

In this study, Clippinger and Alwine present a novel molecular mechanism for mTORC1 activation. They show that the molecular motor protein dynein is involved in mTORC1 activity and that human cytomegalovirus (HCMV) manipulates mTOR subcellular localization. Essentially, dynein moves mTORC1 to locations in the cell where it can be active. In the case of infected cells, HCMV controls this dynein-dependent mTOR activation to localize mTOR to perinuclear regions, where it is constitutively active and resistant to stress inhibition. This localization corresponds to what will become the viral assembly compartment that protects mTOR from inhibitory stress signaling.
Keywords: dynein, HCMV, assembly compartment, mTOR
Abstract
Activation of stress signaling pathways normally leads to inhibition of the mammalian target of rapamycin complex 1 (mTORC1); however, human cytomegalovirus (HCMV) infection maintains mTORC1 activity in the presence of numerous types of stress. We previously demonstrated that HCMV infection maintains mTORC1 activity during amino acid deprivation through a Ras-related GTP-binding (Rag) protein-independent mechanism. This depends on the colocalization of mTOR and its activator, Rheb (Ras homology enriched in brain)-GTP, to a perinuclear position that corresponds to the viral cytoplasmic assembly compartment (AC). The data presented here show that the HCMV-induced, amino acid depletion-resistant perinuclear localization and activation of mTORC1 occurs as early as 8 h post-infection, prior to AC formation. We show that the molecular motor dynein is required for perinuclear localization of mTORC1 in both uninfected and HCMV-infected cells. Association between dynein and mTOR is shown by coimmunoprecipitation, and inhibition of dynein function using RNAi or the small molecule inhibitor ciliobrevin A inhibits mTORC1 activity in both uninfected and HCMV-infected cells. The data suggest that mTORC1 activation requires dynein-dependent transport to a position in the cell where it can be activated. Thus, the HCMV commandeers a cellular dynein-dependent mTORC1 activation mechanism to maintain stress-resistant mTORC1 activity during infection and to form the AC.
The mammalian target of rapamycin (mTOR) kinase is found in two multiprotein complexes: mTOR complex 1 (mTORC1) and mTORC2. A major difference between the two complexes is their substrate specificity factors: raptor is present in mTORC1, and rictor is present in mTORC2; consequently, the two complexes have different substrate specificities (Kim et al. 2002; Sarbassov et al. 2004). Human cytomegalovirus (HCMV), the largest of the human herpesviruses, activates both complexes during infection (Kudchodkar et al. 2006). However, the results of studies using shRNAs specific for raptor or rictor suggest that mTORC1 has a greater effect on viral growth; specifically, raptor depletion is more deleterious to HCMV infection than rictor depletion (Moorman and Shenk 2010; Clippinger et al. 2011a). The role of mTORC1 in translational control has been well-established; mTORC1-mediated phosphorylation of p70 S6 kinase (S6K) and the eukaryotic initiation factor 4E-binding protein 1 (4E-BP1) is necessary for the maintenance of translation (Sarbassov et al. 2005; Mamane et al. 2006; Reiling and Sabatini 2006). Maintaining mTOR activity and translation is vital to the success of an HCMV infection. However, a viral infection induces stress response signaling that normally inhibits mTORC1 and translation. Consequently, HCMV has evolved mechanisms to circumvent these inhibitory stress responses and maintain mTORC1 activity (Kudchodkar et al. 2004, 2006; Walsh et al. 2005; Alwine 2008; Moorman et al. 2008; Clippinger et al. 2011a).
Many cellular stress responses lead to the inhibition of mTORC1 through the activation of the guanosine triphosphatase (GTPase)-activating protein (GAP) function of the tuberous sclerosis complex (TSC) (Fig. 1; for review, see Alwine 2008; Buchkovich et al. 2008b). Activated TSC stimulates the intrinsic GTPase activity of Rheb (Ras homology enriched in brain), converting it from Rheb-GTP to Rheb-GDP. Rheb-GTP is an activator of mTORC1, while Rheb-GDP is not. Thus, when the TSC is inactivated, for example, through phosphorylation by Akt (protein kinase B), Rheb-GTP levels remain high, leading to activation of mTORC1 (Fig. 1; Sancak et al. 2007). Conversely, many cellular stresses activate the TSC, resulting in TSC-dependent inhibition of mTORC1. However, HCMV protects mTORC1 from TSC-dependent inhibition (Kudchodkar et al. 2004, 2007; Moorman et al. 2008). In part, this is accomplished through an interaction between the viral UL38 protein and the TSC, which is believed to inactivate the TSC (Fig. 1; Moorman et al. 2008). Additionally, the levels of Rheb increase in infected cells, which may also overcome the inhibitory effects of TSC activation (Clippinger et al. 2011b). These findings suggest that any stress response that exerts its effect on, or upstream of, the TSC will be circumvented, and Rheb-GTP levels and mTORC1 activity will remain high. However, some types of stress, including oxidative stress and amino acid deprivation, inhibit mTORC1 through a TSC-independent mechanism (Smith et al. 2005; Nobukuni et al. 2007). We showed recently that mTORC1 activity is maintained in HCMV-infected cells under both oxidative stress (Tilton et al. 2011) and amino acid depletion (Clippinger et al. 2011b).
Figure 1.

mTORC1 regulation. Activation of the PI3K (phosphatidylinositol 3-kinase)–Akt–TSC–mTORC1 pathway is critical for the phosphorylation of 4E-BP1 and S6K and the maintenance of translation. Numerous types of stresses inhibit this pathway, halting translation. HCMV has multiple mechanisms to circumvent the effects of stress signaling to maintain translation under stress conditions (see the text for details). The mechanisms for amino acid control of mTORC1 are also shown.
Amino acids signal to mTORC1 through the Ras-related GTP-binding (Rag) proteins by stimulating GTP charging of Rag proteins, thereby promoting the association of the Rag proteins with mTORC1 (Fig. 1; Kim et al. 2008; Sancak et al. 2008, 2010). It has been proposed that the interaction of the Rag proteins with mTORC1 promotes mTORC1 translocation to a membrane-bound compartment that contains Rheb-GTP (Kim et al. 2008; Sancak et al. 2008). More recent work suggests that a protein complex termed Ragulator interacts with the Rag proteins, resulting in the localization of the Rag dimer–mTORC1 complex to lysosomal membranes, which is essential for mTORC1 activation (Sancak et al. 2010). Constitutive targeting of mTORC1 to the lysosomal surface renders mTORC1 activity insensitive to amino acid deprivation and, correspondingly, independent of Rag and Ragulator but not Rheb-GTP (Sancak et al. 2010).
We showed recently that mTORC1 activity is sensitive to amino acid deprivation in mock-infected cells as expected; however, it is resistant in HCMV-infected cells (Clippinger et al. 2011b). This suggested that HCMV infection bypasses normal Rag-dependent amino acid signaling. Indeed, we established that shRNA-mediated depletion of the Rag proteins had little effect on mTORC1 activity in infected cells but inhibited activity in mock-infected cells. However, Rheb was necessary, as its depletion inhibited mTOR activity in both mock- and HCMV-infected cells (Clippinger et al. 2011b). In mock-infected cells, mTOR activity in the presence of amino acids correlated with a perinuclear localization of mTOR with Rheb. This localization dispersed following the depletion of amino acids and correlated with the loss of mTOR activity (Clippinger et al. 2011b). In HCMV-infected cells, mTOR was also found in a perinuclear localization with Rheb but did not disperse upon amino acid depletion, and mTOR activity was maintained (Clippinger et al. 2011b). At late times in HCMV infection, the perinuclear localization of mTOR coincided with the cytoplasmic assembly compartment (AC), a perinuclear, HCMV-specific structure that is needed for virion maturation and egress. Detailed studies of AC structure propose a three-dimensional model of a cylindrical AC composed of organelle-specific vesicles (Golgi, trans-Golgi network, and early endosomes), which form nested cylinders (Das et al. 2007; Das and Pellett 2011). The AC forms on a microtubule-organizing center (MTOC), and microtubules radiate out from the AC (Sanchez et al. 2000). Our previous work and the work of others have shown that formation of the AC on the MTOC is mediated by the molecular motor dynein (Buchkovich et al. 2010; Indran et al. 2010). Thus, we questioned whether the movement of mTOR to its perinuclear location in the AC may be dynein-mediated and, correspondingly, whether dynein is involved in the normal translocation and activation of mTORC1 in uninfected cells.
