

Abstract
Anti-tumour necrosis factor (TNF) biologics have revolutionized therapy of rheumatoid arthritis (RA). We compared the effects of infliximab on numbers of circulating leucocyte subsets in early RA (disease/symptom duration of ≤1 year) and late RA patients (>1 year). A control group consisted of early RA patients treated with a combination of methotrexate (MTX) and methylprednisolone. Blood samples were obtained at baseline (pre-therapy) from all RA patients, divided into three groups: (i) late RA receiving infliximab/MTX, (ii) early RA–infliximab/MTX, (iii) early RA–steroid/MTX, and also from follow-up patients at 2 and 14 weeks. Significant differences in absolute counts of monocytes and granulocytes were observed between healthy controls and RA patients. At baseline CD14bright monocytes and CD16+ granulocytes were increased in both early RA and late RA patients. CD4+ T cells, CD8+ T cells and B cells were all increased at baseline in early RA, but not in late RA. At 2 weeks following infliximab treatment decreased granulocytes were observed in both early and late RA and decreased natural killer (NK) cells in late RA. CD16+ granulocytes and NK cells were also decreased at 14 weeks post-infliximab in early RA. Biotinylated infliximab was used to detect membrane-associated TNF (mTNF)-expressing leucocytes in RA patients. CD16+ granulocytes, NK cells and CD14dim monocytes all expressed higher levels of mTNF in RA patients. In summary infliximab is associated with decreased CD16+ granulocyte and NK cell counts, possibly through binding of mTNF. Differential effects of infliximab between early and late RA suggest that pathogenic mechanisms change as disease progresses.
Keywords: infliximab, leucocyte counts, mTNF, rheumatoid arthritis, TNF
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease affecting up to 1% of adults and characterized by infiltration of inflammatory cells into synovial joints [1]. RA is a heterogeneous disease and there is a developing consensus that it is composed of at least two distinct disease entities, characterized generally by the presence or absence of anti-cyclic-citrullinated peptide antibodies (ACPA) [2]. Non-pathogenic autoimmune responses to citrullinated antigens (ACPA-positive) and rheumatoid factors in genetically susceptible individuals may precede the onset of clinically detectable inflammatory arthritis; this is referred to as the pre-articular or preclinical phase, which may last for a number of years [3]. Adaptive immune responses, dependent on T helper type 17 (Th17), may then lead to epitope spreading, with recognition of an increased number of citrullinated antigens occurring before the onset of clinical RA [4]. Innate-immune driven inflammatory processes facilitate the development of pathological immune responses in early disease [5], which occur locally within the joint. Thus, the development of RA is usually the end result of an evolving process, and evidence exists for a therapeutic ‘window of opportunity’ in the early stage of RA, which may prevent disease progression [6] and, indeed, achieve disease remission [7].
Proliferation of synovial fibroblasts (SF) and inflammatory cell infiltration of the synovial lining, with elevated levels of proinflammatory cytokines, including tumour necrosis factor (TNF), interleukin (IL)-6 and IL-1β, are key to disease progression in RA [8,9]. TNF is produced largely by macrophages, but also by monocytes, neutrophils, T cells, activated SF and natural killer (NK) cells. The TNF-α-converting enzyme (TACE) cleaves membrane-associated TNF (mTNF) resulting in the release of soluble TNF (sTNF) [10]. Infliximab, a chimaeric antibody to TNF, is thought to have multiple mechanisms of action based on its ability to bind circulating sTNF, thereby reducing the effective circulating concentrations of TNF and binding to mTNF, which leads potentially to target cell death [11]. Monocytes, precursors of macrophages, a predominant infiltrating cell in the RA synovium [12], as well as neutrophils, both express mTNF constitutively. The most effective therapies for RA lead to reduced synovial macrophage numbers, potentially through enhanced efflux from the synovial tissues [13,14].
Circulating leucocyte populations may be dysregulated in RA. Decreased chemotactic reactivity of neutrophils has been reported in RA patients [15]. Neutrophils migrate to the joint spaces of RA patients when attracted by chemoattractants, such as C5a and leukotriene B4; exposure to immune complexes and cytokines in the synovial fluid results in the release of proinflammatory products of the arachidonic acid cascade, such as prostaglandins, which may contribute to joint damage. NK cells are also present in the joints of patients with RA and interact with other immune cells to modulate disease pathogenesis [16].
In humans, two major monocyte subpopulations can be defined by their CD14 expression. Approximately 90% of adult peripheral monocytes are CD14bright (classical monocytes), which express variable levels of the intermediate affinity Fcγ receptor IIIa (CD16). The dominant cytokines released by these CD14bright cells are proinflammatory, such as TNF and IL-6, depending on the stimulus, but as they also produce IL-10 they may also have anti-inflammatory roles [17,18]. The remaining 10% of monocytes constitute the TNF- and IL-1β-producing CD14dim monocytes with high CD16 expression, which are thought to be proinflammatory [19]. An increased frequency of CD14+CD16+ monocytes has been reported in the blood of RA patients [20]; in particular, the CD14bright CD16+ subpopulation is increased and associated with expansion of Th17 cells [21].
The current goal in treating RA is to induce very low disease activity, and the clinical outcome can be influenced by early diagnosis and adoption of a ‘treat to target’ strategy to achieve tight disease control and disease remission [22,23]. The aims of this pilot interventional study were to test the hypothesis that there are differences in numbers of circulating leucocyte subsets in early compared with late RA and to also look for infliximab-specific effects after 2 and 14 weeks of therapy. Therefore, total numbers of circulating leucocyte subsets were compared from the peripheral blood of healthy controls and both early and late RA patients, at baseline (prior to infliximab or methylprednisolone treatment) and after 2 and 14 weeks of therapy.
