

Abstract
Type I interferon (IFN) medications cause various adverse reactions, including vascular diseases. Although an association between chemokines and vascular diseases has also been reported, the relationship between type I IFN and chemokines in vascular endothelial cells (VEC) remains unclear. To provide clues to pathogenesis of the diseases, we analysed the effects of type I IFN on chemokine production in human VEC. Type I IFN induced higher CX3CL1 (fractalkine) mRNA expression and protein secretion in pulmonary arterial VEC than in umbilical vein VEC. Type I IFN also induced CCL5 [regulated upon activation normal T cell expressed and secreted (RANTES)] production in VEC, especially in lung micro-VEC. IFN-β induced much higher chemokine production than IFN-α, and Janus protein tyrosine kinase (JAK) inhibitor I prevented type I IFN-induced chemokine secretion. Type I IFN-induced chemokines may be involved in the pathophysiology of pulmonary vascular diseases, and the JAK inhibitor may serve as a therapeutic option for these diseases.
Keywords: CCL5 (RANTES), connective tissue disease, CX3CL1 (fractalkine), pulmonary vascular disease, type I interferon
Introduction
Type I interferon (IFN) has been used as a therapeutic drug for the treatment of various diseases [1]; however, type I IFN-induced adverse reactions, including the development of connective tissue diseases (CTD), have been reported [2]. Furthermore, type I IFN has been reported to be a probable pathogenic factor in CTD, such as systemic lupus erythematosus (SLE) [3] and systemic sclerosis (SSc), through vascular pathology [4]. Pulmonary arterial hypertension (PAH) is a severe vascular disease that develops in patients with CTD and has a high mortality rate. Type I IFN therapies, such as IFN-α for hepatitis C [5] and IFN-β for multiple sclerosis [6], result in PAH development. Therefore, type I IFN may be responsible for vascular pathogenesis.
Increased chemokine levels have been reported in patients with vascular diseases; expression of CCL2 (MCP-1) [7], CCL5 [regulated upon activation normal T cell expressed and secreted (RANTES)][8] and CX3CL1 (fractalkine) [9] is augmented in PAH patients. Increased leucocyte migration promoted by these chemokines may enhance inflammation and be involved in the development of vascular disorders.
Although an association of type I IFN or chemokines with vascular diseases has been reported, the relationship between type I IFN and the chemokines in vascular endothelial cells (VEC) remains unclear. To provide clues to pathogenesis of the diseases, we analysed the effects of type I IFN on chemokine production in various VEC [i.e. human umbilical vein endothelial cells (HUVEC), human pulmonary arterial endothelial cells (HPAEC), human aortic endothelial cells (HAEC) and human lung micro-VEC (HLMVEC)].
Materials and methods
Cell culture
Through different manufacturers, VEC were obtained from at least two donors who had given their informed consent. Pooled HUVEC (Lonza, Portsmouth, NH, USA), HPAEC (Life Technologies, Carlsbad, CA, USA) and HAEC (Life Technologies) were maintained in a conditioned medium (Humedia-EG2, Kurabo, Japan). Highly pure HLMVEC (Lonza) were maintained in the medium EGM®-2 MV BulletKit® (Lonza). VEC were cultured until a confluent monolayer was formed on Cellstar® Multiwell Plates (Greiner Bio-One, Frickenhausen, Germany).
Type I IFN stimulation assay
Human recombinant IFN-α2b and IFN-β were purchased from Peprotech (Rocky Hill, NJ, USA) and dissolved in phosphate-buffered saline (PBS) containing 0·1% BSA (Sigma-Aldrich, St Louis, MO, USA).
All experiments were performed in duplicate. All cells were washed with phosphate-buffered saline (PBS) 24 h before stimulation and placed in the same medium, EBM®-2 basal medium containing 5% fetal bovine serum (FBS) (Lonza). In the stimulation assay, VEC received fresh medium (5% FBS in EBM®-2 basal medium) containing IFN-α or IFN-β (0·1, 1 and 10 ng/ml). After 1, 8 and 24 h, RNA was isolated from VEC using the Nucleospin® RNA 2 kit (Macherey-Nagel, Düren, Germany). The culture supernatant was harvested 72 h after stimulation. Endotoxin contamination was excluded routinely using the ToxinSensor® endotoxin detection system (Genscript, Piscataway, NJ, USA).
Janus protein tyrosine kinase (JAK) inhibition assay
Confluent HPAEC were preincubated with JAK inhibitor I (Calbiochem, La Jolla, CA, USA) in EBM®-2 basal medium containing 5% FBS for 2 h, and type I IFN (10 ng/ml) was added to the medium containing JAK inhibitor I. After 30 h, the culture supernatant was harvested.
