR siRNA (24 hrs). Comparing the collective effect of IGF-IR siRNA on decreasing the viability of the K562 and BV173 cell lines versus KBM-5 demonstrated that P= 0.0002. (B) IGF-IR siRNA increases apoptotic cell death in K562, KBM-5 and BV173 cell lines. The effect of IGF-IR siRNA is pronounced in the KBM-5 cell line at 24 hrs after transfection and, therefore, additional analysis at 72 hrs was not performed. For the K562 and BV173 cell lines, after 72 hrs apoptosis became equally similar to KBM-5.at 24 hrs. *: P < 0.01 and †: P < 0.001 compared with control untreated cells. Statistical significance was not achieved when individual cell lines were compared but comparing the collective effect of IGF-IR siRNA on apoptosis showed a slightly more significant response in KBM-5 cells versus K562 and BV173 (P= 0.048).
Blockade of IGF-IR decreases the viability of imatinib-resistant p210 BCR-ABL-expressing cell lines or primary neoplastic cells
To test the effect of inhibition of IGF-IR on imatinib-resistant p210 BCR-ABL-expressing cells, we employed three different approaches. In the first, we used BaF3 cells permanently transfected with WT p210 BCR-ABL or one of its mutants BCR-ABLE255K or BCR-ABLT315I that are known to be resistant to imatinib. As shown in Fig. 5A, Western blotting confirmed the expression of WT p210 BCR-ABL or one of its mutants, IGF-IR, and pIGF-IR in BaF3. At the other hand, BaF3 cells transfected with empty vector only demonstrated the expression of IGF-IR and pIGF-IR proteins. Only BaF3 cells that expressed WT p210 BCR-ABL demonstrated marked concentration- and time-dependent decrease in cell viability after treatment with imatinib (Fig. 5B). In contrast, BaF3 cells that expressed either BCR-ABLE255K or BCR-ABLT315I or transfected with an empty vector were completely resistant to imatinib (Fig. 5B). Notably, BaF3 cells transfected with BCR-ABLE255K or BCR-ABLT315I mutants demonstrated significant concentration- and time-dependent decrease in their viability when treated with PPP (Fig. 5C).
Fig 5.
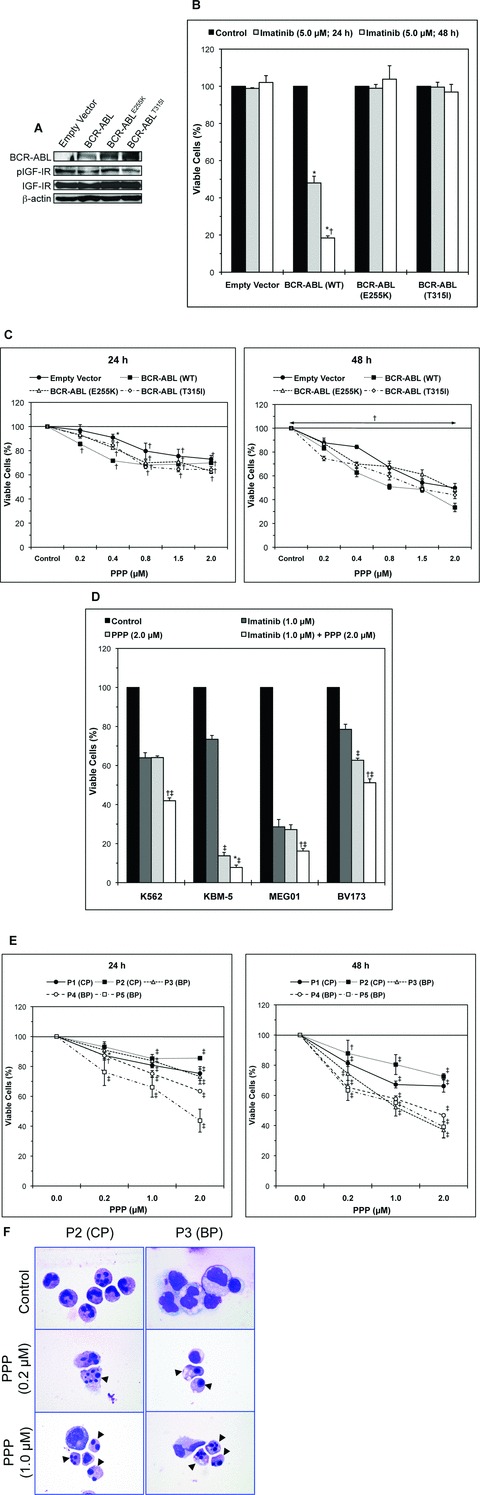
Inhibition of IGF-IR decreases the viability of imatinib-resistant p210 BCR-ABL-expressing cells. (A) Western blotting shows the expression of IGF-IR, pIGF-IR, WT p210 BCR-ABL, BCR-ABLE255K or BCR-ABLT315I in BaF3 cells. BaF3 cells transfected with empty vector lack the expression of BCR-ABL. β-Actin confirms equal loading of the proteins. (B) Only BaF3 cells transfected with WT p210 BCR-ABL demonstrate time-dependent decrease in cell viability after treatment with imatinib (5.0 μM). In contrast, BaF3 cells transfected with BCR-ABL mutants or empty vector are completely resistant to imatinib treatment. *: P < 0.001 compared with control untreated cells and †: P < 0.001 compared with imatinib (5.0 μM; 24 hrs). (C) Targeting IGF-IR by PPP induces concentration- and time-dependent decrease in the viability of BaF3 cells transfected with WT BCR-ABL or the BCR-ABLE255K and BCR-ABLT315I mutants that are known to be resistant to imatinib. The results at 24 hrs are shown in the left panel and those at 48 hrs in the right panel. The results are means ± S.D. of three consistent experiments. *: P < 0.05 and †: P < 0.001 versus control untreated cells. (D) Treatment of KBM-5 or BV173 cell lines with PPP alone is significantly more effective in decreasing the viability of these cells than treatment with imatinib alone. The effect on cell viability of all CML cell lines is significantly enhanced when imatinib was combined with PPP. Notably, the MEG01 cell line demonstrates pronounced response to imatinib. Nonetheless, the effect of imatinib in MEG01 cell line is significantly enhanced when PPP was additionally used. The data represent the means ± S.D. of three experiments. *: P < 0.05 and †: P < 0.001 