

Abstract
Chronic Helicobacter pylori infection is associated with the decreased expression of the gastric tumour suppressor protein p27. Because transcription of the gene p27 may be regulated epigenetically through histone acetylation, which is mediated by G-protein coupled delta opioid receptor (DOR) stimulation, we examined whether H. pylori regulates the DOR/histone acetylation/p27 promoter pathway. The levels of acetylated histone and p300, a gene-specific histone acetyltransferase within the p27 promoter, were measured using ChIP assays. The expression of phospho-DOR was evaluated by Western blot and immunohistochemical analyses. Growth curves were constructed, and cell proliferation was assessed after BrdU incorporation. Low p27 expression in acutely H. pylori-infected AGS gastric epithelial cells and in chronically H. pylori-infected AGS-derived HS3C cells was associated with approximate 20% and 40% decreases in p27 mRNA expression, respectively, when compared to p27 mRNA levels in uninfected AGS parental cells. The low p27 mRNA levels following H. pylori infection were associated with a 15%–60% reduction in p27 promoter histone H4 acetylation. The recruitment of p300 to the p27 promoter was also markedly decreased by H. pylori infection. The expression of phospho-DOR was decreased by H. pylori infection in cell lines in vitro and in H. pylori-infected human gastric mucosa in vivo. The level of cellular p27 inversely correlated with cell proliferation in HS3C cells. These results demonstrate that H. pylori decreases p27 expression by modulating the DOR and thereby inhibiting histone acetylation of the p27 promoter. These findings link low gastric p27 expression levels with increased instances of gastric carcinogenesis associated with H. pylori infection.
Keywords: Helicobacter pylori, p27, Histone Acetylation, Delta Opioid Receptor
Introduction
p27kip1 is a cyclin-dependent kinase inhibitor 1B that binds to cyclinE/cdk2 and thereby blocks the G1/S transition necessary for cell cycle progression [1]. The levels of the protein p27 are upregulated under stress conditions, leading to cell cycle inhibition and apoptosis [1]. Additional roles for p27 have also been recognised, including tumour suppression [2] and the regulation of cell migration [3]. Low p27 expression has been reported in many cancers, including those of the colon, breast, prostate, lung, and brain, and in gastric carcinoma of both intestinal and diffuse subtypes [4,5]. A low p27 level is a poor prognostic indicator for gastric and other cancers [6].
The expression of p27 is regulated at multiple levels. The major mechanism responsible for decreasing p27 during late G1 phase, which allows for physiological cell cycle progression, is thought to be through proteasomal p27 degradation mediated by the SKP2 ubiquitin ligase [7], although p27 degradation by an alternative cytoplasmic ubiquitin ligase, termed KPC, has also been described to operate at the G0–G1 transition [8,9]. The phosphorylation of multiple tyrosine, serine, and threonine sites within p27 have also been shown to regulate the degradation, localisation, and cellular effects of p27 [10,11]. In addition to the post-translational regulation of p27 expression, including by microRNAs in cancer cells [12], the gene p27 may be transcriptionally modulated with epigenetic regulation through histone acetylation from delta opioid receptor (DOR) stimulation [13]. In this mechanism, the phosphorylation of DOR signalling in the cell membrane leads to the translocation of cytoplasmic β-arrestin1 into the nucleus. In the nucleus, β-arrestin1 recruits the histone acetyltransferase p300 to the promoter of p27, resulting in the acetylation of histone H4, chromatin reorganisation and the enhanced transcription of p27.
Helicobacter pylori is a bacterium that colonises the stomachs of approximately half of the world’s population, where it induces a robust and long-lived inflammatory response [14]. Infection by H. pylori is the most significant risk factor for the development of gastric cancer [15,16]. Approximately 70% of all gastric cancer cases worldwide are directly attributable to prior H. pylori infection [17]. The mechanisms responsible for the gastric carcinogenesis induced by H. pylori remain poorly defined. One of the principle mechanisms of H. pylori-related gastric carcinogenesis is an acceleration of normal gastric epithelial cell turnover [18], which provides a biologically plausible link between increased cell proliferation and neoplastic transformation via an increased mutation risk arising stochastically [19]. In patients chronically infected with H. pylori, epithelial cell turnover in the gastric mucosa is increased [20], and the expression of p27 by gastric epithelial cells is decreased. These trends occur in both cases of chronic gastritis and gastric intestinal metaplasia [21,22]. Both the accelerated cell turnover and decrease in p27 can be reversed to normal following the successful eradication of H. pylori in patients with chronic gastritis [21,22]. In animal [23] and cell culture [24] models of chronic H. pylori infection, the long-term exposure of H. pylori to gastric epithelial cells leads to the emergence of epithelial cells that are relatively apoptosis resistant. The levels of p27 and p27 mRNA transcripts in these cells are low when compared to parental AGS cells [24]. These observations led us to hypothesise that the reduction in p27 expression following H. pylori infection may be responsible for inducing a state of accelerated cell turnover in gastric epithelial cells, thereby promoting gastric tumourigenesis [25].
The aims of the current study are to investigate how H. pylori decreases p27 levels in gastric epithelial cells and to examine whether H. pylori regulates DOR signalling to inhibit histone acetylation of the p27 promoter.
2. Materials and methods
2.1. Reagents
The antibodies used in this study were the following: mouse monoclonal anti-p27kip1 (clone 57; BD Biosciences), anti-β-actin (clone AC-74; Sigma-Aldrich), anti-p300 (clone RW105; Novus Biologicals), rabbit polyclonal anti-DOR (ab66318; Abcam), anti-phospho-DOR (Ser363) (ab62152; Abcam), and anti-Ac-H4 (06–866; Millipore). Trichostatin A (Sigma-Aldrich), a histone H4 deacetylase inhibitor, and the DOR agonist DPDPE ([D-Pen2, D-Pen5]-enkephalin) (Sigma-Aldrich) were used to stimulate histone H4 acetylation at the p27 promoter.
