

Abstract
Neuroinflammation is typically observed in neurodegenerative diseases such as Alzheimer’s disease, as well as after traumatic injury and pathogen infection. Resident immune cells, microglia and astrocytes, are activated and joined by blood-borne monocytes that traverse the blood–brain barrier and convert into activated macrophages. The activated cells express various cytokines, chemokines and proteolytic enzymes. To study the role of heparan sulfate proteoglycans in neuroinflammation, we employed a transgenic mouse overexpressing heparanase, an endoglucuronidase that specifically degrades heparan sulfate side chains. Neuroinflammation was induced by systemic challenge with lipopolysaccharide, or by localized cerebral microinjection of aggregated amyloid-β peptide, implicated in Alzheimer’s disease. Lipopolysaccharide-treated control mice showed massive activation of resident microglia as well as recruitment of monocyte-derived macrophages into the brain parenchyma. Microinjection of aggregated amyloid-β elicited a similar inflammatory response, albeit restricted to the injection site, which led to dispersion and clearance of the amyloid. In the heparanase-overexpressing mice, all aspects of immune cell recruitment and activation were significantly attenuated in both inflammation models, as was amyloid dispersion. Accordingly, an in vitro blood–brain barrier model constructed from heparanase-overexpressing cerebral vascular cells showed impaired transmigration of monocytes compared to a corresponding assembly of control cells. Our data indicate that intact heparan sulfate chains are required at multiple sites to mediate neuroinflammatory responses, and further point to heparanase as a modulator of this process, with potential implications for Alzheimer’s disease.
Electronic supplementary material
The online version of this article (doi:10.1007/s00401-012-0997-1) contains supplementary material, which is available to authorized users.
Keywords: Neuroinflammation, Heparan sulfate, Heparanase, Amyloid-β, Clearance, Alzheimer’s disease
Introduction
Neuroinflammation is a prominent feature of several pathological conditions of the central nervous system (CNS), including neurodegenerative diseases such as Alzheimer’s disease (AD) [1, 25], CNS autoimmune diseases [5], traumatic brain injury [63] and pathogen infection [7]. Resident immune cells (microglia and astrocytes) are activated to release various cytokines, chemokines and proteolytic enzymes [38, 58]. Moreover, the cerebral parenchyma is invaded by blood-borne monocytes that traverse the blood–brain barrier (BBB) and convert into activated macrophages [16, 45]. The mechanisms behind these events are only partly understood, and the pathophysiological consequences of neuroinflammation are diverse and complex.
In AD, aggregated 40- or 42-residue peptides of amyloid-β (Aβ) deposit in the brain parenchyma as extracellular senile plaques and typically throughout the vasculature as cerebral amyloid angiopathy.
Seemingly contradictory effects of neuroinflammation have been noted in AD, on the one hand deposits appear to induce inflammatory reactions that recruit immune cells capable of dispersing the plaques, contributing to their clearance from the tissue [3, 52]. The relevance of these phenomena to the disease course is unclear, particularly so since soluble intermediate Aβ aggregates rather than fibrils have been directly implicated with neurotoxicity [20, 32]. Nevertheless, reactions leading to dispersal of Aβ deposits are potentially beneficial and merit characterization. Efficient Aβ phagocytes are believed to be derived from blood-borne monocytes that are recruited into the brain, where they differentiate to macrophages in the course of a complex neuroinflammatory response [9, 16]. The mechanism of cell recruitment across the blood–brain barrier (BBB) is only partly understood.
Previous studies demonstrated a key role for cell-surface heparan sulfate (HS) proteoglycans (HSPGs) in regulating leukocyte transmigration from blood to sites of inflammation [27, 33, 60]. The present study was undertaken to clarify the involvement of HS in neuroinflammation, induced either by systemic challenge with bacterial lipopolysaccharide (LPS) or by local deposition of Aβ fibrils in murine brain. We employed a transgenic mouse overexpressing heparanase (Hpa-tg), an endoglucuronidase that specifically degrades HS [11]. The Hpa-tg mice showed attenuated inflammatory response compared to control (Ctr) mice, and an impaired ability to disperse and clear Aβ aggregates. Moreover, HS and heparanase were directly implicated with regulation of monocyte transmigration across the BBB.
Materials and methods
Mice
The transgenic mouse overexpressing human heparanase (Hpa-tg), under the β-actin promoter, were generated according to Zcharia et al. [62], except that the enzyme was expressed as a chimer containing a chick heparanase signal peptide sequence, which promotes enzyme secretion [18]. The transgenic mice were backcrossed more than 10 generations on a C57BL/6 background and C57BL/6 was used as control (Ctr). The mice were used at 3–20 months of age. All experiments were approved by the regional animal research ethics committee.
Determination of heparanase expression level
Heparanase ELISA was performed as described by Shafat et al. [50].
Isolation and analysis of HSPG
Isolation and analysis of HSPG were performed as previously described [11]. See Supplementary methods for details.
Lipopolysaccharide (LPS) treatment
Mice (4-month-old) were intraperitoneally injected with a single dose (5 mg/kg) of LPS (10 μg/100 μl PBS) (LPS L2280, derived from O55:B5 E.coli, Sigma-Aldrich). After 20 h the mice were deeply anesthetized, transcardially perfused with 50 ml saline, and the brains were dissected. One hemisphere of the brain was fixed in 4 % formaldehyde for cryotome tissue section preparation and the other was rapidly frozen in dry ice for preparation of tissue extract.
Western blotting
Protein samples were separated by 10–20 % SDS-PAGE and then transferred to a nitrocellulose membrane. After blocking with 5 % nonfat dry milk, the membranes were probed with primary antibodies (see Supplementary Table 1 for an account of all antibodies used in the study) followed by the corresponding secondary antibodies. Signals were visualized using SuperSignal West Pico or Dura substrates (Thermo). Quantitative band analysis was performed with ImageJ software.
Intracortical Aβ42 injection
The injection was performed on deeply anesthetized (2.5 % avertin 500 μl/mouse i.p.) mice under stereotaxic guidance with coordinates from the bregma: +2.0 mm anteroposterior, −2.0 mm lateral, and −2.3 mm dorsoventral. Five micrograms of aggregated synthetic human Aβ1-42 (PolyPeptide Laboratories GmbH, Germany) (5 μg/1 μl) was injected into one hemisphere at a rate of 0.2 μl/min, followed by a 2-min pause for absorption of the injected solutions. The brains were dissected as described above, fixed in 4 % formaldehyde overnight and processed according to standard protocols for the preparation of paraffin-embedded tissue blocks.
Histochemistry and immunostaining
Immunostaining with antibodies against CD45, F4/80, CD31 and ICAM-1 were performed on 20 μm cryotome brain tissue sections. All other immunostainings and Congo red histochemistry were performed on 5 μm paraffin sections. After antigen retrieval, primary antibody incubation was carried out overnight at 4 °C followed by incubation with secondary antibody for 30–60 min at room temperature. ABC™ complex and NOVA™ red reagents (Vector Labs) were used to visualize the immunosignals. In another setting, a rat on mouse AP-polymer kit, developed with Vulcan Fast Red Chromogen kit2 (Biocare Medical) was employed. For double immunostaining, primary antibodies were incubated overnight, simultaneously or stepwise at 4 °C, followed by incubation with the appropriate Alexa fluor labeled secondary antibodies (Invitrogen). Hematoxylin and DAPI (4′,6-diamidino-2-phenylindole) were used for counterstaining of nuclei.
Microscopy and image analysis
Microscopy was performed using a Nikon DXM1200F™ instrument (Nikon, Melville, USA). CD45-positive cells associated with microvessel walls or partially exited from vessels were counted under microscope and the number of such cells associated with 10 microvessels of similar length (~300 μm) in each section was summarized. Images of anti-F4/80 and anti-CD45 immunostained sections were captured and the positive cells were quantified with ImageJ software. The images were converted to 8-bit grayscale format and a standard segmentation threshold for positively stained pixels was established. Using the ImageJ particle analysis tool with a size setting of 8–20 pixels, the number of amoeboid and ramified CD45-positive cells was recorded and with a setting of 20–100 pixels, the number of F4/80-positive cells was recorded. Cross-sectional information and z-stacks for selected deposits were obtained by confocal laser scanning microscopy using a Carl Zeiss LSM 510 META™ instrument (Carls Zeiss, Germany).
