

Abstract
We describe massive enlargement of both adrenal glands in 3 newborns, 2 girls and 1 boy. Two had hemihypertrophy and other congenital abnormalities but no identified genetic mutation; the third had genetically proven Beckwith-Wiedemann syndrome. Two had severe Cushing syndrome, the third had hypercortisolemia but no clinical Cushing syndrome. Bilateral adrenalectomy cured Cushing syndrome in the 2 with the severe symptoms; total adrenal weight was 44 and 53 gm in the patients, respectively. Unilateral adrenalectomy was performed in the third patient: the gland weighed 52 g; postoperatively, the patient's hypercortisolemia normalized and, concomitantly, the enlarged contralateral adrenal gland had a 5-fold decrease in size to slight enlargement 6 years postoperatively. Microscopically, the 3 patients had similar pathology: massive adrenal enlargement due to a combination of cytomegaly, persistence of the transient cortex, and hyperplasia of the permanent cortex. The pathological findings were most likely the result of the genetic mutation identified in 1 patient and of an unknown mutation in the remaining 2 patients.
Keywords: Fetal adrenal gland, neonatal Cushing syndrome, cytomegaly, hemihypertrophy, Beckwith-Wiedemann syndrome
Introduction
The human fetal adrenal gland is very different histologically from the adult gland. The fetal gland is composed of a narrow mantle of small cells (destined to persist and become the postpubertal adult cortex with its 3 classic zones) enclosing a large central mass of transient cells (ordained to undergo rapid involution and disappear shortly after birth). The narrow peripheral mantle has been variously referred to as the definitive cortex, the neocortex, and the permanent cortex; the central impermanent cell mass as the X-zone, the fetal cortex, the provisional cortex, and the transient cortex.
The transient cortex is composed of a monotonous patternless sheet of moderately large, poorly outlined, polygonal cells with eosinophilic cytoplasm and vesicular nuclei. As a consequence of its postnatal involution, the adrenal glands each of which weighs around 4 g at birth, weighs about 1 g at 1 year. Scattered giant cells (later termed cytomegalic cells) were described in the zone by Kampmeier in 1927 (12). These cells each had a very large, round, hyperchromatic nucleus and cytoplasm more eosinophilic than that of the transient cortex cells; they stood out because of their great size. Later, these cells were found elsewhere—in adult patients with congenital adrenal hypoplasia (10), in newborns with congenital anomalies (Beckwith-Wiedemann syndrome) (2 Beckwith, 1969), in newborns with Rh incompatibility (1) and, very rarely, in healthy adults (23). None of these patients had Cushing syndrome.
Herein, we describe massive bilateral adrenal enlargement caused by a combination of cytomegaly, persistence of the transient cortex, and hyperplasia of the permanent cortex in 3 newborns. Two of them had Cushing syndrome, and the third had hypercortisolemia but not clinical Cushing syndrome. All had other congenital abnormalities. Findings in the infants were likely resulted likely the result of Beckwith-Wiedemann mutation in 1 patient and unidentified mutation in the remaining 2.
Patients and Methods
Three newborns, 2 girls and 1 boy, comprised the study group. An adrenal abnormality was discovered in 1 at gestational age of 26 weeks and in the other 2 shortly after birth. There was no adrenal abnormality or congenital anomaly in any of the patients' primary relatives.
Blood was obtained for measurement of adrenal steroids and genetic testing for Beckwith-Wiedemann syndrome (patients 1 and 3), and PRKARIA mutations (patient 3). Imaging of the adrenal glands was performed using ultrasound and magnetic resonance technics.
Adrenal glands obtained at surgery were fixed in formalin and embedded in paraffin. Four μm-thick sections were obtained for routine microscopy, histochemical staining, and immunocytochemical staining. Six μm-thick sections were used for digital image analysis and mass spectrometry. Diced fragments of paraffin-embedded tissue were postfixed in 3% phosphate-buffered glutaraldehyde and processed for electron microscopy.
The histochemical stains used were hematoxylin-eosin, periodic acid-Schiff with and without diastase predigestion, reticulin, and Masson trichrome.
Immunostaining was performed with antibodies directed to the following: vimentin (Dual Env; PT link: 1/500 BRD; Dako; V9), synaptophysin (Ventana; CC1 mild; 1/50 BRD; Leica [Novocastro]; 27G12), inhibin-A (Advance; PT link; 1/60 BRD; AbD Serotec; R1), melan A (Advance; PT link; 1/500 BRD; Dako; A103), CD56 (Ventana; CC1 mild; 1/100 BRD; Dako; 123C3), calretinin (Ventana; CC1 mild; 1/50 BRD; Leica [Novocastro]), and b-catenin (Dual Env; PT Link; 1200; Santa Cruz; E-5. A polymer-based detection system was used.
