

Abstract
Anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) is a group of autoimmune diseases, including granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA). It is not known why ANCA develop, but it has been shown that they participate in pathogenesis by activating polymorphonuclear neutrophils (PMNs). In this study we hypothesize that dysregulation of phagocytosis in AAV leads to the accumulation of apoptotic neutrophils seen in association with blood vessels in AAV. These cells progress into secondary necrosis, contributing to tissue damage and autoantibody formation. Peripheral blood cells were counted, and phagocytosis was investigated using monocyte-derived macrophages (MØ) and PMNs from healthy blood donors (HBD), AAV patients and systemic lupus erythematosus (SLE) patients. Furthermore, the effect of serum was assessed. Phagocytosis was measured using flow cytometry. The results showed no deviation in monocyte subpopulations for AAV patients compared to HBDs, although there was a decrease in lymphocyte and pDC (plasmacytoid dendritic cell) populations (4·2 × 106 cells/l versus 10·4 × 106 cells/l, P < 0·001). The number of neutrophils was increased (6·0 × 109 cells/l versus 3·8 × 109 cells/l, P < 0·001). There were no differences found in the ability of MØs to engulf apoptotic cells, nor when comparing apoptotic PMNs to become engulfed. However, serum from AAV donors tended to decrease the phagocytosis ability of MØs (36%) compared to serum from HBDs (43%). In conclusion, there is no intrinsic dysfunction in the MØs or in the PMNs that have an effect on phagocytic activity, but ANCA may play a role by decreasing phagocytic ability.
Keywords: ANCA-associated vasculitis (AAV), autoimmunity, monocyte subpopulations, neutrophil, pDC
Introduction
Anti-neutrophil cytoplasmic antibody (ANCA)-associated small vessel vasculitis (AAV) is a group of autoimmune diseases affecting small blood vessels of mainly kidneys and the respiratory tract [1]. These diseases include granulomatosis with polyangiitis (GPA) (formerly known as Wegener's granulomatosis [2]), microscopic polyangiitis (MPA) and Churg–Strauss syndrome (CSS).
The antigens towards which the ANCAs are directed were discovered in the late 1980s and are either myeloperoxidase (MPO) [3], usually associated with MPA, or proteinase 3 (PR3) [4,5], usually associated with GPA. Both antigens are granule proteins abundant in neutrophils. PR3 shows a bimodal surface expression pattern on resting neutrophils [6]. The surface expression of both autoantigens is increased following degranulation [7]. Apoptosis also increases PR3, but not MPO, surface expression, and this seems to be independent of degranulation [8]. This study also proposes PR3 as a ‘don't eat me’ signal associated with phosphatidylserine exposure during apoptosis. PR3 membrane expression and association with different molecular partners is, however, not fully elucidated [9].
There is extensive evidence for a pathogenic role of ANCA in AAV. ANCAs are able to activate cytokine-primed neutrophils, by binding to its antigen on the neutrophil surface, to release their granules and produce reactive oxygen species (ROS) [10], and are capable of transferring the disease [11]. An animal model for GPA has been missing until recently, when a mouse model with humanized mice developed disease after passive transfer of anti-PR3 ANCA [12]. It is as yet unknown how these autoantibodies arise, although there are many theories. One such theory suggests a connection with infections, and in fact 63% of GPA patients have chronic Staphylococcus aureus nasal carriage, supporting this theory [13]. Furthermore, autoantibodies directed against lysosomal-associated membrane protein 2 (LAMP-2) were discovered recently in active AAV. These cross-react with FimH, a protein common in Gram-negative bacteria, suggesting an infection with Gram-negative bacteria as an initiating factor in the development of AAV [14], although this is still the subject of debate [15,16].
Another proposed mechanism is that dysregulation of apoptotic cell clearance contributes to autoimmunity. Apoptotic cell clearance is normally a non-inflammatory process, but if the system is overwhelmed apoptotic cells can progress into secondary necrosis, an inflammatory process. This might trigger maturation signals in dendritic cells (DCs) which may, in turn, stimulate an immune response towards autoantigens [17]. According to the danger theory first proposed by Matzinger, the immune system would recognize the tissue damage as a danger signal and start to react [18]. Dysregulated clearance of apoptotic cells has also been proposed for other autoimmune diseases, such as systemic lupus erythematosus (SLE) [19].
Neutrophils have been shown to be key players in AAV; in a MPO–ANCA mouse model, neutrophil depletion abrogated the development of necrotizing and crescentic glomerular nephritis (NCGN) [20] and activated neutrophils have been found in renal biopsies of AAV, mediating damage to the vascular wall [21]. Recent data from our group have also shown that polymorphonuclear neutrophils (PMNs) from AAV patients survive longer in vitro compared to healthy blood donors (HBDs) [22]. This might contribute to the accumulation of dying neutrophils seen commonly around vessels in AAV patients [23].
Other causes of this accumulation could be an intrinsic defect in the cells involved in apoptotic cell clearance; for example, in the monocytes/MØs or in the neutrophils. It has been shown previously that the CD14+CD16+ subpopulation of monocytes is increased in several chronic inflammatory diseases, such as rheumatoid arthritis [24]. We have also shown previously that monocytes from patients express more PR3 compared to HBDs [25]. Specific differences in gene expression have also been found for several genes in a gene array study comparing leucocytes from AAV, SLE patients and HBDs. The results from these studies indicate that neutrophils are particularly involved in AAV pathogenesis [26]. This suggests that the monocyte/MØ and/or neutrophil populations in AAV patients deviate to some extent from HBDs.
In this study we hypothesize that there is an intrinsic dysfunctional phagocytosis in AAV patients, either in the ability of MØs to clear apoptotic cells or in the ability of apoptotic neutrophils to become cleared.
Material and methods
Blood samples and patients
Blood from patients and HBDs was drawn in ethylenediamine tetraacetic acid (EDTA) tubes, if not stated otherwise. The blood was used within 2 h. Written informed consent was taken from all donors and these studies were conducted with permission from the Ethical Committee, Lund University, Sweden. AAV patients were, in most cases, in remission, as assessed by Birmingham Vasculitis Activity Score (BVAS). They all had a diagnosis of MPA or GPA. Patient data are summarized in Table 1.
Table 1.