In vitro BBB model and monocyte transmigration
Primary endothelial cells and pericytes were prepared from the brains of Hpa-tg and Ctr mice. The in vitro BBB model was constructed on Falcon™ HTS FluoroBlok™ 3.0 μm colored PET Membrane Inserts for 24 well culture plates (BD). Pericytes (10,000 cells/insert) were grown on the basolateral side and endothelial cells (150,000 cells/insert) on the apical side of the membrane, which was pre-coated with bovine fibronectin. The cells were grown for 3 days prior to experiments. The integrity of the endothelial monolayer was confirmed by establishing that Evans blue-bound BSA did not diffuse from the apical to the basolateral compartment. GFP-monocytes (120,000 cells/insert) were, immediately after isolation, loaded into the apical compartment (blood), and 50 ng/ml of CCL2 was added to the basolateral compartment (brain). Monocytes migration from the apical to the basolateral compartment was monitored with a Zeiss laser scanning microscope LSM 700/CO2 Microscope with a stage incubator (37 °C, 5 % CO2). The number of GFP-monocytes was counted with ImageJ software. For details, see Supplementary methods.
Statistical method
Two-tailed unpaired Student’s t test was used to determine the significance between population means. Statistical significance was set at P < 0.05.
Results
Cerebral expression of heparanase and altered HS structure in transgenic mice
We have previously described a transgenic mouse overexpressing human heparanase, which results in extensive fragmentation of HS chains in vivo [11, 62]. To study the role of HS in neuroinflammation, we generated a new strain (Hpa-tg) that overexpresses a high level of heparanase in the brain. The transgenic enzyme was expressed, albeit to variable extent, in all examined regions of the brain (Fig. 1a, b). Gel chromatography of metabolically radiolabeled HSPG isolated from Hpa-tg brain detected HS fragments that were not present in Ctr brain tissue (Fig. 1c). Disaccharide compositional analysis revealed a decrease in the -GlcA-GlcNS6S- unit, representing the cleavable linkage target for heparanase (Fig. 1d; peak 2), and an increase in trisulfated -IdoA2S-GlcNS6S- disaccharide (Fig. 1d; peak 5), as previously ascribed to heparanase upregulation [11].
Fig. 1.
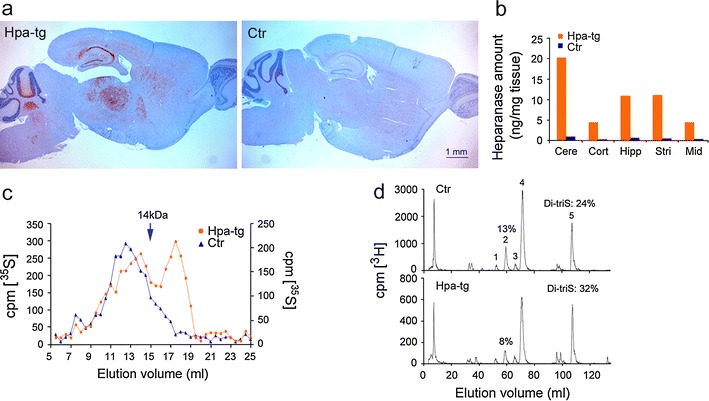
Changed structure of heparan sulfate in mouse brain due to heparanase overexpression. a Paraffin-embedded sagittal brain sections of Hpa-tg (left) and Ctr (right) mice were immunostained with anti-heparanase antibody 733 (red). b Heparanase levels in extracts of Hpa-tg and Ctr brain tissue, analyzed with ELISA. Cere cerebellum, Cort cortex, Hipp hippocampus, Stri striatum, Mid midbrain. c Gel chromatography (Superose 6) of metabolically 35S-labeled HSPG purified from Hpa-tg and Ctr brains. The elution position of heparin (14 kDa) is indicated by the arrow. d Anion-exchange HPLC (Partisil-10 SAX column) of disaccharides generated by deaminative cleavage of HS derived from brain tissue extracts of Hpa-tg and Ctr mice. The resulting disaccharides were radio-endlabeled by reduction with NaB3H4. Numbered peaks represent the following disaccharide units in the intact HS chains: 1 -GlcA2S-GlcNS-, 2 -GlcA-GlcNS6S-, 3 -IdoA-GlcNS6S-, 4 -IdoA2S-GlcNS-, 5 -IdoA2S-GlcNS6S-. The proportions of peaks 2 (the disaccharide cleaved by heparanase) and 5 (Di-triS: hypersulfated disaccharide) are indicated as percentage of total disaccharides
Reduced cerebral inflammatory response to LPS challenge in Hpa-tg mice
LPS is a bacterial endotoxin known to upregulate cerebral expression of pro-inflammatory factors such as interleukin-1β (IL-1β) [22], tumor necrosis factor α (TNF-α) [44] and CCL2 (also designated monocyte chemotactic protein-1) [55]. Once released, the pro-inflammatory factors stimulate recruitment of neutrophils [47], and activation of microglia [44]. We examined the cellular response of the Hpa-tg brain to LPS treatment. Brain tissue sections of 4-month-old, LPS-challenged mice were immunostained with antibodies against CD45, a major protein tyrosine phosphatase located at the leukocyte plasma membrane [56] and F4/80, a glycoprotein cell-surface marker that is amply expressed by murine macrophages [2]. Anti-CD45 antibody strongly stained cells with amoeboid morphology in association with microvasculature (Fig. 2a, e–f) and in the parenchyma (Fig. 2j; left insert). This phenotype is primarily associated with macrophages derived from blood-borne monocytes [19], which was supported by the detection of CD45-positive cells at various stages of transmigration (Fig. 2b; i–iv), including cells attached to the luminal wall (Fig. 2c). The transmigration process is outlined in the graphic in Fig. 2d. To assess transmigration of blood-borne monocytes, we counted the CD45-positive cells associated with microvasculature (Fig. 2e, f) and found significantly more CD45-positive cells in Ctr than in Hpa-tg mice (Fig. 2g). Resident microglia are characterized by highly branched morphology and low levels of CD45 [41, 49] that are upregulated in the course of activation [14, 42]. We counted CD45-positive cells of ramified and amoeboid morphology (Fig. 2h–j; inserts) in whole tissue sections and found, again, significantly increased numbers of CD45-positive cells in Ctr compared to Hpa-tg mice (Fig. 2j). Immunostaining with anti-F4/80 antibody revealed a massive recruitment of activated macrophages in Ctr brains (Supplementary Fig. 1a: insert, b). Again, such cells were less abundant in Hpa-tg brains (Supplementary Fig. 1c). The abundant ramified macrophages presumably include activated resident microglia as well as cells derived from blood-borne monocytes. Mice of similar age, not challenged with LPS showed few CD45- and F4/80-positive cells, irrespective of Ctr or Hpa-tg status (Supplementary Fig. 1d-g). Consistent with these findings, protein levels of the pro-inflammatory cytokine IL-1β were significantly higher in LPS-challenged Ctr brains than in Hpa-tg brains (Fig. 2k). Heparanase overexpression thus attenuates recruitment of inflammatory cells into the brain and reduces activation of resident microglia in response to systemic LPS challenge.
Fig. 2.