2.2. Cell lines and culture conditions
AGS human gastric epithelial cells (CRL-1739; American Type Culture Collection, Manassas, Virginia, USA) were maintained in an atmosphere of 5% CO2 at 37 °C in Ham’s F12 medium supplemented with 10% foetal bovine serum (Gibco-BRL, Carlsbad, CA, USA) without antibiotics in 75 cm2 tissue culture flasks (BD Biosciences, San Jose, California, USA). The HS3C cell line is from a single clone that was derived from AGS gastric epithelial cells by continually exposing those AGS cells to increasing concentrations of H. pylori in co-culture [26]. HS3C cells exhibit resistance to apoptosis induced by several diverse stimuli. p27 and p27 mRNA transcript levels are low in HS3C cells when compared to parental AGS cells, and the apoptosis-resistant phenotype of HS3C cells can be partially reversed by p27 transfection [24]. In the following experiments, HS3C cells and AGS cells were cultured under identical conditions.
2.3. H. pylori strain and culture conditions
H. pylori strain 60190 (ATCC49503; American Type Culture Collection), a cagA- and vacA-positive strain isolated from a patient with dyspepsia, was maintained on trypticase soy agar containing 5% sheep blood (BD Biosciences) and incubated at 37 °C in 10% CO2 for a minimum of 2 and a maximum of 4 passages from frozen stocks. The inocula for co-cultures were diluted from suspensions that had been prepared from 48 h subcultures and were adjusted by a comparison of absorbance to McFarland standards. H. pylori bacteria were added to gastric epithelial cells at a ratio of 200:1 in all experiments.
2.4. Western blotting
Cells were scraped from plates and washed twice in ice-cold PBS and then incubated in a PRO-PREP protein extraction solution (Intron Biotechnology, Kyunggi, Korea). Protein concentrations were determined using the BCA Protein Assay (Pierce Chemical Co., Rockford, IL). For Western blotting, 30 μg of total cell lysate was subjected to SDS-PAGE. The proteins were then transferred to a polyvinylidene difluoride membrane (Pall Corp., Ann Arbor, MI), and the membranes were blocked in 1 × PBS, 0.1% Tween-20, and 5% skim milk. After blocking, the membranes were incubated with primary antibodies diluted 1:1,000 in 1 × PBS, 5% skim milk, and 0.1% Tween-20 overnight at 4 °C. Immunodetection was performed using the Western blotting Luminol Reagent (Santa Cruz Biotechnology Inc. CA, USA). Actin immunoblotting was performed to verify that equal amounts of protein had been loaded in each lane. Quantitative densitometric analysis was performed using NIH Image software.
2.5. mRNA quantification using real-time reverse transcription PCR
Real-time reverse transcription PCR was performed as previously described [22]. The total RNA was extracted using the RNeasy RNA purification kit (Qiagen) according to the manufacturer’s instructions. The reverse transcription of RNA was performed in a final volume of 20 μL containing 4 μL of 5 x reverse transcription buffer (250 mM Tris HCl, 40 mM MgCl2, 150 mM KCl, 5 mM dithiothreitol, pH 8.5), 250 mM each of deoxynucleotide triphosphate, 40 units of RNase inhibitor, 3.2 μg of random hexamers, 20 units of avian myeloblastosis virus reverse transcriptase (all from Roche), and 250 ng/10 μL of total RNA. Real-time quantitative PCR was performed using an iCycler iQ Multi-Color Real Time PCR Detection System (Bio-Rad, Hercules, California, USA) in a 25 μL reaction mix containing 5 μL of diluted reverse transcriptase samples (0.25 ng of equivalent total RNA), 12.5 μL of SYBR Green PCR Master Mix (Bio-Rad), 6.5 μL of deionised distilled water, and 0.5 μL of 10 mM of each p27 primer (forward, 5′-AGGACACGCATTTGGTGGA-3′; reverse, 5′-TAGAAGAATCGTCGGTTGCAGGT-3′). The relative amounts of p27 transcripts were determined using the standard curve method and were normalised to 18S rRNA (forward, 5′-GGACACGGACAGGATTGACA-3′; reverse, 5′-ACCCACGGAATCGAGAAAGA-3′).
2.6. Chromatin immunoprecipitation (ChIP) assay
To measure the amount of acetylated histone H4 and p300 bound to DNA, a ChIP assay kit (Upstate Biotechnology) was used according to manufacturer’s instructions. Cells were fixed with 1% formaldehyde for 10 min, washed twice in ice-cold PBS containing a protease inhibitor mixture (1 mM phenylmethylsulfonyl fluoride, 1 μg/mL aprotinin, and 1 μg/mL pepstatin A) and rapidly collected in ice-cold PBS. Cells were then lysed in a SDS lysis buffer (1% Nonidet P-40, 150 mM NaCl, 50 mM Tris-HCl, supplemented with protease inhibitor mixture), and the chromatin was sheared by sonication. The lysates were cleared by centrifugation, and the supernatants were stored in aliquots at −80 °C. Anti-Ac-H4 and anti-p300 antibodies were used for immunoprecipitation. Immune complexes were collected with salmon sperm DNA-bovine serum albumin-sepharose beads for 60 min and washed twice with RIPA buffer, washed once with high-salt buffer (2 M NaCl, 10 mM Tris, pH 7.5, 1% Nonidet P-40, 0.5% deoxycholic acid, and 1 mM EDTA), washed again with RIPA buffer, and then washed with TE buffer. The immune complexes were extracted in elution buffer (TE buffer/1% SDS). Following digestion with RNase (1 μg/20 μL) for 30 min at 37 °C and proteinase K (1 μg/8 μL for 6 h at 37 °C and 6 h at 65 °C), the DNA was extracted using a PCR purification kit (Qiagen). The p27 promoter DNA was amplified by PCR using the primer sequences shown in Table I. Products were separated by agarose gel electrophoresis and visualised with ethidium bromide staining. The data obtained were normalised to the corresponding DNA input control.