Materials and methods
Patients and samples
Heparinized peripheral blood samples from 63 RA patients and 22 healthy controls were collected for flow cytometric analysis. Ethical approval was obtained from the Leeds (Central and West) Research Ethics Committee and all participants gave informed consent. Details of these patients' demographics are shown in Table 1. Samples from patients were taken pre-infusion at week 0 and again at weeks 2 and 14. The 63 RA patients [45 women, 18 men, mean standard deviation (s.d.) age 55·0 (± 13) years)] fulfilled the revised 1987 American College of Rheumatology (ACR) criteria for RA. For this study ACR/European League Against Rheumatism (EULAR) criteria were used to define early RA as disease being present for less than a year [24,25] and late RA was defined as greater than 12 months duration of symptoms. Eighteen of these patients were classified as late RA patients who had failed at least two disease-modifying anti-rheumatic drugs (DMARDs), including methotrexate (MTX), a disease activity score 28 (DAS28) of >5·1 at baseline [in line with National Institute for Health and Clinical Excellence (NICE) guidelines], and were chosen consecutively from RA out-patients who were about to commence infliximab treatment in combination with concurrent MTX. The 45 patients in the early RA cohort had a disease/symptom duration of ≤1 year, were DMARD and biologics-naive and were entered into a double-blind randomized clinical trial receiving either infliximab (at weeks 0, 2, 6, 14 and 22) or a baseline steroid infusion [250 mg intravenous (i.v.) methylprednisolone] followed by placebo infusion, all in combination with MTX [the Infliximab as Induction Therapy in Early Rheumatoid Arthritis (IDEA) study; http://clinicaltrials.gov/ct2/show/NCT01308255][26]. Infliximab was administered by infusion using a standard regimen of 3 mg/kg for both early and late RA patients. All patients received concomitant MTX, commencing at 10 mg weekly, progressing to 20 mg by week 6. Patients randomized to the comparator arm of the IDEA study (steroid/placebo) received their i.v. infusion of 250 mg methylprednisolone at week 0 and those without an adequate clinical response after 26 weeks received an additional steroid as intramuscular (i.m.) methylprednisolone 120 mg. Patients on this arm received an i.v. placebo infusion of 250 ml of 9 mg/l NaCl at weeks 2, 6, 14 and 22. Twenty-two healthy volunteers, who were gender- (16 women, six men) and age-matched [mean (s.d.) age 57·0 (±10) years], served as controls.
Table 1.
Characteristics of rheumatoid arthritis (RA) patients and healthy controls
| Early RA | ||||||
|---|---|---|---|---|---|---|
| All | Infliximab | Steroid | Late RA | Healthy controls | ||
| Female : male | – | 30F : 15 M | 14F : 5 M | 16F : 10 M | 15F : 3 M | 16F : 6 M |
| Mean age (s.d.) | – | 53·1 (13·6) | 52·4 (12·8) | 53·6 (14·3) | 60·4 (14·0) | 57·0 (10·0) |
| Mean CCP (s.d.) | Baseline | 173 (158) | 157 (153·3) | 212·4 (171·9) | 152 (117) | – |
| Mean DAS28 (s.d.) | Baseline | 5·7 (1·2) | 5·69 (1·33) | 5·51 (1·08) | 6·1 (0·63) | – |
| Week 14 | 3·86 (1·76) | 3·78 (1·61) | 3·85 (1·71) | 4·28 (1·80) | – | |
| Median CRP (s.d.) | Baseline | 16·15 (38·4) | 15·9 (38·4) | 15·15 (37·7) | 16·1 (25·3) | – |
| Week 14 | 5·00 (26·9) | 4·9 (11·9) | 5·0 (30·8) | 1·0 (10·1) | – | |
| Median ESR (s.d.) | Baseline | 32·5 (30·8) | 31·0 (27·4) | 40·0 (33·1) | 42·0 (34·7) | – |
| Week 14 | 18·0 (25·9) | 17·5 (23·8) | 18·5 (27·4) | 28·0 (30·53) | – | |
CCP: cyclic citrullinated peptides; CRP: C-reactive protein; DAS: disease activity score; ESR: erythrocyte sedimentation rate; s.d.: standard deviation.
Flow cytometry
Red cell lysis was used to isolate leucocytes for flow cytometry. A volume of 1 ml of heparinized peripheral blood was mixed with 20 ml of ammonium chloride solution, and incubated for 5–10 min at room temperature. Cells were washed with phosphate-buffered saline (PBS) and resuspended in fluorescence activated cell sorter (FACS) buffer with 5% v/v mouse serum (Sigma Aldrich, Poole, Dorset, UK) and incubated for 30 min on ice. The surface expression of CD3, CD4 and CD8 on T cells, CD3 and CD56 on NK cells, CD19 on B cells, CD16 on granulocytes and CD14 and CD16 on monocytes was analysed on the day of collection by direct staining with appropriate dilutions of the monoclonal antibodies CD4-allophycocyanin (APC), CD8-fluorescein isothiocyanate (FITC), CD14-phycoerythrin (PE), CD16-FITC, CD19-PE (AbD Serotec, Kidlington, UK), CD3-PE-Texas Red (Invitrogen, Paisley, UK), CD56-APC (BD Bioscience, Oxford, UK) or isotype-matched control monoclonal antibodies. The working concentrations of all antibodies were 50 µg/ml, apart from CD3-PE-Texas Red (one in 10 of stock) and CD56-APC (one in 20 of stock). The samples were incubated with the antibodies on ice for 30 min in the dark. Cells were washed and resuspended in PBS. A total volume of 100 µl Caltag counting beads (Invitrogen) was added immediately to each sample before analysis on the LSRII flow cytometer (BD Biosciences). The total number of events collected was 30 000 live cells and the gating was carried out as shown in Fig. 1. Data were analysed using BD FACSDiva version 5.0.2 (BD Biosciences).
Fig. 1.