In the present study, we show that HCMV infection induces a perinuclear localization of mTOR by 8 h post-infection (hpi), which is very early in infection, prior to the formation of the AC. This localization is maintained during amino acid deprivation and corresponds with maintenance of mTORC1 activity. Uninfected, growing human fibroblasts (HFs) show a perinuclear localization of mTOR that is less compact than in infected cells but disperses upon amino acid depletion, corresponding with the loss of mTORC1 activity. We show that the molecular motor dynein is required for the perinuclear localization of mTOR in both uninfected and HCMV-infected cells; association between dynein and mTOR is shown by coimmunoprecipitation. Inhibition of dynein function using RNAi or the small molecule inhibitor ciliobrevin A (CA) inhibits mTORC1 activity in both uninfected and HCMV-infected cells. The data show that in normal, uninfected cells, mTORC1 activation is dependent on dynein-mediated transport. Thus, HCMV commandeers a cellular, dynein-dependent mTOR activation function in order to (1) maintain mTOR in a location where it is constitutively active and resistant to stress inhibition and (2) form the AC, where mTOR may be further protected from inhibitory stress signaling.
Results
The activity and perinuclear translocation of mTORC1 is resistant to amino acid deprivation by 8 h after HCMV infection
Our previously published results demonstrated that in HCMV-infected cells, mTORC1 and Rheb-GTP localize to the cytoplasmic viral AC, where mTORC1 activity is independent of Rag proteins and resistant to inhibition by amino acid deprivation (Clippinger et al. 2011b). The immunofluorescence micrograph in Figure 2A reiterates these data using cells that have been HCMV-infected for 72 h. The AC was visualized using antibodies to the HCMV protein pp28 (Fig. 2A, green), an AC resident protein. The two arrows in Figure 2A indicate two morphologies of mTOR localization (red) in the AC; in one morphology (which is more predominantly seen), mTOR and pp28 colocalize in the AC core. In the other morphology, mTOR is in an outer ring surrounding a core of pp28. We observed similar mixed morphologies for other AC components, suggesting that localization of these components is dynamic (Buchkovich et al. 2008a, 2009, 2010).
Figure 2.

The perinuclear localization and activity of mTORC1 are maintained following amino acid deprivation as early as 8 h after HCMV infection. (A) HCMV-infected HFs (72 hpi) were stained for pp28 or mTOR followed by immunofluorescence microscopy. This shows the characteristics of perinuclear viral cytoplasmic ACs and the enlarged kidney-shaped nuclei around them. (Green) pp28; (red) mTOR; (blue) nuclei; (arrows) ACs. (B) Mock- or HCMV-infected HFs (8 hpi) were maintained in amino acid-containing medium (+AAs), amino acid-starved for 50 min prior to cell fixation (−AAs), or amino acid-starved for 50 min followed by the readdition of amino acids for 30 min prior to cell fixation (−/+AAs). Cells were then stained for mTOR followed by immunofluorescence microscopy. (Red) mTOR; (blue) nuclei. (Top right panel) An example of mTOR staining (red) in quiescent, mock-infected HFs is shown to contrast the more diffuse mTOR localization pattern of quiescent cells with its more concentrated localization in actively growing HFs (+AAs). (C) The phosphorylation of S6K was examined by Western analysis at 3, 6, 8, and 24 h after mock or HCMV infection. HFs were maintained in amino acid-containing medium (+), amino acid-starved for 50 min prior to cell collection (−), or amino acid-starved for 50 min followed by the readdition of amino acids for 30 min prior to cell collection (−/+).
The AC does not begin to form until 24–36 hpi, as measured by the accumulation and perinuclear positioning of viral protein markers of the AC, such as pp28 and glycoprotein B (gB) (Krzyzaniak et al. 2007; Buchkovich 2010). Thus, we examined earlier times in infection to determine whether the perinuclear localization of mTOR and the resulting resistance to amino acid depletion preceded or coincided with AC formation.
In the top panel of Figure 2B, mTOR localization (red) was examined in subconfluent, actively growing, mock-infected HFs. In the presence of complete medium (containing amino acids, +AAs), mTOR is localized in a perinuclear position. A much more diffuse cytoplasmic localization of mTOR is observed after cells are maintained in amino acid-free medium (−AAs) for 50 min; this is similar to the predominant localization of mTOR in confluent, contact-inhibited, quiescent HFs (Fig. 2B, quiescent). In the subconfluent, actively growing HFs, the perinuclear localization of mTOR returns when cells are returned to amino acid-containing medium for 30 min following the 50 min of amino acid withdrawal (Fig. 2B, top panel, −/+AAs). In the left panel of Figure 2C, phosphorylation of p70 S6K was examined as an indicator of mTORC1 activity in uninfected cells at 3, 6, 8, and 24 h after mock infection. At each time point, mTOR is active when cells are maintained in complete medium (+) but is inhibited after 50 min of amino acid depletion (−). mTOR activity is regained when cells are returned to amino acid-containing medium for 30 min following the 50 min depletion (−/+). These data are in agreement with previous reports of mTORC1 activity under amino acid depletion conditions (Sancak et al. 2008; Clippinger et al. 2011b). The bottom panel of Figure 2B shows mTOR localization (red) in HCMV-infected HFs at 8 hpi. In complete medium (+AAs), the perinuclear localization of mTOR becomes more compact and punctate. Remarkably, by 8 hpi, the tight perinuclear localization does not disperse following 50 min of amino acid deprivation (−AAs). The results of Western analysis in the right panel of Figure 2C show that this corresponds to the retention of mTOR activity in the absence of amino acids at 8 h after HCMV infection. Thus, very early in HCMV infection, conditions are induced to maintain active mTOR in a perinuclear position even in the absence of amino acids. This corresponds with the virus-induced maintenance of mTORC1 activity when amino acids are depleted. In addition, the very early perinuclear localization and activation of mTOR at 8 hpi precedes the formation of the AC, suggesting that mTOR perinuclear localization may be a prerequisite for AC formation.
Recent reports suggest that mTORC1, complexed with Rag heterodimers, is recruited to lysosomal membranes, which is essential for mTORC1 activation (Sancak et al. 2010). Supplemental Figure S1A shows that the lysosomal marker LAMP2 colocalizes with perinuclear mTOR in uninfected cells as well as with mTOR in the AC of infected cells; previous studies have established the presence of lysosomal markers in the AC (Das and Pellett 2011). These data suggest that the association of active mTOR with lysosomal membranes is maintained in HCMV-infected cells. Additionally, Supplemental Figure S1B shows that Rag proteins are also located perinuclearly.
Dynein function is needed for mTOR localization in uninfected and HCMV-infected cells
Dynein is a multiprotein complex that functions as the predominant minus end-directed microtubule motor in eukaryotic cells. We and others showed previously that AC formation requires dynein (Buchkovich et al. 2010; Indran et al. 2010). In addition, the formation and maintenance of the characteristic enlarged, kidney-shaped nucleus found wrapped around the AC in HCMV-infected cells depends on dynein function (Buchkovich et al. 2010). Dynein interacts with dynactin, an adaptor complex that allows dynein to bind its cargo. A projecting arm of dynactin, made up of a dimer of the p150Glued subunit, binds directly to dynein (Karki and Holzbaur 1995). Overexpression of p150Glued results in a wide range of motility defects; however, constructs have been made that express only the coiled-coil domain 1 (CC1; amino acid residues 217–548) of p150Glued. Overexpression of CC1 has far fewer adverse effects than overexpression of p150Glued (Quintyne et al. 1999; Kwinter et al. 2008; Maier et al. 2008). CC1 binds dynein directly, acting as a competitive inhibitor for the interaction between dynein and dynactin; thus, dynein cannot load its cargo (Quintyne et al. 1999).