Detection of signal transducer and activator of transcription 1 (STAT-1) activation
HPAEC preincubated with JAK inhibitor I (1 µM) for 2 h were stimulated with type I IFN (10 ng/ml). After 30 min, STAT-1 phosphorylation was analysed using the RayBio® Cell-Based STAT 1 enzyme-linked immunosorbent assay (ELISA) kit (RayBiotech, Norcross, GA, USA) according to the manufacturer's protocol. In brief, stimulated HPAEC were fixed, quenched, blocked and incubated with primary antibody (anti-STAT-1 or anti-phospho-STAT-1 antibody). After incubation with horseradish peroxidase (HRP)-conjugated secondary antibody, 3,3',5,5'-tetramethylbenzidine (TMB) was added and optical density (OD) was measured at 450 nm. Phosphorylation was calculated as the OD ratio of the phosphorylated to total STAT-1 (standardized to non-stimulated HPAEC).
Measurement of mRNA expression
RNA isolated from VEC was treated with DNase contained in the Nucleospin® RNA 2 kit and reverse transcribed to cDNA using the Superscript® VILO cDNA synthesis kit (Invitrogen, Carlsbad, CA, USA). Each cDNA sample was analysed using the Applied Biosystems 7500 real-time polymerase chain reaction (PCR) system (Life Technologies) using SYBR® Premix Ex Taq 2 (TaKaRa Bio, Shiga, Japan), according to the manufacturer's protocol. Oligonucleotide primers were synthesized by Life Technologies (Tokyo, Japan). The forward and reverse primer sequences from 5′ to 3′ were CAGTCGTCTTTGTCACCCGAA and TCCCAAGCTAGGACAAGAGCA for CCL5, GCTGAGGAACCCATCCAT and GAGGCTCTGGTAGGTGAACA for CX3CL1 and ATTGCCGACAGGATGCAGGAA and GCTGATCCACATCTGCTGGAA for β-actin. The amplification conditions were 30 s at 95°C, followed by 40 cycles of 10 s at 95°C and 1 min at 60°C. Relative quantitation was performed using 7500 Software version 2·0.1 (Life Technologies). The expression levels of CCL5 and CX3CL1 mRNA were standardized to the expression level of β-actin.
Measurement of protein secretion
The culture supernatant was measured by ELISA using Quantikine® human CX3CL1 or CCL5 immunoassays (R&D Systems, Minneapolis, MN, USA) and the BD OptEIA® human CCL2 ELISA kit (BD Biosciences, Franklin Lakes, NJ, USA) according to the manufacturer's protocol. The culture supernatant was diluted to 1:200 before CCL2 measurement.
Immunofluorescence
HPAEC were cultured on cover glasses (Matsunami Glass Industries Ltd, Osaka, Japan), placed in EBM-2® basal medium containing 5% FBS for 24 h and stimulated with type I IFN (50 ng/ml) for 28 h. To block protein transport, monensin (BioLegend, Franklin Lakes, NJ, USA) was added for the last 4 h. HPAEC were fixed with 2% paraformaldehyde. Blocking and permeabilization of cells were performed by incubation with PBS containing 10% FBS and 0·3% TritonX-100, followed by incubation for 1 h at room temperature with goat anti-CX3CL1 (R&D Systems) or anti-CCL5 Ab (Abcam, Tokyo, Japan) diluted to 2 µg/ml. The cells were incubated with FITC-conjugated donkey anti-goat IgG secondary Ab (Abcam) for 1 h at room temperature. The nuclei were stained with TO-PRO®-3 (Invitrogen), and fluorescent labelling was analysed using the LSM 5 Pascal laser scanning microscope (Carl Zeiss, Oberkochen, Germany).
Statistical analysis
Data are presented as mean ± standard error (s.e.). Statistical significance of differences was examined by unpaired two-tailed t-test, and P < 0·05 was considered to be significantly different.
Results
Effects of type I IFN on chemokine induction in HPAEC
We analysed type I IFN-induced chemokine secretion in HPAEC, used as a representative VEC. IFN-β induced significant CX3CL1 secretion in a dose-dependent manner (maximum concentration: 5·9 ± 0·5 ng/ml). Although IFN-α tended to induce CX3CL1 secretion, it was not significant (Fig. 1a), whereas IFN-α and IFN-β induced significant CCL5 secretion in a dose-dependent manner (maximum concentration: 77·4 ± 2·7 pg/ml and 230·9 ± 9·4 pg/ml, respectively: Fig. 1b). However, no significant changes were observed in CCL2 secretion (Fig. 1c); thus, we focused on CX3CL1 and CCL5. In an immunofluorescence study, type I IFN also induced intracellular expression of CX3CL1 and CCL5 proteins in HPAEC (Fig. 2).
