

Abstract
Peroxisome proliferator-activated receptor gamma (PPARγ) is member of a family of nuclear receptors that interacts with nuclear proteins acting as coactivators and corepressors. The colon is a major tissue which expresses PPARγ in epithelial cells and, to a lesser degree, in macrophages and lymphocytes and plays a role in the regulation of intestinal inflammation. Indeed, both natural and synthetic PPARγ ligands have beneficial effects in different models of experimental colitis, with possible implication in the therapy of inflammatory bowel disease (IBD). This paper will specifically focus on potential role of PPARγ in the predisposition and physiopathology of IBD and will analyze its possible role in medical therapy.
1. Introduction
The peroxisome proliferator-activated receptor gamma (PPARγ) is a nuclear receptor highly expressed in adipose tissue but also intestine, playing a key role in regulation of insulin resistance and inflammation. Recently its role in intestinal diseases, especially colon cancer and intestinal inflammation, is emerging. The discovery that it is the major functional receptor mediating the aminosalicylate activities in inflammatory bowel diseases (IBD) has further enhanced the interest for the role of this receptor in the regulation of gut homeostasis, with possible implication for newer therapeutic targeting. After an extensive search of medical literature in English language from the PubMed database, we aim in this paper to focus on potential role of PPARγ in the predisposition and physiopathology of IBD and to analyze its role in experimental colitis and potential therapy for IBD.
2. IBD and PPARγ: Friend or Foe
The inflammatory bowel diseases (IBD), Crohn's disease (CD), and ulcerative colitis (UC) are common causes of gastrointestinal illness characterised by chronic, relapsing intestinal inflammation, often presenting in early childhood [1]. The incidence varies according to geographical location and in Northern Europe IBD may affect upto one in two hundred of the population [2]. The division into CD and UC is made on the basis of clinical, radiological, endoscopic, and histological features. Common clinical features of CD include abdominal pain, diarrhea, weight loss, and fever. Rectal blood loss is not always a feature and up to 10% of patients with CD may not have diarrhea. Inflammatory changes are patchy in distribution and may occur anywhere within the gastrointestinal tract from the mouth to the anus. Approximately 40% of patients with CD will have disease involving both small and large bowel, in 30% the disease is limited to the small bowel, and 27% percent will have colonic disease only. A small minority of patients will have involvement of the more proximal gastrointestinal tract. Inflammation is transmural and histological examination of bowel and lymph nodes will demonstrate epithelioid cell granulomas in 60–70% of cases. In contrast, patients with UC usually present with bloody diarrhea [3]. There may be associated abdominal pain, urgency, and tenesmus. The disease is limited to the mucosal layer of the colon; it will always involve the rectum and may extend proximally in a continuous fashion.
Current knowledge of aetiology is incomplete, but increasing evidence points towards a combination of environmental triggers in a genetically susceptible individual. More specifically, the intestinal inflammation is thought to result from an inappropriate immune response to microbial antigens of commensal microorganisms [4]. Both diseases manifest themselves primarily in the gastrointestinal tract yet can, in principle, affect all of the organ systems of the body. IBD is also associated with an increased risk of colorectal cancer, which itself is already the third most common cancer in developed countries [2].
The progress in gene discovery in complex disease genetics has increased rapidly in recent years, boosted by the advent of genomewide association (GWA) studies. Few complex diseases have seen as much rapid progress as CD and UC thanks specially to the international inflammatory bowel disease genetics consortium (IIBDGC) who collected around the world some 20,000 cases for each of CD and UC (http://www.ibdgenetics.org/). The statistical power of such large sample sets has proven highly effective in identifying multiple susceptibility loci, even where these confer only modestly increased risk of disease. To date there are 99 IBD susceptibility loci: 71 associated with Crohn's disease, 47 with ulcerative colitis, and 28 with both CD and UC [5, 6]. Amongst these are multiple genes involved in IL23/Th17 signaling (IL23R, IL12B, JAK2, TYK2, and STAT3), genes involved in autophagy, intracellular bacteria processing and innate immunity (NOD2, IRGM, and ATG16L1), and genes involved in barrier (HNF4A, LAMB1, CDH1, and GNA1e). However, from these studies, included the recently reported data with the immunochip from the IIBDGC at DDW 2012 [7], no striking signal of PPARγ gene polymorphisms is emerged, with P values of tagging SNPs ranging from 0.005 to 0.01 (personal communication). Poliska et al. have investigated the association of four polymorphisms of PPARγ and IBD; they found haplotypes with both protective and increased risk [8]. Other studies, however, lead to conflicting results [9–13]. Accordingly, a meta-analysis of seven studies with over one thousand UC and CD found no significant association of the Pro12 Ala polymorphism of PPARγ with IBD [14].