compared with PPP (2.0 μM) and ‡: P < 0.001 compared with imatinib (1.0 μM). (E) PPP induces concentration- and time-dependent decrease in the viability of primary cells from five imatinib-resistant CML patients (two in CP and three in BP). Results at 24 hrs are shown in the left panel and at 48 hrs in the right panel. When compared with untreated cells, the decrease in cell viability is statistically significant at a PPP low concentration of 0.2 μM in four of the five patients. The results are shown as means ± S.D. of three experiments. *: P < 0.05, †: P < 0.01 and ‡: P <0.001 compared with untreated cells. (F) Inhibition of IGF-IR by PPP induces morphological changes consistent with apoptosis (black arrowheads), including cell shrinkage, nuclear condensation, nuclear fragmentation and cytoplasmic vacuolization, in primary CML cells from patients with resistance to imatinib. Representative results from P2 (CP) and P3 (BP), included in Fig. 5E, are shown. Original magnification is ×1000.
In the second approach, we examined the effects of treatment with imatinib, PPP or combined imatinib and PPP on the viability of CML cell lines (Fig. 5D). At 24 hrs, imatinib decreased the viability of K562, KBM-5 and BV173 cell lines by 36%, 27% and 21%, respectively. PPP alone decreased the viability of these cell lines by 36%, 86% and 37%, respectively. Combining imatinib and PPP was more dramatic because the viability of K562, KBM-5 and BV173 cell lines decreased by 58%, 92% and 49%, respectively. It is worth mentioning that although the MEG01 cell line was generally more sensitive and demonstrated a decreased viability of 71% or 73% after treatment with imatinib or PPP alone, respectively, combined treatment significantly enhanced this effect and resulted in 84% decrease in the viability of these cells (Fig. 5D).
Lastly, we examined the effect of inhibition of IGF-IR by PPP on primary neoplastic cells collected from five imatinib-resistant CML patients (two in CP and three in BP). PPP efficiently induced time- and concentration-dependent decrease in the viability of these cells (Fig. 5E). The decrease in the viability of these cells could be explained at least partially by occurrence of apoptotic cell death as demonstrated by cell shrinkage, nuclear condensation, nuclear fragmentation and cytoplasmic vacuolization (Fig. 5F).
Effects of selective or specific down-regulation of IGF-IR on IGF-IR, BCR-ABL and downstream target molecules
To this end we sought to explore a biochemical explanation for the negative effects observed in CML cells after blockade of IGF-IR signalling. We first tested the effects of PPP on IGF-IR and BCR-ABL tyrosine kinases. Whereas PPP decreased the tyrosine kinase activity of IGF-IR in a concentration-dependent fashion in K562 and KBM-5 cell lines, it did not induce a similar effect on BCR-ABL (Fig. 6A). In addition, Western blotting and co-immunoprecipitation studies (the KBM-5 cell line is depicted as a representative example) showed that PPP decreased the levels of tyrosine phosphorylated IGF-IR in a concentration-dependent manner (Fig. 6B). Of important note is that PPP did not reduce the tyrosine phosphorylation levels of BCR-ABL (Fig. 6B). The decrease in IGF-IR tyrosine kinase activity and pIGF-IR levels was associated with down-regulation of activated/phosphorylated Akt and STAT5. PPP also caused concentration-dependent decreases in Bcl-2, Bcl-XL and caspase-3, which is consistent with apoptotic cell death. Finally, inhibition of IGF-IR induced significant alterations in cell cycle regulatory proteins. Specifically, PPP induced up-regulation of cyclin B1, and simultaneous down-regulation of cyclin E and pCdc2. Changes were not seen in Cdc2 or p16. Collectively, these results are consistent with G2/M-phase cell cycle arrest.