Our previous studies used CC1 to inhibit dynein function and show its critical role in AC formation (Buchkovich et al. 2010). A similar approach was used with CC1 to assess dynein's involvement in mTOR localization. HFs were infected for 24 h and then electroporated with a plasmid expressing GFP-labeled CC1. Forty-eight hours after transfection (72 hpi), the cells were fixed, stained, and examined by immunofluorescence microscopy. In Figure 3A, two HCMV-infected cells are shown; the one on the left, indicated by the arrow, has been transfected and expresses GFP-CC1 (white). Both cells have been infected, as indicated by the staining for HCMV gB (Fig. 3A, red), another viral marker of the AC. In the cell not expressing CC1, gB is in the AC next to an enlarged, kidney-shaped nucleus. However, in the CC1-expressing cell, the nucleus is neither enlarged nor kidney-shaped, and gB has become dispersed throughout the cytoplasm; this is more easily seen in the longer exposure of the gB staining (gB*). These results agree with our previous observations that dynein function is needed for AC formation and nuclear remodeling (Buchkovich et al. 2010). The staining for mTOR (Fig. 3A, green) shows that mTOR colocalizes to the AC in the cell that is not expressing CC1; however, mTOR is dispersed throughout the cytoplasm in the CC1-expressing cell. Examination of 30 CC1-expressing, HCMV-infected cells found none with an intact AC. These results suggest that dynein function is required for the localization of mTOR to the AC.
Figure 3.

Dynein function is required for perinuclear localization of mTOR in mock- and HCMV-infected cells. (A) mTOR (green) and gB (red) staining in HCMV-infected HFs (72 hpi); the cell on the left (indicated by the arrow) is also expressing CC1 (white). (B) mTOR (red) staining in mock-infected U373-MG cells; three cells, indicated by arrows, are expressing CC1 (green). (C) mTOR (green) staining in mock-infected HFs; the cell indicated by the arrow is expressing CC1 (white). (D) mTOR (green) staining in HCMV-infected U373-MG cells; the red cells were also electroporated with siRNA that specifically targets dynein heavy chain and siGLO, a fluorescently labeled, nonspecific siRNA that marks the transfected cells. The cell indicated by the arrow has taken up no siRNA.
In our previous study of HCMV's maintenance of mTORC1 activity under amino acid depletion conditions, we used the glioblastoma cell line U373-MG (Clippinger et al. 2011b). These studies showed that, in U373-MG cells, mTOR is active and predominately perinuclear under normal, uninfected conditions. However, just like in HFs, the perinuclear localization of mTOR in uninfected U373-MG cells was lost upon depletion of amino acids and regained when amino acid-containing medium was restored (Clippinger et al. 2011b). We examined whether the perinuclear localization of mTOR in uninfected U373-MG cells was dynein-dependent. The GFP-CC1-expressing plasmid was electroporated into uninfected U373-MG cells, and 48 h post-electroporation, the cells were fixed, stained, and examined by immunofluorescence microscopy. Figure 3B shows a field of U373-MG cells; three of these cells (indicated by arrows) express GFP-CC1 (green)—two at high levels, and one at a much lower level. The relatively tight perinuclear localization of mTOR (Fig. 3B , red) is noted in all of the cells except the three expressing GFP-CC1, which show a diffuse, cytoplasmic localization of mTOR. These results suggest that dynein is necessary for the perinuclear localization of mTOR observed in uninfected U373-MG cells.
An additional control was performed to rule out the possibility that the dynein-dependent localization of mTOR seen in U373-MG cells was a phenomenon specific to a transformed cell line. We examined the effect of CC1 inhibition on dynein function in normal, growing HFs in complete medium. Figure 3C shows a field of three, subconfluent, actively growing HFs, one of which is expressing GFP-CC1 (white). In the CC1-expressing cell, mTOR localization (Fig. 3C, green) is very diffuse throughout the cytoplasm, while it has a more perinuclear localization in the cells not expressing GFP-CC1. All of the CC1-expressing cells that we examined showed diffuse mTOR staining. These results support the conclusion that dynein is required for perinuclear mTOR localization in uninfected cells.
To further verify the CC1 results in infected cells, we tested siRNAs that specifically target the dynein heavy chain. U373-MG cells were first electroporated with the dynein siRNA and siGLO, a fluorescently labeled, nonspecific siRNA that marks the transfected cells. At 6 h post-electroporation, the cells were infected with HCMV, and at 72 h post-electroporation, the cells were fixed and stained for mTOR (Fig. 3D, green). The left two panels of Figure 3D show the siRNA-containing cells, as indicated by siGLO (red); two exposures are shown, and the lighter one is used in the merge so that details of mTOR staining are not obscured. The longer exposure shows that one cell contains no siRNA fluorescence (Fig. 3D, arrow), and only this cell has mTOR concentrated in the perinuclear AC, while mTOR is much more diffuse in the siRNA transfected cells. In the examination of numerous fields, we found that when siRNA transfection was noted, the AC was either undetectable or very diffuse compared with untransfected, infected cells. The results of this alternative approach for disrupting dynein function confirm the results of the CC1 experiments in Figure 3A and reiterate that dynein is required for perinuclear localization of mTOR.
The data in Figure 3, combined with our previous data, suggest that dynein functions in the localization of mTOR, contributing to its activation in uninfected human cells, and that HCMV commandeers this function to (1) localize mTOR to a perinuclear position very early in infection, where mTOR is active and resistant to stress inhibition, and (2) form the AC later in infection (Buchkovich et al. 2010; Clippinger et al. 2011b).
Coimmunoprecipitation of dynein and mTOR
The data above suggest that dynein is involved in transporting mTOR to a perinuclear region where it can be activated. Thus, we asked whether an interaction between mTOR and dynein could be demonstrated by immunoprecipitation. Extracts from mock-infected and HCMV-infected (72 hpi) HFs were immunoprecipitated using anti-dynein intermediate chain, anti-mTOR, or a nonspecific control IgG. The precipitates were analyzed by Western analysis probed for mTOR and dynein. The results (Fig. 4) show that precipitation of dynein coimmunoprecipitated mTOR (Fig. 4A), and precipitation of mTOR coimmunoprecipitated dynein (Fig. 4B). The specificity of the precipitations is indicated by the control IgG, which precipitated neither mTOR nor dynein (Fig. 4C). The data in Figure 3, A and C, suggest that dynein is involved in the localization of mTOR in mock- and HCMV-infected HFs; the coimmunoprecipitation results support this finding, showing that dynein and mTOR coimmunoprecipitate similarly in mock- and HCMV-infected HFs.
Figure 4.