In contrast, PPARγ is highly expressed in colonic epithelial cells and to a lesser degree into macrophages and lymphocytes [15]. In addition, its expression in the colon is closely linked to intestinal-microbial interaction. Using quantitative PCR, western blot, and immunohistochemical assay, a 60% decreased expression of PPARγ was observed at the mRNA and protein levels in the colon of UC patients, compared with CD and controls [16]. This impaired expression was found in both inflamed and noninflamed areas and limited to epithelial cells, suggesting that this modified expression is not secondary to the inflammatory process (Figure 1). A possible explanation is the occurrence of epigenetic changes [16]; this hypothesis is corroborated by the demonstration of similar levels of PPARγ in peripheral mononuclear cells of IBD patients and controls and lack of significant polymorphisms of PPARγ in UC patients. Another intriguing possibility is that the Toll-like receptor 4 (TLR4) signaling to PPARγ is impaired in UC. The resulting imbalance between elevated levels of TLR4 and reduced expression of PPARγ may lead to loss of mucosal tolerance to luminal LPS, resulting in mucosal inflammation [16]. In contrast, Yamamoto-Furusho et al. reported that the mRNA PPARγ expression was significantly reduced in the mucosa with active UC compared to the mucosa of patients in remission, with a significant correlation with disease activity [17].
Figure 1.
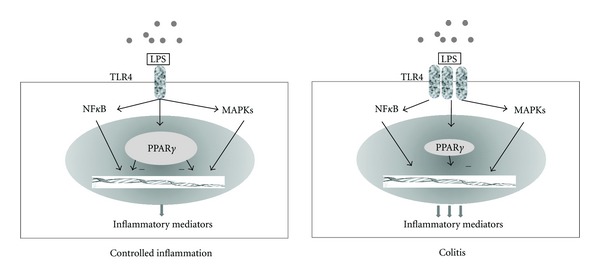
An hypothetical model of influence of PPARγ expression in ulcerative colitis. Induction of PPARγ expression in epithelial cells by bacterial lipopolysaccharide- (LPS-) activated TLR4, in turn leads to break NFκB and MAPK pathways to produce inflammatory mediators. The reduced expression of PPARγ together with TLR4 upregulation might enhance the inflammatory mediators production thus resulting in mucosal damage.
More recently, another important role of PPARγ in the modulation of intestinal inflammation has been put forward. In healthy individual, immune cells and gut mucosa remain largely inactive towards 1014 bacteria of the intestinal microflora. This tolerance is attributed to the prominent presence of regulatory immune cells that may be triggered by the resident microflora and whose function is antagonistic to inflammatory pathways stimulated by pathogenic bacteria [18]. The effector cells are M1 macrophages and dendritic (De) cells secreting inflammatory mediators including factors stimulating additional resting macrophages, dendritic cells precursors (monocytes), and T cells. De and M1 present antigen to resting T cells while secreting cytokines (IL12, IFN-γ, TNF-α, and IL-23) and induce the differentiation to proinflammatory T-helper, specifically Th1 and Th17. The immune response kills the invading bacteria, but may also cause indiscriminate tissue damage. In sterile organ systems, the inflammatory process usually ceases once the antigen population is eliminated. However, in the gut because of the resident microflora, the antigen population cannot be eliminated and the mounted inflammation could be more harmful for the host than the invading bacteria itself, for example, increasing gut permeability and infiltration of bacteria in the lamina propria. In healthy individuals, the gut mucosa contains various regulatory factors such as M2 macrophages, tolerogenic dendritic cells (Dt), and T regulatory cells. This regulatory pathway, by binding to ligands recognized as self to specific receptors, induces the differentiation and switches from M1 to M2 and from De to Dt. One such receptor is PPARγ expressed in T cells, dendritic cells, macrophages, and epithelial cells [19, 20].
3. PPARγ: Structure, Function, and Expression in the Gut
PPARγ belongs to the nuclear receptor family consisting of approximately 50 transcription factors implicated in many biological function. It is an essential nuclear receptor controlling the expression of a large number of regulatory genes in lipid metabolisms, insulin sensitization, inflammation, and cell proliferation [48].