Patient data
| Subpopulations | MØ phagocytosis | PMN edibility | |
|---|---|---|---|
| HBDs, n | 23 | 10 | 11 |
| Age median HBDs (IQR) | 36 (26–48) | 36 (30–45) | 34 (22–40) |
| AAV patients, n | 28 | 10 | 11 |
| MPO ANCA, n | 7 | 4 | 2 |
| PR3 ANCA, n | 20 | 6 | 8 |
| PR3 + MPO, n | 1 | – | – |
| No ANCA, n | – | – | 1 |
| Age median AAV (IQR) | 63 (50–76) | 66 (50–78) | 69 (65–77) |
| BVAS median (IQR) | 0 (0–2) | 0 (0–1·5) | 0 (0–3) |
| Glucocorticoid dose AAV (mg/day) median (IQR) | 6·25 (2·5–14·38) | 11·25 (5–15) | 15 (7·5–20) |
| Rituximab (percentage of patients in the study) | 14 | 20 | 27 |
| Cyclophosphamide (percentage of patients in the study) | 25 | 70 | 64 |
| Azathioprine (percentage of patients in the study) | 25 | 10 | 18 |
| Disease controls, n | 14 (TP) | 10 (SLE) | 12 (SLE) |
| Age median disease controls (IQR) | 57 (47–67) | 41 (28–46) | 59 (45–65) |
| SLEDAI median (IQR) | – | 4 (1·5–4) | 0 (0–2) |
| Glucocorticoid dose disease controls (mg/day) median (IQR) | 2·5 (2·5–5·0) | 1 (0–8·125) | 5 (0–5) |
IQR: interquartile range; BVAS: Birmingham Vasculitis Activity Score; SLEDAI: systemic lupus erythematosus (SLE) disease activity index; PMN: polymorphonuclear neutrophils; TP: transplant patients. Not all donors were present in all variants of the experiments due to too-small cell numbers or other experimental circumstances. In the monocyte-derived macrophages (MØ) phagocytosis experiments donors were missing in the 1:2 MØ : apoptotic cell ratio for healthy blood donors (HBD): three missing, anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV): two missing and SLE: six missing donors. In the PMN edibility experiments, donors were missing for MØ donor 1; 1:2 ratio: HBD: two missing, AAV: three missing. Ratio 1:4: HBD: four missing, AAV: three missing. Missing values for MØ donor 2; 1:2 ratio: HBD: one missing, AAV: two missing. 1:4 ratio: HBD: one missing.
Differential blood cell counts
Six ml blood was taken from AAV patients, patients with a renal transplant (TP) and HBDs and sent to the Clinical Chemistry laboratory at Skåne University Hospital for differential blood cell counts. Monocytes, neutrophils, lymphocytes, eosinophils and basophils were reported in absolute numbers.
Monocyte and plasmacytoid dendritic cells (pDC) counts
Venous blood was drawn in sodium heparin tubes and diluted in 0·9% NaCl before applying it on Lymphoprep™ (Axis-Shield, Oslo, Norway). The buffy coat was isolated and, in cases of red blood cell contamination, lysed using dH2O for 10–15 s. Cells were then washed in staining buffer [0·2% bovine serum albumin (BSA), 0·05% sodium azide in phosphate-buffered saline (PBS), pH adjusted to 7·4] and stained with an antibody cocktail consisting of six different antibodies: CD303-fluorescein isothiocyanate (FITC) (Miltenyi Biotec, Bergisch Gladbach, Germany), mouse anti-human immunoglobulin (Ig)G1 CD123-phycoerythrin (PE) (Becton Dickinson AB, Stockholm, Sweden), human leucocyte antigen D-related peridinin chlorophyll (HLA-DR-PerCP) (Becton Dickinson AB), mouse anti-human IgG2a CD14 PE-cyanin (Cy™)7 (Becton Dickinson AB), CD45-AmCyan (Becton Dickinson AB, Stockholm, Sweden) and mouse anti-human IgG1 CD16 AlexaFluor 647 (Becton Dickinson AB). Different subpopulations of monocytes and DCs were analysed using a fluorescence activated cell sorter (FACS)Canto™II flow cytometer. The results were analysed using FACSDiva version 6.1.3 software. Absolute numbers were calculated based on the results from the same sample sent for differential blood cell counts.
Macrophage preparation
Buffy coat was isolated from whole blood using Lymphoprep™. Monocytes were isolated from buffy coat using magnetic activated cell sorting (MACS®) and positive selection with CD14 MicroBeads (Miltenyi Biotec); 0·5 × 106 monocytes/well were cultured in 24-well plates at 37°C, 5% CO2. These were differentiated for 7–9 days in AIM-V (Invitrogen, Paisley, UK) supplemented with 10% heat-inactivated fetal calf serum (HI FCS) (Invitrogen, UK) and 5 ng/ml macrophage colony-stimulating factor (M-CSF) (Peprotech, London, UK) before they were used in the phagocytosis assay. Medium was changed twice during the differentiation period.
PMN isolation and apoptotic cells
PMNs were isolated from whole blood using Lymphoprep™ and red blood cell lysis with freshly made sterile NH4Cl buffer (8·3 g/l NH4Cl (Sigma-Aldrich, Steinheim, Germany), 37 mg/l EDTA (ICN Biomedicals, Aurora, OH, USA) 1 g/l KHCO3 (Merck, Darmstadt, Germany) in ddH2O, pH 7·4). All steps after density gradient centrifugation were carried out at 4°C in order to prevent cell activation. Isolated PMNs were stained subsequently with CellTracker™ Green (0·2 µM) or Violet (5 µM) (Invitrogen, UK) for 15 min on ice. After washing, they were cultured in 37°C, 5% CO2 for 22 h in AIM-V with 10% HI FCS in HydroCell plates (Thermo Fisher Scientific, Roskilde, Denmark) to render them apoptotic. Jurkat cells were stained with the fluorescent dye CellTracker™ Green, as above, but in 37°C and rendered apoptotic with 2 µM Staurosporine (Sigma-Aldrich) for 3 h. Cells were washed in PBS before use in the phagocytosis assay to remove residual Staurosporine that could affect the phagocytic ability.
Phagocytosis assay
Apoptotic cells (1 × 106 or 2 × 106 cells/well) were added to the macrophages in their 24-well plates at a density of 0·5 × 106 macrophages/well and were incubated at 37°C for 1 h. The assay was performed in the presence of 10% HI FCS except in the serum experiments. Cells were loosened from the plates using 0·5% EDTA (ICN Biomedicals) in PBS (Invitrogen, UK) for 5 min in 37°C. This was performed twice. The cells were then washed in staining buffer, as above, and stained for CD206 using monoclonal antibody (mAB) mouse anti-human CD206 (AbD Serotec, Oxford, UK) and goat anti-mouse IgG AlexaFluor 647-R-phycoerythrin (Invitrogen, Eugene, OR, USA). Phagocytosis was measured using flow cytometry on a BD FACSCanto™ II and analysed with FlowJo version 8·7 software.