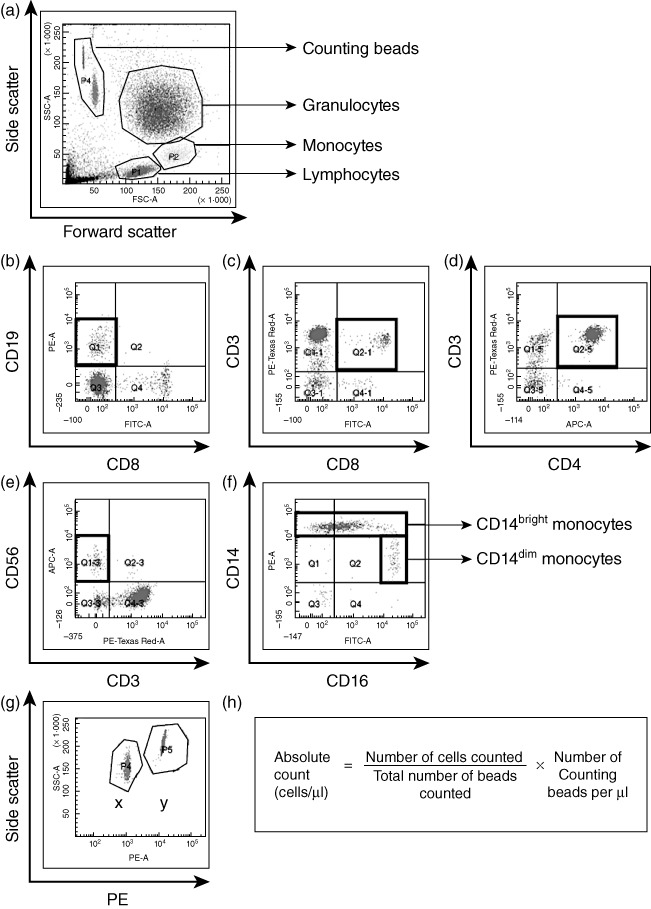
Basic gating strategy for absolute cell count analysis. (a) Cells were gated on forward/side scatter (FSC/SSC). The thick black boxes show cell populations measured. Isotype control antibodies were used to define negative populations and single-stained samples were tested to ensure that fluorochromes were well compensated. (b) CD19+CD8−B cells, (c) CD3+CD8+ T cells, (d) CD3+CD4+ T cells and (e) CD3-CD56+ natural killer (NK) cells were gated within the lymphocyte gate on the FSC/SSC. (f) CD14+ monocytes were gated within the monocyte gate on the FSC/SSC and subdivided into CD14bright and CD14dim. (g) Counting beads were displayed on a SSC/phycoerythrin (PE) plot to distinguish clearly the two populations of beads, labelled as X and Y. The proportion of the bead populations acted as an internal control. (h) Example calculation of absolute cell counts.
Detection of mTNF by conjugated infliximab
Biotinylated infliximab was used to detect cells expressing mTNF in fresh peripheral blood of three RA patients who were infliximab-naive. A 1 mg/ml stock of infliximab was biotinylated using a Sulfo-NHS-LC-Biotin kit (Thermo Scientific, Rockford, IL, USA), according to the manufacturer's protocol. The solution was passed through a PD-10 desalting column (Amersham Biosciences, Piscataway, NJ, USA) to remove unbound biotin, biotin–infliximab collected in 500 µl aliquots and protein concentrations determined on the Nanodrop spectrophotometer. Biotinylation was confirmed by spotting the solution onto polyvinylidene difluoride (PVDF) membrane, and incubating with streptavidin–horseradish peroxidase (HRP) (1:2000 dilution; BD Biosciences, Franklin Lakes, NJ, USA) for 1 h, before detection using enhanced chemiluminescence (ECL) (Amersham Biosciences). Peripheral blood leucocytes were isolated and blocked with 5% mouse serum for 30 min and incubated with 1 mg/ml biotin–infliximab for 30 min on ice. Cells were washed in PBS and resuspended in FACS buffer. Cell surface staining was performed using the monoclonal antibodies listed previously, with the exception of CD14-APC (AbD Serotec) and CD3-Pacific Blue (Invitrogen) being used instead of CD14-PE and CD3-PE-Texas Red, with the addition of streptavidin-PE (AbD Serotec) to detect the biotin–infliximab. Cells were incubated on ice for 30 min, washed in PBS and analysed on the LSRII (BD Biosciences, USA). Biotinylated human and mouse IgG1 were used as negative controls.
Measurement of C-reactive protein (CRP) and cyclic citrullinated peptides (CCP) levels
An immunoturbidometric assay was used to measure levels of CRP (range: 0–0·5 mg/dl) on an ADVIA 1800 (Siemens Healthcare Diagnostics Inc., Camberley, UK). CCP levels (ranges: <7 U/ml neg; 7–10 U/ml equivocal; >10 U/ml positive) were measured by fluoroenzyme immunoassay (Phadia AB, Uppsala, Sweden).
Statistical analysis
Normal distribution of the data was tested with the Shapiro–Wilk test using Prism5 software (GraphPad Software Inc., version 5.0b). The Kruskal–Wallis test was used to test for clinical differences between early RA, late RA and healthy control groups. Differences between values at baseline and weeks 2 or 14 after infliximab infusion were analysed using Wilcoxon's signed-rank test. Absolute counts of the different cell populations were correlated with the presence of systemic inflammation [as measured by CRP/erythrocyte sedimentation rate (ESR)] and disease activity score (DAS28) using Spearman's rank correlation coefficient. Differences were considered to be statistically significant with P values <0·05.
Results
Monocyte and granulocyte counts differ between RA patients at baseline and healthy controls
Absolute leucocyte counts were compared from the peripheral blood of RA patients and healthy controls prior to treatment. CD14bright and CD14brightCD16+ monocyte subsets were increased significantly in both early RA and late RA (both P < 0·001) compared to healthy controls (Fig. 2a,c), whereas the CD14dim monocyte counts were increased significantly in the late RA cohort only when compared to healthy controls (P < 0·001) and early RA patients (P < 0·01) (Fig. 2b). The total numbers of CD16+ granulocytes were increased significantly in both early and late RA cohorts (P < 0·001) compared to healthy controls (Fig. 2d). Absolute counts of NK cells were similar between healthy controls and RA patients at baseline (Fig. 2h).
Fig. 2.