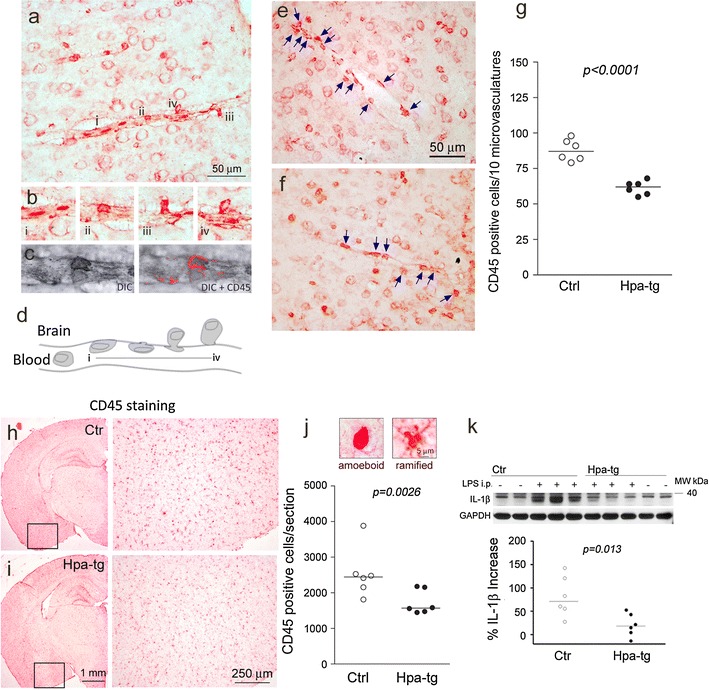
Impaired immune response of Hpa-tg brain to LPS challenge. Four-month-old Hpa-tg and Ctr mice received LPS (5 mg/kg) by intraperitoneal injection, and were killed 20 h later and brain tissue sections were immunostained with anti-CD45 antibody. a–d CD45-positive cells associated with microvascular structures in Ctr brain: a the morphologies of CD45-positive cells associated with the vasculature are consistent with those assumed by monocytes transmigrating from the blood to the brain parenchyma, converting to macrophages in the course of this process. b CD45-positive cells in (i) and (ii) remain intravascular but are associated with the vessel wall, whereas the cells in (iii) and (iv) have largely exited the vessel. c Confocal laser scanning microscopy of the CD45-positive cell in (ii) imaged with differential interference contrast (DIC). d Model of monocyte attachment and diapedesis from blood to brain, typically stimulated in response to CNS-derived chemokines. e–f CD45-positive cells (indicated by arrows) associated with Ctr and Hpa-tg microvasculature, respectively. g Significantly fewer CD45-positive cells are associated with Hpa-tg than with Ctr microvasculature. The data are presented, for each animal, as total number of CD45-positive cells from 10 microvasculatures, each of ~300 μm length. h, i Panels to the right show high-magnification images of the cortical areas indicated by frames in the left panels. j Top representative amoeboid (left) and ramified (right) CD45-positive macrophages from either Ctr or Hpa-tg sections; bottom Significantly fewer CD45-positive macrophages in Hpa-tg whole-brain tissue sections than in Ctr sections. k Western blotting of IL-1β in Hpa-tg and Ctr brain. Densitometry analysis of the bands demonstrated a significantly higher up-regulation of IL-1β in Ctr brains than in Hpa-tg brains following LPS challenge
Delayed clearance of injected Aβ42 and poor recruitment of immune cells in Hpa-tg brain
We also assessed the impact of heparanase overexpression on locally induced neuroinflammation elicited by intracortical injection of aggregated synthetic human Aβ42 (Fig. 3a). Groups of 6–7 Ctr and Hpa-tg mice were killed after 1, 2, and 4 weeks post-injection and brain sections were immunostained using the anti-Aβ antibody 6E10. The newly injected material appeared as compact, 6E10 (Fig. 3b) and Congo red (not shown) positive deposits that were dispersed (Fig. 3c) and ultimately cleared (Fig. 3d) over time. Clearance was markedly delayed in Hpa-tg compared to Ctr mice. Compact residual aggregates thus remained at the injection sites in 10 out of 20 Hpa-tg mice at time points when none were found in any of 18 Ctr mice (Fig. 3e).
Fig. 3.
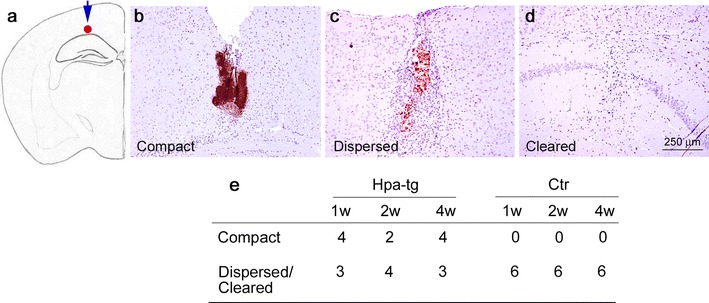
Aβ deposits following intracortical injection of aggregated synthetic human Aβ42 in Ctr and Hpa-tg mice. a Aβ42 injection site. b Representative compact deposit at the injection site from a Hpa-tg mouse. c Dispersed deposit, with scattered 6E10-positive aggregates. d Cleared deposit. e Number of Hpa-tg and Ctr brains presenting with each deposit state in groups killed after 1, 2, and 4 weeks
To elucidate the reason for the impaired elimination of Aβ aggregates in the Hpa-tg brain, we examined injection sites for reactive cells using anti-CD45 antibody for detection of activated macrophages and anti-GFAP antibody for astrocytes. The dispersed Aβ deposits, found in Ctr mice, were heavily infiltrated by CD45-positive macrophages (Fig. 4a), and surrounded by GFAP-positive astrocytes (Fig. 4b), in contrast to the compact deposits in Hpa-tg mice that showed only scant recruitment of cells (Fig. 4c, d). Notably, dispersed deposits were also surrounded by fewer reactive cells in Hpa-tg than in Ctr mice (Supplementary Fig. 2). CD45-positive cells with amoeboid morphology were observed (4e-g), indicative of monocyte origin, similar to those detected after the LPS-induced inflammatory response. Confocal z-scan demonstrated internalization of the injected Aβ42 by CD45-positive macrophages, confirming their phagocytic phenotype (Fig. 4h). These data associate delayed degradation of the injected Aβ with an impaired inflammatory response.
Fig. 4.
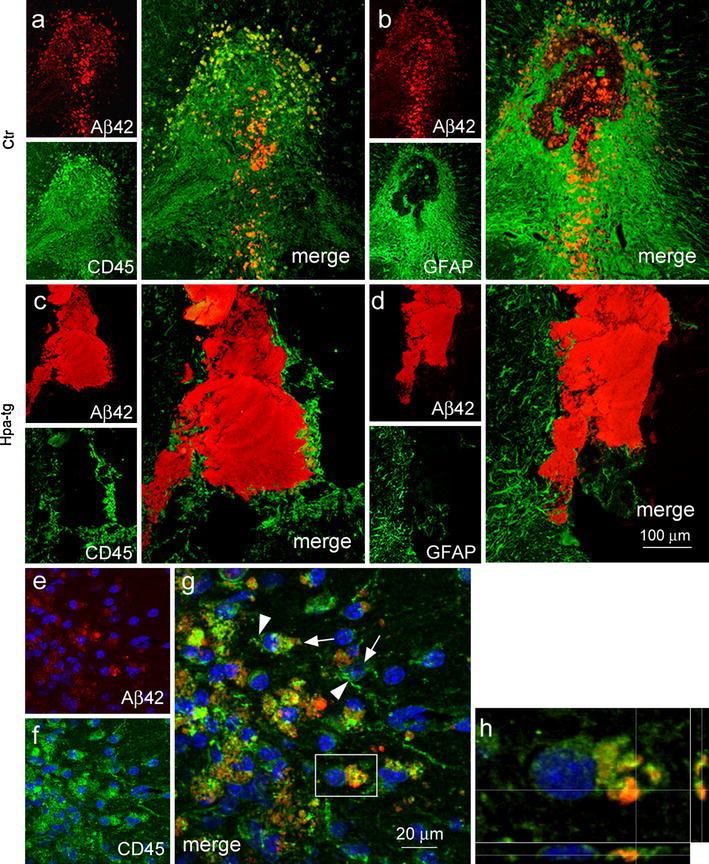
Cellular response to Aβ injection. Adjacent brain sections from mice, 2 weeks after Aβ42 injection were double immunostained with anti-Aβ42 for Aβ42 and anti-CD45 for macrophages or anti-GFAP for astrocytes. a, b Ctr mouse. Anti-Aβ42 stained dispersed Aβ deposits (a, b red), which were heavily infiltrated by CD45-positive macrophages (a, green), and surrounded by GFAP-positive astrocytes (b green). c, d Hpa-tg mouse. Anti-Aβ42 stained compact Aβ deposits (c, d red) with scant recruitment of CD45-positive macrophages (c green) and GFAP-positive astrocytes (d green). Confocal laser scanning microscopy of Aβ42 (e, red) and CD45 (f, green) immunostaining detects cells with amoeboid morphology (g, arrow) and few short processes (g, arrowhead). Aβ was engulfed by several CD45-positive cells. h Confocal microscopy of phagocytosed Aβ in a CD45-positive phagocyte indentified by the frame in g
Degradation of Aβ has been ascribed to proteases expressed by neurons, glial cells, macrophages and cerebral vasculature, in particular neprilysin (NEP) and matrix metalloproteinase-9 (MMP-9) [34]. To assess the impact of these enzymes on the degradation of injected Aβ, we examined the expression of NEP and MMP-9 in brain sections of a Ctr mouse that showed some remaining, dispersed Aβ (6E10-positive material, indicated by arrows in Fig. 5a; insert) and a Hpa-tg mouse retaining compact Aβ deposits (Fig. 5d). Immunosignals of NEP and MMP-9 were restricted to the injection sites, confined to smaller areas in Hpa-tg (Fig. 5e, f) than in Ctr brain (Fig. 5b, c). Double immunostaining of Ctr and Hpa-tg sections revealed expression of NEP mainly in CD45-positive macrophages, whereas MMP-9 appeared primarily associated with astrocytes (Supplementary Fig. 3). In accordance with these data, Ctr brain showed significantly higher NEP and MMP-9 protein levels than Hpa-tg brain, following systemic LPS challenge (Supplementary Fig. 1h, i). The lower expression of these enzymes in the Hpa-tg mouse brain thus correlates with a relative lack of activated macrophages and astrocytes. These data confirm that the delayed Aβ clearance in the heparanase-overexpressing brain is caused by impaired recruitment of inflammatory cells that degrade Aβ aggregates.