For electron microscopy, diced fragments of tissue obtained from the paraffin blocks were post-fixed in 3% glutaraldehyde.
Six μ--thick sections stained with MIB-1 Ki-67 (Ventana; CC1 mild; 1/20BRD; Dako; MIB-1) were used to evaluate proliferation indices. For determination of aneuploidy, nuclear DNA content in Feulgen-stained 6 μ--thick sections was measured with a CAS 200 image analyzer (Bacus Laboratories, Lombard, Illinois).
For mass spectrometry, 6 μ--thick sections were placed on Director™ slides (Expression Pathology Inc, Rockville, Maryland) for laser microdissection. At least 2 different areas were dissected from each slide for analysis.
Results
Clinical findings and follow-up data are presented in Table 1. Results from adrenal imaging and laboratory steroid studies are summarized in Table 2. Treatment, gross pathologic findings, and initial pathological diagnoses are presented in Table 3. Immunocytochemical findings are listed in Table 4. Proliferative indices and ploidy findings are shown in Table 5.
Table 1.
Clinical Findings And Follow-up Data For 3 Neonates With Bilateral Adrenal Enlargement And Hypercortisolemia
| Patient | Sex | Age at Presentation | Age at Diagnosis of Cushing Syndrome | Other Conditions | Status and Age (year) |
|---|---|---|---|---|---|
| 1 | M | Birth | Subclinical (2 wk) | Left hemihypertrophy, enlarged left testis, striated muscle hamartoma, nephromegaly | Alive, 8 |
| 2 | F | Birth | 2 wk | Beckwith-Wiedemann syndrome. Right hemihypertrophy, hemangioendothelioma, epilepsy, mental retardation, virilization | Alive, 3 |
| 3 | F | 26 wk gestation | 2 wk | Right hemihypertropy, short stature, small facial features, café-au-lait spot, hypoglycemia, virilization, hepatic hemangioma, renal cysts | Alive, 4 |
Table 2.
Imaging And Laboratory Results For 3 Neonates With Bilateral Adrenal Enlargement And Hypercortisolemia
| Patient | Imaging | Serum Cortisol | Serum DHEA-S | Plasma Testosterone | Serum Aldosterone | Plasma Corticotropin | Dexamethasone Suppression Test |
|---|---|---|---|---|---|---|---|
| 1 | MRI: bilateral adrenal enlargement | ↑ | ↑ | ND | Normal | Low normal | Not done |
| 2 | US: bilateral adrenal mass | ↑ | ↑ | ↑ | ↑ | ↓ | Cortisol not suppressed |
| 3 | US and MRI (prenatal): bilateral adrenal mass; right, solid and cystic; left, cystic | ↑ | ↓ | ↑ | ↑ | ↓ | Cortisol not suppressed |
Abbreviations: DHEA-S, dehydroepiandrosterone sulphate; MRI, magnetic resonance imaging; ↑, increased, ↓, decreased; ND, test not done; US, ultrasonography.
Table 3.
Treatment, Gross Pathologic Findings, And Initial Pathologic Diagnoses For 3 Neonates With Bilateral Adrenal Enlargement And Hypercortisolemia
| Adrenal Weight, g | ||||||
|---|---|---|---|---|---|---|
| Case | Surgical Treatment | Age at Adrenalectomy, mo | Right | Left | Gross Appearance | Initial Pathologic Diagnosis |
| 1 | Left adrenalectomy and excision of ectopic left adrenal cytomegalic masses (up to 4 × 2.5 × 2 cm) | 1 | Not resected; gland enlarged | 52 | Bulging dark brown nodules, externally. Cut surface showed brown lobules limited by a narrow yellow periphery | Multinodular adrenal with cytomegalic changes |
| 2 | Bilateral adrenalectomy | 2 | 28.7 | 15.4 | Gyriform appearance with yellow and dark blue areas, externally. Cut surface showed flesh-colored, irregularly shaped lobules, many with a brownish center | Fetal adrenal hyperplasia and adult cortex |
| 3 | Bilateral adrenalectomy | 3 | 30.1 | 22.5 | Cerebriform appearance with gray and yellow nodules, externally. Cut surface featured lobules with yellow periphery and central dark red core. Three 0.5-cm pseudocysts | Diffuse adrenocortical hyperplasia with cytomegaly |
Table 4.