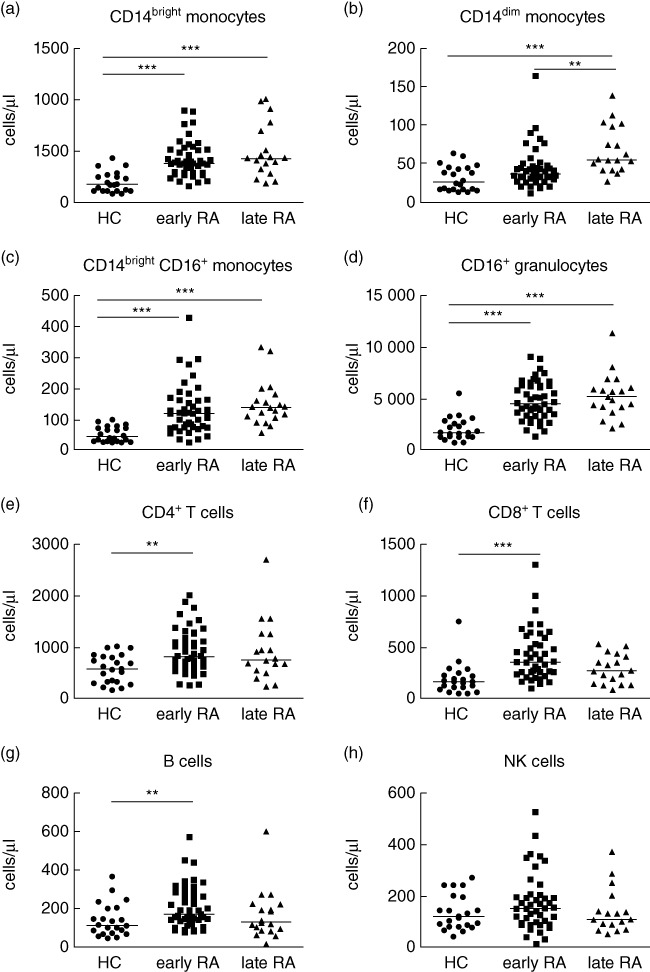
Baseline leucocyte cell counts in early and late rheumatoid arthritis (RA) compared to healthy control. Absolute cell counts for (a) CD14bright monocytes, (b) CD14dim monocytes (c) CD14brighCD16+ monocytes, (d) CD16+ granulocytes, (e) CD4+ T cells, (f) CD8+ T cells, (g) CD19+ B cells and (h) CD3-CD56+natural killer (NK) cells were analysed in healthy controls (n = 22), early RA (n = 45) and late RA (n = 18) patients. **P < 0·01; ***P < 0·001 by Kruskal–Wallis test.
Lymphocyte counts increased in early RA at baseline
Baseline CD4+ T cells, CD8+ T cells and B cells were all increased significantly in early RA patients (CD4+ and B cells P < 0·01, CD8+ T cells P < 0·001), but not in late RA patients, who had similar levels to healthy controls (Fig. 2e–g).
CRP, ESR and DAS28 levels correlated with some leucocyte subsets at baseline in early and late RA
CRP and ESR levels were used to assess the overall degree of systemic inflammation in early RA patients compared to late RA patients, and no significant differences were observed between these three patient groups (Table 1). CRP levels did correlate with CD14bright counts at baseline in early RA (P < 0·01) and also with CD16+ granulocytes in both early RA and late RA (P < 0·01 and P < 0·05, respectively); however, these correlations were not present at either 2 or 14 weeks. ESR levels correlated with CD14bright counts only at baseline in late RA (P < 0·05). Similarly, CD16+ granulocytes correlated with DAS28 (P < 0·05) at baseline in early RA but were not correlated at later time-points. Furthermore, no other cell subsets, including CD14dim monocytes, NK cells, T cells or B cells, were associated with CRP or ESR levels, either at baseline or during therapy.
Infliximab treatment affects innate immune cell counts in the first 2 weeks
Infliximab was administered to early RA and late RA patients at weeks 0, 2, 6, 14 and 22, and the control group of early RA patients were treated with intravenous methylprednisolone followed by placebo infusions, all in combination with MTX. Absolute cell counts were determined at 2 weeks post-therapy and compared to counts in the same patients at week 0 (Fig. 3). These data are from paired samples taken from the same patients at weeks 0 and 2, and again at weeks 0 and 14 (Fig. 4). For this reason, the number of patients in the late RA group at weeks 2 and 14 are fewer than the baseline number of 18 patients – at week 2 paired data were available for 11 patients (61%) and at week 14 paired data were available for seven patients (39%). The dropout rate was due either to lack of response, at which point it was decided to discontinue the therapy, or lack of patient compliance. Despite raised counts seen at baseline, by 2 weeks of treatment neither CD14bright nor CD14dim monocyte subsets were reduced significantly in paired samples in any of the treatment groups (Fig. 3a,b). However, CD16+ granulocytes displayed a significant decrease in both the early RA–infliximab (P = 0·016) and late RA–infliximab (P = 0·002) compared to baseline (Fig. 3c), whereas no significant changes were found in CD16+ granulocytes in the early RA patients receiving methylprednisolone, in agreement with the well-recognized phenomenon that steroids induce neutrophilia by a number of different mechanisms [27]. NK cell numbers were reduced significantly in late RA infliximab patients at 2 weeks compared to baseline (P = 0·006), but not in either the early RA–infliximab or RA–methylprednisolone groups (Fig. 3d); this is in contrast to the baseline results, where similar levels were seen in all groups. No effects were seen in T or B cells despite absolute cell counts of T and B cells being raised at baseline in early RA patients (Fig. 2e–g).
Fig. 3.
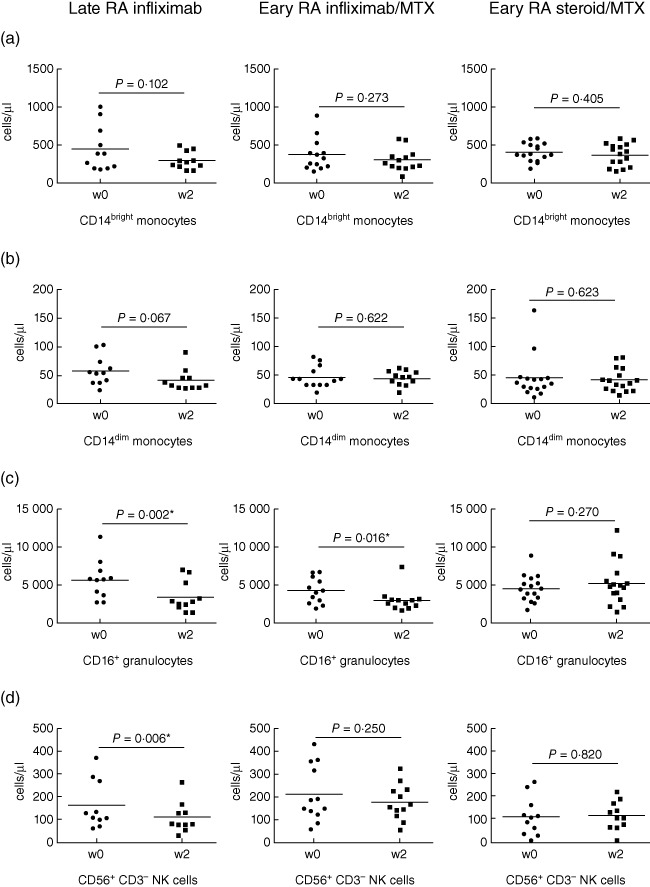
Absolute cell counts at week 2 of infliximab or steroid treatment in early and late disease. The change in cell number between baseline and week 2 was calculated in the late rheumatoid arthritis (RA) group receiving infliximab (n = 11), the early RA–infliximab group (n = 13) and the early RA–steroid group (n = 16). Changes were analysed for (a) CD14bright monocytes, (b) CD14dim monocytes, (c) CD16+ granulocytes and (d) CD3-CD56+ NK cells. Significant differences from baseline to week 2 of treatment were calculated using Wilcoxon's signed-rank test. P < 0·05 was considered significant and marked with an asterish (*).
