

Abstract
Menkes disease is a lethal X-linked recessive neurodegenerative disorder of copper transport caused by mutations in ATP7A, which encodes a copper-transporting ATPase. Early postnatal treatment with copper injections often improves clinical outcomes in affected infants. While Menkes disease newborns appear normal neurologically, analyses of fetal tissues including placenta indicate abnormal copper distribution and suggest a prenatal onset of the metal transport defect. In an affected fetus whose parents found termination unacceptable and who understood the associated risks, we began in utero copper histidine treatment at 31.5 weeks gestational age. Copper histidine (900 μg per dose) was administered directly to the fetus by intramuscular injection (fetal quadriceps or gluteus) under ultrasound guidance. Percutaneous umbilical blood sampling enabled serial measurement of fetal copper and ceruloplasmin levels that were used to guide therapy over a four-week period. Fetal copper levels rose from 17 μg/dL prior to treatment to 45 μg/dL, and ceruloplasmin levels from 39 mg/L to 122 mg/L. After pulmonary maturity was confirmed biochemically, the baby was delivered at 35.5 weeks and daily copper histidine therapy (250 μg sc b.i.d.) was begun. Despite this very early intervention with copper, the infant showed hypotonia, developmental delay, and electroencephalographic abnormalities and died of respiratory failure at 5.5 months of age. The patient’s ATP7A mutation, which severely disrupted mRNA splicing, resulted in complete absence of ATP7A protein on Western blots. These investigations suggest that prenatally initiated copper replacement is inadequate to correct Menkes disease caused by severe loss-of-function mutations, and that postnatal ATP7A gene addition represents a rational approach in such circumstances.
1. Introduction
Menkes disease is a lethal infantile X-linked recessive disorder of copper metabolism caused by mutations in the gene ATP7A [NCBI accession number: NM_000052.5] encoding a copper-transporting ATPase [1]. The condition is characterized by male gender, early-onset cerebral and cerebellar neurodegeneration, failure to thrive, seizures, hypotonia, coarse hair and connective tissue abnormalities. Death typically occurs by 3 years of age. Biochemical features include decreased activities of copper-dependent enzymes such as dopamine-beta-hydroxylase, cytochrome c oxidase, and lysyl oxidase [2]. Affected individuals manifest low copper and ceruloplasmin levels in plasma or serum, as well as in cerebrospinal fluid [3]. Even in healthy newborns, serum copper and ceruloplasmin levels remain low for several weeks and thus are not reliable for diagnosis of the illness until six to eight weeks of age [4]. Prenatally, chorionic villus and amniocyte copper accumulation offer useful biochemical markers of the disease [5].
The only currently available treatment for Menkes disease is daily copper injections, which may improve clinical outcome and prevent death if commenced shortly after birth [6-11]. Patients with mutations in the copper-transporting ATPase ATP7A that permit residual copper transport are more likely to respond to this treatment than those with complete loss-of-function mutations [12,13]. In cases where no residual ATP7A copper-transport activity is predicted, ATP7A gene addition may represent a rational future therapeutic solution [14].
We previously characterized a missense mutation in ATP7A (Q724H) that coincidentally disrupts a splice donor site [12]. The alteration, a G to T transversion, occurs at the final base position of exon 8 in ATP7A. One affected patient with this alteration received prenatal therapy with copper histidine beginning at 31.5 weeks gestation and continued treatment after birth at 35.5 weeks gestation. In this study, we report the details of the in utero copper treatment, as well as the clinical and biochemical outcomes.
2. Material and Methods
2.1 Case Report
The studies were approved by Institutional Review Boards at the National Institute of Child Health and Human Development and Bethesda Naval Hospital. The patient was diagnosed as having Menkes disease at 23.5 weeks gestation by abnormal copper egress in cultured amniocytes (Table 1A) following amniocentesis at 19.5 weeks. The prenatal testing was performed in the context of a family history of 13 cases of Menkes disease born to four obligate carriers over three generations [12]. All untreated affected males showed the classic phenotype and died within the first several years of life. The fetal karyotype was 46,XY. Because pregnancy termination was not acceptable to the parents and they consented to the potential risks, the mother was admitted to the Bethesda Naval Hospital at 31.5 weeks gestation. The interval from diagnosis to initiation of treatment (8 weeks) was related to the complexities of arranging the investigational team, in particular the high-risk obstetrics team, preparing a clinical protocol and obtaining IRB approval at two institutions, obtaining the family’s informed consent, and arranging cross-country travel for the mother.
Table 1.
Prenatal and postnatal diagnostic test results
| A. Prenatal Testinga | Fetus | Normal control | Affected Males |
Unaffected Males |
|---|---|---|---|---|
|
64Cu analyses on amniotic fluid cells (ng 64Cu/mg protein/20h |
25.4, 25.0 | 8.8, 9.3 | 26.2-95.0 (n=20) |
5.8-36.1 (n=60) |
| % 64Cu retained after 20h chase |
61.0, 60.8 | 19.0, 17.5 | 43.6-88.0 (n=6) |
11.8-32.8 (n=10) |
Testing performed on tissues obtained by amniocentesis at 19 wks gestational age.
Over a 24 day period, the fetus received six injections of copper histidine (900 μg per dose) by direct intramuscular injection (either fetal quadriceps or gluteus) under ultrasound guidance. Percutaneous umbilical blood sampling enabled serial measurement of fetal copper and ceruloplasmin levels. The fetus received an average weekly dose of 0.75 mg/kg copper histidine, slightly lower than the dose (1 mg/kg/week) we administered without harmful effects to newborns with Menkes disease [2,8].