Selected Immunocytochemical Results For 3 Neonates With Bilateral Adrenal Enlargement And Hypercortisolemia
| Cortex | Vimentin | Synaptophysin | Inhibin-A | Melan A | CD56 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pos | Neg | Pos | Neg | Pos | Neg | Pos | Neg | Pos | Neg | |
| Permanent | 3 | 0 | 2 | 1 | 3 | 0 | 2 | 1 | 3 | 0 |
| Transient | 1 | 2 | 3 | 0 | 3 | 0 | 3 | 0 | 3 | 0 |
| Cytomegalic | 1 | 2 | 3 | 0 | 3 | 0 | 3 | 0 | 3 | 0 |
Abbreviations: Pos, positive; neg, negative.
Values indicate the number of patients
Table 5.
Proliferative Index and Ploidy Findings For 3 Neonates With Bilateral Adrenal Enlargement And Hypercortisolemia
| Proliferative Index (Ki-67 Positive Nuclei) | Ploidy | |||||
|---|---|---|---|---|---|---|
| Patient | Permanent Cortex | Transient Cortex | Cytomegaly | Permanent Cortex | Transient Cortex | Cytomegalic Cortex |
| 1 | Low (1.9%) | Insufficient for evaluation | Low (2.4%) | Diploid | Insufficient for evaluation | Aneuploid |
| 2 | Low (0.2%) | Low (1.8%) | Low (0.6%) | Diploid | Diploid | Aneuploid |
| 3 | High (6.7%) | Low (1.7%) | Low (0.9%) | Diploid | Diploid | Aneuploid |
Genetic Testing
Testing revealed the Beckwith-Wiedemann mutation in patient 2 who inherited 2 paternal copies of chromosome 11. The mutation was not found in patients 1 and 3. There were no PRKAR1A mutations in patient 3.
Pathology
Gross Findings
The findings are illustrated in Figure 1 and summarized in Table 3.
Figure 1.

Gross appearance of the formalin-fixed left adrenal gland (Patient 1). A, Large, round and oval, brown, tan, and grey nodules protruded from the external surface. B, The cut surface showed discrete and apparently fused tan, black, and orange-tinged nodules; some were separated by grey supporting tissue.
Light Microscopic Findings
The adrenal glands lacked the 3-layer cortical zonation present in the normal postpubertal cortex (the zona reticularis begins to develop at about age 3 years and its development is usually complete at about age 10). Instead, at scanning-power magnification, a very irregularly shaped mass of eosinophilic cells was mantled by a narrow rim of basophilic-appearing cells (higher power magnification showed that the basophilia was due to the high nuclear to cytoplasmic ratio in the cells, not cytoplasmic basophilia). The approximate proportion of the tissue sections occupied by the permanent cortex, the transient cortex, and the cytomegalic cells varied among the patients. The thickness of the permanent cortex was approximately similar in patients 1 and 2 and was considered hyperplastic because of the increased length necessary to accommodate the large core of transient cortex and cytomegalic cells. The cortex was considered to be also hypertrophic in patient 3 because of its increased thickness due to enlargement of its cells. In patient 1, the transient cortex occupied a small area, and the central mass of cytomegalic cells was surrounded almost entirely by permanent cortex through which a focus of cytomegalic cells penetrated into periadrenal fat. The transient cortex in patient 2 showed degeneration of the type seen in normal transient cortex (weak eosinophilia, hypocellularity, vascularity, and much intercellular material, weakly staining with Masson trichrome).
Each gland was surrounded by a thin, sometimes barely perceptible, capsule composed of collagen fibers and scattered hyperchromatic spindle nuclei (Figures 2–4). The capsule contained occasional spherical and small, flattened, elliptical-shaped groups of cells similar to those in the permanent cortex (Figure 3A). A few unencapsulated aggregates of these cells were in the immediate periadrenal fat. In patient 1, some of these cells were larger than those of the permanent cortex rim and were possibly transient cortex type.
Figure 2.

Microscopic appearance of right adrenal gland (Patient 2). A, Scanning-power image showed an irregularly shaped lesion with a barely recognizable adrenal outline; the mass was composed of large, small and ovoid nodules composed of eosinophilic cells often surrounded by a dark rim. B, In this low-power micrograph, a large core of cells with eosinophilic cytoplasm lay between a thin rim of apparently small basophilic cells (left) and a hypocellular vascular area (right) consistent with degeneration of transient cortex. C, Reticulin staining showed narrow columns of cells in the superficial cortex (permanent cortex) (left), reticulin loss deeper in the cortex (transient cortex) (center), and pericellular reticulin in the deepest area (cytomegalic area) (right). D, Intermediate-power micrograph showed some of the features present in panel B. A peripheral rim of small polygonal cells with crowded nuclei was limited by a fibrous capsule. Deep to this were larger cells with eosinophilic cytoplasm and nuclei that varied slightly in size. Still deeper were even larger cells with huge nuclei (cytomegalic cells).
Figure 4.