Fig 6.
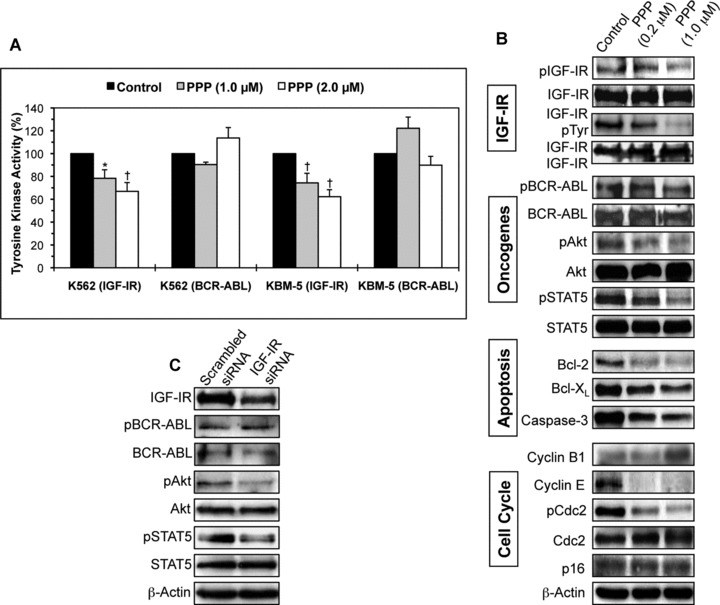
Effects of inhibition of IGF-IR on IGF-IR, BCR-ABL and downstream target proteins in CML cell lines. (A) PPP induces concentration-dependent decrease in IGF-IR tyrosine kinase activity in K562 and KBM-5 cell lines. In contrast, PPP fails to cause similar effect in BCR-ABL tyrosine kinase activity. The results represent the means ± S.D. of three consistent experiments. *: P < 0.01 and †: P < 0.001 compared with control untreated cells. (B) Western blotting and co-immunoprecipitation studies confirm that PPP decreases the tyrosine phosphorylation of IGF-IR in a concentration-dependent fashion (results shown are representative and were obtained from the KBM-5 cell line). The basal levels of IGF-IR did not change after treatment with PPP. The phosphorylation level of BCR-ABL remains unchanged after treatment with PPP. The decrease in pIGF-IR is associated with down-regulation of pAkt and pSTAT5, two oncogenic proteins in CML. Changes are not seen in Akt and STAT5. Also, PPP induces changes consistent with apoptotic cell death including down-regulation of Bcl-2, Bcl-XL and caspase-3. Moreover, treatment with PPP induces up-regulation of cyclin B1 and down-regulation of cyclin E and pCdc2, whereas the levels of Cdc2 and p16 remain unchanged. Overall, the changes in the cell cycle regulatory proteins are consistent with G2/M-phase cell cycle arrest. β-Actin shows equal loading of the proteins. (C) IGF-IR siRNA decreases IGF-IR levels in the KBM-5 cell line and this decrease is associated with down-regulation of pAkt and pSTAT5. Basal levels of these two proteins remain unchanged. IGF-IR siRNA did not induce alterations in BCR-ABL or pBCR-ABL protein. β-Actin confirms equal loading of the proteins.
We also confirmed some of the molecular effects of PPP by using siRNA to specifically down-regulate IGF-IR. Transfection of the KBM-5 cell line with IGF-IR siRNA decreased IGF-IR protein level, whereas BCR-ABL and pBCR-ABL protein levels remained unaltered (Fig. 6C). Similar to the effects observed with PPP, down-regulation of IGF-IR by siRNA was associated with a notable decrease in pAkt and pSTAT5 proteins (Fig. 6C).
Discussion
CML is the most common form of chronic myeloproliferative diseases and is characterized by the BCR-ABL chimeric protein that possesses a constitutively active ABL tyrosine kinase. It is not uncommon that CML evolves into three clinicopathological stages CP, AP and BP. The discovery of imatinib mesylate, a selective inhibitor of ABL, has revolutionized the therapeutic approach and significantly improved the clinical outcome of CML patients [20, 21]. Nonetheless, this therapeutic approach is not always effective because some patients develop BCR-ABL gene mutations that render them resistant to imatinib [23, 32, 33]. Furthermore, imatinib is much less effective when patients evolve into the more aggressive BP stage where sometimes they develop BCR-ABL gene amplifications [23]. Recent studies in mice demonstrated that targeting multiple kinases is more effective in improving treatment of BCR-ABL-expressing leukaemia [34]. In addition, considering the steady increase in the number of CML patients who develop resistance to imatinib, identifying BCR-ABL-independent new therapeutic pathways to be utilized for the treatment of these patients is highly needed [35].