Ingestion/bound assay
Phagocytosis assays were performed as above, but the cells were stained with mouse anti-human IgMκ CD66b-FITC (Becton Dickinson AB) together with mouse anti human IgG1κ CD206, AlexaFluor® 647 conjugated (Biolegend, San Diego, CA, USA) in staining buffer. Mouse anti-human IgG1κ, AlexaFluor® 647 and FITC mouse IgMκ (both from Becton Dickinson AB) were used as isotype controls. Cells were washed before staining and analysed as above. In these experiments MØs and PMNs were isolated from HBDs.
Surface expression of ANCA antigens
Macrophages were stained for the ANCA antigens PR3 and MPO using monoclonal antibodies 4A5 (directed against PR3) and 2B11 (directed against MPO) (both from Wieslab, Lund, Sweden) conjugated directly with AlexaFluor® 647. Mouse anti human IgG1κ, AlexaFluor® 647 (Becton Dickinson AB) were used as an isotype control. Briefly, cells were stained in staining buffer for 20 min at 4°C, washed and resuspended in staining buffer. Cells were measured using a BD FACSCanto™ II machine. Results were analysed with FlowJo version 8·7 software.
Apoptosis
Apoptotic cells used in the phagocytosis assay were analysed for apoptosis and necrosis using flow cytometry. Apoptosis was measured with Cy5™ Annexin V (Becton Dickinson AB) or annexin V AlexaFluor® 488 (Invitrogen, USA), and necrosis with BD Via-Probe™ (Becton Dickinson AB). Cells were measured immediately after staining in FACSCanto™II and analysed using FlowJo version 8·7 software.
Serum samples and IgG depletion
Serum samples were collected and pooled for use in the phagocytosis assay. All sera were handled in the same way. IgG depletion of pooled serum was performed using Protein G Sepharose 4 FastFlow (GE Healthcare, Uppsala, Sweden) and Protein A Sepharose CL-4B (GE Healthcare) using the same volume of Sepharose beads as volume of serum. The sera were incubated with the beads at 4°C with rotation overnight and then spun down at 600 g for 1 min. Supernatant was collected and used as IgG-depleted serum. IgG depletion was confirmed in an enzyme-linked immunosorbent assay (ELISA) coated with sera before and after depletion. Detection was performed with a goat anti-human IgG (Fc-specific) AP antibody (Sigma-Aldrich) and phosphatase substrate tablets (Sigma-Aldrich).
Statistical analyses
Statistical analyses were performed using GraphPad Prism version 5·0b. Phagocytosis experiments were analysed by the Mann–Whitney test. Correlation calculations were analysed by the Spearman correlation test that does not assume a Gaussian distribution. pDC and monocyte data were analysed by the Kruskal–Wallis test and Dunn's multiple comparison test. P values < 0·05 were considered statistically significant.
Results
Plasmacytoid dendritic cells (pDCs), but not monocyte subpopulations, differ quantitatively when comparing AAV patients with HBDs
In this study we investigated if there are inherited or acquired defects in the phagocytosis of dying PMNs. First we wanted to verify that the monocytes did not differ in their pro-/anti-inflammatory differentiation or in numbers. We therefore performed differential blood cell counts on HBD (n = 23), AAV (n = 28) and patients with a renal transplant (TP) (n = 14). Patients were in remission or had a low disease activity (see Table 1 for clinical data of patients used in this study). The total monocyte counts did not differ between the three groups (mean value for HBDs: 0·49 × 109 cells/l, AAV: 0·59 × 109 cells/l and TP: 0·65 × 109 cells/l) (Fig. 1a). There was a significantly increased number of PMNs in AAV patients compared to HBDs (mean value: 6·0 × 109 cells/l compared to 3·8 × 109 cells/l). PMN numbers in TP patients were also increased (mean value: 4·7 × 109 cells/l), although the increases were not significant as for AAV patients. Conversely, the numbers of lymphocytes were decreased in AAV patients compared to HBDs (Fig. 1a) (mean values for AAV: 1·2 × 109 cells/l, HBDs: 1·8 × 109 cells/l and TP: 1·4 × 109 cells/l).
Fig. 1.
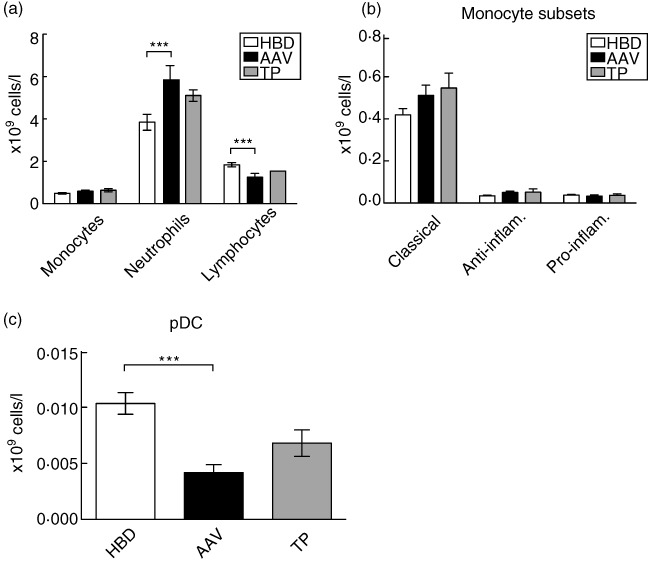
Monocyte and plasmacytoid dendritic cell (pDC) data. (a) Differential cell counts of monocytes, neutrophils and lymphocytes from whole blood from healthy blood donors (HBD), anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) and transplant (TP) patients. (b) Monocyte subsets were measured using flow cytometry and divided into three subsets: classical (CD14highCD16low), anti-inflammatory (CD14highCD16high) and proinflammatory (CD14lowCD16high). (c) pDCs were measured using flow cytometry as CD303 and CD123 positive cells. Where statistical differences were found these are depicted in the figures as *P < 0·05; **0·01 > P < 0·05; and ***P < 0·001. Statistics were calculated using the Kruskal–Wallis test with Dunn's multiple comparison test in all cases. Error bar represents standard error of the mean.