The mother required insulin for gestational diabetes mellitus (class A2) beginning at 33 weeks gestation. When the infant’s pulmonary maturity was confirmed biochemically (mature lecithin-sphingomyelin ratio, trace phosphatidyl glycerol) at 35.5 weeks gestation, induction of labor was begun using Prostin gel 2.5 mg. Rapid cervical dilation to five cm occurred after five hrs. Brief, transient fetal heart rate decelerations were noted and artificial rupture of membranes was performed. The labor was complicated by asynclitism and failure to progress, requiring cesarean delivery. The infant weighed 2150 grams, was cyanotic at birth with Apgar scores of 2, 5 and 7 at 1, 5, and 10 minutes respectively, and required resuscitation with oxygen by mask and vigorous stimulation. He received a 15cc/kg IV bolus of albumin. Blood cultures were drawn after which he received intravenous ampicillin and gentamycin. After less than 24 hrs, blood cultures were positive for Gram-negative rods, later identified at Escherichia coli. Examination of cerebrospinal fluid (CSF) showed 1 red blood cell/mm3 and 7 white blood cells/mm3 with glucose 66 mg/dl and total protein 100 mg/dl. Concurrent immunoelectrophoresis of CSF was negative for common bacterial pathogens and CSF cultures were negative.
The infant began daily copper histidine injections 250 μg sc b.i.d. on postnatal day 1 and was also treated with parenteral antibiotics (either ampicillin/gentamycin or cefotaxime) for ten days. He required several days of phototherapy for hyperbilirubinemia and was weaned from an incubator to an open crib at 10 days of age. He nipple fed slowly at first but improved during the second week of life, receiving breast milk as well as Similac with Iron.
The mother’s postoperative course was complicated by chorioamnionitis requiring intravenous antibiotic therapy with ampicillin, gentamicin and clindamycin for 7 days. The mother and infant were discharged at 15 days of age, and the infant was admitted to the NIH Clinical Center for further testing.
2.2 Cell culture
Human primary fibroblasts were grown in a complete medium containing Dulbecco’s modified Eagle medium (DMEM) supplemented with L-Glutamine, 10% fetal bovine serum, penicillin/streptomycin and fungizone amphotericin B (Gibco, Life Technologies, NY, USA). Fibroblasts were cultured under standard sterile conditions in a 5% CO2 incubator at 37°C.
2.3 Western blot
Total proteins were extracted from fibroblasts harboring the ATP7A Q724H splicing mutation, a deletion of ATP7A exons 2-23, or wild-type ATP7A (CRL-2076; ATCC, Manassas, VA, USA). Protein extraction was performed using RIPA buffer containing 50 mM Hepes buffer, 150 mM NaCl, 0.5% Triton X-100 and 10% glycerol. Supernatants from the lysed cells were collected. The total proteins were denatured by adding 4X NuPage, LDS sample buffer (Invitrogen, Life Technologies, NY, USA) with 1% of β-mercaptoethanol, then heating at 95°C for 5 minutes. Total proteins (30 μg) from each sample were electrophoresed through precast 4–12% NOVEX Tris-glycerin sodium dodecyl sulfate (SDS) polyacrylamide gels (Invitrogen) for 1h at 170V and transferred to polyvinylidine fluoride (PVDF) membranes for 2h at 25V (150 mA). The membranes were then incubated at 4°C overnight in phosphate buffered saline blocking solution containing 0.1% Tween 20 (PBST) and 5% non-fat milk (Bio-Rad, CA, USA). Blots were then washed with PBST, and incubated for 2h at room temperature (RT) with a 1:1000 dilution of a rabbit carboxy-terminal anti-ATP7A antibody [15] or a 1:5000 dilution of anti-β-actin monoclonal antibody coupled with HRP (Abcam, MA, USA). The membrane was washed twice with PBST and next incubated with a 1:2000 dilution of a goat anti-rabbit IgG horseradish peroxidase (HRP)-conjugated secondary antibody (Santa Cruz Biotechnology, CA, USA) for 1h at RT. Membranes were then incubated in SuperSignal West Pico Luminol/Enhancer Solution (Thermo Fisher Scientific, IL, USA) and exposed to a Kodak BioMax MR autoradiography film. The film was developed using a Kodak X-OMAT 2000A processor.
2.4 Catechol Levels
Catechol levels in fetal and postnatal plasma, and amniotic fluid were measured by high performance liquid chromatography with electrochemical detection, as previously described [16].
2.5 Trace metal analyses
Fetal, neonatal, and maternal blood copper, ceruloplasmin, and zinc levels were determined in the Pathology Department, British Columbia Children’s Hospital, Vancouver, Canada as previously described [17,18]. Brain, kidney, liver, and placental tissues were digested in nitric acid and copper levels were measured by inductively coupled plasma mass spectrometry (ICP-MS) at the Biophysical Toxicology Lab, Joint Pathology Center as previously described [13]. Copper levels in cerebrospinal fluid were determined via atomic absorption spectrometry, as previously described [3,13,17].
2.6 Postmortem tissues
Postmortem cerebral frontal cortex, kidney, and liver from the proband were harvested within 24 hrs after expiration. Brain, kidney, and liver tissues from an age-matched normal control (#615) were obtained from the NICHD Brain and Tissue Bank at the University of Maryland. The proband and normal control tissues were each stored at −70°C after harvesting.