Signalling through the IGF-IR tyrosine kinase has recently become a major focus of cancer research and most of this research has been related to solid tumours [7–11, 36]. Despite the stimulatory effects that IGF-IR exerts on haematopoietic cells [12, 37, 38], significantly fewer studies have investigated a role of IGF-IR in haematological malignancies. In the present study we found that the expression of IGF-IR protein is much more pronounced during the advanced BP stage of CML. This finding was further document by using quantitative real-time PCR that showed remarkably higher levels of IGF-IR mRNA in primary cells from the majority of CML patients in BP compared with CP and AP patients. IGF-IR was also expressed in four CML cell lines. It is important to note that these cell lines represent either myeloid – K562, KBM-5, MEG01 – or lymphoid – BV173 – blasts that express p210 BCR-ABL, and were all developed from CML patients during the advanced BP stage of their disease [39].
The significance of IGF-IR as a potential molecular target in CML was stressed when PPP induced negative biological effects in CML cells. PPP is a cyclolignan that induces activation loop-specific inhibition of tyrosine phosphorylation of IGF-IR [28]. The effects of PPP in CML cells included apoptosis, G2/M-phase cell cycle arrest, decreased cell proliferation and abrogation of cell growth in soft agar. Similar to our results in the cell cycle, a recent study showed that PPP induces G2/M-phase cell cycle arrest in plasma cell myeloma cells [14]. In contrast to CML cell lines, PPP did not have significant negative effects on the viability of the human skin fibroblast cells AG01523 suggesting that selective targeting of IGF-IR will probably not affect benign human cells. It is important to emphasize that PPP, despite being able to decrease IGF-IR kinase activity and to down-regulate pIGF-IR, failed to induce similar effects on BCR-ABL. These findings, to a great extent, ruled out the possibility that the effects of PPP were at least partially attributed to nonspecific interactions with BCR-ABL. However, because pharmacologic/selective inhibitors might induce unknown ‘off-target’ effects, we sought to explore specific antagonism of IGF-IR by siRNA. The results were consistent with PPP and demonstrated that IGF-IR siRNA decreases the viability and enhances the apoptosis of CML cell lines.
The negative biological effects of inhibition of IGF-IR could be at least partially explained by several alterations in apoptosis and cell cycle regulatory proteins as well as by the decreases in pSTAT5 and pAkt, two proteins with known significant oncogenic effects in CML. The oncogenic role of pAkt and pSTAT5 in CML has been shown to be even more pronounced when the patients develop resistance to imatinib [40–42]. In our experiments, the decrease in pAkt and pSTAT5 after treatment with PPP or IGF-IR siRNA was significant yet independent from any inhibitory effect on BCR-ABL, a well-documented upstream activator of both Akt and STAT5 [43–45]. These results support the notion that in the advanced and more aggressive clinicopathological stages of CML, BCR-ABL is most likely not the only ‘key player’. Also, these results provide strong evidence to indicate that targeting alternative signalling transduction mechanisms that are independent from BCR-ABL might induce effects superior to targeting BCR-ABL at least in some cases of advanced stage CML patients. Akt is a downstream effector of IGF-IR signalling and, therefore, it was expected that antagonism of IGF-IR signalling would decrease the activation/phosphorylation status of Akt in CML cells [46, 47]. On the other hand, the decrease in pSTAT5 levels after targeting IGF-IR by PPP or siRNA is intriguing. Previous studies exploring the contribution of IGF-I/IGF-IR signalling to the activation of STAT proteins are notably limited. Some of these studies supported the role of IGF-IR in the phosphorylation/activation of STAT3 through a JAK1/JAK3-dependent mechanism but a direct role of IGF-IR in the activation of STAT5 remains debatable [48]. It has been proposed that IGF-I/IGF-IR signalling could function to augment the activation of STAT5 through interaction with other cytokine/growth factor signalling mechanisms such as erythropoietin, for example [49]. It still cannot be completely excluded that the activation of STAT5 by IGF-IR is cell type dependent. In any case, this interesting and important observation needs further exploration.