Permanent, transient, and cytomegalic cortices (patient 2). A, Eosinophilic transient cortical cells occupied the deep portion of the cortex (center and right). The permanent cortical cells lay immediately beneath the thin capsule, which contained an oval group of permanent cortical type cells (arrow). B, Transient cortex cells with eosinophilic cytoplasm and dense nuclei varied slightly in size and were separated by dilated capillaries. C, Cytomegalic and permanent cortical-type cells in the center of the field were flanked by transient cortex cells, above left and below. D, Reticulin surrounded small groups and individual cytomegalic cells.
Figure 3.

Permanent and transient cortices (A and B from patient 2, C and D from patient 3). A, Permanent cortex beneath the capsule featured closely packed small cells with regular, hyperchromatic nuclei. These cells gradually acquired increasing amounts of eosinophilic cytoplasm, a suggestive columnar pattern, and a degree of nuclear enlargement (characteristics of transient cortex). An elongated group of cells similar to those of the permanent cortex was present in the capsule B, Permanent cortex featured round and oval microcysts with wispy content. Deeper cells had increasing amount of cytoplasm. C, Hyperplastic and hypertrophic cortex showed regular subcapsular cells that had pale, weakly eosinophilic cytoplasm and were larger than the permanent cortex-type cells. D, High-power image of cells in panel C showed weakly eosinophilic and clear cytoplasm with some cell borders visible.
Low- and intermediate-power microscopic examination of sections from patients 1 and 2 revealed a subcapsular, thin patternless band of small, poorly defined, polyhedral cells, each with a small, round, hyperchromatic nucleus without a nucleolus (the permanent cortex) (Figure 2B). The cytoplasm was weakly eosinophilic or very finely vacuolated. A rare mitotic figure was seen. The band was separated, sometimes sharply, sometimes poorly, from an underlying zone of larger eosinophilic cells (the transient cortex) (Figure 2B and 2D). Gradual transition between the permanent and the transient cortex was accompanied by loss of the reticulin that demarcated cell columns in the permanent cortex (Figure 2C). Rarely, tongues of the permanent cortex extended deeply into the transient cortex. The usually monotonous permanent cortex occasionally featured scattered, irregularly shaped spaces that were usually round and sometimes occurred in groups (Figure 3B). The spaces were empty or contained red cells or a tangled basophilic web. They are sometimes seen in primary bimorphic adrenocortical disease (McCune-Albright syndrome) (4).
The transient cortex comprised a well-defined or poorly defined, patternless, very vascular zone of cells deep to the permanent cortex (Figures 2B, 2D and 3). Its cells were 2 to 3 times larger than those of the permanent cortex. The amount of transient cortex was much larger in patients 2 and 3 than in patient1 in which it was very small. Occasionally, cells of the transient cortex next to the permanent cortex were arranged in suggestive rows perpendicular to the capsule, a pattern that was absent deeper in the zone. Exceptionally, the transient cortex cells extended to the adrenal capsule.
The transient cortex cells were poorly outlined and had finely granular, eosinophilic cytoplasm (Figure 4) containing scattered basophilic polymorphic granules. The nuclei were vesicular and less chromatic than those of the permanent cortex; a few had 1 or 2 small nucleoli. Mitotic figures were not seen. An occasional very large cell (cytomegalic) with a correspondingly large nucleus stood out among the otherwise uniformly sized cell population. The cells deeper in the zone were increasingly larger; they were huge in patients 2 and 3.
A mass of large to huge eosinophilic cytomegalic cells occupied the center of the glands deep to the transient zone. These cells were sharply outlined and had finely granular, weakly eosinophilic cytoplasm and a fine cell membrane (Figures 2D, 4C, 4D and 5). Some cells seemed to have evolved from cells of the transient zone. Rarely, there was a cytoplasmic spherical or paranuclear zone of increased eosinophilia (Figure 5B). The nuclei were central or eccentric in the cells and each had a small to medium-sized nucleolus. Chromatin was moderately coarse to coarse. Nuclear vacuoles with a granular eosinophilic content similar in appearance to the cytoplasm were common; intranuclear cytoplasmic invaginations were not seen. Mitotic figures were not encountered. Some hyperchromatic nuclei appeared to be degenerative. There was no necrosis. In patient 2, there was a minute focus of “microcysts” in the cytomegalic mass which possibly indicated emerging fatty metaplasia.
Figure 5.

Cytomegalic cortex (patient 1). A, Huge eosinophilic cells with large to very large nuclei were juxtaposed to the permanent cortex with polymorphic cysts, without intervening transient cortex (left). B, Capillaries separated large eosinophilic cells with central and eccentric vesicular nuclei containing 1 or 2 small nucleoli. Some cells had zones of increased eosinophilia (arrows).