Fig. 5.
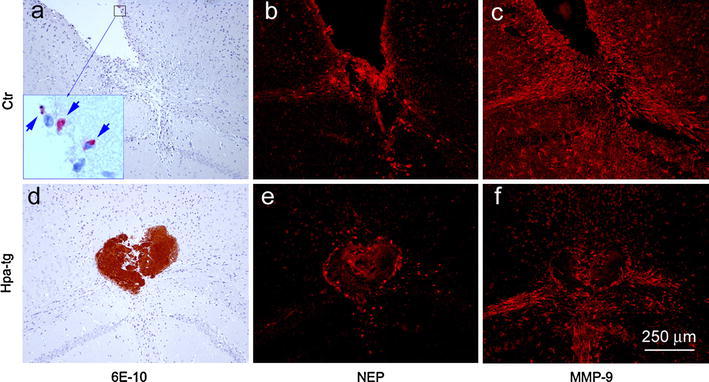
Expression of proteolytic enzymes following intracortical Aβ42 injection. Adjacent brain sections from a Ctr mouse and a Hpa-tg mouse 4 weeks after intracortical injection of Aβ42 were immunostained with the anti-Aβ antibody 6E10 (red in a, indicated by arrows in insert, and red in d), anti-NEP antibody (b, e) and anti-MMP-9 antibody (c, f), respectively. Immunosignals of NEP and MMP-9 were restricted to the injection sites, confined to smaller areas in Hpa-tg (e, f) than in Ctr brain (b, c)
Reduced expression of CCL2 and ICAM-1 in Hpa-tg brain
The above results demonstrated impaired infiltration and activation of immune cells in the Hpa-tg brain. Extravasation of leukocytes from blood to affected tissues in inflammation depends on leukocyte/endothelial cell recognition, which involves soluble chemotactic factors as well as receptors and ligands on cell surfaces [4]. Expression of the chemotactic protein CCL2 was examined in brain sections of a Ctr mouse with some residual, dispersed Aβ (Fig. 6a; insert) and a Hpa-tg mouse retaining compact Aβ deposits (Fig. 6b; insert). CCL2 immunosignals were restricted to the injection site and were much more pronounced in Ctr than in Hpa-tg brain (Fig. 6a, b). CCL2 appeared predominantly associated with CD45-positive macrophages in both Ctr and Hpa-tg animals (Supplementary Fig. 4).
Fig. 6.
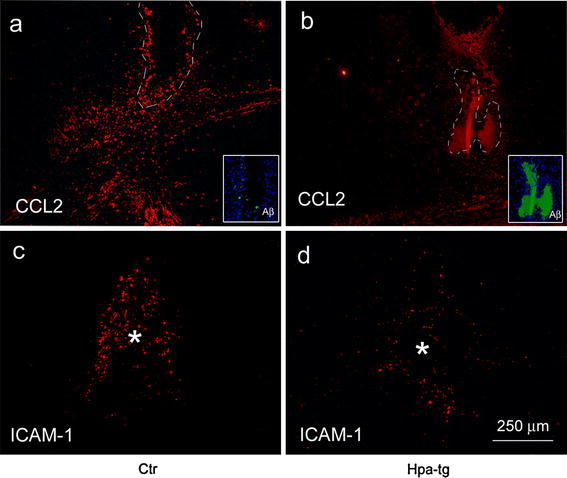
Expression of CCL2 and ICAM-1 at the intracortical Aβ42 injection site. a, b Brain sections from a Ctr mouse 4 weeks and a Hpa-tg mouse 2 weeks after Aβ42 injection were immunostained with anti-CCL2 antibody. The Ctr mouse showed pronounced cell-associated CCL2 immunosignals close to the injection site (a dispersed deposit) compared with scant signals in a Hpa-tg brain section (b compact deposit). The sites of the initial deposits are identified by dashed lines; the inserts show residual Aβ deposits (green) detected by the anti-Aβ antibody 6E10. The red signal surrounded by the dashed line in b is not associated with cells and is likely due to nonspecific fluorescence. c, d Brain tissue sections from a Ctr mouse as shown in Fig. 3c and a Hpa-tg mouse as shown in Fig. 5d were immunostained with anti-ICAM-1 antibody. ICAM-1 immunosignals restricted to the injection site (indicated by asterisk) were pronounced in Ctr brain (c) but scarce in Hpa-tg brain (d)
The transmembrane protein ICAM-1 (intercellular adhesion molecule 1, also known as CD54) is constitutively expressed at low levels on the cell surface of various cells types including endothelial cells and neutrophils, but dramatically upregulated in response to cytokines and LPS [8, 29, 30, 36, 47, 61]. We assessed ICAM-1 expression in brain tissue sections of LPS-challenged mice by immunostaining with anti-ICAM-1 antibody. Ctr brain, with upregulated IL-1β generation (Fig. 2k), showed ICAM-1-positive cells in the parenchyma (Fig. 7a). By contrast, Hpa-tg brain, with attenuated IL-1β response to LPS (Fig. 2k), was essentially devoid of ICAM-1-positive cells in the parenchyma (Fig. 7b). Double immunostaining with anti-CD31 antibody for blood vessel and ICAM-1 antibody showed ICAM-positive cells associated with microvasculature in Ctr parenchyma (Fig. 7g–h, k–l), but few such cells were found associated with Hpa-tg microvasculature (Fig. 7i–j, m–n). Double immunostaining with anti-CD45 and anti-ICAM-1 antibodies revealed that the ICAM-1-positive cells in the parenchyma (Fig. 7c–f) and the ICAM-1 positive cells associated with blood vessel walls (Fig. 7o–r) were also CD45-positive; hence the parenchymal cells are likely derived from blood-borne monocytes. These data are consistent with the view that ICAM-1 expression remains low until monocytes are in the process of leaving the circulation [37]. We did not detect clear upregulation of ICAM-1 on endothelial cells. It has been reported that ICAM-1 expression on monocytes mediates trans-endothelial migration [54]; thus the inhibited expression of ICAM-1 may be attributed to the impaired recruitment of CD45-positive macrophages in Hpa-tg mouse. CD45-associated ICAM-1 expression was also observed in brain sections of mice that had received intracortical Aβ42 injection (Supplementary Fig. 5) with more pronounced ICAM-1 immunosignals around the injection site in Ctr mice (Fig. 6c) than in Hpa-tg mice (Fig. 6d).