3. Results
3.1 Biochemical effects of in utero copper treatment
Fetal copper and ceruloplasmin levels rose in conjunction with fetal copper treatment (Figure 1A,B). While control data for age-matched unaffected fetuses was not available, comparison to the reference range for serum copper levels in normal neonates aged 0-5 days (9 to 46 μg/dl) [17] indicated that the proband’s baseline pre-treatment level (17μg/dl) was in the lowest quartile. The six in utero copper injections over 3.5 weeks resulted in a rise of the patient’s plasma copper level to the top quartile (45 μg/dl) by the time of birth. Except for the baseline measurement (39 mg/L), fetal serum ceruloplasmin levels fell within the range found in newborns aged 0-5 days (50-260 mg/L) [18]. The copper concentration in amniotic fluid, which represents fetal urine, showed a large spike (462 μg/L) after the initial copper dose (Figure 1C). Thereafter, amniotic fluid copper levels stabilized closer to the baseline, pre-treatment value (159 μg/L) for the remainder of fetal copper therapy (Figure 1C). Copper level in the cerebrospinal fluid at birth was 7.1 μg/L (Table 1C; normal pediatric range: 7.26 ± 3.1, and in untreated Menkes disease: 2.39 ± 1.62) [3].
Figure 1.

Fetal biochemical response to copper injections. A. Fetal copper levels rose in correlation with fetal copper injections (arrows). B. Fetal serum ceruloplasmin levels correlated with copper levels, as expected. In contrast, amniotic fluid copper (C), representing fetal urinary excretion of copper, were mainly unchanged except for a spike following the first injection. D. Fetal plasma zinc levels declined slightly during copper treatment. See text for normal reference values for plasma copper and zinc [17], serum ceruloplasmin [18].
| C. Cerebrospinal Fluid Copper (μg/L) | |||
|---|---|---|---|
| Age | Birth | 2.5 weeks | 8 weeks |
| Proband | 7.1 | 5.6 | 2.8 |
Reference Ranges [3]: Apparently healthy infants: 7.26 ± 3.1 μg/L
Untreated Menkes disease: 2.39 ± 1.62 μg/L
As a secondary measure of improved trace metal homeostasis, the fetal plasma zinc:copper (Zn:Cu) ratio progressively declined, from 7.0 at baseline to 2.0 at one week before birth (normal Zn:Cu ratio in newborns aged 0-5 days = 0.5 to 1.1) [17]. The distinct trend toward normalization of this ratio resulted not only from rising fetal copper levels, but also from a concurrent decline in fetal zinc levels (Figure 1D)
Pre-treatment fetal plasma neurochemical analysis (Table 2A) showed elevated levels of dihydroxyphenylacetic acid (DOPAC), and low levels of norepinephrine (NE) and dihydroxyphenylglycol (DHPG) in comparison to pediatric controls [16]. Ratios of dihydroxyphenylalanine (DOPA) to DHPG and DOPAC to DHPG were markedly elevated, an abnormal pattern found in postnatal plasma and cerebrospinal fluid from Menkes disease patients [4,16]. Amniotic fluid (fetal urine) neurochemical values during fetal copper treatment (Table 2B) showed elevated ratios of dopamine to norepinephrine compared to urine from apparently healthy infants younger than 1 year of age [19].
Table 2.
Neurochemical response to in utero copper treatment
| A. Fetal Plasma Neurochemical Findings (pg/ml) | |||||||
|---|---|---|---|---|---|---|---|
| Age (wks) | DOPA | DOPAC | DA | NE | DHPG | DOPA:DHPG | DOPAC:DHPG |
| 31.5a | 3456 | 4975 | - | 273 | 466 | 7.4 | 10.7 |
| 32 | 4319 | 9003 | - | 18 | 461 | 9.4 | 19.5 |
| 33 | 3913 | 5294 | - | 31 | 370 | 10.6 | 14.3 |
| 34 | 3701 | 5774 | - | 24 | 413 | 9.0 | 14.0 |
| Normal ranges [16] |
1573- 4512 |
1329- 2936 |
0-22 | 500- 1056 |
826- 1399 |
1.7-3.3 | 1.5-3.2 |
Baseline (pre-treatment)
| B. Fetal Urine (Amniotic fluid) Neurochemical Findings (in pg/ml) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Age (wks) | DOPA | DOPAC | DA | NE | DHPG | DA:NE | DOPA:DHPG | DOPAC:DHPG |
| 32 (n=2) | 922 | 14036 | 1760 | 84 | 422 | 20.9 | 2.2 | 33.3 |
| 33 | 1050 | 12129 | 2130 | 97 | 377 | 22.0 | 2.8 | 32.2 |
| 34 | 726 | 14704 | 1649 | 129 | 473 | 12.8 | 1.5 | 31.1 |
| Normal | 4.5b | |||||||
Calculated based on upper reference limits for urine dopamine and norepinephrine (reported as mmol/mol creatinine), in pediatric subjects under 1 year of age (n=70) [19].
DOPA=dihydroxyphenylalanine; DOPAC=dihydroxyphenylacetic acid; DA=dopamine; NE=norepinephrine; DHPG=dihydroxyphenylglycol
There was no clear influence of fetal copper injections on maternal copper distribution. Maternal plasma copper levels (Figure 2A) correlated with serum ceruloplasmin levels (Figure 2B) during in utero copper treatment. Ceruloplasmin is an estrogen-sensitive plasma protein, levels of which double in late pregnancy [20]. This fact also accounts for the increased maternal levels of plasma copper, since ceruloplasmin accounts for 90-95% of circulating copper [1]. Maternal plasma zinc levels and 24 hr urine copper excretion varied during the period of fetal copper treatment without distinctive patterns (Figure 2C,D).