Thus, cell size and degree of eosinophilia gradually increased from the subcapsular permanent cortex, through the intermediately located transient cortex, to the deep cytomegalic zone.
In patient 1, a pale gray zone was visible grossly; microscopically, this was composed of stellate and spindle cells with regular nuclei and pale cytoplasm that formed papillary projections into an empty cystic space (Figure 6).
Figure 6.

Mesenchymal mass (patient 1). A, Undifferentiated mesenchymal cells surrounding an empty cavity with papillary infoldings were flanked on both sides by adrenal cortical tissue. B, The mesenchymal cells had oval and round nuclei. The area was bordered by transient cortex cells (left). Several cytomegalic cells (arrows) were located in an area of hemorrhage.
Immunostaining Findings
The results (Table 4) varied among the patients by site of cell staining (cell membrane staining compared with diffuse or localized cytoplasmic staining), type of staining (fine or coarse cytoplasmic granularity), and intensity of staining (strong or weak) (Figure 7). A rare focus of cytomegalic cells stained strongly for inhibin-A. Strong b-catenin cell membrane and punctate cytoplasmic staining decreased progressively from the permanent cortex, through absent cytoplasmic and weaker cell membrane staining in the transient cortex, to minimal or absent staining cell membrane staining in cytomegalic cells. Normally, the transient and permanent fetal and neonatal cortices stain with synaptophysin, inhibin-A and melan A.
Figure 7.
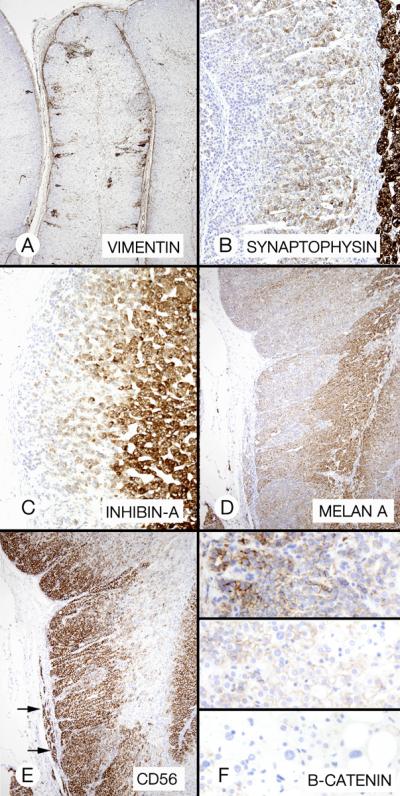
Cortical hyperplasia and hypertrophy (patient 3). A, The cortex featured zones of permanent cortical cells (asterisks) that flanked an area of larger cells with partly clear cytoplasm. B, Vimentin staining emphasized the difference between the cell types in panels A. C and D, Micrographs of approximately the same cortical area as in A showed different inhibin-A and melan A staining patterns.
Electron Microscopic Results
Electron microscopy showed that the differences between the permanent, transient, and cytomegalic cells were quantative (cell and nuclear size) rather than qualitative (organelle content). The round to oval nuclei each had a single nucleolus. The nucleus occupied most of the area of the permanent cortex cells and measured about 1/3 the diameter of the cytomegalic cells. Heterochromatin was more prominent in the nuclei of the permanent cortex. The cytoplasm was composed mainly of matrix; the most frequent organelles were mitochondria that varied more in shape in the permanent cortex (round, oval and curved) than in the cytomegalic cells (round). Stacked rough endoplasmic reticulum was more prominent in the cytomegalic cells. The findings in the latter cells were considered similar to previous descriptions (3, 25).
Proliferative Index And Ploidy Findings
The proliferation index in the permanent cortex was high in patient 3 (Table 5). The permanent cortex and the transient cortex cells were diploid. Cytomegalic cells were aneuploid (Table 5).
Discussion
Cushing syndrome is an uncommon condition (22). Up to 80% of cases of the disorder among all ages groups are caused by an ACTH-secreting tumor, almost always in the pituitary gland, rarely in an ectopic location. The majority of the remaining cases result from a primary unilateral adrenal neoplasm, usually a cortisol-secreting adenoma or less frequently a carcinoma. In the very small residual group of patients who are largely infants and young individuals, the epidemiology is different; the syndrome stems from a primary bilateral adrenal disorder that is sometimes familial and sometimes associated with nonendocrine conditions. The latter include spotty skin pigmentation, myxomas, and testicular large-cell calcifying Sertoli cell tumor (Carney complex) (6), and polyostotic fibrous dysplasia, precocious puberty, and café-au-lait spots (McCune-Albright syndrome)(7). Exceptionally, the Cushing syndrome is congenital (16,19), as in the cases we describe.