Table I.
p27 promoter-specific primers.
| Locus | Primer sequence | Length (bp) | Range |
|---|---|---|---|
| chip1 | 5′-CTGTCACATTCTGGAGCGTA-3′ 5′-AGTGGATCTTCAACTGCCTC-3′ |
230 | −1523~−1294 |
| chip2 | 5′-CCTGCTCATCGTCCTACTTT-3′ 5′-CCAGATTTCACTGCTCCAAC-3′ |
254 | −1273~−1020 |
| chip3 | 5′-GAAGGAGCTGCTGTATTTGG-3′ 5′-ACTCAAGCTCTCCCTCAATG-3′ |
291 | −997~−707 |
| chip4 | 5′-CTGAGCGAACCATTGCCCA-3′ 5′-AACAAACTAGCCAAACGGCC-3′ |
331 | −592~−262 |
| chip5 | 5′-GCTCCCGCCGCCGCAACCAAT-3′ 5′-CGAACCCAGCCGCTCTCCAAACC-3′ |
220 | −74~+145 |
2.7. Patients and tissue collection
A total of 36 patients with peptic ulcers who had successfully been treated for H. pylori in Uijongbu St. Mary’s Hospital, Korea, were included in this study. The mean age of the patients was 60 years (range 23–87 years), and 30 of the patients were male. In all, 24 patients had gastric ulcers, and 12 had duodenal ulcers. Two endoscopic antral biopsies from each patient were extracted before and 2 months after the eradication of H. pylori. Tissues were fixed in neutral-buffered formalin and embedded in paraffin blocks for immunohistochemical staining. H. pylori infection was diagnosed from the biopsies using the Warthin-Starry silver stain. H. pylori were eradicated using a proton pump inhibitor (40 mg of omeprazole, 30 mg of lansoprazole, or 20 mg of rabeprazole) with 1 g of amoxicillin and 500 mg of clarithromycin twice a day for 7 days; eradication was confirmed with a negative urea breath test. This study using human samples was approved by the institutional review board of the above hospital (UC11SISI0015).
2.8. Immunohistochemistry
Immunohistochemical staining was performed as previously described with some modifications [27]. After the deparaffinisation of 4 μM thick tissue sections, heat-induced epitope retrieval was conducted by immersing the slides in Coplin jars filled with 10 mmol/L citrate buffer (pH 6.0) and boiling them for 30 min in a pressure cooker (Nordic Ware, Minneapolis, MN) inside a microwave oven at 700 W. Following epitope retrieval, the slides were treated with 0.3% H2O2 in PBS for 15 min at room temperature to abolish endogenous peroxidase activity and were placed in TNB buffer (0.1 mol/L Tris-HCl, pH 7.4, 0.15 mol/L NaCl and 0.5% blocking reagent). Sections were then incubated overnight at 4 °C with the primary antibody for p27 (1:100 dilution) or for phospho-DOR (Ser363; 1:50 dilution). Reaction products were developed using the ABC avidin-biotin-peroxidase method (Invitrogen, Camarillo, CA, USA) and counterstained with hematoxylin. Immunohistochemical evaluation was performed by a single pathologist who provided a score in which the intensity of the staining (no staining = 0, very weak staining = 1, weak staining = 2, medium staining = 3, and strong staining = 4) and the percentage of stained cells (0% = 0, 1%–24% = 1, 25%–49% = 2, 50%–74% = 3 and > 75% = 4) were multiplied. With this system, the maximum score was 16 (> 75% of the cells showing strong staining), and the minimum score was 0 (negative staining). As a negative control, the primary antibody was replaced by the blocking reagent.
2.9. Transfection of plasmid and siRNA
A pcDNA-p27 plasmid expression construct was used as previously described [26]. To silence endogenous p27, validated siRNAs for p27kip1 (sc-29429) and control siRNA (sc-37007) were obtained from Santa Cruz. Human gastric epithelial cells were transfected with pcDNA3.1, p27-pcDNA3.1, or control siRNA or p27-siRNA using Lipofectamine (Invitrogen) according to the manufacturer’s protocol. A total of 1.5 × 105 cells were plated in 6-well plates containing Ham’s F12 with 10% FBS 24 h before transfection. Transfections were performed with serum-free Ham’s F12 containing 4 μg of plasmid or siRNA and 5 μL of Lipofectamine. After 6 h, 2 mL of fresh Ham’s F12 containing 10% FBS was added. The cells were collected 48 h post-transfection and were used to prepare protein extracts or to perform the cell proliferation assay. A transfection efficiency of at least 40% was achieved for all of the experiments.
2.10. Assessment of cellular proliferation
For the cell counting analyses, the numbers of transfected or DPDPE-treated cells were counted using a hemocytometer at 24 h, 48 h, 72 h, 96 h, 120 h, and 144 h. The viable cells were identified with the trypan blue exclusion assay. BrdU incorporation was measured at 48 h using a cell proliferation ELISA BrdU colorimetric kit (Millipore) according to the manufacturer’s instructions. Absorbance at 450 nm was measured using a Model 680 Microplate Reader (Bio-Rad).
2.11. Statistical analyses
All cell culture experiments were performed with a minimum of 3 replicates. The quantitative data are expressed as the mean ± standard deviation (SD), and statistical significance was determined with paired t tests. The expression of phospho-DOR and p27 in association with H. pylori eradication were analysed with the Wilcoxon-signed ranks test and Spearman’s correlation test (SPSS statistics). P-values of less than 0.05 were considered significant.
3. Results
3.1. Altered p27 mRNA and p27 expression levels following H. pylori infection and the effects of stimulating histone acetylation or the delta opioid receptor on gastric epithelial cells
H. pylori regulates the expression of p27 mRNA at a transcriptional level. As previously reported, the expression levels of p27 mRNA and p27 were decreased in HS3C cells compared to AGS cells (40% and 70% less, respectively) (Fig. 1A and B). Following 6 h of exposure of H. pylori to AGS cells, p27 and p27 mRNA levels were also reduced by 30% and 20%, respectively (Fig. 1C and D). Following treatment of HS3C cells with the histone deacetylase inhibitor trichostatin A (TSA) for 6 h and the DOR agonist DPDPE for 1 h, p27 mRNA and p27 expression was increased in a dose-dependent manner. Following TSA treatment, p27 levels were increased from 28% to a maximum of 74%, and p27 mRNA was increased from 61% to 88% of the amount in AGS cells (Fig. 1E and G). Following treatment with DPDPE, p27 and p27 mRNA levels were increased from 31% to a maximum of 60% and from 61% to 84%, respectively (Fig. 1F and G).