Figure 2.

Maternal biochemical response to copper injections. Maternal plasma copper levels (A) correlated with serum ceruloplasmin (B) levels during in utero copper treatment and did not appear to be influenced by the timing of fetal copper injections (arrows). Maternal plasma zinc levels (C) and 24 hr urine copper excretion (D) varied during the period of fetal copper treatment, without distinctive patterns. See text for discussion of increased blood copper and ceruloplasmin levels in pregnancy.
3.2 Placental pathology and biochemical findings
The placenta showed inflammation most severe in the fetal chorionic plate vessels and the umbilical cord showed acute funisitis, (data not shown), consistent with a fetal inflammatory response [21]. Tissue staining with rhodanine for copper documented accumulations in placental Hofbauer cells and the endothelia of stem vessels and villous capillaries (data not shown). Copper content in the placenta and placental membranes, measured by inductively coupled plasma mass spectrometry, was markedly elevated in comparison to two controls studied concurrently (Table 1B).
| Cu content (μg/g dry weight) | ||
|---|---|---|
| B. Postnatal Testing |
Placenta | Placental membranes |
| Proband | 58.3 | 65.1 |
| Control A | 1.97 | 3.7 |
| Control B | 2.26 | 4.92 |
3.3 Clinical findings
On admission to the NIH Clinical Center at 16 days of age, the proband’s weight was 2.16 kg, length was 48.5 cm, and head circumference was 34 cm. His skin was dry and wrinkled with a post-mature quality, and loose and redundant on the back and trunk. The hair was light brown and normal in appearance. On neurological exam, the infant was alert and had a strong cry and suck. He fixed visually to the examiner’s face. The following infant reflexes were elicited: rooting, arm recoil, palmar and plantar grasps, magnet response, crossed extension, withdrawal, Bauer, Galant (R>L), placing and stepping, and Moro. There was a head lag on pulling-to-sit, with fair head control when held sitting or in ventral suspension. His truncal tone was slightly decreased and appendicular tone appeared normal. The remainder of the physical exam was unremarkable.
Copper level in the cerebrospinal fluid was 5.6 μg/L (Table 1C; normal pediatric range: 7.26 ± 3.1, and in untreated Menkes disease: 2.39 ± 1.62) [3]. The CSF cell count showed 1 RBC/mm3 and 2 WBC/mm3 with total protein 86 and glucose 57. Serum ceruloplasmin (194 mg/L) and plasma neurochemical ratios were higher than during prenatal treatment [12]. Urine beta-2-microglobulin concentration was 6.68 mg/L (normal ≤ 0.3 mg/L), reflecting renal tubular injury from copper accumulation [8].
At 6.5 weeks of age, the patient underwent bilateral inguinal hernia repairs at his local hospital, without complications.
When admitted to the NIH Clinical Center for at 2 months of age for additional follow-up, the patient’s admission physical was notable for an axillary temperature of 40°C. A septic workup including blood cultures, sterile straight catheter urine and lumbar puncture was obtained and he defervesced spontaneously within several hours (36.6°C). All cultures were ultimately negative.
His weight and length were the 5th centile for age corrected for prematurity, whereas head circumference was at the 50th centile. The hair was sparse and coarse in contrast to the finer textured hair present at birth and observed at 16 days of age. The anterior fontanelle was patent. The abdomen showed a small umbilical hernia and well-healed herniorrhaphy scars in the inguinal region. On neurological examination, he smiled, fixed and tracked visually, turned his head from side to side and lifted his head from a prone position. He had a strong cry and suck. Head control was poor on pull-to-sit and fair in sitting and in ventral suspension. Truncal tone was normal with good shoulder girdle strength. Appendicular tone was mildly increased (upper extremities greater than lower). The Moro reflex was faintly evident. The crossed-extensor reflex was extinguished. The Bauer, placing, and stepping reflexes were present. The Babinski response was negative.
He underwent a number of laboratory and clinical diagnostic tests. Serum copper was 146 μg/dl (normal 70-150 μg/dl) with corresponding serum ceruloplasmin level of 396 mg/L (normal 70-487 mg/L) [12]. A spinal tap revealed clear CSF with 1 WBC and 0 RBC, glucose 69 mg/dL and total protein 24 mg/dL. CSF copper concentration was 2.8 μg/L (Table 1C; normal pediatric range: 7.26 ± 3.1 μg/L, and for untreated Menkes disease: 2.39 ± 1.62 μg/L) [3]. Both plasma and CSF neurochemical ratios were markedly higher than normal [12], denoting persistent deficiency of dopamine-beta-hydroxylase (DBH), a copper dependent enzyme. CSF ratio of DOPA:DHPG was 4.5 (normal range 0.3-0.7) and DOPAC:DHPG was 9.7 (normal range 0.1-0.4).
Urinary copper excretion averaged 58 μg per 24 hrs. The fractional excretion of uric acid was 47.9% and urine beta-2-microglobulin concentration was 29.96 mg/L, nearly five-fold higher than at the prior visit. A brain MRI was considered normal, with no ventricular enlargement or vascular dilatation.
Between 2 and 4 1/2 months of age, the infant showed progressive failure to thrive and underwent gastrostomy tube placement. He exhibited hypotonia, delayed development, multiple rib fractures, and possible clinical seizures with documented EEG abnormalities (right central and left temporal spike and wave discharges, poorly organized posterior rhythm). He was started on anti-convulsant treatment (carbamazapine). A series of pulmonary infections led to several hospitalizations, and he required oxygen (0.1L by nasal cannula) at home.