Fig. 1.
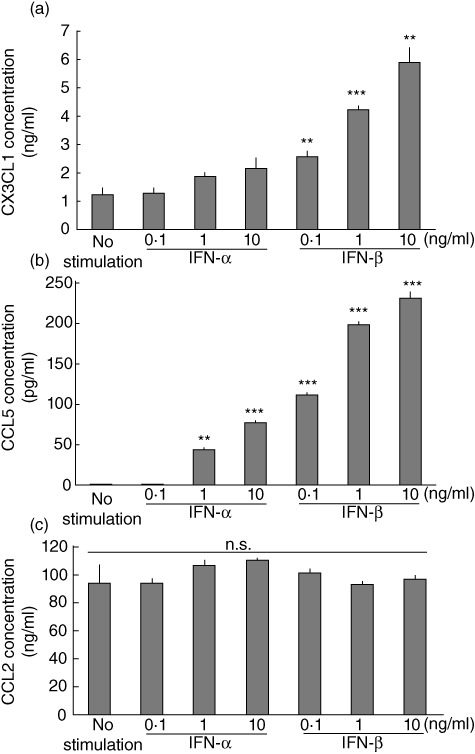
Type I interferon (IFN) increases chemokine concentration in the culture supernatant of human pulmonary arterial endothelial cells (HPAEC). HPAEC were stimulated with IFN-α or IFN-β at the indicated concentrations. Protein levels of (a) CX3CL1 (fractalkine), (b) CCL5 [regulated upon activation normal T cell expressed and secreted (RANTES)] or (c) CCL2 (MCP-1) in the culture supernatant of HPAEC at 72 h. Data from three independent experiments are presented as mean ± standard error. **P < 0·01; ***P < 0·001 in comparison with non-stimulated controls.
Fig. 2.
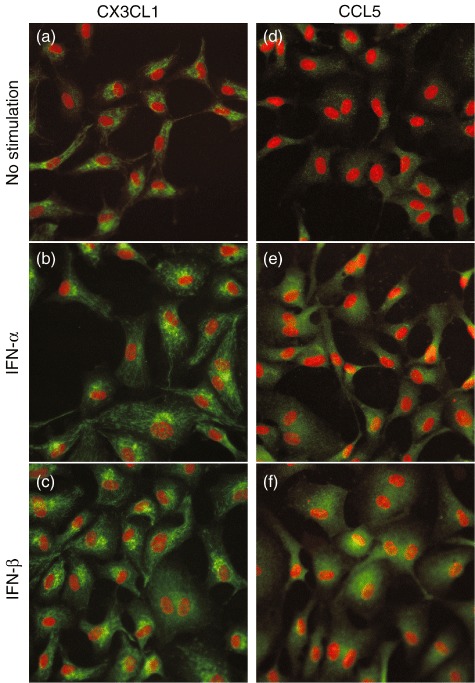
Type I interferon (IFN) induces chemokine protein production in human pulmonary arterial endothelial cells (HPAEC). (a,d) Unstimulated; (b,e) IFN-α-stimulated; and (c,f) IFN-β-stimulated HPAEC stained by anti-CX3CL1 or anti-CCL5 antibody, respectively (green). The nuclei were stained with TO-PRO®-3 (red). Representative immunofluorescence images from three independent experiments are shown.
Type I IFN induces CX3CL1 in VEC
IFN-α stimulation induced CX3CL1 mRNA expression in HPAEC, HAEC, HLMVEC and HUVEC, and maximum expression was achieved at 8 h (Fig. 3a). IFN-β induced CX3CL1 mRNA expression intensely in VEC, and maximum expression was observed at 8 h (Fig. 3b). Dose-dependent induction of CX3CL1 mRNA by both IFN-α and IFN-β was observed at 8 h. A quantitative difference was also observed among different VEC. Induction of mRNA expression in HAEC (maximum induction: 25·3 ± 0·9; P < 0·05) and HPAEC (maximum induction: 22·5 ± 2·6; P < 0·05) was significantly higher than that in HUVEC (maximum induction: 9·1 ± 3·1); induction in HLMVEC (maximum induction: 20·1 ± 8·9) maintained this tendency (Fig. 3c). Preincubation of anti-IFN-α or anti-IFN-β antibody with corresponding cytokine blocked IFN-α- or IFN-β-induced CX3CL1 mRNA expression, respectively (Fig. S1a).
Fig. 3.