Fig. 1.
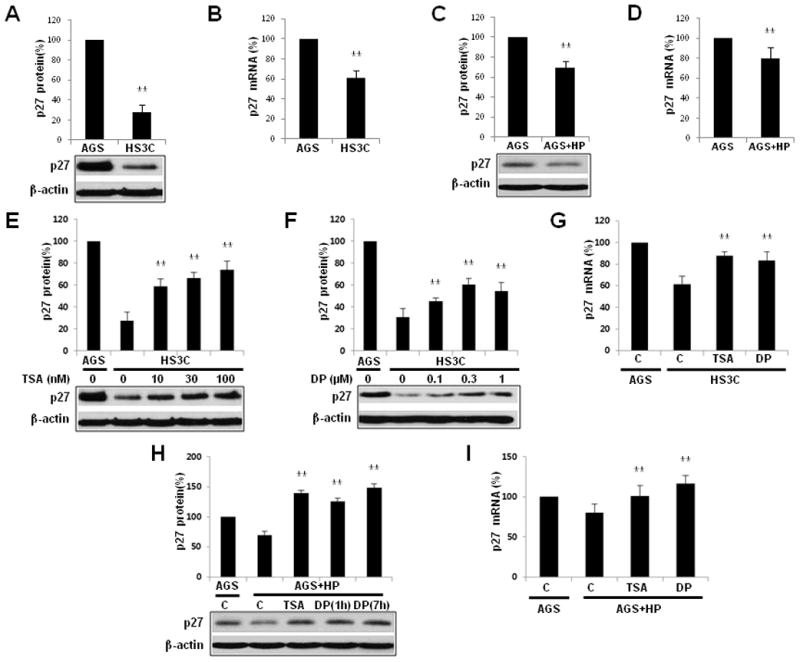
Downregulation of p27 mRNA and p27 expression by H. pylori infection and by treatment with trichostatin A (TSA) and DPDPE (DP). A comparison of steady-state p27 (A) and p27 mRNA (B) expression in HS3C cells relative to AGS cells. The effects of H. pylori infection on p27 (C) and p27 mRNA (D) in AGS cells that were acutely infected with H. pylori. Following treatment with increasing doses of TSA (E) for 6 h and DPDPE (F) for 1 h, p27 expression levels in HS3C cells were measured by immunoblot analyses. The expression of p27 mRNA was analysed by real-time quantitative PCR in HS3C cells with or without treatment of TSA and DPDPE (G). AGS cells that were infected with H. pylori for 6 h were treated with 100 nM TSA and 0.3 μM DPDPE for the times indicated. The immunoblot analysis for endogenously expressed p27 was conducted using an anti-p27 antibody (H). The expression of p27 transcripts in AGS cells following H. pylori infection and treatment with TSA for 6 h and DPDPE for 1 h was analysed by real-time quantitative PCR (I). The levels of p27 were quantified and normalised to actin via Western blotting. 18S rRNA was used as a control in the RT-qPCR experiments. The data represent the mean ± SD of 3 independent experiments with **P < 0.01 versus the corresponding control.
In AGS cells infected with H. pylori, treatment with TSA increased the expression of p27 by 140% compared to uninfected AGS cells. Following DPDPE treatment, p27 increased to 120%–150% of the levels observed in uninfected AGS cells (Fig. 1H). Treatment with TSA and DPDPE increased p27 mRNA in AGS cells infected with H. pylori by 21% and 36%, respectively, as compared to nontreated H. pylori-infected AGS cells (Fig. 1I). Taken together, these data indicate that the expression levels of p27 and p27 mRNA are decreased by H. pylori infection; however, they can be restored by treatment with TSA and DPDPE.
3.2. Changes in the levels of acetylated histone H4 and p300 that are recruited to p27 promoter following H. pylori infection
To determine whether the decrease in p27 mRNA expression is associated with a decrease in histone H4 acetylation of the p27 promoter following H. pylori infection, the level of acetylated histone H4 was measured with ChIP assays. In HS3C cells, the level of acetylated histone H4 was decreased by 15%–60% when compared to AGS cells in each of the 5 regions of the p27 promoter (Fig. 2A). For the AGS cells that were acutely infected with H. pylori, the level of acetylated histone H4 was also decreased by 26%–48% when compared to uninfected AGS cells (Fig. 2B).
Fig. 2.
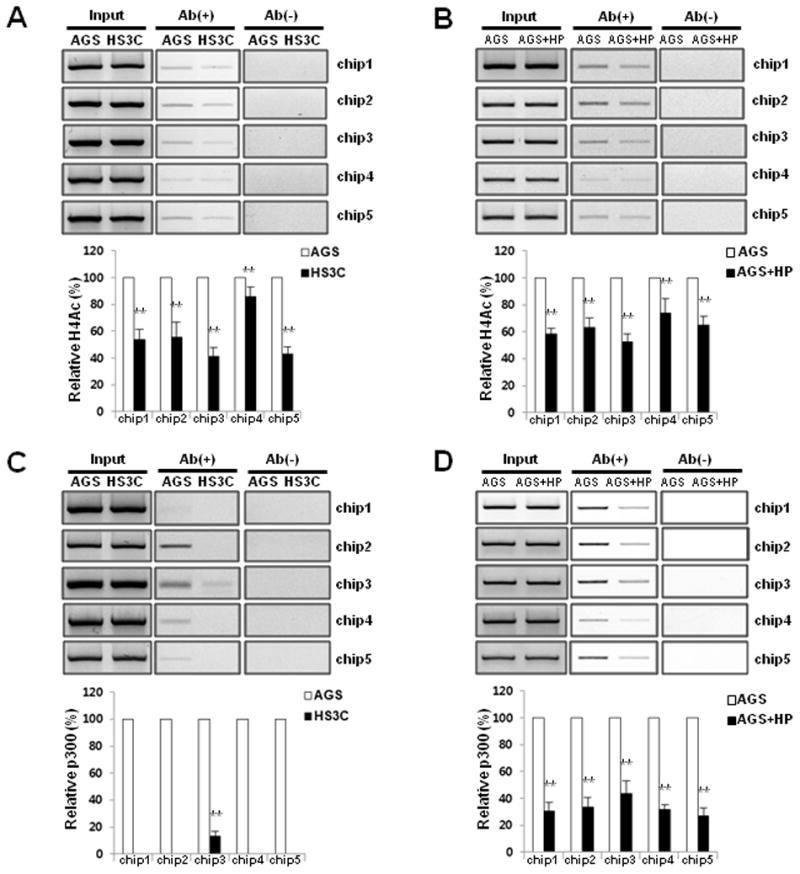
Decrease in acetylated histone H4 and the binding level of p300 to the promoter of p27 H. pylori infected cells. The acetylation status of histone H4 at the p27 promoter was monitored with ChIP assays using an anti-acetyl H4 antibody in HS3C (A) and AGS cells that were infected with H. pylori for 6 h (B). The ChIP experiments were conducted using an antibody against p300 in HS3C cells (C) and following acute H. pylori infection in AGS cells (D). The endogenous p27 promoter DNA that co-precipitated with the anti-acetyl H4 and p300 antibody was quantified by PCR and normalised to the corresponding input controls. Antibody negative (Ab−) controls are shown in the right panel of each figure. The summary data are shown in the graphs at the bottom. The data represent the mean ± SD of 3 independent experiments with **P < 0.01 versus the corresponding control.