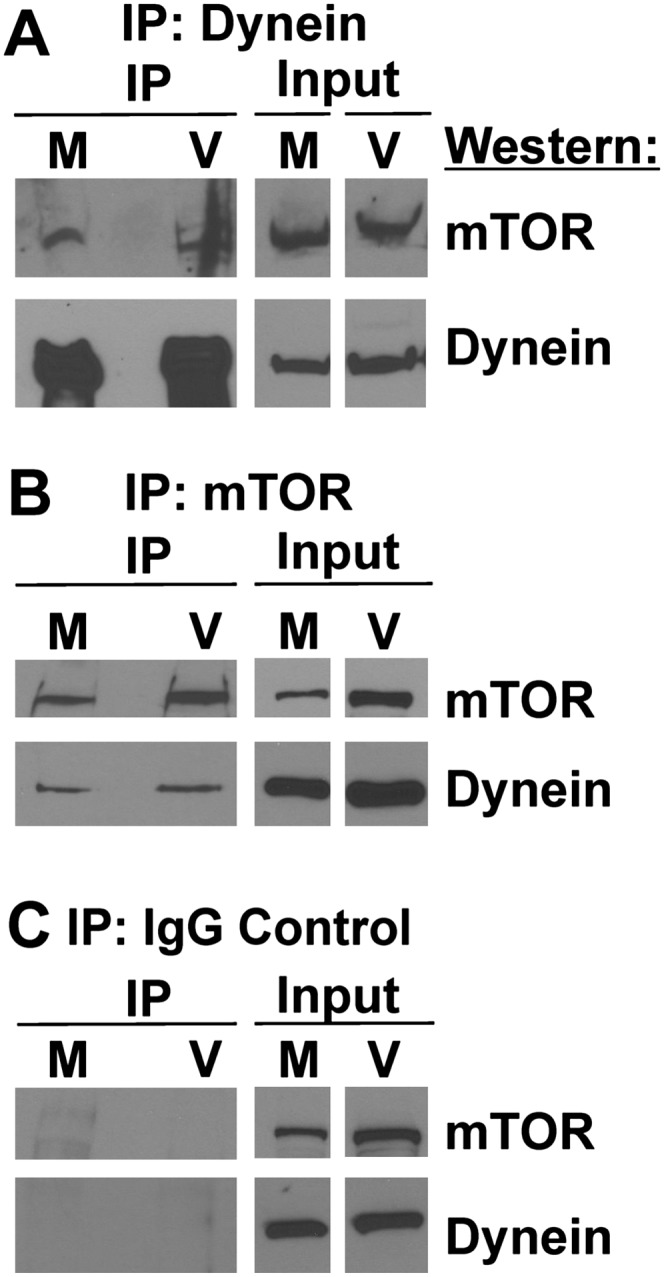
Coimmunoprecipitation of dynein and mTOR. Mock- or HCMV-infected HF lysates (72 hpi) were immunoprecipitated with mouse anti-dynein intermediate chain (A), goat anti-mTOR (B), or a nonspecific goat IgG control (C); a mouse IgG control gave the same result as the goat IgG control (not shown). Western analysis was then performed for mTOR and dynein heavy chain. (M) Mock-infected; (V) HCMV-infected.
It is important to note that, in comparison with the input, the amount of dynein–mTOR complexes is relatively low; the input represents ∼5% of the lysate. This may indicate that the complexes are unstable. Alternatively, if the dynein–mTOR association occurs only during transport, one would expect that only a small amount of mTOR would be in active transport, and therefore associated with dynein, at any given time. In sum, the data show an interaction between dynein and mTOR; this would be expected if dynein function is needed to move and localize mTOR to a perinuclear position in normal and infected HFs. Also of note in the input lanes in Figure 4, A–C, is the observation that dynein levels are not notably altered by HCMV infection.
mTOR activity is not needed for mTOR localization to the AC in an established HCMV infection
We next examined whether the dynein-mediated AC localization of mTOR required mTOR kinase activity. To inhibit mTOR kinase, we used Torin1, a highly-specific, small molecule inhibitor that binds the kinase domain (Thoreen and Sabatini 2009). We and others previously characterized Torin1 as an effective mTOR inhibitor during HCMV infection (Moorman and Shenk 2010; Clippinger et al. 2011a). For these experiments, we used confluent HFs; the mTOR staining of these cells (Fig. 5A, Mock) provides a very good example of the diffuse staining of mTOR seen in confluent, contact-inhibited, quiescent, uninfected cells (also described in Fig. 2B). Untreated HFs were HCMV-infected and fixed at 24 or 48 hpi for immunofluorescence staining. Other HCMV-infected cultures were treated with Torin1 from 2 to 48 hpi or from 24 to 48 hpi and then fixed for examination. Figure 5A shows that pp28 (red) and mTOR (green) are localized to the perinuclear AC region by 24 hpi and become more concentrated in the AC by 48 hpi. It is known that Torin1 significantly delays the course of HCMV infection if added from the start of the infection (at 2 hpi) (Clippinger et al. 2011a). This can be seen in the sample treated with Torin1 from 2 to 48 hpi, where pp28 is not detected due to the delayed infection. In addition, mTOR localization to the perinuclear region is more diffuse in the samples treated from 2 to 48 hpi than it is at 48 hpi in untreated cells; however, mTOR localization is still more perinuclear than in confluent, mock-infected cells. Thus, inhibition of mTOR activity at 2 hpi had only a partial effect on mTOR localization by 48 hpi; this could be due to pleiotropic effects resulting from the delay of the lytic cycle caused by Torin1 (Clippinger et al. 2011a). Therefore, we took the alternative approach of allowing the infection to establish for 24 h before adding Torin1. Figure 5A shows that the addition of Torin1 between 24 and 48 hpi had little effect on the perinuclear AC localization of pp28 and mTOR. The Western analysis in Figure 5B shows that Torin1 inhibition of mTOR's ability to phosphorylate S6K and 4E-BP was very effective during the 24–48 hpi treatment. Taken together, the data in Figure 5 suggest that once the infection is established (by 24 hpi), mTOR activity is not necessary for the maintenance of the AC.
Figure 5.

mTOR activity is not necessary for its perinuclear localization in an established infection. (A) Immunofluorescence analysis of mock- or HCMV-infected HFs at 24 or 48 hpi. Where applicable, HCMV-infected cells were treated with the mTOR kinase inhibitor Torin1 from 2 to 48 hpi (Torin 2-48hpi) or from 24 to 48 hpi (Torin 24-48hpi). (Red) pp28; (green) mTOR; (blue) nuclei. (B) Western analysis of phosphor S6K (P-S6K), total S6K (T-S6K), and total 4E-BP1 (T-4E-BP1) shows the effectiveness of Torin1 inhibition of mTORC1. Note also the expected decrease in pp28 detected in HCMV-infected cells treated with Torin1 from 2 to 48 and 24 to 48 hpi compared with untreated cells at 48 hpi.
Dynein depletion inhibits mTORC1 activity
We next asked whether the depletion of functional dynein causes a loss of mTOR activity under conditions in which it is usually active. HFs were infected with lentiviruses expressing shRNAs specific for luciferase (a control) or the dynein heavy chain, followed by mock or HCMV infection at 24 h after shRNA addition. At 72 h after HCMV infection, the cells were lysed and proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) followed by Western analysis for total 4E-BP1, phospho-4E-BP1, dynein, or actin. Figure 6 shows that dynein knockdown resulted in a decrease in mTORC1 activity, as demonstrated by loss of the more hyperphosphorylated forms of 4E-BP1 in both mock- and HCMV-infected HFs. This decrease is seen despite an incomplete depletion of dynein heavy chain by shRNAs; it is likely that a more substantial loss of mTORC1 activity could be observed if more complete dynein depletion could be accomplished. Nevertheless, these results suggest that dynein function is necessary to maintain mTORC1 activity in both normal and HCMV-infected HFs.
Figure 6.