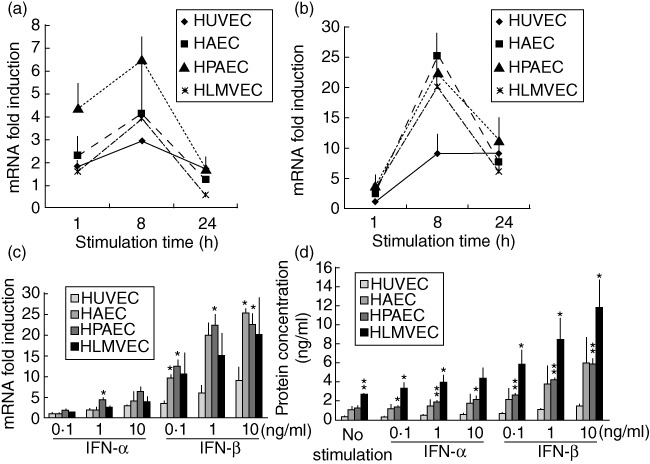
Type I interferon (IFN) augments CX3CL1 expression in various vascular endothelial cells (VEC). CX3CL1 mRNA expression in VEC stimulated with (a) IFN-α (10 ng/ml) or (b) IFN-β (10 ng/ml). (c) CX3CL1 mRNA in VEC stimulated with IFN-α or IFN-β at the indicated concentration for 8 h. (d) Concentration of CX3CL1 protein in the culture supernatant of VEC stimulated with IFN-α or IFN-β for 72 h. Induced mRNA expression is indicated as the relative quantity against non-stimulated VEC. Data from three or four independent experiments are presented as mean ± standard error. *P < 0·05; **P < 0·01 in comparison with equally stimulated HUVEC.
Both IFN-α and IFN-β induced CX3CL1 protein secretion from various VEC in a dose-dependent manner. Secretion of CX3CL1 by HLMVEC (maximum secretion: 11·8 ± 2·9 ng/ml; P < 0·05) and HPAEC (maximum secretion: 5·9 ± 0·5 ng/ml; P < 0·01) was significantly higher than HUVEC (maximum secretion: 1·4 ± 0·2 ng/ml). Although higher levels of CX3CL1 were secreted by HAEC (maximum secretion: 5·9 ± 2·7 ng/ml), this induction was not statistically significant in comparison with that by HUVEC (Fig. 3d).
Type I IFN induces CCL5 in VEC
IFN-α stimulation induced CCL5 mRNA expression in HPAEC, HAEC, HLMVEC and HUVEC, and maximum expression was observed at 24 h (Fig. 4a). IFN-β induced CCL5 mRNA expression intensely in VEC, and maximum expression was also observed at 24 h (Fig. 4b), but the expression levels decreased after 24 h (data not shown). While dose-dependent mRNA induction by both IFN-α and IFN-β was observed at 24 h, no significant differences were observed among different VEC (Fig. 4c). Anti-type I IFN antibodies inhibited type I IFN-induced CCL5 mRNA expression (Fig. S1b).
Fig. 4.
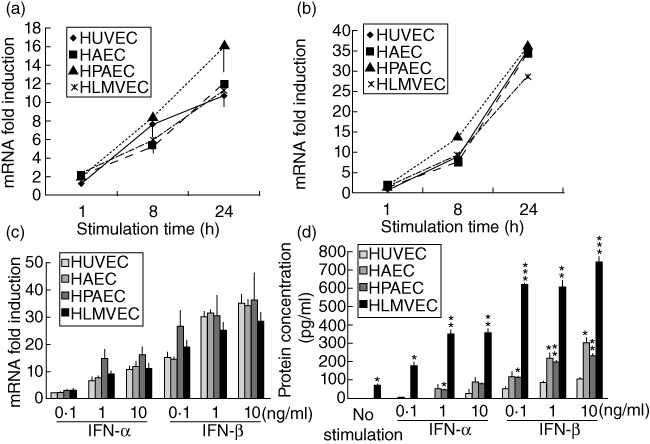
Type I interferon (IFN) induces CCL5 expression in various vascular endothelial cells (VEC). CCL5 mRNA expression in VEC stimulated with (a) IFN-α (10 ng/ml) or (b) IFN-β (10 ng/ml). (c) CCL5 mRNA expression in VEC stimulated with IFN-α or IFN-β for 24 h. (d) CCL5 protein levels in the culture supernatant of VEC stimulated with IFN-α or IFN-β for 72 h. Induced mRNA expression is indicated as the relative quantity against non-stimulated VEC. Data from three or four independent experiments are presented as mean ± standard error. *P < 0·05; **P < 0·01, ***P < 0·001 in comparison with equally stimulated human umbilical vein endothelial cells (HUVEC).