The decreased levels of histone H4 acetylation in the promoter region of p27 were associated with the decreased binding of the histone acetyltransferase p300 to the p27 promoter. In HS3C cells, the binding levels of p300 were markedly decreased by > 90% in each of the 5 promoter regions of p27 when compared to AGS cells (Fig. 2C). In addition, the binding levels of the p300 in the p27 promoter were decreased by acute H. pylori infection in AGS cells (Fig. 2D). These results suggest that decreased p27 levels following H. pylori infection are associated with decreased histone H4 acetylation in the p27 promoter, which was caused by the reduced binding of p300.
3.3. Changes in the levels of acetylated histone H4 at the p27 promoter by TSA and DPDPE
The increases in the expressions of p27 and p27 mRNA following treatment with DPDPE or TSA were associated with increased acetylated histone H4 at the p27 promoter. The level of acetylated histone H4 at the p27 promoter was markedly increased in HS3C cells and in AGS cells infected with H. pylori following treatment with TSA and DPDPE (Fig. 3A and B).
Fig. 3.

Changes in the acetylated histone H4 level at the p27 promoter following treatment with TSA and DPDPE. The levels of acetylated histone H4 at the p27 promoter were examined with ChIP assays following treatment with 100 nM TSA for 6 h and 0.3 μM DPDPE (DP) for 1 h in HS3C cells (A) and in AGS cells following acute H. pylori infection for 6 h (B). The p27 promoter sequences in the input DNA that was recovered from antibody-bound chromatin segments were quantified by PCR and normalised to the corresponding input controls. Antibody negative (Ab−) controls are shown in the right panel of each figure. The summary data are shown in the graphs on the right. The data represent the mean ± SD of 3 independent experiments with *P < 0.05 and **P < 0.01 versus the corresponding control.
The increase in acetylated histone H4 levels at the p27 promoter following DPDPE treatment was associated with the altered binding of p300. In HS3C cells, the binding levels of p300 were increased following treatment with DPDPE in chips 1, 2, and 4 but were not significantly altered in chips 3 and 5 (Fig. 4A). The binding levels of p300 at the p27 promoter were also increased following DPDPE treatment in a time-dependent manner in AGS cells that had been acutely infected with H. pylori (Fig. 4B). Taken together, these data confirm that the restoration of p27 and p27 mRNA expression following treatment with DPDPE or TSA in cells infected with H. pylori was associated with increased acetylated histone H4 and increased p300 binding to the p27 promoter.
Fig. 4.

Changes in the binding of p300 at the p27 promoter following treatment with DPDPE. The binding levels of p300 to the p27 promoter were monitored with ChIP assays using an anti-p300 antibody and following treatment with 0.3 μM DPDPE (DP) for 1 h in HS3C cells (A) and AGS cells following acute H. pylori infection for 6 h (B). The endogenous p27 promoter that co-precipitated with the anti-p300 antibody was quantified by PCR, and the data were normalised to the corresponding input controls. The panels on the right are antibody negative (Ab−) controls. The data represent the mean ± SD of 3 independent experiments with **P < 0.01 versus the corresponding control.
3.4. Effects of H. pylori infection on the delta opioid receptor
In the DOR signalling pathway, DOR phosphorylation modulates the recruitment of the histone acetyltransferase p300 to the p27 promoter. We therefore investigated whether H. pylori infection directly regulates the phosphorylation of DOR in the cell membrane. In HS3C cells and AGS cells that were infected with H. pylori for 6 h, the total protein expression of phospho-DOR, DOR, p300, and acetylated histone H4 was evaluated by immunoblotting using specific antibodies. H. pylori infection significantly decreased the phospho-DOR (S363) protein levels in HS3C cells and AGS cells infected with H. pylori when compared to AGS control cells. In contrast, the cellular levels of DOR, p300, and acetylated histone H4 were not altered by H. pylori infection (Fig. 5A). Therefore, the decreased expression of p27 during H. pylori infection is associated with decreased DOR phosphorylation at S363, and H. pylori infection decreases the recruitment of p300 to the p27 promoter rather than altering overall cellular p300 levels.
Fig. 5.

Effects of H. pylori infection on phospho-DOR and p27. Western blot analyses of phospho-DOR, DOR, p300, and Ac-H4 protein levels in whole-cell lysates of HS3C cells and AGS cells that were infected with H. pylori for 6 h (A). Actin was used as a loading control. Immunohistochemical staining of phospho-DOR and p27 was conducted on the gastric mucosa of 36 patients before and after the eradication of H. pylori. The mean expression scores of phospho-DOR and p27 in 36 patients before and after H. pylori eradication (B). Here, *P < 0.05 and **P < 0.01 versus the corresponding control. Representative immunohistochemical staining of phospho-DOR and p27 expression in gastric epithelial cells (C). Magnification X400. The correlation between phospho-DOR and p27 expression was analysed (D).