We have recently identified a pathogenetic role of IGF-IR in T-cell ALK+ anaplastic large cell lymphoma [50]. Our results in this lymphoma demonstrate the physical association and reciprocal functional collaboration between IGF-IR and NPM-ALK, a chimeric protein with constitutive tyrosine kinase activity that carries significant biological similarities with BCR-ABL. Therefore, we questioned whether the effect of IGF-IR in CML could be attributable to its interaction with BCR-ABL. Although the results of inhibition of IGF-IR by PPP or siRNA do not support this interaction, we further explored this point. To this end, we used different antibody combinations to perform co-immunoprecipitation experiments that failed to demonstrate the physical association between IGF-IR and BCR-ABL (data not shown). It is possible that BCR-ABL indirectly enhances IGF-IR signalling through stimulation of the signalling loop of IGF-I, the primary ligand of IGF-IR, as has been recently reported by Lakshmikuttyamma et al.[51]. In agreement with Lakshmikuttyamma et al.[51] we also found that the K562 cell line secretes considerable amounts of IGF-I, but in contrast, our experiments demonstrated a minimal release of IGF-I by the other three CML cell lines used in the present study (data not shown). Based on these observations, we propose that the increased expression and activation of IGF-I/IGF-IR signalling loop in CML could also be due to additional factors that might not be completely dependent on BCR-ABL. These factors appear to be more pronounced during the BP stage. Our findings carry significant clinical implications as they suggest that IGF-IR signalling in CML might be independent from BCR-ABL and thus targeting IGF-IR is expected to be more therapeutically effective for the treatment of imatinib-resistant patients.
Not surprisingly the effect of inhibition of IGF-IR was not equal among the CML cell lines. In general, the cell lines KBM-5 and MEG01 were more sensitive to IGF-IR inhibition when compared with K562 and BV173. We cannot completely exclude that the cell lines used in our study have different rate of PPP efflux pump but the similarities in the relative sensitivities of the cell lines when treated with PPP or IGF-IR siRNA makes this possibility questionable. Most probably the difference in the sensitivities of these cell lines to IGF-IR inhibition is, at least in part, due to differences in the phosphorylation level and tyrosine kinase activity of IGF-IR. In support of this notion, we found that the more sensitive KBM-5 and MEG01 cell lines possess less phosphorylation levels and tyrosine kinase activity of IGF-IR in comparison with K562 and BV173. We have elected to measure the levels of pIGF-IR by using an ELISA-based method because, in our experience, the commercially available anti-pIGF-IR antibodies are nonspecific and interact with the insulin receptor. According to the manufacturer, the antibody included in the ELISA kit is specific for pIGF-IR with no apparent cross-reactivity with the insulin receptor. Similar to the variability in their response to PPP, we also found the K562, KBM-5 and BV173 cell lines to be more resistant to imatinib than the MEG01 cell line. Although this variability reflects the heterogeneity of the cell lines, similar observations could also be seen in the patients’ response to therapeutic interventions, which is in agreement with the current concept that a ‘personalized’ therapeutic approach rationally tailored based on a common molecular background could be significantly more effective in eradicating cancer. Regardless, a combined treatment of these cell lines with both PPP- and imatinib-induced synergetic effect and a more pronounced decrease in cell viability. The ability of IGF-IR inhibitor to overcome the resistance to imatinib was also illustrated when PPP caused marked decrease in the viability of BaF3 cells permanently transfected with BCR-ABLE255K or BCR-ABLT315I, two imatinib-resistant BCR-ABL mutants that are known to have significant clinical impact in CML patients [23, 32, 33]. The clinical potential of the inhibition of IGF-IR in imatinib-resistant CML was further emphasized in primary cells from CML patients with known resistance to imatinib. Although the level of IGF-IR activation was not known in the cells collected from these patients, PPP decreased the viability and induced significant apoptosis in these cells.
In conclusion, in this paper we show markedly increased expression levels of IGF-IR with the advancement of CML to the BP stage. Also, inhibition of IGF-IR signalling induces negative biological impact in CML cells. Importantly, blockade of IGF-IR appears to overcome imatinib-resistance of CML cells. These findings suggest that targeting IGF-IR might become a potential therapeutic strategy for the treatment of CML patients particularly during the more aggressive BP stage or when resistance to therapy with imatinib evolves. New selective and specific IGF-IR inhibitors are currently being developed and utilized in clinical trials [52, 53]. Some of these inhibitors have demonstrated promising results with minimal untoward effects in patients with aggressive solid tumours [54], which make these inhibitors reasonable alternative to be evaluated for the treatment of CML patients.
Acknowledgments
We thank E. Lin, M.S., from Department of Biostatistics at M. D. Anderson Cancer Center for her valuable help with statistical analysis. This work is supported by CA114395 grant from the National Institutes of Health and by the Physician Scientist Program Award from M. D. Anderson Cancer Center to H.M.A.