At 5 1/2 months of age, the patient was admitted with tachypnea and an increased oxygen requirement. A CXR revealed a hazy L heart border and R upper lobe density. A nasopharyngeal culture was positive for respiratory syncytial viral (RSV). He was treated with aerosolized bronchodilator treatments, a short course of steroids, and chest physiotherapy. His condition worsened and necessitated transfer to the pediatric intensive care unit where he subsequently developed cardiopulmonary failure and died.
3.4 Molecular
The G to T transversion at the final base of ATP7A exon 8, predicting severely aberrant RNA splicing, and a glutamine to histidine substitution (Q724H), was previously described [12]. In wild-type ATP7A, the splice donor site where this mutation occurs contains a gc rather than gt dinucleotide at the +1 and +2 intronic positions and is thus nonconforming [22]. Western blots of protein from cultured fibroblasts harboring the Q724H splicing mutation indicated no detectable ATP7A (Figure 3).
Figure 3.
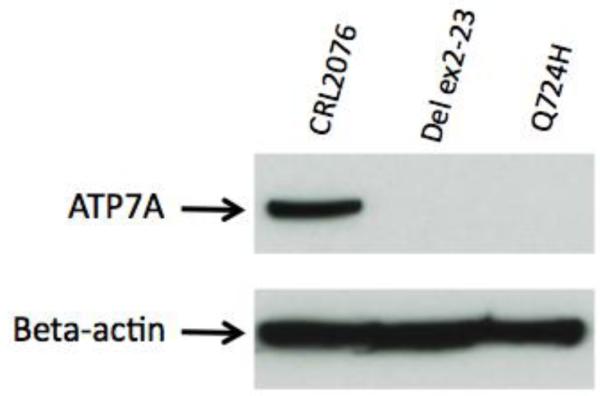
Western blot analysis of fibroblast protein. Western blotting with a robust antibody against the carboxyl-terminus of ATP7A [15] revealed no discernible ATP7A in fibroblasts harboring the Q724H splicing mutation. Positive control (apparently normal fibroblast cell line CRL2076) and negative control (fibroblasts from a Menkes patient with deletion of ATP7A exons 2 through 23) for ATP7A are indicated. Hybridization with an antibody against beta-actin confirmed equivalent loading.
3.5 Postmortem pathological analyses
Upon death at 5.5 months of age, the proband weighed 3.44 kg. Length was 55 cm, and head circumference was 42 cm. Crown-rump length was 37 cm and heel-toe length was 7.5 cm. The wet weight of brain was 670g. Brain copper level was low (20 to 30% of normal), compared to an age-matched control and 21 adult controls (Table 1D). The leptomeninges were thin and translucent. Gyri and sulci were well formed. There was no evidence of atrophy, deformity or herniation. The cerebellum was well formed without evidence of atrophy. The brain stem was normal in appearance. The vasculature was well formed without abnormalities. Microscopic examination revealed no abnormalities of the midbrain, pons, medulla, hippocampus, basal ganglia, thalamus, cingulated gyrus, cerebellar vermis, and cerebellum. No neuronal dropout, gliosis, or architectural abnormalities. Age-appropriate myelination was present. The cerebellum showed a thin external granular cell layer with a normal internal granular cell component. Purkinje cells were present throughout with an even distribution and without abnormalities on hematoxylin and eosin (H&E) or Bielschowsky silver staining. No atrophy of the granular cell layers or astrocytosis of the cerebellum was noted. Electron microscopic analysis of postmortem cerebellum showed preserved cellular structure and detail. Mitochondria were present with normal numbers and architecture. No nuclear or cytoplasmic abnormalities were noted.
| D. Postmortem Testing |
Liver (μg/g dry weight) |
Kidney (μg/g dry weight) |
Brain (μg/g dry weight) |
|---|---|---|---|
| Proband | 19.1 | 634.7 | 2.9 |
| Age-matched control | 11.8 | 9.8 | 8.9 |
| Normal Adult Range | 9.2-46.8 (n=24) | 5.1-35.7 (n=23) | 13.1-39.4 (n=21) |
The liver weighed 258g, spleen 18g, thymus 5g, R lung 44g, L lung 42g, right kidney and adrenal 26g, left kidney and the adrenal 28g, heart 38g. Microscopic sections of the lungs showed immature parenchyma with substantial regions of atelectasis and the alveoli were filled with foamy macrophages. Hepatic copper level was increased in comparison to an age-matched control and within the normal adult range, whereas renal copper concentration was 20- to 60-fold elevated above normal (Table 1D).
4. Discussion
Menkes disease is a neurogenetic disorder of copper metabolism in which certain biochemical abnormalities are evident prenatally [23]. However, clinical signs and symptoms are typically absent at birth [1], which indicates a therapeutic window of opportunity. The present case provided a circumstance in which the effects of prenatal treatment of Menkes disease with copper replacement could be assessed for the first time. This aggressive medical intervention necessitated careful and realistic discussion with the parents in terms of the associated risks and potential outcomes [24].
Our major findings were that prenatal copper treatment successfully raised circulating fetal copper levels, but did not improve the activity of a copper-dependent enzyme, dopamine-beta-hydroxylase, which relies upon ATP7A. The prenatal treatment was complicated by fetal chorioamnionitis and neonatal sepsis, from which the patient fully recovered. Biochemical responses to infection or inflammation include decreased serum zinc levels and elevated serum ceruloplasmin levels [25], which may have affected some of the values we obtained for these analytes (Table 1 and 2).