Fig. 4.
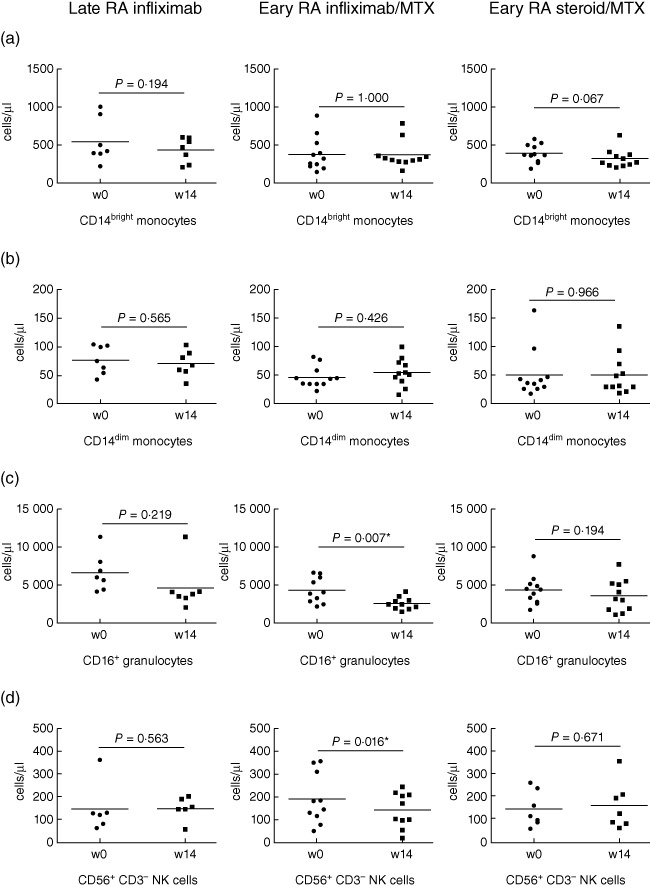
Absolute cell counts at week 14 after infliximab or steroid treatment in early and late disease. The change in cell number between baseline and week 14 was calculated in the late rheumatoid arthritis (RA) group receiving infliximab (n = 7), the early RA–infliximab group (n = 11) and the early RA–steroid group (n = 11). Changes were analysed for (a) CD14bright monocytes, (b) CD14dim monocytes, (c) CD16+ granulocytes and (d) CD3-CD56+ natural killer (NK) cells. Significant differences from baseline to week 14 of treatment were calculated using Wilcoxon's signed rank test. P < 0·05 was considered significant and marked with a star (*).
Decreased CD16+ granulocyte counts are maintained at 14 weeks post-infliximab in early RA patients
When absolute cell counts were examined at 14 weeks post-treatment, CD16+ granulocyte counts remained low in the early RA–infliximab group (P = 0·007) whereas in the late RA–infliximab group the levels had recovered (Fig. 4c) when compared to the 2-week time-point, with levels similar to those seen at baseline. NK cells were decreased significantly only in the early RA–infliximab group (P = 0·016 Fig. 4d). The NK cell counts in the late RA–infliximab group had increased from week 2 back to levels seen at baseline. The monocyte cell counts were not changed in either of the infliximab groups (Fig. 4a,b). No significant differences in any of the cell subsets were observed in the early RA–steroid group (Fig. 4a–d). None of the changes in cell subsets were associated with CRP, ESR or DAS 28 at 14 weeks (Table 1). A total of 66% of the early RA patients had CCP levels in the positive range and all the late RA patients were ACPA-positive.
Infliximab binds monocytes, granulocytes, NK cells and CD4+ T cells
Biotinylated mouse or human IgG did not show any significant binding to cell subsets (Fig. 5). Biotinylated infliximab bound strongly to CD14dim monocytes (Fig. 6a), granulocytes (Fig. 6c) and CD3-CD56+ NK cells (Fig. 6d), with lower levels of binding to CD14bright monocytes (Fig. 6a) and CD4+ T cells (Fig. 6b) (Fig. 6d) indicating mTNF expression on these cell types. Infliximab did not bind to CD8+ T cells or B cells in this assay (Fig. 6b).
Fig. 5.
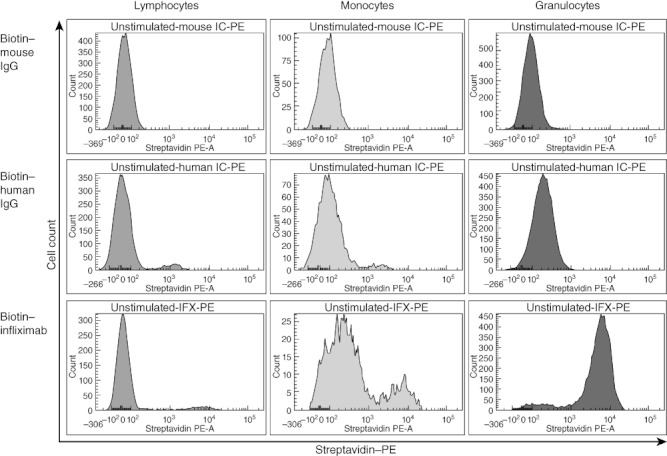
Biotinylated immunoglobulin (Ig)G isotype control binding on leukocyte populations. Human IgG, mouse IgG and infliximab were biotinylated. Binding of each IgG class to the cell surface of leucocyte populations was determined using healthy control (HC) blood. The lymphocyte, monocyte and granulocyte populations were gated on forward/side scatter (FSC/SSC), as depicted in Fig. 1. Each histogram represents streptavidin-phycoerythrin (PE) binding to IgG in each cell population. Data were analysed using BD FACSDiva version 5.0.2 (BD Biosciences, Oxford, UK).