References
- 1.Ullrich A, Gray A, Tam AW, et al. Insulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 1986;5:2503–12. doi: 10.1002/j.1460-2075.1986.tb04528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baserga R, Sell C, Porcu P, et al. The role of the IGF-I receptor in the growth and transformation of mammalian cells. Cell Prolif. 1994;27:63–71. doi: 10.1111/j.1365-2184.1994.tb01406.x. [DOI] [PubMed] [Google Scholar]
- 3.Sell C, Rubini M, Rubin R, et al. Simian virus 40 large tumor antigen is unable to transform mouse embryonic fibroblasts lacking type 1 insulin-like growth factor receptor. Proc Natl Acad Sci USA. 1993;90:11217–21. doi: 10.1073/pnas.90.23.11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopez T, Hanahan D. Elevated levels of the IGF-I receptor convey invasion and metastatic capability in a mouse model of pancreatic islet tumourigenesis. Cancer Cell. 2002;1:339–53. doi: 10.1016/s1535-6108(02)00055-7. [DOI] [PubMed] [Google Scholar]
- 5.Mauro L, Bartucci M, Morelli C, et al. IGF-I receptor-induced cell-cell adhesion of MCF-7 breast cancer cells requires the expression of junction protein ZO-1. J Biol Chem. 2001;276:39892–7. doi: 10.1074/jbc.M106673200. [DOI] [PubMed] [Google Scholar]
- 6.van Golen CM, Schwab TS, Kim B, et al. Insulin-like growth factor-I receptor expression regulates neuroblastoma metastasis to bone. Cancer Res. 2006;66:6570–8. doi: 10.1158/0008-5472.CAN-05-1448. [DOI] [PubMed] [Google Scholar]
- 7.Adachi Y, Lee CT, Coffee K, et al. Effects of genetic blockade of the insulin-like growth factor receptor in human colon cancer cell lines. Gastroenterology. 2002;123:1191–204. doi: 10.1053/gast.2002.36023. [DOI] [PubMed] [Google Scholar]
- 8.Burfeind P, Chernicky CL, Rininsland F, et al. Antisense RNA to the type I insulin-like growth factor receptor suppresses tumor growth and prevents invasion by rat prostate cancer cells in vivo. Proc Natl Acad Sci USA. 1996;93:7263–8. doi: 10.1073/pnas.93.14.7263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunn SE, Ehrlich M, Sharp NJ, et al. A dominant negative mutant of the insulin-like growth factor-I receptor inhibits the adhesion, invasion, and metastasis of breast cancer. Cancer Res. 1998;58:3353–61. [PubMed] [Google Scholar]
- 10.Hongo A, Kuramoto H, Nakamura Y, et al. Antitumor effects of a soluble insulin-like growth factor I receptor in human ovarian cancer cells: advantage of recombinant protein administration in vivo. Cancer Res. 2003;63:7834–9. [PubMed] [Google Scholar]
- 11.Lee CT, Park KH, Adachi Y, et al. Recombinant adenoviruses expressing dominant negative insulin-like growth factor-I receptor demonstrate antitumor effects on lung cancer. Cancer Gene Ther. 2003;10:57–63. doi: 10.1038/sj.cgt.7700524. [DOI] [PubMed] [Google Scholar]
- 12.McCubrey JA, Steelman LS, Mayo MW, et al. Growth-promoting effects of insulin-like growth factor-1 (IGF-1) on hematopoietic cells: overexpression of introduced IGF-1 receptor abrogates interleukin-3 dependency of murine factor-dependent cells by a ligand-dependent mechanism. Blood. 1991;78:921–9. [PubMed] [Google Scholar]
- 13.Menu E, Jernberg-Wiklund H, Stromberg T, et al. Inhibiting the IGF-1 receptor tyrosine kinase with the cyclolignan PPP: an in vitro and in vivo study in the 5T33MM mouse model. Blood. 2006;107:655–60. doi: 10.1182/blood-2005-01-0293. [DOI] [PubMed] [Google Scholar]
- 14.Stromberg T, Ekman S, Girnita L, et al. IGF-1 receptor tyrosine kinase inhibition by the cyclolignan PPP induces G2/M-phase accumulation and apoptosis in multiple myeloma cells. Blood. 2006;107:669–78. doi: 10.1182/blood-2005-01-0306. [DOI] [PubMed] [Google Scholar]
- 15.Walsh PT, Smith LM, O’Connor R. Insulin-like growth factor-1 activates Akt and Jun N-terminal kinases (JNKs) in promoting the survival of T lymphocytes. Immunology. 2002;107:461–71. doi: 10.1046/j.1365-2567.2002.01525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faderl S, Talpaz M, Estrov Z, et al. The biology of chronic myeloid leukemia. New Engl J Med. 1999;341:164–72. doi: 10.1056/NEJM199907153410306. [DOI] [PubMed] [Google Scholar]
- 17.Ben-Neriah Y, Daley GQ, Mes-Masson AM, et al. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science. 1986;233:212–4. doi: 10.1126/science.3460176. [DOI] [PubMed] [Google Scholar]