The expression of phospho-DOR and p27 was then investigated with the immunohistochemical staining of 36 human gastric tissues before and after the eradication of H. pylori. In gastric epithelial cells, phospho-DOR and p27 levels were increased following H. pylori eradication (Fig. 5B). The mean expression scores of phospho-DOR before and after H. pylori eradication were 4.7 ± 3.4 and 7.9 ± 4.2, respectively (P = 0.001), and the scores of p27 before and after H. pylori eradication were 5.1 ± 3.5 and 7.1 ± 3.8, respectively (P = 0.012). Examples of the patterns and ranges of phospho-DOR and p27 expression in gastric mucosa before and after H. pylori eradication are shown in Fig. 5C. A significant correlation between the expression scores of phospho-DOR and those of p27 in gastric epithelial cells (r = 0.479; P = 0.001) was observed (Fig. 5D). Taken together, these data suggest that H. pylori induces the dephosphorylation of DOR in the cell membrane, leading to decreased p27 expression.
3.5. H. pylori induces the proliferation of gastric epithelial cells by inhibiting the expression of p27
Growth curves and a BrdU incorporation assay were performed to investigate whether altered p27 expression levels affect the growth and proliferation of gastric epithelial cells. The growth of HS3C cells was markedly increased compared to parental AGS cells, but the growth of HS3C cells could be reduced by transfecting exogenous p27. Treatment of HS3C cells with DPDPE, which stimulates p27 expression, led to a reduction in proliferation. In AGS cells, the suppression of p27 by siRNA-p27 transfection resulted in a marked increase in the rate of proliferation (Fig. 6A and B). These data indicate that the reduction of p27 by long-term H. pylori infection can promote the growth and proliferation of gastric epithelial cells.
Fig. 6.
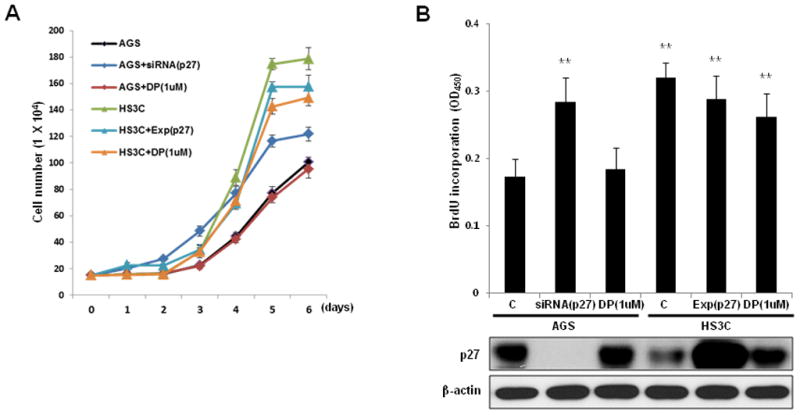
Effects of p27 on gastric epithelial cell proliferation. AGS cells were transfected with a control siRNA, p27 siRNA, or treated with 1 uM DPDPE (DP). HS3C cells were transfected with a control plasmid, p27 expression plasmid, or treated with 1 uM DP. The monolayer growth rates of cells were determined with cell counting (A) and a BrdU incorporation assay (B). The cell number was counted on the indicated day, and the BrdU incorporation assays were conducted at 48 h after cellular transfections. The levels of cellular p27 in association with BrdU incorporation are shown at the bottom of (B). The data represent the mean ± SD of 3 independent experiments with **P < 0.01 versus the corresponding control.
4. Discussion
We have demonstrated in HS3C cells that the downregulation of p27 mRNA by H. pylori is caused by histone H4 acetylation of the p27 promoter via the reduced phosphorylation of the DOR and that this process is reversible. Additionally, the reduced acetylation of histone H4 in the p27 promoter is associated with a decreased recruitment of p300, a p27-specific histone acetyltransferase. Similar events occur during the acute infection of AGS cells by H. pylori in vitro. The resultant reductions in p27 mRNA expression are, in part, responsible for the reduced p27 levels observed in these models.
Histone acetylation is a key epigenetic mechanism that modulates gene transcription. Histone acetylation of the p27 promoter has been previously shown to be regulated by the DOR pathway [13]. Following the phosphorylation of the G-protein coupled DOR, β-arrestin1 is translocated from the cytoplasm to the nucleus, and p300 accumulates at the p27 promoter. These events lead to histone H4 acetylation and the increased expression of p27 mRNA and p27. Similar mechanisms serve to regulate the activity of c-fos [13]. In the current study, H. pylori infection directly decreased the expression of phosphorylated DOR in the cell membrane, thereby reducing the acetylation of histone H4 at the p27 promoter by reducing the recruitment of the histone acetyltransferase p300. The levels of phosphorylated DOR were reduced in HS3C cells and in AGS cells infected with H. pylori in vitro. Decreased expression of phosphorylated DOR and p27 was also confirmed in the gastric mucosa of patients with infections of H. pylori. In the gastric epithelium of patients with peptic ulcers, the expression of phosphorylated DOR was restored after H. pylori eradication, and a statistically significant correlation between the reduced expression of phosphorylated DOR and reduced cellular p27 levels was observed. These results indicate that H. pylori decreases p27 through the regulation of the DOR. In cells that were treated with DPDPE, a DOR stimulator, the recruitment of p300 and histone H4 acetylation in the p27 promoter was increased, and eventually, the expression of p27 mRNA transcription was also increased, suggesting that p27 is regulated by the DOR pathway in these cell lines. Trichostatin A, a histone H4 deacetylase inhibitor, was used to directly stimulate histone acetylation in cells to determine whether p27 is regulated epigenetically by histone modifications [13].