Fig. 6.
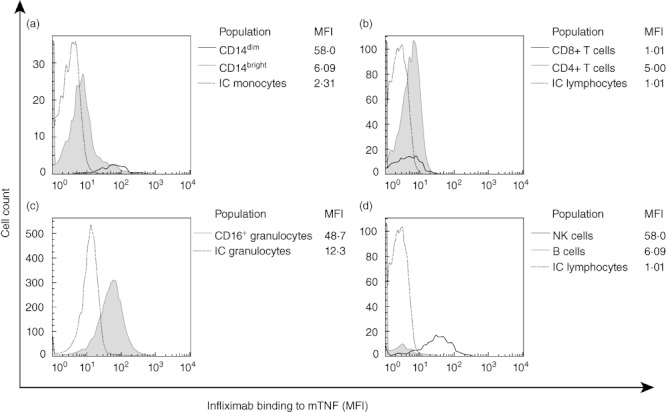
Expression of membrane-associated tumour necrosis factor (mTNF) on the surface of different leucocyte populations. The expression of mTNF was detected by biotinylated infliximab in three rheumatoid arthritis (RA) patients who were infliximab-naive. Histograms showing binding of infliximab on (a) CD14dim and CD14bright monocytes, (b) CD4+ and CD8+ T cells, (c) CD16+ granulocytes and (d) natural killer (NK) cells and B cells. Median fluorescence intensity (MFI) was analysed using FlowJo (Tree Star, Inc.). The respective isotype controls (IC) were shown as dotted lines.
Discussion
This study demonstrates significant differences in circulating leucocyte numbers between RA patients and healthy controls, and also differential effects of infliximab on absolute circulating blood leucocyte counts of innate immune cells in early and late RA. The mechanisms responsible for altered circulating cellular phenotype in RA are complex, and include stage of disease, types of therapy, degree of systemic inflammation and disease severity. We correlated absolute counts of different cell populations with systemic inflammation (CRP/ESR) to determine whether observed differences were specific to either different stages of the disease (early or late) or different therapies (infliximab/MTX or methylprednisolone/MTX), or a consequence of chronic inflammation inherent in late RA in particular.
There is an increasing awareness of the contribution of neutrophils to RA pathophysiology, particularly by localized production of prostaglandins, reactive oxygen intermediates and proteases in the inflamed synovium [28,29]. In this study CD16+ granulocyte counts were increased significantly at baseline in both early and late RA and a significant decrease in this cellular subset was observed after 2 weeks infliximab therapy in both these cohorts, which was maintained at 14 weeks in the early RA patients. The total numbers of CD16+ granulocytes were correlated with CRP levels in both early and late RA at baseline but not after infliximab treatment. As multiple comparisons were made in this study it could be argued that Bonferroni's correction should be applied despite the fact that we were testing the a priori hypothesis that there are differences in numbers of circulating leucocyte subsets in early RA compared with late RA patients. A statistically significant association for differences in the absolute leucocyte counts using a Bonferroni-adjusted P-value threshold given 12 independent tests is set at P < 0·004. When this adjustment is applied, then only CD16+ granulocytes (Fig. 3, weeks 0 and 2) would remain significant (corrected P = 0·024). The finding of mTNF expression on CD16+ granulocytes, NK cells and CD14dim monocytes in RA patients provides a potential explanation for the observed differences.
CD14bright monocytes and the CD14brightCD16+ monocyte subset, which is the major TNF-producing population [30], were also increased significantly at baseline in both early RA and late RA, supporting the reported expansion of the CD14brightCD16+ subpopulation in blood of RA patients [21]. CD14dim monocytes were increased at baseline only in the late RA cohort, suggesting that CD14dim monocytes may contribute to the chronic inflammatory state and disease progression of RA. CD14bright monocytes correlated with CRP in the early RA cohort only at baseline and, unlike granulocytes, the monocyte subsets were not reduced after 2 weeks or 14 weeks in any treatment groups. There were no significant correlations with CRP or ESR, for any other cell type either at baseline or 14 weeks. These data suggest that systemic inflammation, as measured by CRP or ESR, may contribute to some of the differences in granulocytes and monocyte subsets observed between healthy controls and RA patients at baseline, prior to initiation of infliximab or methylprednisolone therapy. However, there would appear to be an infliximab-dependent mechanism of decrease in granulocytes, in particular, as the reduction in CD16+ granulocytes after 2 and 14 weeks of infliximab therapy were not associated with concomitant falls in CRP, ESR or DAS 28. Furthermore, the CD16+ granulocyte population expresses high levels of mTNF and infliximab may target mTNF-expressing cells for depletion.
The higher numbers of circulating granulocytes and monocytes at baseline, prior to either infliximab or methylprednisolone therapy in both early RA and late RA patients, support the importance of innate immunity in RA pathogenesis, particularly as these two cell subsets are major producers of key proinflammatory cytokines such as TNF and IL-1β. NK cells also have an important role in innate immune responses; these cells were not increased in RA patients at baseline but were decreased significantly at 2 weeks after infliximab in late RA and also at 14 weeks in early RA patients. These changes may reflect a therapeutic response to infliximab, as we found that NK cells also have high mTNF expression. NK cells produce large amounts of proinflammatory cytokines, including TNF in the RA synovium [16], and modulate the adaptive immune response in autoimmunity [31]. Significantly increased T and B cells were also found at baseline in early RA but not in late RA, which may point to a particular role for adaptive immune cells in early RA. T cells and B cells, which were found to express very low levels of mTNF, were not depleted by infliximab, and in most cases counts were increased at 2 weeks post-baseline infusion.
In this regard, it is important to note that no significant changes were seen in any cell subsets after 2 weeks and 14 weeks of methylprednisolone/MTX in this study, suggesting that the observed effects in cell counts in infliximab-treated patients are more related to the effects of this biological response modifier than to previous DMARD or concomitant MTX therapy, or decreased levels of systemic inflammation. All late RA patients had failed to respond to at least two DMARDs at the time of initiation of infliximab therapy, whereas all early RA patients were DMARD-naive. The cellular changes in the blood of late RA patients are therefore less likely to be the pure product of RA pathogenesis than changes observed in early RA patients.