CCL5 protein secretion from various VEC was induced by both IFN-α and IFN-β in a dose-dependent manner. Secretion of CCL5 by HLMVEC (maximum secretion: 742·2 ± 30·0 pg/ml; P < 0·001), HAEC (maximum secretion: 299·5 ± 26·9 pg/ml; P < 0·05) and HPAEC (maximum secretion: 230·9 ± 9·4 pg/ml; P < 0·01) was significantly higher than that by HUVEC (maximum secretion: 101·4 ± 8·6 pg/ml; Fig. 4d).
JAK inhibitor I prevents type I IFN-induced chemokine secretion
JAK inhibitor I is a reversible inhibitor that inhibits JAK1, 2, 3 and Tyk2. To investigate the involvement of JAK in chemokine production we performed experiments using JAK inhibitor I, which prevented type I IFN-induced CX3CL1 (Fig. 5a) and CCL5 (Fig. 5b) secretion from HPAEC in a dose-dependent manner. JAK inhibitor I also inhibited type I IFN-induced STAT 1 activation in HPAEC (Fig. 5c).
Fig. 5.
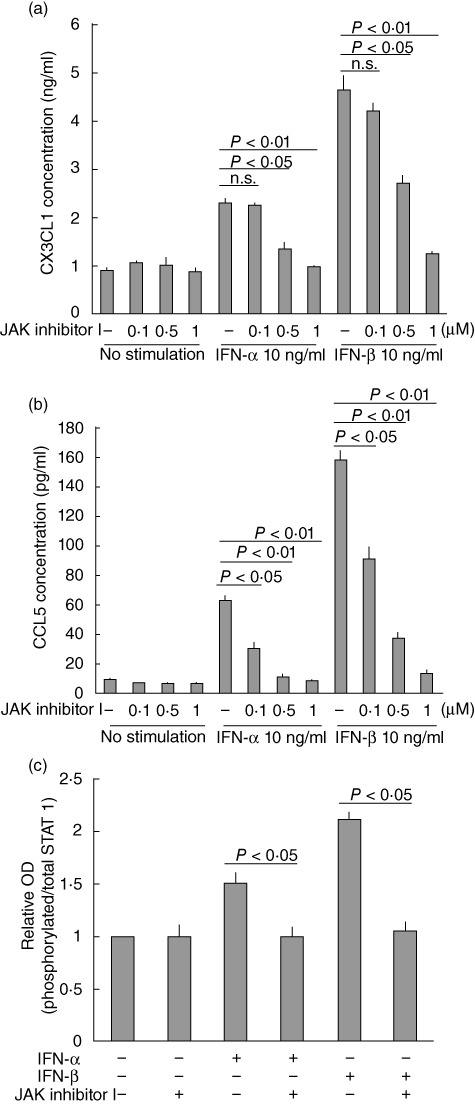
Janus protein tyrosine kinase (JAK) inhibitor I prevents augmentation of type I interferon (IFN)-induced chemokine secretion and signal transducer and activator of transcription (STAT) activation. (a) CX3CL1 or (b) CCL5 concentration in the culture supernatant of human pulmonary arterial endothelial cells (HPAEC) stimulated for 30 h. (c) STAT 1 activation in HPAEC stimulated for 30 min. Data from two independent experiments are presented as mean ± standard error.
Discussion
To the best of our knowledge, this is the first report that clearly reveals the induction capacity of type I IFN for CX3CL1 in pulmonary VEC. CX3CL1 is a unique chemokine, which captures leucocytes independently of selectin and integrin [10]. CX3CL1 has a higher affinity for its receptor than other chemokine-receptor pairs [11,12] and mediates preferentially the migration of CD16+ monocytes [13], which produce high TNF-α[14,15] and IL-1 levels [14]. CX3CL1 production in VEC is induced by TNF-α, IL-1 [16], lipopolysaccharide (LPS) [17] and IFN-γ[18]. We showed that type I IFN also induces CX3CL1 expression in VEC.
CX3CL1 expression is augmented in pulmonary vessels obtained from SSc patients [19], and frequencies of mutations in CX3CR1 (a CX3CL1 receptor) alleles are increased in SSc patients with PAH [20]. CX3CL1 not only has chemotactic abilities, but also proliferative activities in vascular smooth muscle cells (SMC) [21,22]. Proliferation of SMC and/or the accumulation of mononuclear cells in lung vessels are the major pathological findings in PAH [23]. In addition, an activated type I IFN system has also been reported in SSc [4]. Therefore, type I IFN-induced CX3CL1 may be involved in severe SSc complications such as PAH. Moreover, type I IFN and CX3CL1 levels are augmented in SLE [3,24]; thus, the type I IFN–CX3CL1 axis may be involved in the pathophysiology of various CTD. Although it may be more preferable for determining the type I IFN-mediated pulmonary injury in CTD to use HPAEC derived from patients with CTD and/or PAH, the present study suggested that type I IFN have a stimulatory effect on pulmonary VEC even from non-CTD individuals.