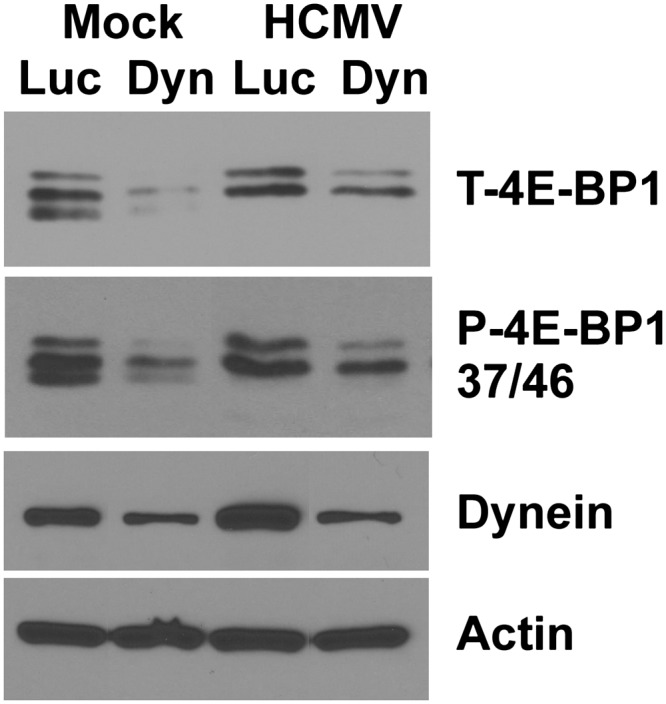
Dynein depletion inhibits mTORC1 activity. HFs were infected with lentiviral vectors expressing shRNA specific for luciferase (Luc), a control, or dynein heavy chain (Dyn) for 24 h followed by mock or HCMV infection. Seventy-two hours post HCMV infection, cells were lysed and proteins were separated by SDS-PAGE followed by Western analysis for total 4E-BP (T-4E-BP1), phospho-4E-BP (P-4-EBP1 37/46), dynein, and actin.
Dynein inhibition by CA inhibits mTORC1 activity and disturbs the formation of the AC
Recent studies have shown that the small molecule inhibitor CA is a specific dynein inhibitor due to its ability to interfere with dynein's ATPase activity and dynein-dependent microtubule gliding (Firestone et al. 2012). Thus, we further analyzed the role of dynein in mTORC1 activation using CA inhibition. For these experiments, we allowed the mock infection and the HCMV infection to establish for 48 h and then added 60 μg/mL CA or a solvent control for 24 h and then extracted or fixed the cells at 72 hpi. Figure 7 shows the activity of mTORC1 as measured by S6K and 4E-BP1 phosphorylation. CA treatment between 48 and 72 h after HCMV infection or mock infection decreased the hyperphosphorylation of 4E-BP1 and the phosphorylation of S6K in both mock- and HCMV-infected cells in comparison with the levels in untreated cells 72 h after HCMV infection or mock infection. In addition, the levels of the viral protein pp28 are lowered by the CA treatment; this may be due to inhibition of translation by hypophosphorylated 4E-BP1. Figure 8 shows the immunofluorescence examination of AC formation as indicated by pp28 (green) and mTOR (red) staining; we see a more compact and intense AC at 72 hpi as compared with 48 hpi. The panels in Figure 8 showing GFP indicate the infected cells, since the virus carries an inserted GFP gene. This is shown because the 48 and 72 hpi samples have a few uninfected cells that provide an internal, uninfected control; these cells are indicated by the white arrows in Figure 8. In contrast to the AC formation at 48 and 72 hpi, dynein inhibition by CA between 48 and 72 hpi results in malformation of the AC, shown by a more diffuse staining of mTOR and pp28.
Figure 7.

Inhibition of dynein function using CA causes loss of mTORC1 activity. HFs were mock- or HCMV-infected for 48 or 72 h and then extracted, or treated with the dynein inhibitor CA (60 μg/mL) between 48 and 72 h after mock or HCMV infection and then harvested. Western analysis was performed for total 4E-BP1 (T-4E-BP1), phospho-S6K (P-S6K), total p70 S6K (T-S6K), pp28, and actin.
Figure 8.

Inhibition of dynein function using CA affects AC formation. HFs were mock- or HCMV-infected for 48 or 72 h and then fixed, or treated with the dynein inhibitor CA (60 μg/mL) between 48 and 72 h and then fixed. Immunofluorescence analysis for pp28 (green) and mTOR (red) is shown; nuclei were stained with 4′6′-diamidino-2-phenylindole (DAPI) (blue). The HCMV used encodes GFP; thus, infected cells are indicated in the GFP panels (green). This allows uninfected cells (arrows) to be seen as internal, uninfected controls.
As mentioned above, we used uninfected U373-MG cells to study the perinuclear localization of mTOR because mTOR is predominately perinuclear under normal growth conditions; this localization was lost upon depletion of amino acids, correlating with the loss of mTORC1 activity, and was regained when amino acid-containing medium was restored, corresponding with the return of mTORC1 activity (Clippinger et al. 2011b). In Figure 3, we show that inhibition of dynein using CC1 disrupted mTOR localization to the perinuclear position in U373-MG cells. Figure 9A shows that inhibition of dynein by CA also disrupts the perinuclear localization of mTOR in U373-MG cells. Figure 9B shows the effect of CA on mTORC1 activity by examining phosphorylation of 4E-BP1; in this experiment, samples at 48 and 72 h after HCMV infection or mock infection were compared with samples treated with CA for 24 h between 48 and 72 h after HCMV infection or mock infection. Again, we observed that hyperphosphorylation of 4E-BP1 is lost in both cases. Additionally, the total level of 4E-BP1 is reduced, particularly in the mock-infected cells, in agreement with the recent observation that severely hypophosphorylated 4E-BP1 is degraded by the KLHL25-CUL3 ubiquitin ligase (Yanagiya et al. 2012).
Figure 9.

Inhibition of dynein function using CA affects the activity and perinuclear localization of mTOR in uninfected U373-MG cells. (A) Monolayers of U373-MG cells were treated with CA or the solvent control for 24 h and then stained for mTOR (red) and the nuclei (blue). (B) Monolayers of U373-MG cells were mock-infected or HCMV-infected for 48 and 72 h or treated with CA between 48 and 72 h after mock or HCMV infection. Cells were extracted for Western analysis of 4E-BP1, pp28, and actin. A lighter exposure of the 4E-BP1 analysis is provided in order to see the details in the HCMV-infected cell samples.
In sum, our data show that dynein function is needed for mTORC1 activity, apparently to move mTORC1 to locations in the cell where it will be active. HCMV has commandeered this function of dynein to insure the activation of mTORC1 and the later formation of the AC.
Discussion
During HCMV infection, mTORC1 activity is maintained even in the face of stress response signaling, which would inhibit it in uninfected cells. Existing data show that this is in part accomplished during the first 12 h of infection by transient activation of Akt, leading to the inactivation of the TSC (Fig. 1; Yu and Alwine 2002; Kudchodkar et al. 2004). However, by 12 hpi, the viral UL38 protein is produced and binds to the TSC, which is believed to inactivate the TSC, bypassing the need for Akt (Fig. 1; Moorman et al. 2008). Thus, any stress signaling that normally inactivates mTORC1 through activation of the TSC, such as hypoxia and energy deprivation, is potentially blocked (Kudchodkar et al. 2004, 2007). In addition, in HCMV-infected cells, the level of Rheb, the mTORC1 activator, is increased, potentially offering an additional means of bypassing the effects of the TSC (Clippinger et al. 2011b). However, stress responses that inhibit mTORC1 in a TSC-independent manner, such as amino acid depletion, must also be dealt with during infection. Our previous studies showed that HCMV infection induces mTOR colocalization with its activator, Rheb-GTP, in a perinuclear region, where it is maintained in an active state regardless of the presence of amino acids (Clippinger et al. 2011b). This contrasts with uninfected cells where the localization and activity of mTORC1 are lost following amino acid depletion. At later times in infection, the perinuclear localization of active mTOR coincides with the viral cytoplasmic AC, a perinuclear body that forms on an MTOC and is important in virion maturation. The nucleus and the AC are well integrated; the nucleus is tightly pulled around the AC in a kidney shape. Both the formation of the AC and the reshaping of the nucleus around it require movement of the molecular motor dynein toward the MTOC at the center of the AC (Buchkovich et al. 2010; Indran et al. 2010). In the work presented here, we show that dynein also mediates the movement of mTOR to the AC. Additionally, our data establish that dynein-mediated perinuclear localization of mTOR is induced very early in HCMV infection (by 8 hpi), well before the formation of the AC.