- 18.Muller AJ, Young JC, Pendergastg A-M, et al. BCR first exon sequences specifically activate the BCR/ABL tyrosine kinase oncogene of Philadelphia chromosome-positive human leukemias. Mol Cell Biol. 1991;11:1785–92. doi: 10.1128/mcb.11.4.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rowley JD. A new consistent chromosomal abnormality in chronic myelogenous leukaemia. Nature. 1973;243:290–3. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- 20.Druker BJ, Talpaz M, Rest DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. New Engl J Med. 2001;344:1031–7. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 21.Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–6. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 22.Schindler T, Bornmann W, Pellicena P, et al. Structural mechanism for STI-571 inhibition of Abelson tyrosine kinase. Science. 2000;289:1938–42. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 23.Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–80. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 24.Lahaye T, Riehm B, Berger U, et al. Response and resistance in 300 patients with BCR-ABL positive leukemias treated with imatinib in a single center: a 4.5-year follow-up. Cancer. 2005;103:1659–69. doi: 10.1002/cncr.20922. [DOI] [PubMed] [Google Scholar]
- 25.Sureda A, Carrasco M, De Miguel M, et al. Imatinib mesylate as treatment for blastic transformation of Philadelphia chromosome positive chronic myelogenous leukemia. Haematologica. 2003;88:1213–20. [PubMed] [Google Scholar]
- 26.Sell C, Baserga R, Rubin R. Insulin-like growth factor I (IGF-I) and the IGF-I receptor prevent etoposide-induced apoptosis. Cancer Res. 1995;55:303–6. [PubMed] [Google Scholar]
- 27.Gorre ME, Ellwood-Yen K, Chiosis G, et al. BCR-ABL point mutants isolated from patients with imatinib mesylate-resistant chronic myeloid leukemia remain sensitive to inhibitors of the BCR-ABL chaperone heat shock protein 90. Blood. 2002;100:3041–4. doi: 10.1182/blood-2002-05-1361. [DOI] [PubMed] [Google Scholar]
- 28.Vasilcanu D, Girnita A, Girnita L, et al. The cyclolignan PPP induces activation loop-specific inhibition of tyrosine phosphorylation of the insulin-like growth factor-1 receptor. Link to phosphatidyl inositol-3 kinase/Akt apoptotic pathway. Oncogene. 2004;23:7854–62. doi: 10.1038/sj.onc.1208065. [DOI] [PubMed] [Google Scholar]
- 29.Amin HM, Hoshino K, Yang H, et al. Decreased expression level of SH2 domain-containing protein tyrosine phosphatase-1 (Shp1) is associated with progression of chronic myeloid leukaemia. J Pathol. 2007;212:402–10. doi: 10.1002/path.2178. [DOI] [PubMed] [Google Scholar]
- 30.Ban K, Gao Y, Amin HM, et al. BCR-ABL1 mediates up-regulation of Fyn in chronic myelogenous leukemia. Blood. 2008;111:2904–8. doi: 10.1182/blood-2007-05-091769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qiu L, Lai R, Lin Q, et al. Autocrine release of interleukin-9 promotes Jak3-dependent survival of ALK+ anaplastic large-cell lymphoma cells. Blood. 2006;108:2407–15. doi: 10.1182/blood-2006-04-020305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Branford S, Rudzki Z, Walsh S, et al. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood. 2002;99:3472–5. doi: 10.1182/blood.v99.9.3472. [DOI] [PubMed] [Google Scholar]
- 33.Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–25. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 34.Hu Y, Swerdlow S, Duffy TM, et al. Targeting multiple kinase pathways in leukemic progenitors and stem cells is essential for improved treatment of Ph+ leukemia in mice. Proc Natl Acad Sci USA. 2006;103:16870–5. doi: 10.1073/pnas.0606509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giles FJ, DeAngelo DJ, Baccarani M, et al. Optimizing outcomes for patients with advanced disease in chronic myelogenous leukemia. Semin Oncol. 2008;35:S1–17. doi: 10.1053/j.seminoncol.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 36.Samani AA, Yakar S, LeRoith D, et al. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev. 2007;28:20–47. doi: 10.1210/er.2006-0001. [DOI] [PubMed] [Google Scholar]