It has been reported that infliximab induces a decrease in monocyte numbers within the first few weeks of treatment [32]; however, that particular report did not differentiate between CD14dim and CD14bright monocyte subsets. In our study the lack of differences in monocyte subsets between baseline and 2 and 14 weeks after infliximab may be due to a number of reasons. First, compared to NK and granulocyte populations, the CD14dim cell count is very low, so significant changes in that particular subset may be difficult to detect. Differences in synovial fluid cytokine profiles between early RA and late RA have also been published [33], and these differences may be due to a combination of changing RA pathogenesis at different disease stages and the consequence of previous treatment in late RA patients.
The mechanisms by which infliximab depletes mTNF-expressing cells remain to be determined. Infliximab has been shown to reduce the influx of inflammatory cells into the joints of RA patients [34], but we found no evidence of circulating apoptotic cells and previous reports also suggest that apoptosis is unlikely to have occurred directly in the bloodstream [35]. Severe neutropenia has also been reported following administration of infliximab [36], indicating that infliximab, or lack of TNF, may have a direct effect on haematopoietic cell development.
Granulocyte counts were decreased at 2 weeks following infliximab treatment in both early and late RA, and NK cells were also decreased significantly at the 2-week time-point in late RA and at 14 weeks in the early RA group. T cells and B cells were not altered significantly post-infliximab. These data suggest that cells of the innate immune system, which express high levels of mTNF (granulocytes, CD14dim monocytes and NK cells), may be targeted by infliximab, thereby decreasing these cell populations in RA, possibly through reverse signalling which enables two-way communication between cells; it is conceivable that this bidirectional signal exchange may contribute to the removal of mTNF-expressing cells [37].
There is an unavoidable dropout rate in all longitudinal studies of therapies in RA, which may be due to lack of therapeutic response, toxicity or patient unavailability, due mainly to lack of compliance or occasionally concomitant illness or death. This reduction in numbers at the follow-up time-points has potential implications regarding the power of the study and may introduce potential bias. However, there were no significant differences in baseline values between those patients who did and did not drop out. Larger longitudinal observational studies of inception cohort of patients with early RA are in progress and should help to address some of these problems. Nevertheless, this study suggests that cellular phenotyping may be a useful tool in monitoring disease in RA, and further follow-up work is in progress to define its utility in long-term management of RA and the mechanisms of action of infliximab.
Acknowledgments
The authors thank Arthritis Research UK (grant number 19269), the MRC and the NIHR-Leeds Musculoskeletal Biomedical Research Unit (NIHR-LMBRU) for their support. We thank the IDEA investigators and coordinators who collected data used in this study, as well as participants and their families, who made this work possible.
Disclosure
P.E. has received research grants and honoraria from Abbott, Bristol-Myers Squibb, Roche, MSD, UCB Pharma, Pfizer and Wyeth. M.H.B. has received honoraria from Abbott, Roche, UCB Pharma and Pfizer.
References
- 1.Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet. 2010;376:1094–108. doi: 10.1016/S0140-6736(10)60826-4. [DOI] [PubMed] [Google Scholar]
- 2.Simon M, Girbal E, Sebbag M, et al. The cytokeratin filament-aggregating protein filaggrin is the target of the so-called ‘antikeratin antibodies,’ autoantibodies specific for rheumatoid arthritis. J Clin Invest. 1993;92:1387–93. doi: 10.1172/JCI116713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deane KD, Norris JM, Holers VM. Preclinical rheumatoid arthritis: identification, evaluation, and future directions for investigation. Rheum Dis Clin North Am. 2010;36:213–41. doi: 10.1016/j.rdc.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Woude D, Rantapää-Dahlqvist S, Ioan-Facsinay A, et al. Epitope spreading of the anti-citrullinated protein antibody response occurs before disease onset and is associated with the disease course of early arthritis. Ann Rheum Dis. 2010;69:1554–61. doi: 10.1136/ard.2009.124537. [DOI] [PubMed] [Google Scholar]
- 5.Gierut A, Perlman H, Pope RM. Innate immunity and rheumatoid arthritis. Rheum Dis Clin North Am. 2010;36:271–96. doi: 10.1016/j.rdc.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Dongen H, van Aken J, Lard LR, et al. Efficacy of methotrexate treatment in patients with probable rheumatoid arthritis: a double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 2007;56:1424–32. doi: 10.1002/art.22525. [DOI] [PubMed] [Google Scholar]
- 7.Emery P, Breedveld FC, Hall S, et al. Comparison of methotrexate monotherapy with a combination of methotrexate and etanercept in active, early, moderate to severe rheumatoid arthritis (COMET): a randomised, double-blind, parallel treatment trial. Lancet. 2008;372:375–82. doi: 10.1016/S0140-6736(08)61000-4. [DOI] [PubMed] [Google Scholar]
- 8.Firestein GS, Alvaro-Gracia JM, Maki R. Quantitative analysis of cytokine gene expression in rheumatoid arthritis. J Immunol. 1990;144:3347–53. [PubMed] [Google Scholar]
- 9.Arend WP. Cytokine imbalance in the pathogenesis of rheumatoid arthritis: the role of interleukin-1 receptor antagonist. Semin Arthritis Rheum. 2001;30(Suppl 2):1–6. doi: 10.1053/sarh.2001.23693. [DOI] [PubMed] [Google Scholar]
- 10.Black RA, Rauch CT, Kozlosky CJ, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–33. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 11.Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008;117:244–79. doi: 10.1016/j.pharmthera.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 12.Burmester GR, Stuhlmüller B, Keyszer G, Kinne RW. Mononuclear phagocytes and rheumatoid synovitis. Mastermind or workhorse in arthritis? Arthritis Rheum. 1997;40:5–18. doi: 10.1002/art.1780400104. [DOI] [PubMed] [Google Scholar]