In uninfected cells, we show that mTOR localization to perinuclear sites also requires dynein function. Significantly, we found that mTORC1 activity, as measured by 4E-BP1 and S6K phosphorylation, is dependent on dynein function. Thus, the movement and activity of mTOR are dependent on a normal cellular function of dynein; HCMV commandeers this to ensure the constitutive activation of mTOR, the protection of mTOR from stress inhibition, and the formation of the AC. The use of molecular motors for transport of virion components, nucleocapsids, and virions in the assembly and egress process has long been known (for review, see Dodding and Way 2011); our studies extend these findings, showing that viral manipulation of molecular motors modulates fundamental cellular signaling mechanisms.
The dynein-mediated formation of the perinuclear AC on the MTOC is reminiscent of perinuclear aggresome formation, which provides a means to dispose of misfolded proteins that exceed the degradative capacity of ubiquitin–proteasome and autophagy–lysosome systems. Previous studies have shown that decreased dynein function impairs autophagic clearance of aggregate-prone proteins (Ravikumar et al. 2005). These studies suggest that loss of dynein function results in premature aggregate formation and impaired autophagosome–lysosome fusion. Thus, one interpretation of HCMV's utilization of dynein in AC formation would be to promote aggregate formation and autophagosome–lysosome fusion. However, the AC has characteristics that are distinctly different from aggresomes. A significant amount of evidence suggests that the highly vesicular AC results from HCMV-induced remodeling of the membrane transport apparatus, resulting in new functional profiles for the transport apparatus (Das and Pellett 2007, 2011; Das et al. 2007). Such functional modification may affect the lysosomal component of the AC, since there is no clear link between the AC and autophagosome–lysosome fusion. In this regard, HCMV has been shown to modulate autophagy differentially. Very early in infection, HCMV stimulates autophagy (McFarlane et al. 2011) by a mechanism that is independent of de novo viral protein synthesis. However, at later times of infection, coinciding with AC formation, HCMV blocks autophagy (Chaumorcel et al. 2008) by a mechanism that requires de novo viral protein expression. One viral protein synthesized during this period is TRS1, which has been shown to block autophagosome biogenesis (Chaumorcel et al. 2012). Interestingly, TRS1 has been shown to be present in the AC (Buchkovich et al. 2009). Additionally, it has been shown that active Rheb disrupts the interaction between dynein and misfolded protein cargos and therefore blocks aggresome formation by inhibiting dynein-dependent transportation of misfolded proteins (Zhoua et al. 2009). As mentioned above, Rheb levels are greatly increased during HCMV infection (Clippinger et al. 2011b); this may aid in uncoupling dynein from misfolded protein cargos in order to direct it to AC formation and block aggresome formation.
Recent studies have shown that targeting mTORC1 to lysosomal surfaces maintains mTORC1 activity (Sancak et al. 2010). Additionally, the involvement of lysosomes in the activation of mTORC1 and in degrading autophagic substrates has been associated with lysosomal positioning (Korolchuk et al. 2010). In these HeLa cell studies, activation of mTORC1 by nutrients (serum and amino acids) correlated with its association with peripheral lysosomes, whereas starvation (removal of serum and amino acids) caused perinuclear clustering of lysosomes and mTOR, which correlated with loss of mTOR activity. In contrast, our present and previous experiments (Clippinger et al. 2011b), conducted in U373-MG cells and HFs, as well as studies done in HEK-293T cells (Sancak et al. 2008) show that the diffuse cytoplasmic localization of mTOR correlates with inactivity (for example, in the absence of amino acids) and that the perinuclear localization correlates with activity. Additionally, our present data show that the perinuclear localization of mTOR in HCMV-infected cells correlates with mTORC1 activity. While these experiments use different cells and conditions, the contrasting results suggest that the status of mTOR activity in either the perinuclear or diffuse cytoplasmic locations may be influenced by other cellular conditions or signaling. For example, the use of serum is quite different between the experiments; therefore, growth status or the availability of serum growth factors may alter whether mTORC1 is active or inactive in the perinuclear or diffuse cytoplasmic locations. Such control would allow localized activity of mTORC1, providing, for example, localized translation. Localized translation mediated by localized changes in mTORC1 activity has been demonstrated in axons (Lin and Holt 2008; Richter and Klann 2009)
Overall, our data suggest that HCMV commandeers a normal dynein function in mTOR localization and activation in order to ensure the constitutive activation of mTORC1. In this process, the virus bypasses the Rag protein requirement in mTORC1 activation (Clippinger et al. 2011b) and constitutively localizes mTOR in a perinuclear position, where it is constitutively activated and protected from inhibitory stress response signaling. The data also suggest that HCMV's takeover of dynein function is prerequisite for the formation of the AC and the reshaping of the nucleus around it. Most importantly, our data establish that dynein function is necessary for mTORC1 activity under normal cellular conditions.
Materials and methods
Cell culture
Life-extended foreskin HFs (Bresnahan et al. 2000) or the human glioblastoma–astrocytoma cell line U373-MG were cultured at 37°C in 5% CO2 in Delbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS), 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM Gluta-Max (Gibco, 35050). HFs were used between passages 2 and 10 after thawing. For amino acid starvation experiments, cells were incubated in RPMI 1640 medium without amino acids (US Biological, R8999-04A) for 50 min.
Reagents, plasmids, and lentiviral vectors
Torin1 was kindly provided by David Sabatini (Whitehead Institute, Cambridge, MA) and was used at a concentration of 250 nM. The CC1-GFP expression plasmid, which expresses the CC1 (amino acids 217–548) of p150Glued, was a gift from Erika Holzbaur (University of Pennsylvania, Philadelphia, PA) (Quintyne et al. 1999; Kwinter et al. 2008; Maier et al. 2008). The siRNA directed against the dynein heavy chain (GenBank NM_001376; 5′-GAGAGGAGGUUAUGUUUAAUU-3′) and the siGLO transfection indicator were purchased from Thermo Scientific Dharmacon. CA (Sigma Aldrich, HPI-4) was used at a concentration of 60 μM in DMSO, which was used as the solvent control (Firestone et al. 2012).
Primary antibodies used in this study were mTOR, total 4E-BP1 and phospho-4E-BP1 (Thr37/46), and total S6K and phospho-S6K (T389) (Cell Signaling); Rag A and Rag C (Cell Signaling); LAMP2 (Abcam); pp28 (Santa Cruz Biotechnology); gB (East Coast Bio); and β-actin (Millipore). A dynein intermediate chain antibody (Millipore) or an mTOR antibody (Santa Cruz Biotechnology, N-19) was used for immunoprecipitation experiments.
Plasmids encoding lentiviruses that express shRNAs against dynein heavy chain (NM_001376; TRCN0000116326) were purchased from Open Biosystems. The lentivirus containing the luciferase shRNA, used as a control, was constructed in this laboratory (Yu et al. 2011). Lentiviruses were prepared in 293T cells as previously described (Yu and Alwine 2008). Where applicable, lentiviruses were incubated with cells for 7 h in the presence of 10 μg/mL polybrene. Cells were then maintained in serum-containing medium for 24 h before infection. At 72 h after HCMV infection, cells were collected and lysed for Western blot analysis.
Virus preparation, titration, and infection
HCMV (Towne strain) stocks were prepared and purified as previously described (Kudchodkar et al. 2004). Titers were determined using the 50% tissue culture infective dose method (TCID50). All experiments were performed using a multiplicity of infection (MOI) of 3.