CCL5 secretion from HPAEC was induced by IFN-β and, to a lesser extent, by IFN-α. Type I IFN-induced CCL5 secretion from HLMVEC was higher than that from other VEC. This finding may be consistent with the observation that leucocytes tend to accumulate in pulmonary microvessels in the presence of IFN-inducible factors such as viruses and immune complexes containing DNA or RNA [25,26]. Although HPAEC used in our study include pulmonary artery VEC alone, HLMVEC contain VEC derived from micro-pulmonary artery, vein and capillary. Future studies will be necessary to determine the molecular mechanisms of chemokine production in response to type I IFN in both cells. Meanwhile, augmented CCL5 expression has been reported in pulmonary vessels obtained from PAH patients [8] and in plasma from SLE patients [27]. The type I IFN–CCL5 axis may be involved in the pathophysiology of PAH or SLE.
In contrast to CCL5 protein secretion, although CCL5 mRNA expression was induced by type I IFN, no significant difference was observed in the levels of mRNA induction among the various types of VEC. CCL5 mRNA silencing has been reported in T cells [28]. Differences of CCL5 protein secretion among VEC may be caused by various mRNA silencing mechanisms.
JAK inhibitor I prevented type I IFN-induced chemokine secretion from HPAEC and STAT 1 activation. In the present study, we focused on the regulatory effect of JAK inhibitor I on HPAEC, because pulmonary artery sometimes shows vascular remodelling in patients with severe PAH. JAK inhibitor, such as tofacitinib, has been proposed as an anti-rheumatic drug [29] and may serve as a therapeutic alternative for vascular diseases. JAK inhibitor I regulates JAK family members non-selectively [30]. Further studies are needed to determine the usefulness of JAK inhibitor as a therapeutic medication in patients with CTD and PAH.
Type I IFN-induced CCL2 secretion from HPAEC was not observed in our experiments, similar to another study regarding human coronary arterial VEC [31]. We also analysed CCL2 mRNA in type I IFN-stimulated VEC. Maximum expression of CCL2 mRNA was observed in the early phase; although there seems to be minor dose dependency, no statistically significant induction was observed (data not shown). IFN-β-induced CCL2 secretion from human dermal micro-VEC has been reported [32]. Conversely, CCR2, a CCL2 receptor, was expressed on VEC [33]; thus, CCL2 secreted in the early phase may be utilized in an autocrine mechanism.
IFN-β induced much higher chemokine secretion than IFN-α, which was consistent with the finding that all type I IFN bind to the same receptor [34] with different properties [35,36]. These differences are explained by their respective receptor recognition [37]. IFN-α and IFN-β production is augmented by nucleic acids of various pathogens [38]. IFN-β is also induced by LPS via Toll-like receptor (TLR)-4 and involved in LPS-induced mortality [39]. These findings may partially explain the mechanism of Gram-negative sepsis and the involvement of the IFN-β–chemokine axis in this mechanism.
In conclusion, type I IFN induces CX3CL1 secretion from HPAEC and can be prevented by JAK inhibitor I. Type I IFN also induces CCL5 production in pulmonary VEC, especially micro-VEC. The type I IFN–chemokine axis may be an important target for the treatment of patients with pulmonary vascular diseases.
Acknowledgments
The authors thank Dr Kosaku Murakami for his critical comments regarding our experiments and Drs Naho Ayuzawa, Masaki Katayama and Natsuki Yamamoto for their helpful advice. This work was supported by the Grant-in-Aid for Scientific Research from the Japanese Ministry of Education, Culture, Sports, Science and Technology (to T.F.); a Grant for Intractable Diseases from the Japanese Ministry of Health, Labor and Welfare (to T.F.); and a grant from the Smoking Research Foundation (to A.N.).