The infant’s subsequent postnatal clinical course declined; he manifested developmental delay, failure to thrive, and EEG evidence of seizure activity, all typical signs of untreated classic Menkes disease [1,2,26]. In sum, in utero copper replacement therapy, in the context of a severe ATP7A mutation (Q724H splice junction defect) [2], failed to significantly improve the expected clinical course.
The mechanisms that underlie placental transport of copper are not entirely known, however, this study helps to illuminate some aspects. As observed previously by others [23], we found that the placenta in this case showed marked accumulation of copper (Table 1B]. The syncytiotrophoblast layer, which represents polarized epithelia covering the placental chorionic villi [27] express ATP7A at their basolateral (fetal-facing) aspect [28]. ATP7A is also expressed in endothelial cells of fetal blood vessels [28]. Thus, in the context of a dysfunctional or absent ATP7A, these placental cells would be expected to retain excess copper, supported by our detection of rhodanine-positive cells on histopathology.
Interestingly, postnatal CSF copper levels at birth and 2.5 weeks of age fell within the normal pediatric range (Table 1C) [3]. By 2 months of age, however, the CSF copper level was subnormal (Table 1C), and postmortem brain copper levels were clearly subnormal (Table 1D) despite peripheral copper injections that maintained serum copper and ceruloplasmin within normal ranges. This dichotomy is consistent with a defect in copper transport at the level of the blood-CSF barrier [3] and/or blood-brain barrier that is exacerbated postnatally. The intracellular tight junctions which comprise the mammalian blood-brain and blood-CSF barriers develop early in gestation, and generic transporters are expressed in embryonic choroid plexus epithelia [29]. However, other evidence suggests that the fetal cerebral vasculature is somewhat fragile and may be more permeable than at later developmental stages [29], consistent with our results.
The patient’s brain MRI examination at 2 months of age and postmortem gross and microscopic evaluation of the brain at nearly 6 months of age indicated no overt pathology. These latter findings, in conjunction with the clinical outcome, suggest that the detrimental effects of brain copper deficiency involve metabolic derangements related to copper enzyme deficiencies and a drastic impact on normal neurophysiology, potentially mediated by broad alterations in gene expression [13]. Our findings support the hypothesis that at least some ATP7A activity is crucial for therapeutic response in this disorder [7,8]. Better, though still suboptimal, treatment outcome in the context of early postnatal copper treatment was noted in a third cousin of this patient, with the identical mutation and aberrant transcript formation described previously [2], consistent with intrafamilial variability in Menkes disease [30].
The present case also highlights the inability of copper replacement alone to counter deficiency of dopamine-beta-hydroxylase in Menkes disease, and accumulation of copper by the kidneys, resulting in renal tubular injury. Persistently elevated neurochemical ratios in plasma, CSF, and even amniotic fluid (fetal urine) indicated profound deficiency of DBH, which normally acquires copper ions as enzymatic co-factors, via transmembrane transport of copper into the trans-Golgi compartment of cells by ATP7A [2]. The renal copper accumulation (Table D) exposes another major function of ATP7A, removal of copper from polarized epithelial and other cells via transport across plasma membranes. In healthy kidneys, the proximal renal tubules mediate the first step of copper reabsorption from urine. Copper ions are then transferred back to the blood circulation via ATP7A at the basolateral aspect of these polarized cells [31]. In Menkes disease, and in mouse models of the disease, uptake of copper from urine into the renal tubule epithelia is not disturbed, but the mechanism of pumping copper back from the cell into the circulatory system is impaired, due to low copper ATPase activity [32]. As a result, copper ions accumulate in proximal tubule epithelial cells and cause cell damage [32], also reflected in urinary spillage of beta-2-microglobulin, a sensitive marker of renal tubule injury [8].
Copper supplementation in Menkes disease mouse models may worsen these renal effects, but may also improve survival. The latter outcome is typical when mutations in the homologous copper transporter (Atp7a) do not cause complete loss-of-function [2,32]. We speculate that in utero copper treatment for Menkes disease would be more successful in instances of missense mutations that retain partial ATP7A activity, as documented for early postnatal copper replacement [8,9,11].
This investigation illustrates the potential risks (e.g., fetal hemorrhage and sepsis) associated with aggressive prenatal intervention. While in utero approaches in murine models provide useful proof-of-principle [33], the translational applications of this approach may be limited. Indeed, the success of early copper treatment in some Menkes disease patients [6-11] implies that prenatal approaches are not necessarily required for dramatically improved neurodevelopmental outcomes. Recent studies of postnatal viral gene therapy in the mottled-brindled mouse, a Menkes disease mouse model, [14] are also consistent with this principle. The failure to thrive, persistent DBH deficiency, and brain copper deficiency in the patient reported here were both circumvented in mottled-brindled animals rescued by recombinant adeno-associated virus serotype 5 (rAAV5)-mediated gene therapy [14].
ATP7A addition via systemic administration of a rAAV serotype, such as rAAVrh10, which crosses the blood-CSF and/or blood-brain barrier(s), has tropism for neuronal cells, and also transduces the kidney [34] represents a novel and rational treatment approach for Menkes disease, especially when caused by a severe loss of function ATP7A mutation. The biochemical parameters outlined in this report (CSF copper, plasma and CSF neurochemical levels and ratios, urine beta-2-microglobulin concentration) represent candidate biomarkers for treatment monitoring in a future first-in-human gene therapy trial for this difficult illness.
Highlights.
We treated a 3rd trimester Menkes disease fetus with in utero copper injections.
Treatment increased fetal copper levels but did not improve copper transport.
The patient developed signs and symptoms of the disease and died at 5.5 months.