Monocyte subsets were examined further using flow cytometry. Identified by forward- and side-scatter and CD45 positive expression, monocytes were subdivided further based on their expression of CD14 and CD16 into classical (CD14highCD16low), anti-inflammatory (CD14highCD16high) and proinflammatory (CD14lowCD16high) monocytes [27,28]. As seen in Fig. 1b, there were no significant differences in monocyte subsets between HBDs, AAV or TP patients.
pDC subsets were also examined by flow cytometry. From peripheral blood mononuclear cells (PBMCs) as identified by CD45- and HLA-DR-positive cells, pDCs were identified further as CD123- and CD303-positive cells. In AAV patients, the number of pDCs was decreased significantly compared to HBDs (mean value: 4·2 × 106 cells/l compared to 10·4 × 106 cells/l). In TP, we saw the same tendency as for AAV (mean value: 6·8 × 106 cells/l), but this was not significant (Fig. 1c).
Macrophages from AAV and SLE patients have similar phagocytic ability as macrophages from HBDs
In order to compare phagocytic ability of MØs, cells were prepared from HBDs, AAV and SLE patients (see Table 1) and were allowed to engulf apoptotic cells (PMNs from HBD donors or Jurkat cells) during 1 h; the apoptotic cells were labelled fluorescently with CellTracker™ Green. Phagocytosis was measured as a percentage of CellTracker™ Green-stained CD206-positive MØs using flow cytometry (Fig. 2). These experiments were performed in the presence of 10% HI FCS to enable intrinsic differences between the different groups to be detected.
Fig. 2.
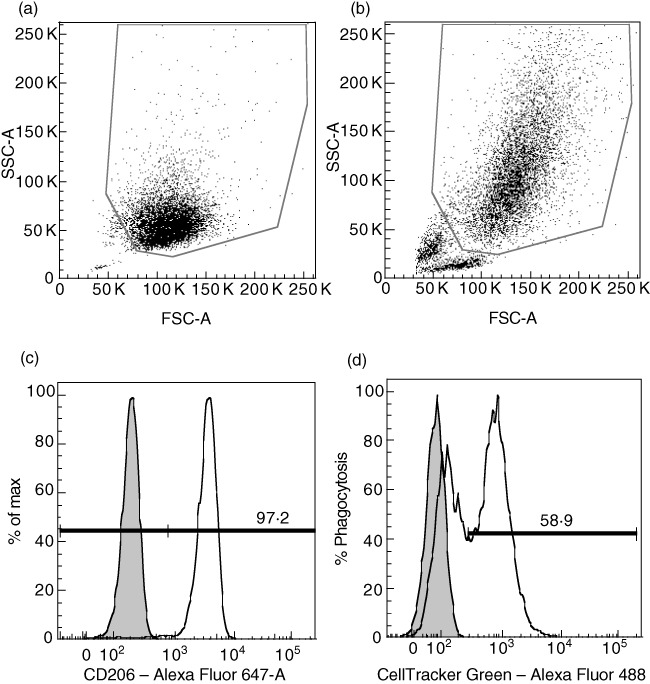
Example of flow cytometry results for phagocytosis experiments. (a) Forward-/side-scatter (FSC/SSC) gate showing control with only monocyte-derived macrophages (MØs). (b) FSC/SSC gate showing MØs that have engulfed apoptotic Jurkat cells. (c) From the FSC/SSC gate, cells are further gated for the MØ-specific marker CD206. Filled histogram represents unstained cells without apoptotic cells. (d) Percentage of CellTracker™ Green-stained cells out of CD206-positive cells is used as a measure of phagocytosis. This represents an experiment where healthy blood donors (HBD) MØs were allowed to feed on apoptotic Jurkat cells at a MØ : Jurkat ratio of 1:2 during 1 h. Filled histogram represents control cells that did not engulf any apoptotic cells and were single-stained for CD206.
We used Jurkat cells as a control in order to compare results more effectively. Because this is a cell line, we could expect these experiments to be more stable on a day-to-day basis compared to PMNs. We thus calculated relative phagocytosis by dividing the phagocytosis value taken from one PMN experiment with the value from the corresponding Jurkat experiment performed on the same day and with the same donor.
At a MØ : apoptotic cell ratio of 1:4, we did not detect any significant differences between any of the groups (mean value of relative phagocytosis: HBD: 77% AAV: 73%, SLE: 69%) (see Fig. 3a, right panel). At a ratio of 1:2, the mean values were, for HBD: 67%, AAV: 64%, SLE: 55%, and we could see a tendency for AAV MØ phagocytic ability to be decreased compared to HBDs, although its effect was not statistically significant (P = 0·072) (see Fig. 3a, left panel). The difference between the two MØ : apoptotic cell ratios might be explained by a saturation effect when increasing the amount of apoptotic material for the MØs. When comparing the phagocytic activity of apoptotic Jurkat cells only, there were still no significant differences between HBD MØs and AAV MØs. There was a small yet significant decrease in phagocytosis of MØs from SLE patients compared to HBDs at a MØ : Jurkat cell ratio of 1:4 (P = 0·037) (data not shown).
Fig. 3.