Amino acid starvation
HFs grown in six-well plates were mock- or HCMV-infected (MOI = 3). At the indicated time points, cells were washed three times with 1× phosphate-buffered saline (PBS) and incubated for 50 min in RPMI 1640 medium without amino acids. Following the 50-min incubation, cells were harvested or incubated for an additional 30 min in complete, amino acid-containing medium prior to harvest.
Viral infections and Western analysis
HFs grown in six-well plates were serum-starved for 24 h followed by mock or HCMV infection (MOI = 3). At various time points after infection, cells were collected and resuspended in cold RIPA lysis buffer (1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 0.15 M NaCl, 0.01 M sodium phosphate at pH 7.2, 2mM EDTA, 50 mM sodium fluoride, 200 μM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 1.5 μg/mL aprotinin, 1 μg/mL leupeptin). Lysates were then centrifuged for 10 min at 4°C at maximum speed in a Beckman microcentrifuge. The supernatant was transferred to a fresh tube and stored at −20°C until ready for Western analysis. Loading dye (3×) (187.5 mM Tris-HCl at pH 6.8, 6% sodium dodecyl sulfate, 30% glycerol, 0.3% bromophenol blue, 467 mM β-mercaptoethanol) was added to lysates, and the samples were boiled for 5 min. Proteins were separated by SDS-PAGE on an 8% or 12% gel and transferred to nitrocellulose membranes. The membranes were blocked for 1 h in 5% nonfat dry milk in Tris-buffered saline with 0.1% Tween 20 (TBST) and then incubated in primary antibody overnight at 4°C with the appropriate antibody diluted in 2% bovine serum albumin in TBST. Membranes were then incubated for 1 h with horseradish peroxidase-conjugated secondary antibodies (Thermo Scientific) diluted in 5% milk and visualized with ECL reagents (Roche).
Indirect immunofluorescence
Collagen-coated coverslips (BD Biosciences, BD BioCoat, 354089) containing HFs or U373-MG cells were mock- or HCMV-infected (MOI = 3). At the indicated time point, cells were washed three times with PBS and fixed for 20 min in 4% paraformaldehyde at room temperature. Cells were permeabilized in PBS containing 0.5% Triton X-100 and blocked in PBS containing 10% human serum (blocking buffer) (Buchkovich et al. 2009). Primary and secondary antibodies (Alexa Fluor 594 and Alexa Fluor 647, Invitrogen) were diluted in blocking buffer. Coverslips were washed three times in PBS, rinsed in H2O, and mounted on slides using VectaShield mounting medium containing 4′6′-diamidino-2-phenylindole (DAPI) (Vector Laboratories). Slides were examined using a Nikon Eclipse E600 (40×) microscope, and pictures were taken using a Hamamatsu camera.
For the CC1 experiments, cells were infected for 24 h and then electroporated with the CC1-GFP plasmid. Briefly, at 24 hpi, cells grown on 6-cm plates were trypsinized and centrifuged to pellet the cells. The cell pellet was resuspended in 250 μL of DMEM plus the CC1 plasmid and electroporated at 260 V/950 μF using a Bio-Rad Gene Pulser II. Cells were then replated on collagen-coated coverslips in six-well plates. Forty-eight hours after transfection (72 hpi), the cells were fixed and stained for immunofluorescence.
For the sidynein experiments, cells were first electroporated, as described above, with siRNA directed against the dynein heavy chain (GenBank NM_001376; 5′-GAGAGGAGGUUAUGUUUAAUU-3′; Dharmacon) and then infected with HCMV 6 h post-electroporation. The sidynein was coelectroporated with siGLO transfection indicator (Thermo Scientific Dharmacon) to monitor which cells took up the siRNA. At 72 h post-electroporation, the cells were fixed and stained for mTOR.
Immunoprecipitation
Three 10-cm plates of HFs were mock-infected, and three 10-cm plates were HCMV-infected. At 72 hpi, cells were washed once in PBS and lysed in 300 μL of immunoprecipitation lysis buffer (100 mM NaCl, 1 mM EGTA, 50 mM Trip at pH 8.0, 1% Igepal, PMSF, leupeptin, sodium orthovanadate, aprotinin). Following a 5-min incubation on ice, cells were centrifuged at maximum speed in a microfuge for 10 min at 4°C. Dynabead Protein G beads (Invitrogen) were used according to the manufacturer's protocol. Briefly, the dynabeads were conjugated to 10 μg of dynein intermediate chain antibody (Millipore), mTOR/FRAP antibody (Santa Cruz Biotechnology), or a nonspecific control antibody (IgG; Santa Cruz Biotechnology). The antibody–bead complexes and cell lysate were incubated overnight at 4°C with rotation. The next day, the complex was washed with immunoprecipitation wash buffer (100 mM NaCl, 1 mM EGTA, 50 mM Tris at pH 8.0, PMSF, leupeptin, sodium orthovanadate, aprotinin), transferred to a fresh tube, and resuspended in 3× load dye for Western analysis.
Acknowledgments
We thank the members of the Alwine laboratory for support and helpful suggestions. This work was supported by NIH grants R01CA157846 and R01CA157679 awarded to J.C.A. from the National Cancer Institute.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.196147.112.
References
- Alwine JC. 2008. Modulation of host cell stress responses by human cytomegalovirus. In Current topics in microbiology and immunology, human cytomegalovirus (ed. TE Shenk and MF Stinski), pp. 263–279. Springer, New York. [Google Scholar]
- Bresnahan WA, Hultman GE, Shenk T 2000. Replication of wild type and mutant human cytomegalovirus in life-extended human diploid fibroblasts. J Virol 74: 10816–10818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchkovich NJ. 2010. “The role of the unfolded protein response regulator BiP in HCMV virion assembly and egress.” PhD thesis, University of Pennsylvania, Philadelphia, PA. [Google Scholar]
- Buchkovich NJ, Maguire TG, Paton AW, Paton JC, Alwine JC 2008a. Human cytomegalovirus specifically controls the levels of the endoplasmic reticulum chaperone BiP/GRP78 which is required for virion assembly. J Virol 82: 31–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchkovich NJ, Yu Y, Zampieri CA, Alwine JC 2008b. The TORrid affairs of viruses: Effects of mammalian DNA viruses on the PI3K–Akt–mTOR signaling pathway. Nat Rev Microbiol 6: 265–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchkovich NJ, Maguire TG, Paton AW, Paton JC, Alwine JC 2009. The endoplasmid reticulum chaperone BiP/GRP78 is important in the structure and function of the HCMV assembly compartment. J Virol 83: 11421–11428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchkovich NJ, Maguire TG, Alwine JC 2010. Role of the endoplasmic reticulum chaperone BiP, SUN domain proteins, and dynein in altering nuclear morphology during human cytomegalovirus infection. J Virol 84: 7005–7017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaumorcel M, Souquere S, Pierron G, Codogno P, Esclatine A 2008. Human cytomegalovirus controls a new autophagy-dependent cellular antiviral defense mechanism. Autophagy 4: 1–8 [DOI] [PubMed] [Google Scholar]
- Chaumorcel M, Lussignol A, Mouna L, Cavignac Y, Fahie K, Cotte-Laffitte J, Geballe A, Brune W, Beau I, Codogno P, et al. 2012. The human cytomegalovirus protein TRS1 Inhibits autophagy via its interaction with Beclin 1. J Virol 86: 2571–2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clippinger AJ, Maguire TG, Alwine JC 2011a. The changing role of mTOR kinase in the maintenance of protein synthesis during human cytomegalovirus infection. J Virol 85: 3930–3939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clippinger AJ, Maguire TG, Alwine JC 2011b. Human cytomegalovirus infection maintains mTOR activity and its perinuclear localization during amino acid deprivation. J Virol 85: 9369–9376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Pellett PE 2007. Members of the HCMV US12 family of predicted heptaspanning membrane proteins have unique intracellular distributions, including association with the cytoplasmic virion assembly complex. Virology 361: 263–273 [DOI] [PubMed] [Google Scholar]