We suggest that postnatal viral gene therapy is needed for severe Menkes disease.
Acknowledgements
We especially acknowledge the patient and his mother for participating in this study. We thank Jay Beckstead, Randal Nixon, and Anthony D’Agostino for postmortem analyses, Richard Miller and Edward Wolf for expert perinatal care, John Clemons for expert neonatal care, Gillian Lockitch for helpful advice on infant trace metal analyses, Tonne Tonnesen for expert prenatal diagnosis, and Neil Buist for referring the family.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kaler SG, Packman S. Inherited Disorders of Human Copper Metabolism. In: Rimoin DL, Connor JM, Pyeritz RE, Korf BR, editors. Emery and Rimoin’s Principles and Practice of Medical Genetics. 6th edition. Churchill Livingstone/Elsevier; New York: 2012. (in press) [Google Scholar]
- 2.Kaler SG. The neurology of ATP7A copper transporter disease: emerging concepts and future trends. Nat. Rev. Neurol. 2011;7:15–29. doi: 10.1038/nrneurol.2010.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Donsante A, Johnson P, Jansen LA, Kaler SG. Somatic mosaicism in Menkes disease suggests choroid plexus-mediated copper transport to the developing brain. Am. J. Med. Genet. A. 2010;152A:2529–2534. doi: 10.1002/ajmg.a.33632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaler SG, Gahl WA, Berry SA, Holmes CS, Goldstein DS. Predictive value of plasma catechol levels in neonatal detection of Menkes disease. J. Inher. Metab. Dis. 1993;16:907–908. doi: 10.1007/BF00714295. [DOI] [PubMed] [Google Scholar]
- 5.Kaler SG, Tumer Z. Invited commentary: the prenatal diagnosis of Menkes disease. Prenat. Diagn. 1998;18:287–289. [PubMed] [Google Scholar]
- 6.Kaler SG, Das S, Levinson B, Goldstein DS, Holmes CS, Patronas NJ, Packman S, Gahl WA. Successful early copper therapy in Menkes disease associated with a mutant transcript containing a small in-frame deletion. Biochem. Mol. Med. 1996;57:37–46. doi: 10.1006/bmme.1996.0007. [DOI] [PubMed] [Google Scholar]
- 7.Kaler SG. Menkes disease mutations and response to early copper histidine treatment. Nat. Genet. 1996;13:21–22. doi: 10.1038/ng0596-21. [DOI] [PubMed] [Google Scholar]
- 8.Kaler SG, Holmes CS, Goldstein DS, Tang JR, Godwin SC, Donsante A, Liew CJ, Sato S, Patronas N. Neonatal diagnosis and treatment of Menkes disease. N. Engl. J. Med. 2008;358:605–614. doi: 10.1056/NEJMoa070613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang J, Donsante A, Desai V, Patronas N, Kaler SG. Clinical outcomes in Menkes disease patients with a copper-responsive ATP7A mutation, G727R. Mol. Genet. Metab. 2008;95:174–181. doi: 10.1016/j.ymgme.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaler SG, Tang JR, Donsante A, Kaneski C. Translational read-through of a nonsense mutation in ATP7A. Ann. Neurol. 2009;65:108–113. doi: 10.1002/ana.21576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaler SG, Liew CJ, Donsante A, Hicks JD, Sato S, Greenfield JC. Molecular correlates of epilepsy in early diagnosed and treated Menkes disease. J. Inher. Metab. Dis. 2010;33:583–589. doi: 10.1007/s10545-010-9118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaler SG, Buist NRM, Holmes CS, Goldstein DS, Miller RC, Gahl WA. Early copper therapy in classic Menkes disease patients with a novel splicing mutation. Ann. Neurol. 1995;38:921–928. doi: 10.1002/ana.410380613. [DOI] [PubMed] [Google Scholar]
- 13.Liu PC, Chen YW, Centeno J, Quesado M, Lem KE, Kaler SG. Downregulation of myelination, energy, and translational genes in Menkes disease brain. Molec. Genet. Metab. 2005;85:291–300. doi: 10.1016/j.ymgme.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 14.Donsante A, Yi L, Zerfas P, Brinster L, Sullivan P, Goldstein DS, Prohaska J, Centeno JA, Kaler SG. ATP7A gene addition to the choroid plexus results in long-term rescue of the lethal copper transport defect in a Menkes disease mouse model. Molec. Ther. 2011;19:2114–2123. doi: 10.1038/mt.2011.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yi L, Donsante A, Kennerson ML, Mercer JFB, Garbern JY, Kaler SG. Altered intracellular localization and valosin-containing protein (p97 VCP) interaction underlie ATP7A-related distal motor neuropathy. Hum. Mol. Genet. 2012;21:1794–1807. doi: 10.1093/hmg/ddr612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaler SG, Goldstein DS, Holmes C, Salerno JA, Gahl WA. Plasma and cerebrospinal fluid neurochemical pattern in Menkes disease. Ann. Neurol. 1993;33:171–175. doi: 10.1002/ana.410330206. [DOI] [PubMed] [Google Scholar]
- 17.Lockitch G, Halstead AC, Wadsworth L, Quigley G, Reston L, Jacobson B. Age- and sex-specific pediatric reference intervals and correlations for zinc, copper, selenium, iron, vitamins A and E, and related proteins. Clin. Chem. 1988;34:1625–1628. [PubMed] [Google Scholar]