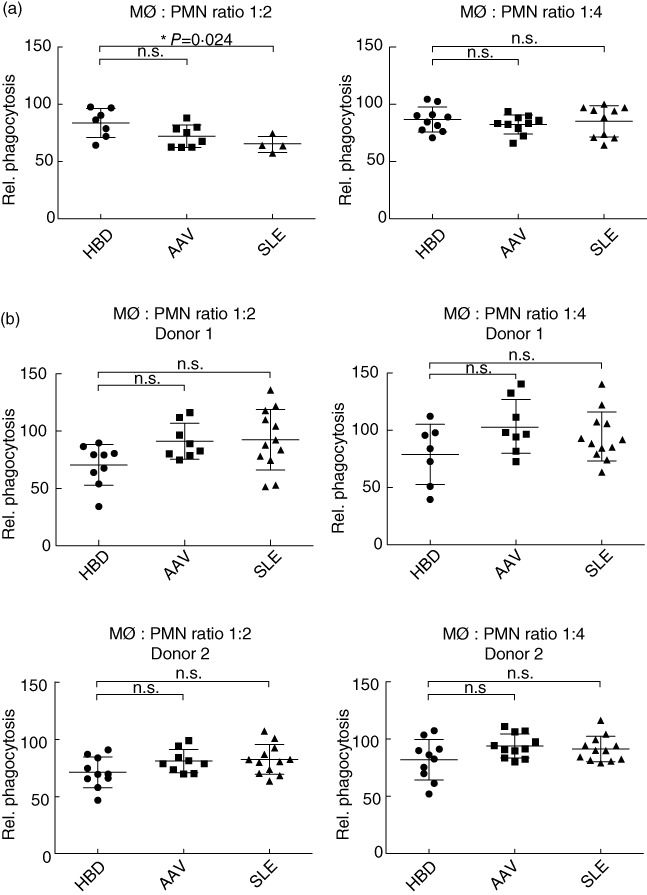
Phagocytosis results. Phagocytosis of apoptotic polymorphonuclear neutrophils (PMNs) by monocyte-derived macrophages (MØ). Phagocytosis is measured as % CellTracker™ Green-positive MØ (CD206-positive cells). Relative phagocytosis represents phagocytosis values normalized to phagocytosis of apoptotic Jurkat cells. Values are corrected by subtracting values from a control sample stained only for CD206. (a) MØ from healthy blood donors (HBD), anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) and systemic lupus erythematosus (SLE) patients compared for their ability to engulf apoptotic PMNs from HBD donors. Left panel corresponds to a MØ/PMN ratio of 1:2 (i.e. 0·5 × 106 MØ incubated with 1 × 106 PMNs). Number of MØ donors: HBD: seven, AAV: eight and SLE: four. Right panel corresponds to a ratio of 1:4 (i.e. 0·5 × 106 MØ incubated with 2 × 106 PMNs). There were 10 MØ donors in each group. (b) Apoptotic PMNs from HBDs, AAV and SLE patients compared for their ability to become engulfed by MØ derived from two different HBD donors; the results from the different donors are shown below each other. Left panel corresponds to a MØ/PMN ratio of 1:2; right panel corresponds to a ratio of 1:4. The number of PMN donors in the 1:2 ratio: HBD: nine, AAV: eight, SLE: 12 for donor 1 and HBD: 10, AAV: nine, SLE: 12 for donor 2. The number of PMN donors in the 1:4 ratio: HBD: seven, AAV: eight, SLE: 12 for donor 1 and HBD: 10, AAV: 11, SLE: 12 for donor 2. Each point represents a single value or in some cases a mean value of two replicates. Error bar represents standard deviation and statistics are calculated using the Mann–Whitney test. P-values for significant results are given in the figure; non-significant values are annotated n.s.
The level of apoptosis of PMNs and Jurkat cells was measured in each experiment by flow cytometry. Due to variation in our results, we correlated the level of apoptosis with phagocytosis to rule out the possibility that different levels of apoptosis affected the phagocytosis results in experiments performed on different days. We did not detect any such correlation, with one exception: PMN apoptosis in MØ : PMN at a ratio of 1:2 in the experiments investigating MØs, r = 0·56 (data not shown).
PMNs from AAV and SLE patients have similar ability to become eaten by macrophages as PMNs from HBDs
We did not detect any significant differences in the ability of MØs to engulf apoptotic cells when comparing HBDs with AAV patients; we therefore turned our focus to PMNs and investigated whether apoptotic PMNs from AAV patients have a lower tendency to become engulfed compared to HBD PMNs. This would support the theory that apoptotic cells accumulate in AAV. In these experiments we used two HBD monocyte donors for all assays. The experiments were normalized to phagocytosis of apoptotic Jurkat cells, as described above. We did not detect any significant differences in either AAV or SLE patients compared to HBDs, irrespective of the MØ donor or MØ : PMN ratio (see Fig. 3b).
As a control experiment, we stained the cells in the phagocytosis assay with a neutrophil-specific antibody directed against CD66b after ingestion, in addition to CellTracker® staining of PMNs and CD206 staining of MØ. The results show that CD66b can be detected on only a small subset of the CellTracker-stained MØ[mean of three experiments: 9·7% ± 3·5 standard deviations (s.d.)]. This indicates that we mainly detect ingested apoptotic cells in our assay, but also some bound or partly ingested cells.
Sera from AAV patients lower the phagocytic ability of macrophages
We did not detect any intrinsic differences in phagocytosis in the immune cells; we therefore investigated if serum factors affect phagocytic ability using MØ taken from four HBDs and Jurkat cells as apoptotic cells. After induction of apoptosis, we added 10% serum from pools of eight HBDs, AAV or SLE patients in the phagocytosis assay. In the experiments with AAV serum, we could see that phagocytosis tends to be decreased (mean value: 36%) compared to HBD serum (mean value: 43%), but this did not reach significance. This effect was not seen for SLE serum (mean value: 45%) (see Fig. 4a).
Fig. 4.
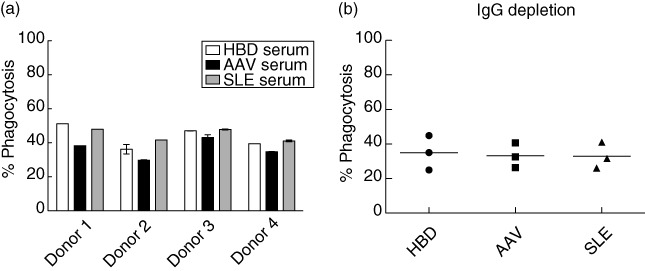
Phagocytosis in the presence of serum. (a) Four healthy blood donors (HBD) monocyte-derived macrophages (MØ) donors were incubated with CellTracker™ Green-stained apoptotic Jurkat cells at a MØ : Jurkat ratio of 1:2 in the presence of 10% pooled serum from HBDs, anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) or systemic lupus erythematosus (SLE) patients with eight donors in each pool. A decrease in phagocytosis was seen when adding AAV serum compared to HBD- and SLE serum, although this did not reach significance. This graph represents one experiment for each donor, but with duplicate samples for all except MØ donor 1. (b) Immunoglobulin (Ig)G depletion of serum from the same serum pools as in (a). This experiment was performed as in (a), but with three HBD MØ donors. Percentage phagocytosis is measured as % CellTracker™ Green-stained CD206-positive MØ. Statistics were calculated using the Mann–Whitney test and error bar represents standard deviation in cases where duplicate samples exist.