Disclosure
The authors have no competing interests.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Anti-type I interferon (IFN) antibody inhibits type I IFN-induced chemokine mRNA expression. To verify the specificity of type I IFN-induced chemokine induction, we performed an antibody blocking assay. IFN-α or IFN-β was preincubated with anti-IFN-α antibody (RELIATech GmbH, Germany) or anti-IFN-β antibody (Peprotech) over-night and added to the human pulmonary arterial endothelial cells (HPAEC) monolayer. The expression of (a) CX3CL1 or (b) CCL5 mRNA in HPAEC stimulated for 6 h was analysed. The induced mRNA expression is indicated as the relative quantity against non-stimulated HPAEC. Data from three independent experiments are presented as mean ± standard error.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Borden EC, Sen GC, Uze G, et al. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov. 2007;6:975–90. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ioannou Y, Isenberg DA. Current evidence for the induction of autoimmune rheumatic manifestations by cytokine therapy. Arthritis Rheum. 2000;43:1431–42. doi: 10.1002/1529-0131(200007)43:7<1431::AID-ANR3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 3.Rönnblom L, Eloranta ML, Alm GV. The type I interferon system in systemic lupus erythematosus. Arthritis Rheum. 2006;54:408–20. doi: 10.1002/art.21571. [DOI] [PubMed] [Google Scholar]
- 4.Eloranta ML, Franck-Larsson K, Lovgren T, et al. Type I interferon system activation and association with disease manifestations in systemic sclerosis. Ann Rheum Dis. 2010;69:1396–402. doi: 10.1136/ard.2009.121400. [DOI] [PubMed] [Google Scholar]
- 5.Dhillon S, Kaker A, Dosanjh A, et al. Irreversible pulmonary hypertension associated with the use of interferon alpha for chronic hepatitis C. Dig Dis Sci. 2010;55:1785–90. doi: 10.1007/s10620-010-1220-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ledinek AH, Jazbec SS, Drinovec I, et al. Pulmonary arterial hypertension associated with interferon beta treatment for multiple sclerosis: a case report. Mult Scler. 2009;15:885–6. doi: 10.1177/1352458509104593. [DOI] [PubMed] [Google Scholar]
- 7.Sanchez O, Marcos E, Perros F, et al. Role of endothelium-derived CC chemokine ligand 2 in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2007;176:1041–7. doi: 10.1164/rccm.200610-1559OC. [DOI] [PubMed] [Google Scholar]
- 8.Dorfmüller P, Zarka V, Durand-Gasselin I, et al. Chemokine RANTES in severe pulmonary arterial hypertension. Am J Respir Crit Care Med. 2002;165:534–9. doi: 10.1164/ajrccm.165.4.2012112. [DOI] [PubMed] [Google Scholar]
- 9.Balabanian K, Foussat A, Dorfmüller P, et al. CX3C chemokine fractalkine in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2002;165:1419–25. doi: 10.1164/rccm.2106007. [DOI] [PubMed] [Google Scholar]
- 10.Fong AM, Robinson LA, Steeber DA, et al. Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J Exp Med. 1998;188:1413–9. doi: 10.1084/jem.188.8.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imai T, Hieshima K, Haskell C, et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–30. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 12.Haskell CA, Cleary MD, Charo IF. Unique role of the chemokine domain of fractalkine in cell capture. Kinetics of receptor dissociation correlate with cell adhesion. J Biol Chem. 2000;275:34183–9. doi: 10.1074/jbc.M005731200. [DOI] [PubMed] [Google Scholar]
- 13.Ancuta P, Rao R, Moses A, et al. Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J Exp Med. 2003;197:1701–7. doi: 10.1084/jem.20022156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thieblemont N, Weiss L, Sadeghi HM, et al. CD14lowCD16high: a cytokine-producing monocyte subset which expands during human immunodeficiency virus infection. Eur J Immunol. 1995;25:3418–24. doi: 10.1002/eji.1830251232. [DOI] [PubMed] [Google Scholar]
- 15.Belge KU, Dayyani F, Horelt A, et al. The proinflammatory CD14+CD16+DR++ monocytes are a major source of TNF. J Immunol. 2002;168:3536–42. doi: 10.4049/jimmunol.168.7.3536. [DOI] [PubMed] [Google Scholar]
- 16.Bazan JF, Bacon KB, Hardiman G, et al. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385:640–4. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- 17.Harrison JK, Jiang Y, Wees EA, et al. Inflammatory agents regulate in vivo expression of fractalkine in endothelial cells of the rat heart. J Leukoc Biol. 1999;66:937–44. doi: 10.1002/jlb.66.6.937. [DOI] [PubMed] [Google Scholar]
- 18.Matsumiya T, Imaizumi T, Fujimoto K, et al. Soluble interleukin-6 receptor alpha inhibits the cytokine-induced fractalkine/CX3CL1 expression in human vascular endothelial cells in culture. Exp Cell Res. 2001;269:35–41. doi: 10.1006/excr.2001.5300. [DOI] [PubMed] [Google Scholar]