To investigate the effects of chronic H. pylori infection on gastric epithelial cells, we used HS3C cell lines. HS3C cells are single clones that were derived from AGS gastric carcinoma cells by continually exposing those cells to increasing concentrations of H. pylori in co-cultures [24,26]. HS3C cells have a characteristic resistance to apoptosis caused by several stimuli, such as 5-fluorouracil, VP16, irradiation, and H. pylori [26]. p27 and p27 mRNA transcripts of these cells are decreased when compared to parental AGS cells, and their apoptosis-resistant phenotype can be partially reversed by transfecting back p27 [24]. However, the mechanism by which p27 expression is reduced in H. pylori infections was not clarified in these reports. We suggest in the current study that H. pylori regulates p27 expression epigenetically through the modulation of histone H4 acetylation by decreasing the phosphorylation of cell membrane DORs. We also showed that the decrease in p27 in H. pylori infections has a significant role in accelerated cell turnover. This cell turnover may contribute to the development of gastric cancer. This finding was not consistent with a previous report [26]. We believe that this discrepancy may be due to the different cell types used, HS3 cells, which are not single cloned cells but are rather contaminated with cells containing various levels of p27 expression, and thereby demonstrate various proliferation levels.
Recent studies have demonstrated that H. pylori bacteria may directly effect on the structure or function of host cells’ chromatin [28,29]. One study reported that H. pylori increased histone H4 acetylation in the promoter region of p21 (a gene that is partially homologous to p27), thereby increasing p21 expression [28]. In NCI-N87 gastric epithelial cell lines, infections with H. pylori led to the acetylation of histone H3 and H4 together with increased histone H4 acetylation in the p21 promoter. In the current study, we measured the total acetylated histone H3 and H4 via Western blotting; however, the levels of acetylated histone H3 or H4 were not increased in the AGS cell line infected with H. pylori, nor was total p300 altered. These results suggest that the reduced acetylation of histone H4 in the promoter of p27 in H. pylori infection does not occur through total p300 reduction but through the reduced recruitment of that protein to the p27 promoter.
In recent years, appreciation for the epigenetic regulation role of gastric epithelial gene expression by H. pylori during gastric carcinogenesis has increased. E-cadherin methylation by H. pylori may be of particular significance in diffuse gastric carcinogenesis given that germline mutations in E-cadherin are responsible for the syndrome of hereditary diffuse gastric cancer and are commonly acquired in sporadic diffuse type gastric cancer associated with H. pylori infection [30]. The methylation of the E-cadherin promoter can be reversed with the eradication of H. pylori [31]. p27 levels can also be regulated post-translationally by proteasome-mediated degradation following H. pylori infection [32]. The current experiments confirm that the expression of p27 and p27 mRNA are decreased in HS3C cells and in AGS cells that were acutely infected with H. pylori when compared to uninfected AGS cells. These results suggest that H. pylori can modulate p27 expression through at least 2 distinct mechanisms: the epigenetic regulation of gene transcription by histone acetylation and the control of the rate of p27 degradation [32,33]. In addition, H. pylori can alter the subcellular localisation of p27 from the nucleus to the cytoplasm [34]. Which mechanism is more significant in vivo, particularly during the gradual progression of chronically inflamed gastric mucosa towards gastric cancer, remains to be determined.
Acknowledgments
This work was supported by the U.S. National Institutes of Health [CA111533 to S.F.M.] and by the Basic Science Research Program through the National Research Foundation of Korea, funded by the Ministry of Education, Science and Technology [2010-0024303].
Footnotes
Conflict of Interest Statement: No conflicts of interest were declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8:253–264. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- 2.Fero M, Randel E, Gurley KE, Roberts JM, Kemp CJ. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature. 1998;396:177–180. doi: 10.1038/24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004;18:862–876. doi: 10.1101/gad.1185504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mori M, Mimori K, Shiraishi T, Tanaka S, Ueo H, Sugimachi K, Akiyoshi T. p27 expression and gastric carcinoma. Nat Med. 1997;3:593. doi: 10.1038/nm0697-593. [DOI] [PubMed] [Google Scholar]
- 5.Ohtani M, Isozaki H, Fujii K, Nomura E, Niki M, Mabuchi H. Impact of the expression of cyclin-dependent kinase inhibitor p27Kip1 and apoptosis in tumor cells on the overall survival of patients with non-early stage gastric carcinoma. Cancer. 1999;85:1711–1718. [PubMed] [Google Scholar]
- 6.Sgambato A, Migaldi M, Leocata P, Ventura L, Criscuolo M, Di Giacomo C, Capelli G, Cittadini A, De Gaetani C. Loss of p27Kip1 expression is a strong independent prognostic factor of reduced survival in N0 gastric carcinomas. Cancer. 2000;89:2247–2257. doi: 10.1002/1097-0142(20001201)89:11<2247::aid-cncr13>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 7.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 8.Hara T, Kamura T, Nakayama K, Oshikawa K, Hatakeyama S, Nakayama K. Degradation of p27(Kip1) at the G(0)– G(1) transition mediated by a Skp2-independent ubiquitination pathway. J Biol Chem. 2001;276:48937–48943. doi: 10.1074/jbc.M107274200. [DOI] [PubMed] [Google Scholar]