Serum IgG was depleted from the serum pools and phagocytic ability was subsequently lowered, as expected, as the Fc-receptor mediated phagocytosis would be lost after depletion of IgG. Unexpectedly, the difference seen before depletion was lost and all three depleted sera stimulated phagocytosis to a similar extent (mean value for HBDs: 35%, AAV: 33% and SLE: 33%) (see Fig. 4b).
ANCA antigens are expressed on the surface of macrophages
Because we used Jurkat cells in the serum experiments, we speculate that AAV serum could affect the MØs, rather than Jurkat cells, as they do not express ANCA antigens. We therefore measured PR3 and MPO surface expression on monocyte-derived MØ from a HBD. The results show a high expression of both antigens (91% of cells were PR3-positive, 97% were MPO-positive when isotype control had been subtracted). Whether or not the surface expression of the ANCA antigens on MØs exhibit individual variation and whether this has any impact on phagocytosis ability remains to be established.
Discussion
The purpose of this study was to investigate whether there is a cellular defect in AAV that could explain an impaired clearance of apoptotic cells as an inducer of autoimmunity. If apoptotic cells are not cleared properly, this could lead to an accumulation of these cells that progresses eventually to secondary necrosis. This has been proposed as a pathogenic mechanism for autoimmune diseases [17,29]. Our first approach was to determine if the cell numbers of different immune cells are normal in AAV patients compared to HBDs. In peripheral blood we detected an increased number of PMNs in AAV patients. This could be explained partly by the treatment with glucocorticoids that most of these patients were taking, because glucocorticoids are known to prolong PMN survival [30,31]. Therefore, we used patients on renal transplant therapy as a treatment control; their PMN numbers were also increased, although not as greatly as for AAV patients. This could indicate that PMNs overwhelm the phagocytic system in AAV leading to an initiation of the immune response and eventually autoimmunity. The decreased number of lymphocytes in AAV patients could also be a treatment-related effect, as these patients are treated with a varying amount of immunosuppressive drugs as well as cytotoxic drugs, all of which dampens the immune system in different ways. However, we did not detect any significant difference between AAV patients in the active state of their disease versus patients in remission, and no differences were found between different treatment regimens.
In our studies, pDCs were found to be decreased in AAV compared to both transplant patients and HBDs. pDCs are important for fighting viral infections and also have regulatory effects on other immune cells [32]. Recently, Rimbert et al. have described a decrease in pDCs in AAV patients, and this could reflect migration of these cells to tissues [33]. Interestingly, Rimbert et al. also showed an increased expression of CD62L on AAV pDCs, indicating that these pDCs could be recruited more easily to lymph nodes, where they could initiate an immune response.
We looked for clearance defects either in phagocytic ability of MØs or ability of apoptotic PMNs to be engulfed, and we did not detect any such intrinsic defect in AAV PMNs in the presence of heat-inactivated FCS. In theory, PMNs from AAV patients would lower the phagocytosis ability of MØs, as they generally express more PR3 on their surface compared to HBDs [34], and PR3 surface exposure during apoptosis seems to be a ‘don't eat me’ signal [8]; however, in that study they used PR3 transfected RBL cells and not PMNs. Thus, this does not have to apply for PMNs.
There was, however, a small decrease, although not statistically significant, in the phagocytic ability of MØs in AAV patients and SLE patients (P = 0·02) compared to HBDs when looking at the 1:2 MØ : PMN ratio. No differences were detected in the 1:4 ratio; this effect can possibly be explained by a saturation effect when increasing the amount of apoptotic cells. Both the AAV patients and SLE patients were generally taking glucocorticoids that could possibly skew these results, as glucocorticoids are known to augment phagocytosis [35]. In our study, however, we did not find any correlation between glucocorticoid dosage and phagocytic ability, nor did we find any correlation between phagocytosis ability and administration of rituximab, cyclophosphamide or azathioprine, although there were low numbers of patients in each treatment group (data not shown).
The age of AAV patients was significantly higher than HBDs, but no correlation was found between age of MØ donor and their phagocytosis capacity (data not shown). Studies are inconclusive with regard to phagocytic ability and ageing individuals [36]. However, it is clear that the innate immune system is altered with age in various ways [37].
We show that we detected mainly ingested cells in the phagocytosis assay, because the phagocyting MØs were positive for the neutrophil specific marker CD66b only to a small extent. We believe that these CD66b-positive MØs represent partly ingested PMNs, and as phagocytosis is a continuous process there will always be partly ingested cells as long as there is enough apoptotic material.
When adding serum from HBDs, AAV and SLE patients, AAV serum tended to stimulate phagocytosis less well, but this did not reach significance; however, the numbers were small. These results contradict the general idea that clearance is impaired in SLE, although whether this is due to cellular defects or other factors is still under debate [19,38,39]. These contradictory results could both be due to individual variations in patients and differences in experimental protocols. Our finding that AAV sera seems to lower the phagocytic ability of MØs was surprising, as it has been reported previously by Moosig et al. that apoptotic PMNs preincubated with ANCA lead to an increased uptake by MØs compared to preincubation with control IgG [40]. However, Moosig et al. detected phagocytosis by measuring MPO activity in MØ lysates after ingestion. Furthermore, we speculate that the ANCA might affect and bind to MØs rather than the apoptotic Jurkat cells used in our experiments, as they lack the ANCA antigens and FcRs on their surface [41]. To support this, we show that ANCA antigens are expressed readily on the surface of MØs; however, if this has any impact on phagocytosis ability remains to be established. Little is known about how ANCAs affect the monocyte/macrophage population, although it has been shown that the isolation procedure as well as tumour necrosis factor (TNF)-α priming affects the level of surface expression of ANCA antigens in monocytes [42]. Furthermore, there are reports showing that incubating monocytes with ANCA-positive sera leads to increased production of ROS [43] and proinflammatory cytokines [42], indicating that the ANCAs can affect the monocytes to a more inflammatory phenotype. We speculate that ANCA might bind to the MØs, affecting its ability to engulf cells in our serum experiments either by binding to the ANCA antigens on the MØ surface, via Fc receptors, or both, but this requires further experiments to clarify fully.
In conclusion, this study does not confirm our hypothesis that there is a cellular phagocytic defect in MØs or apoptotic PMNs in AAV. The monocyte number, subclass distribution as well as phagocytic ability did not differ between AAV and HBDs when investigating patients in remission, nor was any difference seen in the edibility of apoptotic PMNs. However, PMN numbers were increased in circulation, which could lead possibly to an accumulation of apoptotic PMNs around the vessels and an increased antigen exposure. The fact that the number of pDCs is decreased in circulation at the same time could indicate that these have migrated to lymph nodes to initiate an immune response against the ANCA antigens PR3 and MPO. Furthermore, we found that AAV serum seems to lower the phagocytic ability of MØs, and this could be caused by ANCA. This does not explain how AAV develop in the first place, but might indicate that dysfunctional phagocytosis in AAV is a consequence of ANCA rather than the initiating factor of these.