- 19.Hasegawa M, Sato S, Echigo T, et al. Up regulated expression of fractalkine/CX3CL1 and CX3CR1 in patients with systemic sclerosis. Ann Rheum Dis. 2005;64:21–8. doi: 10.1136/ard.2003.018705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marasini B, Cossutta R, Selmi C, et al. Polymorphism of the fractalkine receptor CX3CR1 and systemic sclerosis-associated pulmonary arterial hypertension. Clin Dev Immunol. 2005;12:275–9. doi: 10.1080/17402520500303297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chandrasekar B, Mummidi S, Perla RP, et al. Fractalkine (CX3CL1) stimulated by nuclear factor kappaB (NF-kappaB)-dependent inflammatory signals induces aortic smooth muscle cell proliferation through an autocrine pathway. Biochem J. 2003;373:547–58. doi: 10.1042/BJ20030207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perros F, Dorfmüller P, Souza R, et al. Fractalkine-induced smooth muscle cell proliferation in pulmonary hypertension. Eur Respir J. 2007;29:937–43. doi: 10.1183/09031936.00104706. [DOI] [PubMed] [Google Scholar]
- 23.Pietra GG, Capron F, Stewart S, et al. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol. 2004;43:S25–S32. doi: 10.1016/j.jacc.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 24.Yajima N, Kasama T, Isozaki T, et al. Elevated levels of soluble fractalkine in active systemic lupus erythematosus: potential involvement in neuropsychiatric manifestations. Arthritis Rheum. 2005;52:1670–5. doi: 10.1002/art.21042. [DOI] [PubMed] [Google Scholar]
- 25.Vallin H, Blomberg S, Alm GV, et al. Patients with systemic lupus erythematosus (SLE) have a circulating inducer of interferon-alpha (IFN-α) production acting on leucocytes resembling immature dendritic cells. Clin Exp Immunol. 1999;115:196–202. doi: 10.1046/j.1365-2249.1999.00772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lenert P. Nucleic acid sensing receptors in systemic lupus erythematosus: development of novel DNA- and/or RNA-like analogues for treating lupus. Clin Exp Immunol. 2010;161:208–22. doi: 10.1111/j.1365-2249.2010.04176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shah D, Wanchu A, Bhatnagar A. Interaction between oxidative stress and chemokines: possible pathogenic role in systemic lupus erythematosus and rheumatoid arthritis. Immunobiology. 2011;216:1010–7. doi: 10.1016/j.imbio.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 28.Swanson BJ, Murakami M, Mitchell TC, et al. RANTES production by memory phenotype T cells is controlled by a posttranscriptional, TCR-dependent process. Immunity. 2002;17:605–15. doi: 10.1016/s1074-7613(02)00456-9. [DOI] [PubMed] [Google Scholar]
- 29.Fleischmann R, Cutolo M, Genovese MC, et al. Phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) or adalimumab monotherapy versus placebo in patients with active rheumatoid arthritis with an inadequate response to disease-modifying antirheumatic drugs. Arthritis Rheum. 2012;64:617–29. doi: 10.1002/art.33383. [DOI] [PubMed] [Google Scholar]
- 30.Thompson JE, Cubbon RM, Cummings RT, et al. Photochemical preparation of a pyridone containing tetracycle: a Jak protein kinase inhibitor. Bioorg Med Chem Lett. 2002;12:1219–23. doi: 10.1016/s0960-894x(02)00106-3. [DOI] [PubMed] [Google Scholar]
- 31.Krishnaswamy G, Smith JK, Mukkamala R, et al. Multifunctional cytokine expression by human coronary endothelium and regulation by monokines and glucocorticoids. Microvasc Res. 1998;55:189–200. doi: 10.1006/mvre.1998.2079. [DOI] [PubMed] [Google Scholar]
- 32.Buttmann M, Goebeler M, Toksoy A, et al. Subcutaneous interferon-beta injections in patients with multiple sclerosis initiate inflammatory skin reactions by local chemokine induction. J Neuroimmunol. 2005;168:175–82. doi: 10.1016/j.jneuroim.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 33.Salcedo R, Ponce ML, Young HA, et al. Human endothelial cells express CCR2 and respond to MCP-1: direct role of MCP-1 in angiogenesis and tumor progression. Blood. 2000;96:34–40. [PubMed] [Google Scholar]
- 34.Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. 2004;202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- 35.Leaman DW, Chawla-Sarkar M, Jacobs B, et al. Novel growth and death related interferon-stimulated genes (ISGs) in melanoma: greater potency of IFN-β compared with IFN-α2. J Interferon Cytokine Res. 2003;23:745–56. doi: 10.1089/107999003772084860. [DOI] [PubMed] [Google Scholar]
- 36.van Boxel-Dezaire AHH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25:361–72. doi: 10.1016/j.immuni.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 37.Thomas C, Moraga I, Levin D, et al. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell. 2011;146:621–32. doi: 10.1016/j.cell.2011.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 39.Thomas KE, Galligan CL, Newman RD, et al. Contribution of interferon-beta to the murine macrophage response to the toll-like receptor 4 agonist, lipopolysaccharide. J Biol Chem. 2006;281:31119–30. doi: 10.1074/jbc.M604958200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.