- 9.Hara T, Kamura T, Kotoshiba S, Takahashi H, Fujiwara K, Onoyama I, Shirakawa M, Mizushima N, Nakayama KI. Role of the UBL-UBA protein KPC2 in degradation of p27 at G1 phase of the cell cycle. Mol Cell Biol. 2005;25:9292–9303. doi: 10.1128/MCB.25.21.9292-9303.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vervoorts J, Lüscher B. Post-translational regulation of the tumor suppressor p27(KIP1) Cell Mol Life Sci. 2008;65:3255–3264. doi: 10.1007/s00018-008-8296-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borriello A, Cucciolla V, Oliva A, Zappia V, Della Ragione F. p27Kip1 metabolism: a fascinating labyrinth. Cell Cycle. 2007;6:1053–1061. doi: 10.4161/cc.6.9.4142. [DOI] [PubMed] [Google Scholar]
- 12.le Sage C, Nagel R, Egan DA, Schrier M, Mesman E, Mangiola A, Maira G, Mercatelli N, Ciafrè SA, Farace MG, Agami R. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007;26:3699–3708. doi: 10.1038/sj.emboj.7601790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang J, Shi Y, Xiang B, Qu B, Su W, Zhu M, Zhang M, Bao G, Wang F, Zhang X, Yang R, Fan F, Chen X, Pei G, Ma L. A nuclear function of beta-arrestin1 in GPCR signaling: regulation of histone acetylation and gene transcription. Cell. 2005;123:833–847. doi: 10.1016/j.cell.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 14.Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med. 2002;347:1175–1186. doi: 10.1056/NEJMra020542. [DOI] [PubMed] [Google Scholar]
- 15.IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Schistosomes, liver flukes and Helicobacter pylori. IARC Monogr Eval Carcinog Risks Hum. 1994;61:1–241. [PMC free article] [PubMed] [Google Scholar]
- 16.Peek RM, Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- 17.Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006;118:3030–3044. doi: 10.1002/ijc.21731. [DOI] [PubMed] [Google Scholar]
- 18.Shirin H, Weinstein IB, Moss SF. Effects of H. pylori infection of gastric epithelial cells on cell cycle control. Front Biosci. 2001;6:E104–E118. doi: 10.2741/shirin. [DOI] [PubMed] [Google Scholar]
- 19.Preston-Martin S, Pike MC, Ross RK, Jones PA, Henderson BE. Increased cell division as a cause of human cancer. Cancer Res. 1990;50:7415–7421. [PubMed] [Google Scholar]
- 20.Moss SF, Sordillo EM, Abdalla AM, Makarov V, Hanzely Z, Perez-Perez GI, Blaser MJ, Holt PR. Increased gastric epithelial cell apoptosis associated with colonization with cagA+ Helicobacter pylori strains. Cancer Res. 2001;61:1406–1411. [PubMed] [Google Scholar]
- 21.Yu J, Leung WK, Ng EK, To KF, Ebert MP, Go MY, Chan WY, Chan FK, Chung SC, Malfertheiner P, Sung JJ. Effect of Helicobacter pylori eradication on expression of cyclin D2 and p27 in gastric intestinal metaplasia. Aliment Pharmacol Ther. 2001;15:1505–1511. doi: 10.1046/j.1365-2036.2001.01038.x. [DOI] [PubMed] [Google Scholar]
- 22.Kim SS, Meitner P, Konkin TA, Cho YS, Resnick MB, Moss SF. Altered expression of Skp2, c-Myc and p27 proteins but not mRNA after H. pylori eradication in chronic gastritis. Mod Pathol. 2006;19:49–58. doi: 10.1038/modpathol.3800476. [DOI] [PubMed] [Google Scholar]
- 23.Peek RM, Jr, Wirth HP, Moss SF, Yang M, Abdalla AM, Tham KT, Zhang T, Tang LH, Modlin IM, Blaser MJ. Helicobacter pylori alters gastric epithelial cell cycle events and gastrin secretion in Mongolian gerbils. Gastroenterology. 2000;118:48–59. doi: 10.1016/s0016-5085(00)70413-6. [DOI] [PubMed] [Google Scholar]
- 24.Eguchi H, Carpentier S, Kim SS, Moss SF. P27kip1 regulates the apoptotic response of gastric epithelial cells to Helicobacter pylori. Gut. 2004;53:797–804. doi: 10.1136/gut.2003.032144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 26.Shirin H, Sordillo EM, Kolevska TK, Hibshoosh H, Kawabata Y, Oh SH, Kuebler JF, Delohery T, Weghorst CM, Weinstein IB, Moss SF. Chronic Helicobacter pylori infection induces an apoptosis-resistant phenotype associated with decreased expression of p27(kip1) Infect Immun. 2000;68:5321–5328. doi: 10.1128/iai.68.9.5321-5328.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim JS, Shin OR, Kim HK, Cho YS, An CH, Lim KW, Kim SS. Overexpression of protein phosphatase non-receptor type 11 (PTPN11) in gastric carcinomas. Dig Dis Sci. 2010;55:1565–1569. doi: 10.1007/s10620-009-0924-z. [DOI] [PubMed] [Google Scholar]
- 28.Fehri LF, Rechner C, Janssen S, Mak TN, Holland C, Bartfeld S, Bruggemann H, Meyer TF. Helicobacter pylori-induced modification of the histone H3 phosphorylation status in gastric epithelial cells reflects its impact on cell cycle regulation. Epigenetics. 2009;4:577–586. doi: 10.4161/epi.4.8.10217. [DOI] [PubMed] [Google Scholar]
- 29.Xia G, Schneider-Stock R, Diestel A, Habold C, Krueger S, Roessner A, Naumann M, Lendeckel U. Helicobacter pylori regulates p21(WAF1) by histone H4 acetylation. Biochem Biophys Res Commun. 2008;369:526–531. doi: 10.1016/j.bbrc.2008.02.073. [DOI] [PubMed] [Google Scholar]
- 30.Hamilton JP, Meltzer SJ. A review of the genomics of gastric cancer. Clin Gastroenterol Hepatol. 2006;4:416–425. doi: 10.1016/j.cgh.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 31.Chan AO, Peng JZ, Lam SK, Lai KC, Yuen MF, Cheung HK, Kwong YL, Rashid A, Chan CK, Wong BC. Eradication of Helicobacter pylori infection reverses E-cadherin promoter hypermethylation. Gut. 2006;55:463–468. doi: 10.1136/gut.2005.077776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eguchi H, Herschenhous N, Kuzushita N, Moss SF. Helicobacter pylori increases proteasome-mediated degradation of p27(kip1) in gastric epithelial cells. Cancer Res. 2003;63:4739–4746. [PubMed] [Google Scholar]
- 33.Slingerland J, Pagano M. Regulation of the cdk inhibitor p27 and its deregulation in cancer. J Cell Physiol. 2000;183:10–17. doi: 10.1002/(SICI)1097-4652(200004)183:1<10::AID-JCP2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 34.Wen S, So Y, Singh K, Slingerland JM, Resnick MB, Zhang S, Ruiz V, Moss SF. Promotion of cytoplasmic mislocalization of p27 by Helicobacter pylori in gastric cancer. Oncogene. 2012;31:1771–1780. doi: 10.1038/onc.2011.362. [DOI] [PMC free article] [PubMed] [Google Scholar]