Acknowledgments
The authors thank the nurses at the Nephrology and Rheumatology departments at Skåne University Hospital for their assistance in collecting blood samples and Lena Gunnarsson for skilful technical assistance. This work was funded by the Crafoord foundation, Thure Carlssons Minnesfond, Birgit o Sven Håkan Ohlssons foundation and the Swedish Research Council 65x-15152.
Disclosure
The authors declare no conflict of interest.
References
- 1.Kallenberg CG, Heeringa P, Stegeman CA. Mechanisms of disease: pathogenesis and treatment of ANCA-associated vasculitides. Nat Clin Pract Rheumatol. 2006;2:661–70. doi: 10.1038/ncprheum0355. [DOI] [PubMed] [Google Scholar]
- 2.Falk RJ, Gross WL, Guillevin L, et al. Granulomatosis with polyangiitis (Wegener's): an alternative name for Wegener's granulomatosis. Ann Rheum Dis. 2011;70:704. doi: 10.1136/ard.2011.150714. [DOI] [PubMed] [Google Scholar]
- 3.Falk RJ, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies with specificity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotizing and crescentic glomerulonephritis. N Engl J Med. 1988;318:1651–7. doi: 10.1056/NEJM198806233182504. [DOI] [PubMed] [Google Scholar]
- 4.Ludemann J, Utecht B, Gross WL. Detection and quantitation of anti-neutrophil cytoplasm antibodies in Wegener's granulomatosis by ELISA using affinity-purified antigen. J Immunol Methods. 1988;114:167–74. doi: 10.1016/0022-1759(88)90169-x. [DOI] [PubMed] [Google Scholar]
- 5.Niles JL, McCluskey RT, Ahmad MF, Arnaout MA. Wegener's granulomatosis autoantigen is a novel neutrophil serine proteinase. Blood. 1989;74:1888–93. [PubMed] [Google Scholar]
- 6.Halbwachs-Mecarelli L, Bessou G, Lesavre P, Lopez S, Witko-Sarsat V. Bimodal distribution of proteinase 3 (PR3) surface expression reflects a constitutive heterogeneity in the polymorphonuclear neutrophil pool. FEBS Lett. 1995;374:29–33. doi: 10.1016/0014-5793(95)01073-n. [DOI] [PubMed] [Google Scholar]
- 7.Rarok AA, Limburg PC, Kallenberg CG. Neutrophil-activating potential of antineutrophil cytoplasm autoantibodies. J Leukoc Biol. 2003;74:3–15. doi: 10.1189/jlb.1202611. [DOI] [PubMed] [Google Scholar]
- 8.Kantari C, Pederzoli-Ribeil M, Amir-Moazami O, et al. Proteinase 3, the Wegener autoantigen, is externalized during neutrophil apoptosis: evidence for a functional association with phospholipid scramblase 1 and interference with macrophage phagocytosis. Blood. 2007;110:4086–95. doi: 10.1182/blood-2007-03-080457. [DOI] [PubMed] [Google Scholar]
- 9.Witko-Sarsat V, Reuter N, Mouthon L. Interaction of proteinase 3 with its associated partners: implications in the pathogenesis of Wegener's granulomatosis. Curr Opin Rheumatol. 2010;22:1–7. doi: 10.1097/BOR.0b013e3283331594. [DOI] [PubMed] [Google Scholar]
- 10.Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA. 1990;87:4115–9. doi: 10.1073/pnas.87.11.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao H, Heeringa P, Hu P, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest. 2002;110:955–63. doi: 10.1172/JCI15918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Little MA, Al-Ani B, Ren S, et al. Anti-proteinase 3 anti-neutrophil cytoplasm autoantibodies recapitulate systemic vasculitis in mice with a humanized immune system. PLoS ONE. 2012;7:e28626. doi: 10.1371/journal.pone.0028626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stegeman CA, Tervaert JW, Sluiter WJ, Manson WL, de Jong PE, Kallenberg CG. Association of chronic nasal carriage of Staphylococcus aureus and higher relapse rates in Wegener granulomatosis. Ann Intern Med. 1994;120:12–7. doi: 10.7326/0003-4819-120-1-199401010-00003. [DOI] [PubMed] [Google Scholar]
- 14.Kain R, Exner M, Brandes R, et al. Molecular mimicry in pauci-immune focal necrotizing glomerulonephritis. Nat Med. 2008;14:1088–96. doi: 10.1038/nm.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kain R, Tadema H, McKinney EF, et al. High prevalence of autoantibodies to hLAMP-2 in anti-neutrophil cytoplasmic antibody-associated vasculitis. J Am Soc Nephrol. 2012;23:556–66. doi: 10.1681/ASN.2011090920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roth AJ, Brown MC, Smith RN, et al. Anti-LAMP-2 antibodies are not prevalent in patients with antineutrophil cytoplasmic autoantibody glomerulonephritis. J Am Soc Nephrol. 2012;23:545–55. doi: 10.1681/ASN.2011030273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rovere P, Sabbadini MG, Fazzini F, et al. Remnants of suicidal cells fostering systemic autoaggression. Apoptosis in the origin and maintenance of autoimmunity. Arthritis Rheum. 2000;43:1663–72. doi: 10.1002/1529-0131(200008)43:8<1663::AID-ANR1>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 18.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 19.Katsiari CG, Liossis SN, Sfikakis PP. The pathophysiologic role of monocytes and macrophages in systemic lupus erythematosus: a reappraisal. Semin Arthritis Rheum. 2010;39:491–503. doi: 10.1016/j.semarthrit.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 20.Xiao H, Heeringa P, Liu Z, et al. The role of neutrophils in the induction of glomerulonephritis by anti-myeloperoxidase antibodies. Am J Pathol. 2005;167:39–45. doi: 10.1016/S0002-9440(10)62951-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brouwer E, Huitema MG, Mulder AH, et al. Neutrophil activation in vitro and in vivo in Wegener's granulomatosis. Kidney Int. 1994;45:1120–31. doi: 10.1038/ki.1994.149. [DOI] [PubMed] [Google Scholar]