The bilateral infant and childhood disorders likely originate in the adrenal primordia each of which arises from 2 groups of coelomic epithelial cells (18). An initial aggregate of cells (destined to become the transient cortex) is joined by a second group of cells (the antecedent of the permanent cortex), and the 2 become enclosed by a loose band of spindle cells that forms a capsule around the primordium. At about the 8th week of gestation, the permanent cortex becomes recognizable as a narrow band of small basophilic-appearing cells between the capsule and the deeper transient cortex which is composed of larger cells with eosinophilic cytoplasm. Mitotic division in the permanent cortex (cellular hyperplastia) permits lateral growth of the zone and addition of cells in its depths that evolve into the transient cortex. The latter is mitotically inactive; its subsequent growth occurs through enlargement of its cells (cellular hypertrophy). As a result of this, the transient cortex eventually comes to dwarf the permanent cortex that remains as a narrow band. It is unclear whether fibroblast-like cells or rare undifferentiated cells in the capsule give rise to the cells of the outer permanent cortex (14). Starting at birth, apoptotic involution of the transient cortex (12) results in its rapid disappearance with consequent collapse of the adrenal glands.
Scattered giant cells with huge nuclei, later termed cytomegalic cells, were described in the zona fasciculata (subsequently recognized to be the transient cortex) of fetal adrenal gland by Kampmeier in 1927 (13). Later, the cells were found in pediatric autopsies (8), in adult patients with congenital adrenal hypoplasia (20) and, very rarely, in healthy adult individuals (25). A small focus of the cells was described in the noninvoluted fetal cortex of a 4-month old infant with congenital Cushing syndrome (16). Ultrastructurally, the cells have features consistent with steroidogenesis (3, 25).
Aterman (1, 10) suggested that the giant nuclei of cytomegalic cells were polyploid; they were later found to contain up to 25 times the normal amount of DNA (9). At a time when the transient zone is normally largely degenerated, DNA synthesis was still occurring in the cytomegalic nuclei (Ki-67 positivity) in our patients as late as 2 months postpartum,. Interestingly, although polyploidy is a major positive evolutionary mechanism in plants, in humans it usually causes defects in affected organs. The 3 newborns we report all had congenital abnormalities.
In connection with fetal adrenal steroidogenesis, the normal permanent cortex is capable of glucocorticoid synthesis and there is no adrenocortical insufficiency at birth (18). The transient cortex produces dehydroepiandrosterone (DHEA), an androgenic steroid, but not cortisol (18). Cytomegalic cells have no known hormonal product. There were 3 possible sources of the hypercortisolism in the 3 infants we report, the permanent cortex, the transitional cortex, and the cytomegalic cortex. The permanent cortex is the obvious first choice—in our cases, the zone showed 2 abnormalities that might explain its hyperfunction, an increase in the number of its cells of normal size (hyperplasia) that resulted from an increase in length (not thickness) of the cortex (patients 1 and 2) and a cortex of increased length and also of thickness (the latter due to hypertrophy of its cells) (patient 3).
The evolution of the findings in patient1 in whom unilateral adrenalectomy only was performed raised another, albeit remote, possibility. Following the adrenalectomy, there was a gradual restoration of normal cortisol production, accompanied by a 5-fold decrease in size of the remaining enlarged adrenal gland, during the 3 years following surgery. The gland presumably had the same pathology as the excised gland, massive cytomegaly. Assuming this, the loss of mass was presumably due to involution of its large cytomegalic core, the only component with bulk enough to explain the loss of mass. If certain cytomegalic cells were able to synthesize and secrete cortisol, this would offer an alternative explanation for the resumption of eucortisolemia. Alternatively, if the permanent cortex decreased in length simultaneously with the decrease of the cytomegalic core, this might offer another yet another explanation for the decrease of the patient's hypercortisolism.
From the foregoing, it is clear that several pathological processes were operational in the cases we describe, cell hyperplasia, cell hypertrophy, and cell involution and degeneration (the latter in patient 2). Centripedal proliferation of the permanent cortex resulted in an internal mass of cells individually larger in size than those of the parent permanent cortex. To accommodate this mass and maintain its lateral integrity there must have been addition of cells to the permanent cortex. Later, the normal involution and shrinkage of the transient cortex was delayed in our cases: its cells persisted, acquired additional cytoplasm and enlarged nuclei, multiplied, and evolved into huge multinodular masses. The findings indicate a persistence of DNA synthesis in the transient cortex (synthesis was also present in the cytomegalic cortex), delay of the programmed rapid decay of the fetal transient cortex, and transformation of many transient cortical cells into giant cells.