Fig. 7.
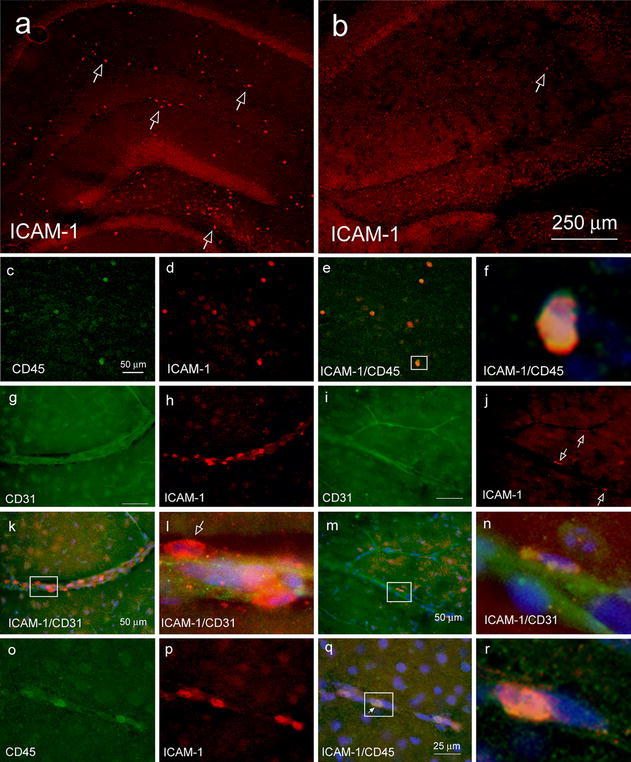
Expression of ICAM-1 by CD45-positive macrophages in brains of mice 20 h after intraperitoneal LPS challenge. a–f Parenchymal CD45-positive macrophages expressing ICAM-1. Brain tissue sections of a Ctr and a Hpa-tg mouse challenged by LPS were immunostained with anti-ICAM-1 (a–b) or double immunostained with anti-ICAM-1/CD45 antibodies (c–f). a Abundant cell-associated ICAM-1 immunosignals in the hippocampus of the Ctr mouse (some of the cells are indicated by arrows), b scant ICAM-1-positive cells in the hippocampus of the Hpa-tg mouse. Note: Similar nonspecific background fluorescence from the CA regions and dentate gyrus. c amoeboid CD45-positive macrophages, d ICAM-1 immunosignals, e overlay of ICAM-1 immunosignals with CD45-positive macrophages. f Enlarged image of a CD45-positive macrophage expressing ICAM-1 as framed in e. g–n Expression of ICAM-1 in parenchymal microvasculature. Brain tissue sections of the same animals as above were double immunostained with anti-CD31/anti-ICAM-1 antibodies. g, i Microvasculature stained by CD31 in Ctr and Hpa-tg mouse, respectively. h ICAM-1 immunosignals in Ctr mouse microvasculature. j ICAM-1 immunosignals in Hpa-tg mouse microvasculature (arrows point ICAM-1 immunosignals). k Abundant ICAM-1-positive cells associated with a micro-blood vessel in Ctr brain. m In contrast, a Hpa-tg micro-blood vessel is almost devoid of ICAM-1-positive cells. l, n Enlarged image of the frame in k and m, respectively. An arrow in l points to an ICAM-1-positive cell associated with the parenchymal abluminal surface of the blood vessel. o–r Expression of ICAM-1 by blood-borne macrophages associated with microvasculature. o CD45-positive blood-borne monocytes/macrophages. p ICAM-1 immunosignals. q Overlay of ICAM-1 immunosignals with CD45-positive monocyte/macrophages. Arrow indicates expression of ICAM-1 by a CD45-positive cell still within the blood vessel. r Enlarged image of a CD45 positive macrophage expressing ICAM-1 as framed in q
Impaired transmigration of blood-borne monocytes across an in vitro model of the Hpa-tg blood–brain barrier
Endothelial HS is essential for leukocyte trafficking, as demonstrated by impaired neutrophil infiltration in various inflammation models due to deficient HS sulfation [60]. We therefore examined whether the scant recruitment of CD45-positive cells into the brain of Aβ-injected Hpa-tg mice could be caused by derangement of HS chains on cerebral microvascular endothelial cells. To this end, we prepared primary cerebral endothelial cells and pericytes from Hpa-tg and Ctr mice (Supplementary Fig. 6a-c) and assembled an in vitro BBB model to study monocyte transmigration (Fig. 8a). Both endothelial cells and pericytes from Hpa-tg mice showed distinct overexpression of heparanase (Fig. 8b–e). Similar to the HS isolated from whole brain (Fig. 1c), the Hpa-tg endothelial HS was truncated compared to Ctr HS chains (Fig. 8f). Monocytes, isolated from Ctr mice expressing green fluorescent protein (GFP) were added to the apical compartment of the transmigration device, and their migration across the cellular barrier was determined in response to CCL2 added to the basolateral compartment (Fig. 8a). The number of GFP monocytes settling in the basolateral chamber was significantly reduced in the Hpa-tg compared to the Ctr BBB model (Fig. 8g; Supplementary Fig. 7). Control experiments performed without addition of CCL2 showed minimal numbers of monocytes crossing the insert (data not shown), proving the active role of the chemoattractant in the transmigration event. These data demonstrate that perturbation of endothelial HS by heparanase significantly impedes transmigration of blood-borne monocytes across the BBB.
Fig. 8.
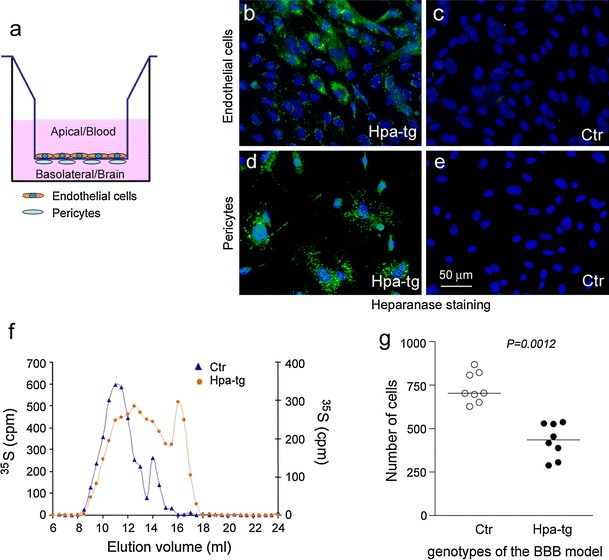
Monocyte transmigration assay using an in vitro BBB model. a Diagram of the BBB model assembled in cell-culture transwell inserts with primary endothelial cells (apical site) and pericytes (basolateral site) isolated from mouse brains. Heparanase overexpression in Hpa-tg endothelial cells (b) and pericytes (d) compared with Ctr cells (c, e) was verified by immunostaining with anti-heparanase antibody 733 (green). f Gel chromatography (Superose 12) showed a pronounced reduction in size of metabolically 35S-labeled HS free chains purified from Hpa-tg primary endothelial cells compared with HS from Ctr endothelial cells. g Number of monocytes transmigrated from the apical to the basolateral compartment containing CCL2 (50 ng/ml)
Discussion
Proliferation and recruitment of activated immune cells in the brain parenchyma, including macrophages derived from microglia or from blood-borne monocytes, is a hallmark of neuroinflammation. The inflammatory state may serve to eliminate pathogens, dead cells and tissue debris after various acute assaults, but are also conspicuous in several neurodegenerative diseases, including AD [15, 35, 46]. An inflammatory stimulus typically induces cytokines such as IL-1β or TNF-α, which in turn activates a variety of cells (endothelial cells, microglia/macrophages, astrocytes) to release cell-attractant chemokines such as CCL2 [6, 23], and up-regulate expression of cell adhesion molecules such as ICAM-1 [29].