- 22.Abdgawad M, Pettersson A, Gunnarsson L, et al. Decreased neutrophil apoptosis in quiescent ANCA-associated systemic vasculitis. PLoS ONE. 2012;7:e32439. doi: 10.1371/journal.pone.0032439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Travis WD, Hoffman GS, Leavitt RY, Pass HI, Fauci AS. Surgical pathology of the lung in Wegener's granulomatosis. Review of 87 open lung biopsies from 67 patients. Am J Surg Pathol. 1991;15:315–33. doi: 10.1097/00000478-199104000-00001. [DOI] [PubMed] [Google Scholar]
- 24.Ziegler-Heitbrock L. The CD14+ CD16+ blood monocytes: their role in infection and inflammation. J Leukoc Biol. 2007;81:584–92. doi: 10.1189/jlb.0806510. [DOI] [PubMed] [Google Scholar]
- 25.Ohlsson S, Hellmark T, Pieters K, Sturfelt G, Wieslander J, Segelmark M. Increased monocyte transcription of the proteinase 3 gene in small vessel vasculitis. Clin Exp Immunol. 2005;141:174–82. doi: 10.1111/j.1365-2249.2005.02819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alcorta DA, Barnes DA, Dooley MA, et al. Leukocyte gene expression signatures in antineutrophil cytoplasmic autoantibody and lupus glomerulonephritis. Kidney Int. 2007;72:853–64. doi: 10.1038/sj.ki.5002371. [DOI] [PubMed] [Google Scholar]
- 27.Skrzeczynska-Moncznik J, Bzowska M, Loseke S, Grage-Griebenow E, Zembala M, Pryjma J. Peripheral blood CD14high CD16+ monocytes are main producers of IL-10. Scand J Immunol. 2008;67:152–9. doi: 10.1111/j.1365-3083.2007.02051.x. [DOI] [PubMed] [Google Scholar]
- 28.Mikolajczyk TP, Skrzeczynska-Moncznik JE, Zarebski MA, et al. Interaction of human peripheral blood monocytes with apoptotic polymorphonuclear cells. Immunology. 2009;128:103–13. doi: 10.1111/j.1365-2567.2009.03087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140:619–30. doi: 10.1016/j.cell.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 30.Heasman SJ, Giles KM, Ward C, Rossi AG, Haslett C, Dransfield I. Glucocorticoid-mediated regulation of granulocyte apoptosis and macrophage phagocytosis of apoptotic cells: implications for the resolution of inflammation. J Endocrinol. 2003;178:29–36. doi: 10.1677/joe.0.1780029. [DOI] [PubMed] [Google Scholar]
- 31.Zahuczky G, Kristof E, Majai G, Fesus L. Differentiation and glucocorticoid regulated apopto-phagocytic gene expression patterns in human macrophages. Role of Mertk in enhanced phagocytosis. PLoS ONE. 2011;6:e21349. doi: 10.1371/journal.pone.0021349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blanco P, Palucka AK, Pascual V, Banchereau J. Dendritic cells and cytokines in human inflammatory and autoimmune diseases. Cytokine Growth Factor Rev. 2008;19:41–52. doi: 10.1016/j.cytogfr.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rimbert M, Hamidou M, Braudeau C, et al. Decreased numbers of blood dendritic cells and defective function of regulatory T cells in antineutrophil cytoplasmic antibody-associated vasculitis. PLoS ONE. 2011;6:e18734. doi: 10.1371/journal.pone.0018734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muller Kobold AC, Kallenberg CG, Tervaert JW. Leucocyte membrane expression of proteinase 3 correlates with disease activity in patients with Wegener's granulomatosis. Br J Rheumatol. 1998;37:901–7. doi: 10.1093/rheumatology/37.8.901. [DOI] [PubMed] [Google Scholar]
- 35.McColl A, Michlewska S, Dransfield I, Rossi AG. Effects of glucocorticoids on apoptosis and clearance of apoptotic cells. ScientificWorldJournal. 2007;7:1165–81. doi: 10.1100/tsw.2007.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gomez CR, Nomellini V, Faunce DE, Kovacs EJ. Innate immunity and aging. Exp Gerontol. 2008;43:718–28. doi: 10.1016/j.exger.2008.05.0168.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Devitt A, Marshall LJ. The innate immune system and the clearance of apoptotic cells. J Leukoc Biol. 2011;90:447–57. doi: 10.1189/jlb.0211095. [DOI] [PubMed] [Google Scholar]
- 38.Schulze C, Munoz LE, Franz S, et al. Clearance deficiency – a potential link between infections and autoimmunity. Autoimmun Rev. 2008;8:5–8. doi: 10.1016/j.autrev.2008.07.049. [DOI] [PubMed] [Google Scholar]
- 39.Moosig F, Graf D, Knorr-Spahr A, Zeuner RA, Bottcher S, Schroder JO. Monocyte and granulocyte phagocytosis of Escherichia coli particles in systemic lupus erythematosus is not reduced when compared to healthy controls. Lupus. 2003;12:490–2. doi: 10.1191/0961203303lu415xx. [DOI] [PubMed] [Google Scholar]
- 40.Moosig F, Csernok E, Kumanovics G, Gross WL. Opsonization of apoptotic neutrophils by anti-neutrophil cytoplasmic antibodies (ANCA) leads to enhanced uptake by macrophages and increased release of tumour necrosis factor-alpha (TNF-alpha) Clin Exp Immunol. 2000;122:499–503. doi: 10.1046/j.1365-2249.2000.01410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boonyarattanakalin S, Martin SE, Sun Q, Peterson BR. A synthetic mimic of human Fc receptors: defined chemical modification of cell surfaces enables efficient endocytic uptake of human immunoglobulin-G. J Am Chem Soc. 2006;128:11463–70. doi: 10.1021/ja062377w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hattar K, Bickenbach A, Csernok E, et al. Wegener's granulomatosis: antiproteinase 3 antibodies induce monocyte cytokine and prostanoid release-role of autocrine cell activation. J Leukoc Biol. 2002;71:996–1004. [PubMed] [Google Scholar]
- 43.Weidner S, Neupert W, Goppelt-Struebe M, Rupprecht HD. Antineutrophil cytoplasmic antibodies induce human monocytes to produce oxygen radicals in vitro. Arthritis Rheum. 2001;44:1698–706. doi: 10.1002/1529-0131(200107)44:7<1698::AID-ART294>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]