The pathological changes resulted in remarkably shaped gross specimens that were excessively folded, convoluted, and nodular in appearance. Microscopically, however, the glands lacked monomorphic nodules; the appearances must somehow have resulted from the manner of development of the abnormal cell hyperplasia and hypertrophy. The remarkable and unexpected shrinkage of the unresected gland in patient 1, presumably from eventual involution of the cytomegalic zone, would be expected to result in a redundant, excessively folded permanent cortex unless it lost length in parallel with the shrinkage of the cytomegalic zone.
In recent years, a group of Cushing syndrome-associated, genetically-mediated, bilateral adrenal disorders have recently been described in young patients. These include primary pigmented nodular adrenocortical disease (PPNAD) caused by mutations of PRKAR1A (15), PPNAD and other adrenal pathology caused by mutation of PDE11a (5), and bimorphic adrenocortical disease resulting from GNAS mutation (7). In fact, our patient 2 had Beckwith-Wiedemann syndrome, a genetic disorder, increasing the probability that the other 2 infants also had a similar type of abnormality, as yet unidentified. The disorder we describe herein is a candidate for addition to the group.
We have described the clinical, imaging, and pathologic findings from 3 newborn infants with hypercortisolemia: 2 had florid Cushing syndrome and the third had hypercortisolemia: 1 had Beckwith-Wiedemann syndrome, the 2 other congenital abnormalities. The resected adrenal glands were greatly enlarged due to a combination of tumefactive cytomegaly, transient cortex persistence, and permanent cortical hyperplasia and hypertrophy. Bilateral adrenalectomy cured the Cushing syndrome in the 2 patients thus treated. Unilateral adrenalectomy in the third was followed by a gradual reduction in size of the the remaining gland to approximately normal size, and eventual return to eucortisolemia. The adrenal disorder in the 3 infants was likely a developmental disorder manifested by delayed maturation of the fetal adrenal gland and hyperplasia of the permanent cortex.
Figure 8.

Immunostaining of permanent cortex hyperplasia (patient 2). A, Strong but patchy staining with vimentin. B, Weak staining of transient cortex (center) with synaptophysin. The adrenal medulla (right) was heavily stained. C, Strong inhibin-A staining of the transient cortex. The permanent cortex was very weakly stained. D, With melan A, there was moderately strong staining of the transient cortex (right) and weak staining of the permanent cortex (center). E, The permanent cortex showed strong staining with CD56. Extracortical permanent--type cells showed similar staining (arrows). F, B-catenin showed strong punctate and membranous staining of permanent cortex (top panel), absent punctate and reduced membranous staining of transient cortex (center panel) and no punctate or membranous staining of cytomegalic cells (bottom panel).
Table 6.
Selected Findings In Three Infants Reported With Hemihypertrophy and Cushing Syndrome.
Acknowledgement
We acknowledge Michael Christensen, BS, and Thomas J. Sebo, MD, PhD, for assistance with the digital image analysis; Hidenobu Soejima, MD for the molecular genetic study in patient 2, and Noriyuki Takubo, MD for assistance with clinical findings in case 2.
Dr Stratakis has no conflict of interest and was supported by Intramural Program, NICHD, NIH, project: Z01 HD000642-04i
Footnotes
Drs Carney, Ho, Kitsuda and Young have no conflict of interest or funding to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aterman K, Kerenyi N, Lee M. Adrenal cytomegaly. Virchows Arch Abt A Path Anat. 1972;355:105–122. doi: 10.1007/BF00556313. [DOI] [PubMed] [Google Scholar]
- 2.Beckwith JB. Macroglossia, omphalocele, adrenal cytomegaly, gigantism, and hyperplastic visceromegaly. Birth Defects. 1969;5:188–196. [Google Scholar]
- 3.Borit A, Kosek J. Cytomegaly of the adrenal cortex: Electron microscopy in Beckwith's syndrome. Arch Pathol Lab Med. 1969;88:58–64. [PubMed] [Google Scholar]
- 4.Carney JA. Adrenal Gland. In: Mills SE, editor. Histology for Pathologists. 4th edition. Lippincott, Williams and Wilkins; Philadelphia, PA: 2012. in press. [Google Scholar]