Previous studies have implicated endothelial HS in various inflammation models [60]. HS may function as a ligand for leukocyte L-selectin, as a carrier in chemokine transcytosis, and as a scaffold for presenting chemokines at the luminal surface of the endothelium. In the present work, we have specifically studied the various roles of HS in neuroinflammation by employing Hpa-tg mice that overexpress heparanase in multiple regions of the CNS, thus having HSPGs with truncated HS side chains (Fig. 1). Two inflammation models were assessed, one involving systemic LPS challenge, the other a local microinjection of fibrillar Aβ42. Ctr mice subjected to intraperitoneal LPS injection showed increased levels of cerebral IL-1β (Fig. 2k) and an abundance of monocytes adhering to and migrating across the wall of cerebral microvasculature (Fig. 2a–g), where they differentiated into CD45/ICAM-1 positive macrophages (Fig. 7). Moreover, there was massive activation of microglia to macrophage-like state as shown by up-regulated CD45 expression and ramified morphology (Fig. 2h–j). All of these effects were significantly attenuated in Hpa-tg mice (shown in the corresponding figures).
The effects of injected fibrillar Aβ, as expected, were essentially confined to brain areas in close vicinity to the protein deposition. Ctr mice showed massive infiltration of activated immune cells, including macrophages (presumably derived from both resident microglia and blood-borne monocytes) and astrocytes (Fig. 4). These CD45-positive macrophages expressed CCL2 (Supplementary Fig. 4), a chemoattractant with a central role in the recruitment of blood-borne monocytes to sites of tissue injury. Expression of the proteolytic enzymes, NEP and MMP-9 (Fig. 5; Supplementary Fig. 3), together with phagocytosis of Aβ by macrophages (Fig. 4e–h) presumably contributed to efficient dispersion of the Aβ deposits following intracortical injection (Fig. 4). Again, heparanase expression strongly attenuated all of these responses to challenge.
A schematic overview of relevant steps of the neuroinflammatory process is provided in Fig. 9a, along with the consequences of heparanase overexpression in Fig. 9b. Such effects, presumably due to truncation of HS chains, are discerned at several stages. Interaction of endothelial HSPGs with L-selectin contributes to the initial stages of monocyte tethering to the vascular endothelium [60]. HS chains of sufficient length are required for presentation of chemokines to monocyte receptors to arrest the cells, and ensure firm adhesion of the cells to the endothelium through interaction of ICAM-1 on the endothelium with ICAM-1 receptors on monocytes, a key step in diapedesis. Moreover, HSPGs serve as carriers of chemokines in transcytosis from the parenchymal abluminal to the luminal aspect of the vascular endothelium [40]. Activation of resident microglia may be induced by pro-inflammatory molecules [21]), but also through direct interaction between Aβ and cell-surface HSPG [17]. Notably, LPS-induced upregulation of TNF-α and IL-1β was inhibited in Hpa-tg primary microglia compared with Ctr cells (our unpublished data). The poor response of Hpa-tg mice to either systemic LPS challenge or local Aβ injection, with scant recruitment of activated macrophages from blood-borne monocytes and resident microglia is thus due to failing HSPG support. Our data implicate heparanase as a potential key modulator of neuroinflammation.
Fig. 9.
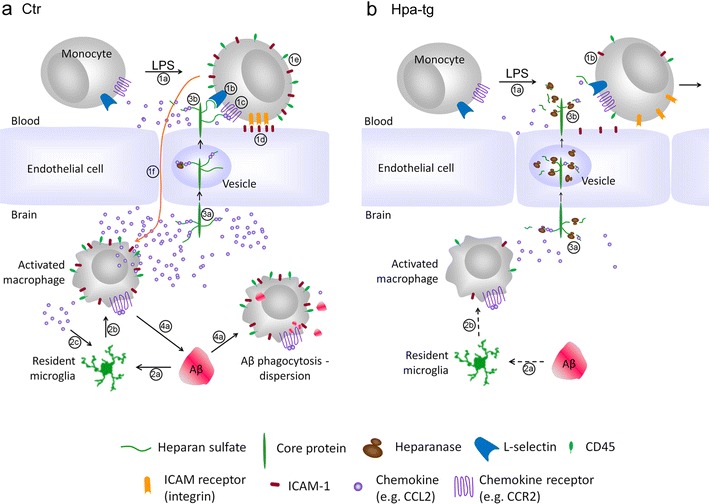
Schematic illustration of immune cell recruitment in response to LPS challenge and to microinjection of aggregated Aβ, illustrating the proposed inhibitory effect of heparanase. a Ctr: 1a–1f transmigration of blood-borne monocytes across the endothelial barrier in response to systemic LPS challenge. 1a LPS challenge; 1b monocyte tethering through interactions of cell-surface L-selectin with HS for cell rolling; 1c monocyte activation by HS-bound chemokine oligomers (e.g. CCL2) binding to specific receptors (CCR2); 1d monocyte adhesion to endothelium through interaction of endothelial ICAM-1 with monocyte ICAM-1 receptors; 1e monocytes upregulate CD45 and ICAM-1; 1f monocytes transmigrate through the endothelial barrier into the brain, where they differentiate into activated macrophages and release cytokines (e.g. IL-1β) and chemokines (e.g. CCL2). 2a–2c Activation of resident microglia in response to Aβ injection: 2a Aβ deposit stimulates differentiation of resident microglia (2b) into activated macrophages; 2c Cytokines released by the macrophages promote further activation and differentiation of resident microglia. Similar transition may be elicited by cytokines/chemokines released due to LPS stimulation. 3a–3b Chemokines bind to HS chains of HSPGs at the abluminal surface of the endothelium, and are relayed by transcytosis to the luminal surface where they are presented to their receptors on tethered monocytes. 4a Activated macrophages, presumably derived mainly from blood-borne monocytes, disperse the Aβ deposits through phagocytosis and proteolytic degradation. b Hpa-tg: Contrary to Ctr mice, Hpa-tg mice showed reduced expression of (1b) CD45 and ICAM-1 in response to LPS challenge (1a), as well as reduced response of resident microglia to Aβ injection (dashed arrows 2a, 2b). 3a–3b Fragmentation of HS chains in the Hpa-tg brain downregulates binding of chemokines to HSPGs. These monocytes fail to attach and transmigrate, resulting in impaired Aβ clearance
The critical role for endothelial HS in trans-BBB diapedesis, suggested by the attenuating effects of heparanase, was supported by experiments involving an in vitro model of monocyte transmigration across a BBB using primary cerebral vascular cells. Cells derived from Hpa-tg mice overexpressed heparanase (Fig. 8b–e) and displayed truncated HS chains compared to Ctr cells (Fig. 8f). The passage of monocytes through the Hpa-tg BBB in response to CCL2 was significantly reduced relative to the Ctr BBB. Notably, chemokines, including CCL2, bind to HS as oligomers [26, 43] that occupy extended domains of the saccharide chain. For example, a dimer of the chemokine IL-8 requires a minimal sequence of 18 monosaccharide units for interaction [53]. The marked susceptibility of CCL2 function to HS cleavage is thus readily explained.