- 13.Herenius MM, Thurlings RM, Wijbrandts CA, et al. Monocyte migration to the synovium in rheumatoid arthritis patients treated with adalimumab. Ann Rheum Dis. 2011;70:1160–2. doi: 10.1136/ard.2010.141549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bresnihan B, Pontifex E, Thurlings RM, et al. Synovial tissue sublining CD68 expression is a biomarker of therapeutic response in rheumatoid arthritis clinical trials: consistency across centers. J Rheumatol. 2009;36:1800–2. doi: 10.3899/jrheum.090348. [DOI] [PubMed] [Google Scholar]
- 15.Bültmann B, Geitner R, Seibold H, Kratsch G, Haferkamp O. Interaction of circulating immune complexes with granulocyte function in patients with rheumatoid arthritis. Klin Wochenschr. 1980;58:727–32. doi: 10.1007/BF01478460. [DOI] [PubMed] [Google Scholar]
- 16.Ahern DJ, Brennan FM. The role of natural killer cells in the pathogenesis of rheumatoid arthritis: major contributors or essential homeostatic modulators? Immunol Lett. 2011;136:115–21. doi: 10.1016/j.imlet.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 17.Skrzeczyńska-Moncznik J, Bzowska M, Loseke S, Grage-Griebenow E, Zembala M, Pryjma J. Peripheral blood CD14high CD16+ monocytes are main producers of IL-10. Scand J Immunol. 2008;67:152–9. doi: 10.1111/j.1365-3083.2007.02051.x. [DOI] [PubMed] [Google Scholar]
- 18.Cooper DL, Martin SG, Robinson JI, et al. FcγRIIIa expression on monocytes in rheumatoid arthritis: role in immune-complex stimulated TNF production and non-response to methotrexate therapy. PLoS ONE. 2011;7:e28918. doi: 10.1371/journal.pone.0028918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belge KU, Dayyani F, Horelt A, et al. The proinflammatory CD14+CD16+DR++ monocytes are a major source of TNF. J Immunol. 2002;168:3536–42. doi: 10.4049/jimmunol.168.7.3536. [DOI] [PubMed] [Google Scholar]
- 20.Kawanaka N, Yamamura M, Aita T, et al. CD14+, CD16+ blood monocytes and joint inflammation in rheumatoid arthritis. Arthritis Rheum. 2002;46:2578–86. doi: 10.1002/art.10545. [DOI] [PubMed] [Google Scholar]
- 21.Rossol M, Kraus S, Pierer M, Baerwald C, Wagner U. The CD14(bright) CD16+ monocyte subset is expanded in rheumatoid arthritis and promotes Th17 expansion. Arthritis Rheum. 2012;64:671–7. doi: 10.1002/art.33418. [DOI] [PubMed] [Google Scholar]
- 22.Smolen JS, Aletaha D, Bijlsma JW, et al. Treating rheumatoid arthritis to target: recommendations of an international task force. Ann Rheum Dis. 2010;69:631–7. doi: 10.1136/ard.2009.123919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saleem B, Keen H, Goeb V, et al. Patients with RA in remission on TNF blockers: when and in whom can TNF blocker therapy be stopped? Ann Rheum Dis. 2010;69:1636–42. doi: 10.1136/ard.2009.117341. [DOI] [PubMed] [Google Scholar]
- 24.Aletaha D, Neogi T, Silman AJ, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis. 2010;69:1580–8. doi: 10.1136/ard.2010.138461. [DOI] [PubMed] [Google Scholar]
- 25.Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/ European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62:2569–81. doi: 10.1002/art.27584. [DOI] [PubMed] [Google Scholar]
- 26.Nam JL, Villeneuve E, Conaghan PG, et al. A preliminary report of remission induction with two therapeutic strategies with infliximab or high dose intravenous steroids for the treatment of rheumatoid arthritis. Ann Rheum Dis. 2011;70:121. [Google Scholar]
- 27.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205–19. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 28.Cascão R, Rosário HS, Souto-Carneiro MM, Fonseca JE. Neutrophils in rheumatoid arthritis: more than simple final effectors. Autoimmun Rev. 2010;9:531–5. doi: 10.1016/j.autrev.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 29.Cross A, Barnes T, Bucknall RC, Edwards SW, Moots RJ. Neutrophil apoptosis in rheumatoid arthritis is regulated by local oxygen tensions within joints. J Leukoc Biol. 2006;80:521–8. doi: 10.1189/jlb.0306178. [DOI] [PubMed] [Google Scholar]
- 30.Cros J, Cagnard N, Woollard K, et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. 2010;33:375–86. doi: 10.1016/j.immuni.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flodström-Tullberg M, Bryceson YT, Shi FD, Höglund P, Ljunggren HG. Natural killer cells in human autoimmunity. Curr Opin Immunol. 2009;21:634–40. doi: 10.1016/j.coi.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 32.Ohshima S, Saeki Y, Mima T, et al. Long-term follow-up of the changes in circulating cytokines, soluble cytokine receptors, and white blood cell subset counts in patients with rheumatoid arthritis (RA) after monoclonal anti-TNF alpha antibody therapy. J Clin Immunol. 1999;19:305–13. doi: 10.1023/a:1020543625282. [DOI] [PubMed] [Google Scholar]
- 33.Raza K, Falciani F, Curnow SJ, et al. Early rheumatoid arthritis is characterized by a distinct and transient synovial fluid cytokine profile of T cell and stromal cell origin. Arthritis Res Ther. 2005;7:R784–95. doi: 10.1186/ar1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.den Broeder AA, Wanten GJ, Oyen WJ, et al. Neutrophil migration and production of reactive oxygen species during treatment with a fully human anti-tumor necrosis factor-alpha monoclonal antibody in patients with rheumatoid arthritis. J Rheumatol. 2003;30:232–7. [PubMed] [Google Scholar]
- 35.Wijbrandts CA, Remans PH, Klarenbeek PL, et al. Analysis of apoptosis in peripheral blood and synovial tissue very early after initiation of infliximab treatment in rheumatoid arthritis patients. Arthritis Rheum. 2008;58:3330–9. doi: 10.1002/art.23989. [DOI] [PubMed] [Google Scholar]
- 36.Vidal F, Fontova R, Richart C. Severe neutropenia and thrombocytopenia associated with infliximab. Ann Intern Med. 2003;139:W–W63. doi: 10.7326/0003-4819-139-3-200308050-00021-w4. [DOI] [PubMed] [Google Scholar]
- 37.Geiler J, Buch M, McDermott MF. Anti-TNF treatment in rheumatoid arthritis. Curr Pharm Des. 2011;17:3141–54. doi: 10.2174/138161211798157658. [DOI] [PubMed] [Google Scholar]