- 5.Carney JA, Gaillard RC, Bertherat J, Stratakis CA. Familial micronodular adrenocortical disease, Cushing syndrome, and mutations of the gene encoding phosphodiesterase 11A4 (PDE11A) Am J Surg Pathol. 2010;34:547–555. doi: 10.1097/PAS.0b013e3181d31f49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carney JA, Gordon H, Carpenter PC. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine. 1985;64:270–283. doi: 10.1097/00005792-198507000-00007. [DOI] [PubMed] [Google Scholar]
- 7.Carney JA, Young WF, Jr, Stratakis CA. Primary bimorphic adrenocortical disease: Cause of hypercortisolism in McCune-Albright syndrome. Am J Surg Pathol. 2011;35:1311–1326. doi: 10.1097/PAS.0b013e31821ec4ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Craig JM, Landing BH. Anaplastic cells in the fetal adrenal cortex. Am J Clin Pathol. 1951;21:940–949. doi: 10.1093/ajcp/21.10.940. [DOI] [PubMed] [Google Scholar]
- 9.Favara BE, Steele A, Grant JH, et al. Adrenal cytomegaly: Quantitative assessment by image analysis. Fetal Pediatric Pathol. 1991;11:521–536. doi: 10.3109/15513819109064788. [DOI] [PubMed] [Google Scholar]
- 10.Gau GS, Bennett MJ. Fetal adrenal cytomegaly. J Clin Pathol. 1979;32:305–306. doi: 10.1136/jcp.32.3.305. Letter to the Editor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hay ID, Smail PJ, Forsyth CC. Familial cytomegalic adrenocortical hypoplasia: an X-linked syndrome of pubertal failure. Arch Dis Child. 1981;56:715–721. doi: 10.1136/adc.56.9.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ishimoto H, Jaffe RB. Development and function of the human fetal adrenal cortex; a key component in the feto-placental unit. Endocrinol Rev. 2011;32:317–355. doi: 10.1210/er.2010-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kampmeier O. Giant epithelial cells of the human fetal adrenal. Anat Rec. 1927;37:102. [Google Scholar]
- 14.Kim AC, Hammer GD. Adrenocortical cells with stem/progenitor cell properties: recent advances. Mol Cell Endo. 2007;10-16:265–266. doi: 10.1016/j.mce.2006.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirscher LS, Carney JA, Pack SD, et al. Mutations of the gene encoding the protein kinase A type1-α regulatory subunit in patients with the Carney complex. Nat Genet. 2000;26:89–92. doi: 10.1038/79238. [DOI] [PubMed] [Google Scholar]
- 16.Klevit HD, Campbell RA, Blair HR, et al. Cushing's syndrome with nodular hyperplasia in infancy. J Pediatr. 1966;68:912–920. doi: 10.1016/s0022-3476(66)80210-x. [DOI] [PubMed] [Google Scholar]
- 17.Loridan L, Senior B. Cushing's syndrome in infancy. J Pediatr. 1969;75:349–359. doi: 10.1016/s0022-3476(69)80258-1. [DOI] [PubMed] [Google Scholar]
- 18.Mesiano S, Jaffe RB. Development and functional biology of the primate fetal adrenal cortex. Endo Rev. 1997;18:378–403. doi: 10.1210/edrv.18.3.0304. [DOI] [PubMed] [Google Scholar]
- 19.O'Bryan RM, Smith RW, Jr, Fine G, et al. Congenital adrenocortical hyperplasia with Cushing's syndrome. JAMA. 1964;187:257–261. doi: 10.1001/jama.1964.03060170011002. [DOI] [PubMed] [Google Scholar]
- 20.Prader A, Zachmann M, Illig R. Lutenizing hormone deficiency in hereditary congential adrenal hypoplasia. J Pediatr. 1975;86:421–422. doi: 10.1016/s0022-3476(75)80978-4. [DOI] [PubMed] [Google Scholar]
- 21.Robyn JA, Koch CA, Montalto J, et al. Cushing's syndrome in childhood and adolescence. J Paediatr Child Health. 1997;33:522–527. doi: 10.1111/j.1440-1754.1997.tb01663.x. [DOI] [PubMed] [Google Scholar]
- 22.Steffensen C, Bak AM, Rubeck KZ, et al. Epidemiology of Cushing's syndrome. Neuroendocrinol. 2010;92(suppl 1):1–5. doi: 10.1159/000314297. [DOI] [PubMed] [Google Scholar]
- 23.Viljoen D, Pearn J, Beighton P. Manifestations and natural history of idiopathic hemihypertrophy; a review of eleven cases. Clin Genet. 1984;26:81–86. doi: 10.1111/j.1399-0004.1984.tb00797.x. [DOI] [PubMed] [Google Scholar]
- 24.Winter JSD. Fetal and neonatal adrenocortical physiology. In: Polin RA, Fox WW, editors. Fetal and neonatal physiology. 2nd edition WB Saunders; Philadelphia: 1998. pp. 2447–2459. [Google Scholar]
- 25.Yamashina M. Focal adrenocortical cytomegaly observed in two adult cases. Arch Pathol Lab Med. 1986;110:1072–1075. [PubMed] [Google Scholar]