AD is a chronic disease, with Aβ deposition as a hallmark in the brain that is not simply modeled by microinjection of aggregated Aβ. Nevertheless, the effects of cerebral heparanase have potential bearing on various aspects of the disease. HS binds Aβ, supports fibril formation, and is typically part of Aβ deposits [39, 59]. Cell-surface HSPGs have been implicated in Aβ uptake and cytotoxicity [17, 28, 48]. These effects are, by and large, associated with disease pathogenesis, whereas our present findings expose HS also as potentially beneficial, contributing to recruitment and activation of immune cells that eliminate Aβ deposits. The HS/heparanase system is likely of key importance in the inflammatory response to AD. Endogenous heparanase is detected in the vasculature of the AD brain and of the Tg-2576 mouse model of Aβ deposition (Supplementary Fig. 8), thus it is present at the BBB and potentially capable of interfering with transmigration of phagocytes into the injured brain. In AD and the transgenic mouse models of AD, blood-derived macrophages associate with Aβ deposits [13, 31]. Such macrophages, recruited in response to cerebral injection of pre-aggregated Aβ, are thought to be better Aβ phagocytes than brain resident immune cells [10, 12, 24, 51], and poor infiltration of blood-borne macrophages may therefore contribute to Aβ accumulation [57]. Overexpressing heparanase in the brain of an AβPP-transgenic mouse by cross-breeding with a Hpa-tg mouse may clarify the relative roles of HS and heparanase in the Aβ pathology of AD.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by grants from the Swedish Research Council: K2009-67X-21128-01-3, K2012-67X-21128-04-4, K2010-61X-15383-06-3, #2009-4567, Swedish Cancer Foundation, Swedish Heart–Lung Foundation (20110131), The European Commission (EURAMY), the Mizutani Foundation for Glycoscience, Polysackaridforskning AB (Uppsala), Alzheimerfonden, Stohnes Stiftelse, Stiftelsen för Gamla Tjänarinnor, Demensförbundet, The Swedish Brain Foundation, Bertil Hållstens Forskningsstiftelse. The Israel Science Foundation (549/06), the National Cancer Institute, NIH (RO1-CA106456). I. Vlodavsky is a Research Professor of the ICRF.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Footnotes
B. Wang and P. O’Callaghan contributed equally to this work.
References
- 1.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21(3):383–421. doi: 10.1016/S0197-4580(00)00124-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Austyn JM, Gordon S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol. 1981;11(10):805–815. doi: 10.1002/eji.1830111013. [DOI] [PubMed] [Google Scholar]
- 3.Bates KA, Verdile G, Li QX, Ames D, Hudson P, Masters CL, Martins RN. Clearance mechanisms of Alzheimer’s amyloid-beta peptide: implications for therapeutic design and diagnostic tests. Mol Psychiatry. 2009;14(5):469–486. doi: 10.1038/mp.2008.96. [DOI] [PubMed] [Google Scholar]
- 4.Butcher EC. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 1991;67(6):1033–1036. doi: 10.1016/0092-8674(91)90279-8. [DOI] [PubMed] [Google Scholar]
- 5.Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunol Rev. 2006;213:48–65. doi: 10.1111/j.1600-065X.2006.00441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Haas AH, van Weering HR, de Jong EK, Boddeke HW, Biber KP. Neuronal chemokines: versatile messengers in central nervous system cell interaction. Mol Neurobiol. 2007;36(2):137–151. doi: 10.1007/s12035-007-0036-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Vries HE, Kuiper J, de Boer AG, Van Berkel TJ, Breimer DD. The blood-brain barrier in neuroinflammatory diseases. Pharmacol Rev. 1997;49(2):143–155. [PubMed] [Google Scholar]
- 8.Dustin ML, Rothlein R, Bhan AK, Dinarello CA, Springer TA. Induction by IL 1 and interferon-gamma: tissue distribution, biochemistry, and function of a natural adherence molecule (ICAM-1) J Immunol. 1986;137(1):245–254. [PubMed] [Google Scholar]
- 9.El Khoury J, Luster AD. Mechanisms of microglia accumulation in Alzheimer’s disease: therapeutic implications. Trends Pharmacol Sci. 2008;29(12):626–632. doi: 10.1016/j.tips.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 10.El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13(4):432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- 11.Escobar Galvis ML, Jia J, Zhang X, Jastrebova N, Spillmann D, Gottfridsson E, van Kuppevelt TH, Zcharia E, Vlodavsky I, Lindahl U, Li JP. Transgenic or tumor-induced expression of heparanase upregulates sulfation of heparan sulfate. Nat Chem Biol. 2007;3(12):773–778. doi: 10.1038/nchembio.2007.41. [DOI] [PubMed] [Google Scholar]
- 12.Fiala M, Cribbs DH, Rosenthal M, Bernard G. Phagocytosis of amyloid-beta and inflammation: two faces of innate immunity in Alzheimer’s disease. J Alzheimers Dis. 2007;11(4):457–463. doi: 10.3233/jad-2007-11406. [DOI] [PubMed] [Google Scholar]
- 13.Fiala M, Liu QN, Sayre J, Pop V, Brahmandam V, Graves MC, Vinters HV. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer’s disease brain and damage the blood-brain barrier. Eur J Clin Invest. 2002;32(5):360–371. doi: 10.1046/j.1365-2362.2002.00994.x. [DOI] [PubMed] [Google Scholar]
- 14.Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J Immunol. 1995;154(9):4309–4321. [PubMed] [Google Scholar]
- 15.Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol Neurodegener. 2009;4:47. doi: 10.1186/1750-1326-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gate D, Rezai-Zadeh K, Jodry D, Rentsendorj A, Town T. Macrophages in Alzheimer’s disease: the blood-borne identity. J Neural Transm. 2010;117(8):961–970. doi: 10.1007/s00702-010-0422-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giulian D, Haverkamp LJ, Yu J, Karshin W, Tom D, Li J, Kazanskaia A, Kirkpatrick J, Roher AE. The HHQK domain of beta-amyloid provides a structural basis for the immunopathology of Alzheimer’s disease. J Biol Chem. 1998;273(45):29719–29726. doi: 10.1074/jbc.273.45.29719. [DOI] [PubMed] [Google Scholar]
- 18.Goldshmidt O, Zcharia E, Aingorn H, Guatta-Rangini Z, Atzmon R, Michal I, Pecker I, Mitrani E, Vlodavsky I. Expression pattern and secretion of human and chicken heparanase are determined by their signal peptide sequence. J Biol Chem. 2001;276(31):29178–29187. doi: 10.1074/jbc.M102462200. [DOI] [PubMed] [Google Scholar]
- 19.Guillemin GJ, Brew BJ. Microglia, macrophages, perivascular macrophages, and pericytes: a review of function and identification. J Leukoc Biol. 2004;75(3):388–397. doi: 10.1189/jlb.0303114. [DOI] [PubMed] [Google Scholar]
- 20.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8(2):101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 21.Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40(2):140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- 22.Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun. 2009;23(3):309–317. doi: 10.1016/j.bbi.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hesselgesser J, Horuk R. Chemokine and chemokine receptor expression in the central nervous system. J Neurovirol. 1999;5(1):13–26. doi: 10.3109/13550289909029741. [DOI] [PubMed] [Google Scholar]
- 24.Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci. 2008;28(33):8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hickman SE, El Khoury J. Mechanisms of mononuclear phagocyte recruitment in Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2010;9(2):168–173. doi: 10.2174/187152710791011982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoogewerf AJ, Kuschert GS, Proudfoot AE, Borlat F, Clark-Lewis I, Power CA, Wells TN. Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry. 1997;36(44):13570–13578. doi: 10.1021/bi971125s. [DOI] [PubMed] [Google Scholar]
- 27.Johnston B, Butcher EC. Chemokines in rapid leukocyte adhesion triggering and migration. Semin Immunol. 2002;14(2):83–92. doi: 10.1006/smim.2001.0345. [DOI] [PubMed] [Google Scholar]