- 37.Bertrand FE, Steelman LS, Chappell WH, et al. Synergy between an IGF-1R antibody and Raf/MEK/ERK and PI3K/Akt/mTOR pathway inhibitors in suppressing IGF-1R-mediated growth in hematopoietic cells. Leukemia. 2006;20:1254–60. doi: 10.1038/sj.leu.2404217. [DOI] [PubMed] [Google Scholar]
- 38.Reiss K, Porcu P, Sell C, et al. The insulin-like growth factor 1 receptor is required for the proliferation of hemopoietic cells. Oncogene. 1992;7:2243–8. [PubMed] [Google Scholar]
- 39.Drexler HG. Guide to leukemia-lymphoma cell lines. Braunschweig: 2005. eBook on CD. [Google Scholar]
- 40.Burchert A, Wang Y, Cai D, et al. Compensatory PI3-kinase/Akt/mTor activation regulates imatinib resistance development. Leukemia. 2005;19:1774–82. doi: 10.1038/sj.leu.2403898. [DOI] [PubMed] [Google Scholar]
- 41.Deutsch E, Maggiorella L, Wen B, et al. Tyrosine kinase inhibitor AG1024 exerts antileukaemic effects on STI571-resistant Bcr-Abl expressing cells and decreases AKT phosphorylation. Br J Cancer. 2004;91:1735–41. doi: 10.1038/sj.bjc.6602190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y, Cai D, Brendel C, et al. Adaptive secretion of granulocyte-macrophage colony-stimulating factor (GM-CSF) mediates imatinib and nilotinib resistance in BCR/ABL+ progenitors via JAK-2/STAT-5 pathway activation. Blood. 2007;109:2147–55. doi: 10.1182/blood-2006-08-040022. [DOI] [PubMed] [Google Scholar]
- 43.Shuai K, Halpern J, ten Hoeve J, et al. Constitutive activation of STAT5 by the BCR-ABL oncogene in chronic myelogenous leukemia. Oncogene. 1996;18:247–54. [PubMed] [Google Scholar]
- 44.de Groot RP, Raaijmakers JAM, Lammers J-WJ, et al. STAT5 activation by BCR-Abl contributes to transformation of K562 leukemic cells. Blood. 1999;94:1108–12. [PubMed] [Google Scholar]
- 45.Skorski T, Bellacosa A, Nieborowska-Skorska M, et al. Transformation of hematopoietic cells by BCR/ABL requires activation of PI-3K/Akt-dependent pathway. EMBO J. 1997;16:6151–61. doi: 10.1093/emboj/16.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salatino M, Schillaci R, Proietti CJ, et al. Inhibition of in vivo breast cancer growth by antisense oligodeoxynucleotides to type I insulin-like growth factor receptor mRNA involves inactivation of ErbBs, PI-3K/Akt and p42/p44 signaling pathways but not modulation of progesterone receptor activity. Oncogene. 2004;23:5161–74. doi: 10.1038/sj.onc.1207659. [DOI] [PubMed] [Google Scholar]
- 47.Martin MJ, Melnyk N, Pollard M, et al. The insulin-like growth factor I receptor is required for Akt activation and suppression of anoikis in cell transformed by ETV6-NTRK3 chimeric tyrosine kinase. Mol Cell Biol. 2006;26:1754–69. doi: 10.1128/MCB.26.5.1754-1769.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zong CS, Chan J, Levy DE, et al. Mechanism of STAT3 activation by insulin-like growth factor I receptor. J Biol Chem. 2000;275:15099–105. doi: 10.1074/jbc.M000089200. [DOI] [PubMed] [Google Scholar]
- 49.Okajima Y, Matsumura I, Nishiura T, et al. Insulin-like growth factor-I augments erythropoietin-induced proliferation through enhanced tyrosine phosphorylation of STAT5. J Biol Chem. 1998;273:22877–83. doi: 10.1074/jbc.273.36.22877. [DOI] [PubMed] [Google Scholar]
- 50.Shi P, Lai R, Lin Q, et al. IGF-IR tyrosine kinase interacts with NPM-ALK oncogene to induce survival of T-cell ALK+ anaplastic large-cell lymphoma cells. Blood. 2009;114:360–70. doi: 10.1182/blood-2007-11-125658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lakshmikuttyamma A, Pastural E, Takahashi N, et al. Bcr-Abl induces autocrine IGF-1 signaling. Oncogene. 2008;27:3831–44. doi: 10.1038/onc.2008.8. [DOI] [PubMed] [Google Scholar]
- 52.De Meyts P, Whittaker J. Structural biology of insulin and IGF1 receptors: implications for drug design. Nat Rev Drug Discov. 2002;1:769–83. doi: 10.1038/nrd917. [DOI] [PubMed] [Google Scholar]
- 53.Hofmann F, Garcia-Echeverria C. Blocking the insulin-like growth factor-I receptor as a strategy for targeting cancer. Drug Discov Today. 2005;10:1041–7. doi: 10.1016/S1359-6446(05)03512-9. [DOI] [PubMed] [Google Scholar]
- 54.Haluska P, Shaw HM, Batzel GN, et al. Phase I dose escalation study of the anti insulin-like growth factor-I receptor monoclonal antibody CP-751,871 in patients with refractory solid tumors. Clin Cancer Res. 2007;13:5834–40. doi: 10.1158/1078-0432.CCR-07-1118. [DOI] [PubMed] [Google Scholar]