- 18.Lockitch G, Halstead AC, Quigley G, MacCallum C. Age- and sex-specific pediatric reference intervals: study design and methods illustrated by measurement of serum proteins with the Behring LN Nephelometer. Clin. Chem. 1988;34:1618–1621. [PubMed] [Google Scholar]
- 19.Fitzgibbon MC, FitzGerald RJ, Tormey WP, O’Meara A, Kenny D. Reference values for urinary HMMA, HVA, noradrenaline, adrenaline, and dopamine excretion in children using random urine samples and HPLC with electrochemical detection. Ann Clin Biochem. 1992;29:400–404. doi: 10.1177/000456329202900405. [DOI] [PubMed] [Google Scholar]
- 20.Laurell CB, Rannevik GA. A comparison of plasma protein changes induced by danazol, pregnancy, and estrogens. J. Clin. Endocrinol. Metab. 1979;49:719–725. doi: 10.1210/jcem-49-5-719. [DOI] [PubMed] [Google Scholar]
- 21.Pacora P, Chaiworapongsa T, Maymon E, Kim YM, Gomez R, Yoon BH, Ghezzi F, Berry SM, Qureshi F, Jacques SM, Kim JC, Kadar N, Romero R. Funisitis and chorionic vasculitis: the histological counterpart of the fetal inflammatory response syndrome. J. Matern. Fetal Neonatal Med. 2002;11:18–25. doi: 10.1080/jmf.11.1.18.25. [DOI] [PubMed] [Google Scholar]
- 22.Senapathy P, Shapiro MB, Harris NL. Splice junctions, branch point sites, and exons: sequence statistics, identification, and applications to genome project. Methods Enzymol. 1990;183:252–278. doi: 10.1016/0076-6879(90)83018-5. [DOI] [PubMed] [Google Scholar]
- 23.Nooijen JL, DeGroot CJ, Van Den Hamer CJA, Monnens LA, Willemse J, Niermeijer MF. Trace element studies in 3 patients and a fetus with Menkes’ disease. Effect of copper therapy. Pediatr. Res. 1981;15:284–289. doi: 10.1203/00006450-198103000-00017. [DOI] [PubMed] [Google Scholar]
- 24.Sheela SR, Manoj L, Liu P-C, Lem KE, Kaler SG. Copper replacement treatment for symptomatic Menkes disease: ethical considerations. Clin. Genet. 2005;68:278–283. doi: 10.1111/j.1399-0004.2005.00496.x. [DOI] [PubMed] [Google Scholar]
- 25.Cernat RI, Mihaescu T, Vornicu M, Vione D, Olariu RI, Arsene C. Serum trace metal and ceruloplasmin variability in individuals treated for pulmonary tuberculosis. Int. J. Tuberc. Lung Dis. 2011;15:1239–1245. doi: 10.5588/ijtld.10.0445. [DOI] [PubMed] [Google Scholar]
- 26.White SR, Reese K, Sato S, Kaler SG. Spectrum of EEG findings in Menkes disease, Electroenceph. Clin. Neurophysiol. 1993;87:57–61. doi: 10.1016/0013-4694(93)90175-u. [DOI] [PubMed] [Google Scholar]
- 27.Kulanthaivel P, Furesz TC, Moe AJ, Smith CH, Mahesh VB, Leibach FH, Ganapathy V. Human placental syncytiotrophoblast expresses two pharmacologically distinguishable types of Na(+)-H+ exchangers, NHE-1 in the maternal-facing (brush border) membrane and NHE-2 in the fetal-facing (basal) membrane. Biochem. J. 1992;284(Pt 1):33–38. doi: 10.1042/bj2840033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hardman B, Luff S, Ackland ML. Differential intracellular localisation of the Menkes and Wilson copper transporting ATPases in the third trimester human placenta. Placenta. 2011;32:79–85. doi: 10.1016/j.placenta.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 29.Saunders NR, Liddelow SA, Dziegielewska KM. Barrier mechanisms in the developing brain. Front. Pharmacol. 2012;3:46. doi: 10.3389/fphar.2012.00046. doi: 10.3389/fphar.2012.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Donsante A, Tang JR, Godwin SC, Holmes CS, Goldstein DS, Bassuk A, Kaler SG. Differences in ATP7A gene expression underlie intra-familial variability in Menkes disease/occipital horn syndrome. J. Med. Genet. 2007;44:492–497. doi: 10.1136/jmg.2007.050013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greenough M, Pase L, Voskoboinik I, Petris MJ, O’Brien AW, Camakaris J. Signals regulating trafficking of Menkes (MNK; ATP7A) copper-translocating P-type ATPase in polarized MDCK cells. Am. J. Physiol. Cell Physiol. 2004;287:C1463–1471. doi: 10.1152/ajpcell.00179.2004. [DOI] [PubMed] [Google Scholar]
- 32.Lenartowicz M, Windak R, Tylko G, Kowal M, Styrna J. Effects of copper supplementation on the structure and content of elements in kidneys of mosaic mutant mice. Biol. Trace Elem. Res. 2010;136:204–220. doi: 10.1007/s12011-009-8533-4. [DOI] [PubMed] [Google Scholar]
- 33.Haddad MR, Zerfas P, Donsante A, Kaler SG. In utero brain-directed AAV5 gene therapy results in rapid, robust, and specific transduction of choroid plexus epithelia: implications for rescue of prenatal lethal mouse models of neurometabolic disease. Mol. Ther. 2012 in press. [Google Scholar]
- 34.Hu C, Busuttil RW, Lipshutz GS. RH10 provides superior transgene expression in mice when compared with natural AAV serotypes for neonatal gene therapy. J Gene Med. 2010;12:766–778. doi: 10.1002/jgm.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]