- 28.Kanekiyo T, Zhang J, Liu Q, Liu CC, Zhang L, Bu G. Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-beta uptake. J Neurosci. 2011;31(5):1644–1651. doi: 10.1523/JNEUROSCI.5491-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Long EO. ICAM-1: getting a grip on leukocyte adhesion. J Immunol. 2011;186(9):5021–5023. doi: 10.4049/jimmunol.1100646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maio M, Tessitori G, Pinto A, Temponi M, Colombatti A, Ferrone S. Differential role of distinct determinants of intercellular adhesion molecule-1 in immunologic phenomena. J Immunol. 1989;143(1):181–188. [PubMed] [Google Scholar]
- 31.Malm TM, Koistinaho M, Parepalo M, Vatanen T, Ooka A, Karlsson S, Koistinaho J. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. 2005;18(1):134–142. doi: 10.1016/j.nbd.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 32.Martins IC, Kuperstein I, Wilkinson H, Maes E, Vanbrabant M, Jonckheere W, Van Gelder P, Hartmann D, D’Hooge R, De Strooper B, Schymkowitz J, Rousseau F. Lipids revert inert Abeta amyloid fibrils to neurotoxic protofibrils that affect learning in mice. EMBO J. 2008;27(1):224–233. doi: 10.1038/sj.emboj.7601953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Massena S, Christoffersson G, Hjertstrom E, Zcharia E, Vlodavsky I, Ausmees N, Rolny C, Li JP, Phillipson M. A chemotactic gradient sequestered on endothelial heparan sulfate induces directional intraluminal crawling of neutrophils. Blood. 2010;116(11):1924–1931. doi: 10.1182/blood-2010-01-266072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S. Abeta-degrading enzymes in Alzheimer’s disease. Brain Pathol. 2008;18(2):240–252. doi: 10.1111/j.1750-3639.2008.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Minghetti L. Role of inflammation in neurodegenerative diseases. Curr Opin Neurol. 2005;18(3):315–321. doi: 10.1097/01.wco.0000169752.54191.97. [DOI] [PubMed] [Google Scholar]
- 36.Morise Z, Eppihimer M, Granger DN, Anderson DC, Grisham MB. Effects of lipopolysaccharide on endothelial cell adhesion molecule expression in interleukin-10 deficient mice. Inflammation. 1999;23(2):99–110. doi: 10.1023/A:1020232826906. [DOI] [PubMed] [Google Scholar]
- 37.Most J, Neumayer HP, Dierich MP. Cytokine-induced generation of multinucleated giant cells in vitro requires interferon-gamma and expression of LFA-1. Eur J Immunol. 1990;20(8):1661–1667. doi: 10.1002/eji.1830200807. [DOI] [PubMed] [Google Scholar]
- 38.Nakanishi H. Microglial functions and proteases. Mol Neurobiol. 2003;27(2):163–176. doi: 10.1385/MN:27:2:163. [DOI] [PubMed] [Google Scholar]
- 39.O’Callaghan P, Sandwall E, Li JP, Yu H, Ravid R, Guan ZZ, van Kuppevelt TH, Nilsson LN, Ingelsson M, Hyman BT, Kalimo H, Lindahl U, Lannfelt L, Zhang X. Heparan sulfate accumulation with Abeta deposits in Alzheimer’s disease and Tg2576 mice is contributed by glial cells. Brain Pathol. 2008;18(4):548–561. doi: 10.1111/j.1750-3639.2008.00152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parish CR. Heparan sulfate and inflammation. Nat Immunol. 2005;6(9):861–862. doi: 10.1038/ni0905-861. [DOI] [PubMed] [Google Scholar]
- 41.Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol. 2007;7(2):161–167. doi: 10.1038/nri2015. [DOI] [PubMed] [Google Scholar]
- 42.Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, Weiner HL. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-alpha-PU.1 pathway. Nat Med. 2011;17(1):64–70. doi: 10.1038/nm.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Proudfoot AE, Handel TM, Johnson Z, Lau EK, LiWang P, Clark-Lewis I, Borlat F, Wells TN, Kosco-Vilbois MH. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci USA. 2003;100(4):1885–1890. doi: 10.1073/pnas.0334864100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55(5):453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rezai-Zadeh K, Gate D, Town T. CNS infiltration of peripheral immune cells: D-Day for neurodegenerative disease? J Neuroimmune Pharmacol. 2009;4(4):462–475. doi: 10.1007/s11481-009-9166-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rogers J, Mastroeni D, Leonard B, Joyce J, Grover A. Neuroinflammation in Alzheimer’s disease and Parkinson’s disease: are microglia pathogenic in either disorder? Int Rev Neurobiol. 2007;82:235–246. doi: 10.1016/S0074-7742(07)82012-5. [DOI] [PubMed] [Google Scholar]
- 47.Rummel C, Inoue W, Poole S, Luheshi GN. Leptin regulates leukocyte recruitment into the brain following systemic LPS-induced inflammation. Mol Psychiatry. 2010;15(5):523–534. doi: 10.1038/mp.2009.98. [DOI] [PubMed] [Google Scholar]
- 48.Sandwall E, O’Callaghan P, Zhang X, Lindahl U, Lannfelt L, Li JP. Heparan sulfate mediates amyloid-beta internalization and cytotoxicity. Glycobiology. 2010;20(5):533–541. doi: 10.1093/glycob/cwp205. [DOI] [PubMed] [Google Scholar]
- 49.Sedgwick JD, Schwender S, Imrich H, Dorries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci USA. 1991;88(16):7438–7442. doi: 10.1073/pnas.88.16.7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shafat I, Zcharia E, Nisman B, Nadir Y, Nakhoul F, Vlodavsky I, Ilan N. An ELISA method for the detection and quantification of human heparanase. Biochem Biophys Res Commun. 2006;341(4):958–963. doi: 10.1016/j.bbrc.2006.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron. 2006;49(4):489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 52.Solito E, Sastre M. Microglia function in Alzheimer’s disease. Front Pharmacol. 2012;3:14. doi: 10.3389/fphar.2012.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Spillmann D, Witt D, Lindahl U. Defining the interleukin-8-binding domain of heparan sulfate. J Biol Chem. 1998;273(25):15487–15493. doi: 10.1074/jbc.273.25.15487. [DOI] [PubMed] [Google Scholar]
- 54.Steidl U, Haas R, Kronenwett R. Intercellular adhesion molecular 1 on monocytes mediates adhesion as well as trans-endothelial migration and can be downregulated using antisense oligonucleotides. Ann Hematol. 2000;79(8):414–423. doi: 10.1007/s002770000168. [DOI] [PubMed] [Google Scholar]
- 55.Thompson WL, Karpus WJ, Van Eldik LJ. MCP-1-deficient mice show reduced neuroinflammatory responses and increased peripheral inflammatory responses to peripheral endotoxin insult. J Neuroinflamm. 2008;5:35. doi: 10.1186/1742-2094-5-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tonks NK, Charbonneau H, Diltz CD, Fischer EH, Walsh KA. Demonstration that the leukocyte common antigen CD45 is a protein tyrosine phosphatase. Biochemistry. 1988;27(24):8695–8701. doi: 10.1021/bi00424a001. [DOI] [PubMed] [Google Scholar]
- 57.Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14(6):681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. J Neuroinflamm. 2005;2:24. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Horssen J, Wesseling P, van den Heuvel LP, de Waal RM, Verbeek MM. Heparan sulphate proteoglycans in Alzheimer’s disease and amyloid-related disorders. Lancet Neurol. 2003;2(8):482–492. doi: 10.1016/S1474-4422(03)00484-8. [DOI] [PubMed] [Google Scholar]
- 60.Wang L, Fuster M, Sriramarao P, Esko JD. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol. 2005;6(9):902–910. doi: 10.1038/ni1233. [DOI] [PubMed] [Google Scholar]
- 61.Wong D, Dorovini-Zis K. Upregulation of intercellular adhesion molecule-1 (ICAM-1) expression in primary cultures of human brain microvessel endothelial cells by cytokines and lipopolysaccharide. J Neuroimmunol. 1992;39(1–2):11–21. doi: 10.1016/0165-5728(92)90170-P. [DOI] [PubMed] [Google Scholar]
- 62.Zcharia E, Metzger S, Chajek-Shaul T, Aingorn H, Elkin M, Friedmann Y, Weinstein T, Li JP, Lindahl U, Vlodavsky I. Transgenic expression of mammalian heparanase uncovers physiological functions of heparan sulfate in tissue morphogenesis, vascularization, and feeding behavior. FASEB J. 2004;18(2):252–263. doi: 10.1096/fj.03-0572com. [DOI] [PubMed] [Google Scholar]
- 63.Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics. 2010;7(1):22–30. doi: 10.1016/j.nurt.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.