- Das S, Pellett PE 2011. Spatial relationships between markers for secretory and endosomal machinery in human cytomegalovirus-infected cells versus those in uninfected cells. J Virol 85: 5864–5879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Vasanji A, Pellett PE 2007. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J Virol 81: 11861–11869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodding MP, Way M 2011. Coupling viruses to dynein and kinesin-1. EMBO J 30: 3527–3539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestone AJ, Weinger JS, Maldonado M, Barlan K, Langston LD, O'Donnell M, Gelfand VI, Kapoor TM, Chen JK 2012. Small-molecule inhibitors of the AAA+ ATPase motor cytoplasmic dynein. Nature 484: 125–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indran SV, Ballwstas ME, Britt WJ 2010. Bicaudal D1-dependent trafficking of HCMV tegument protein pp150 in virus infected cells. J Virol 84: 3162–3177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki S, Holzbaur EL 1995. Affinity chromatography demonstrates a direct binding between cytoplasmic dynein and the dynactin complex. J Biol Chem 270: 28806–28811 [DOI] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM 2002. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110: 163–175 [DOI] [PubMed] [Google Scholar]
- Kim E, Goraksha-Hicks P, Li L, Neufeld T, Guan K-L 2008. Regulation of TORC1 by Rag GTPases in nutrient response. Nature Cell Biology 10: 935–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolchuk VI, Saiki S, Lichtenberg M, Siddiqi FH, Roberts EA, Imarisio S, Jahreiss L, Sarkar S, Futter M, Menzies FM, et al. 2010. Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol 13: 453–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzyzaniak M, Mach M, Britt WJ 2007. The cytoplasmic tail of glycoprotein M (gpUL100) expresses trafficking signals required for human cytomegalovirus assembly and replication. J Virol 81: 10316–10328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudchodkar S, Yu Y, Maguire T, Alwine JC 2004. Human cytomegalovirus infection induces rapamycin insensitive phosphorylation of downstream effectors of mTOR kinase. J Virol 78: 11030–11039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudchodkar SB, Yu Y, Maguire TG, Alwine JC 2006. Human cytomegalovirus infection alters the substrate specificities and rapamycin sensitivities of raptor- and rictor-containing complexes. Proc Natl Acad Sci 103: 14182–14187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudchodkar SB, Del Prete GQ, Maguire TG, Alwine JC 2007. AMPK-mediated inhibition of mTOR kinase is circumvented during immediate-early times of human cytomegalovirus infection. J Virol 81: 3649–3651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwinter DM, Lo K, Mafi P, Silverman MA 2008. Dynactin regulates bidirectional transport of dense-core vesicles in the axon and dendrites of cultured hippocampal neurons. Neuroscience 162: 1001–1010 [DOI] [PubMed] [Google Scholar]
- Lin AC, Holt CE 2008. Function and regulation of local axonal translation. Curr Opin Neurobiol 18: 60–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier KC, Godfrey JE, Echeverri CJ, Cheong FK, Schroer TA 2008. Dynamitin mutagenesis reveals protein-protein interactions important for dynactin structure. Traffic 9: 481–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamane Y, Petroulakis E, LeBacquer O, Sonenberg N 2006. mTOR, translation initiation and cancer. Oncogene 25: 6416–6422 [DOI] [PubMed] [Google Scholar]
- McFarlane S, Aitken J, Sutherland JS, Nicholl MJ, Preston VG, Preston CM 2011. Early induction of autophagy in human fibroblasts after infection with human cytomegalovirus or herpes simplex virus 1. J Virol 85: 4212–4221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman NJ, Shenk T 2010. Rapamycin-resistant mTORC1 kinase activity is required for herpesvirus replication. J Virol 84: 5260–5269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman NJ, Cristea IM, Terhune SS, Rout MP, Chait BT, Shenk T 2008. Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe 3: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobukuni T, Kozma SC, Thomas G 2007. hvps34, an ancient player, enters a growing game: mTOR complex1/S6K1 signaling. Curr Opin Cell Biol 19: 135–141 [DOI] [PubMed] [Google Scholar]
- Quintyne NJ, Gill SR, Eckley DM, Crego CL, Compton DA, Schroer TA 1999. Dynactin is required for microtubule anchoring at centrosomes. J Cell Biol 147: 321–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Acevedo-Arozena A, Imarisio S, Berger Z, Vacher C, O'Kane CJ, Brown SDM, Rubinsztein DC 2005. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat Genet 37: 771–776 [DOI] [PubMed] [Google Scholar]
- Reiling JH, Sabatini DM 2006. Stress and mTORture signaling. Oncogene 25: 6373–6383 [DOI] [PubMed] [Google Scholar]
- Richter JD, Klann E 2009. Making synaptic plasticity and memory last: Mechanisms of translational regulation. Genes Dev 23: 1–11 [DOI] [PubMed] [Google Scholar]
- Sancak Y, Thoreen C, Peterson T, Lindquist R, Kang S, Spooner E, Carr S, Sabatini D 2007. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell 25: 903–915 [DOI] [PubMed] [Google Scholar]
- Sancak Y, Peterson T, Shaul Y, Lindquist R, Thoreen C, Bar-Peled L, Sabatini D 2008. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320: 1496–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM 2010. Ragulator–Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141: 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez V, Greis KD, Sztul E, Britt WJ 2000. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: Characterization of a potential site of virus assembly. J Virol 74: 975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM 2004. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 14: 1296–1302 [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sabatini DM 2005. Growing roles for the mTOR pathway. Curr Opin Cell Biol 17: 596–603 [DOI] [PubMed] [Google Scholar]
- Smith EM, Finn SG, Tee AR, Browne GJ, Proud CG 2005. The tuberous sclerosis protein TSC2 is not required for the regulation of the mammalian target of rapamycin by amino acids and certain cellular stresses. J Biol Chem 280: 18717–18727 [DOI] [PubMed] [Google Scholar]
- Thoreen CC, Sabatini DM 2009. Rapamycin inhibits mTORC1, but not completely. Autophagy 5: 725–726 [DOI] [PubMed] [Google Scholar]
- Tilton C, Clippinger AJ, Maguire T, Alwine JC 2011. Human cytomegalovirus induces multiple means to combat reactive oxygen species. J Virol 85: 12585–12593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D, Perez C, Notary J, Mohr I 2005. Regulation of the translation initiation factor eIF4F by multiple mechanisms in human cytomegalovirus-infected cells. J Virol 79: 8057–8064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagiya A, Suyama E, Adachi H, Svitkin YV, Aza-Blanc P, Imataka H, Mikami S, Martineau Y, Ronai ZA, Sonenberg N 2012. Translational homeostasis via the mRNA cap-binding protein, eIF4E. Mol Cell 46: 847–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Alwine JC 2002. Human cytomegalovirus major immediate-early proteins and simian virus 40 large T antigen can inhibit apoptosis through activation of the phosphatidylinositide 3′-OH kinase pathway and cellular kinase Akt. J Virol 76: 3731–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Alwine JC 2008. Interaction between simian virus 40 large T antigen and insulin receptor substrate 1 is disrupted by the K1 mutation, resulting in the loss of large T antigen-mediated phosphorylation of Akt. J Virol 82: 4521–4526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Maguire TG, Alwine JC 2011. Human cytomegalovirus activates glucose transporter 4 expression to increase glucose uptake during infection. J Virol 85: 1573–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhoua X, Ikenouea T, Chena X, Lib L, Inokia K, Guana K-L 2009. Rheb controls misfolded protein metabolism by inhibiting aggresome formation and autophagy. Procatl Acad Sci USA 102: 8923–8928 [DOI] [PMC free article] [PubMed] [Google Scholar]