Similarly to other nuclear hormone receptors, PPARγ displays a central DNA-binding domain, a C-terminal ligand-binding domain, and two transcription-activation function motifs (AF-1 and AF-2) [49]. Binding of ligands to PPARγ leads to a conformational change in the receptor which allows recruitment of co-activator proteins to then induce transcriptional activation. The transcriptional activity of PPARγ is regulated by post-translational changes such as phosphorylation or ubiquitination. The activation requires heterodimerization within the nucleus with another nuclear receptor named retinoid X receptor α (RXRα), leading to bind a specific DNA sequence elements known as peroxisome proliferator elements (PPREs) [50]. PPARγ interferes with inflammatory pathways by interactions with transcription factors such as nuclear factor kappa B (NF-κB), activating protein-1 (AP-1), signal transducer and activator of transcription (STAT), and nuclear factor-activated T cell (NFAT). For example, PPARγ is able to form a complex with the NF-κB subunit p65 at a nuclear level and this complex is exported from the nucleus leading to an altered expression of proinflammatory NF-κB-mediated gene expression. Inhibition of NF-κB in response to the activity of PPARγ ligands attenuates the expression of various cytokines in colonic epithelial cells such as IL-1β, COX-2, IL-6, IL-8, TNF-α, INF-γ, iNOS, and chemokines [51, 52]. Its expression has been initially investigated in adipose tissue where it plays a key role in adipocyte differentiation and insulin responses. More recently the colon has been found to highly express PPARγ in epithelial cells but also macrophages and lymphocytes [16, 52, 53]. Regulation of expression is incompletely understood; in vivo mRNA expression is negatively influenced by a long-term hypocaloric diet and fasting and positively by obesity and a rich in fatty acids diet. More recently a close link between intestinal microbial flora and PPARγ expression has been demonstrated. The stimulation of expression is probably multifactorial and involves the LPS recognition by the toll-like receptor (TLR), especially LPS of gram-negative bacteria and TLR4. Another alternative way of stimulation is the production through the bacteria of volatile fatty acid butyrate [15].
4. Experimental Model of Colitis
The initial evidence of the involvement of PPARγ in the regulation of intestinal inflammation derives from the observation of the use of PPARγ synthetic agonist thiazolidinedione (TZD) in mice dextran sodium sulfate- (DSS-) induced colitis. In the study by Su CG et al., both troglitazone and rosiglitazone dramatically reduced the colonic inflammation in mice and in addition significantly attenuated cytokine gene expression in colon cancer cell lines through NF-κB inhibition [21]. This first evidence was subsequently confirmed in another model of experimental colitis induced in mice by intrarectal administration of 2,4,6-trinitrobenzene sulfonic acid (TNBS) [51]. TZD given preventively significantly reduced mortality, severity of macroscopic and histological lesions, and markers of inflammation. So far several studies have reported similar prophylactic and therapeutic efficacy of PPARγ agonists in different animal models (mice, rats, and pigs) with different models of colitis induced by chemical compounds [22–34], ischaemia [35–38], bacteria [39], or genetically modified animals [43–47, 65, 66] (Table 1) [40–42]. Moreover a beneficial effect of PPARγ ligands has been demonstrated in colon carcinogenesis. Of interest, the use of probiotics (VSL#3), conjugated linoleic acid, n-3 polyunsaturated fatty acids, cannabidiol, punic acid, α-eleostearic acid, and a polyphenolic compound has prove beneficial effect on animal model of intestinal inflammation through the activation of PPARγ [67] (Table 1).
Table 1.
Anti-inflammatory properties of PPARγ agonists in experimental models.
| Model | PPARγ modulator | Effect | Authors |
|---|---|---|---|
| Acute colitis | |||
|
| |||
| DSS | Troglitazone | ↓ Colonic inflammation | Su et al. 1999 [21] |
| ↓ Cytokine gene expression | |||
| Rosiglitazone | Reduced inflammation | Saubermann et al. 2002 [22] | |
| More severe colitis | Ramakers et al. 2007 [23] | ||
| Pioglitazone | Prevention colitis | Takagi et al. 2002 [24] | |
| Recovery from colitis | Hontecillas et al. 2011 [25] | ||
| Reduced CXCL10 level | Schaefer et al. 2005 [26] | ||
| PUFA | Accelerated remission | ||
| CLA | Delayed onset of colitis | Bassaganya-Riera 2006 [27] | |
| CLA + VSL#3 | Improvement of colitis | Bassaganya-Riera et al. 2012 [28] | |
| α-Eleostearic acid | Improvement of colitis | Lewis et al. 2011 [29] | |
|
| |||
| TNBS | Troglitazone | Reduced inflammation | Desreumaux et al. 2001 [30] |
| Rosiglitazone | Reduced inflammation | ||
| Reduced inflammation | Sànchez-Hidalgo et al. 2007 [31] | ||
| Pioglitazone | Reduced CXCL10 level | Schaefer et al. 2005 [26] | |
| FMOC-L-leu | Reduced inflammation | Rocchi et al. 2001 [32] | |
| 5-ASA | Reduced inflammation | Rousseaux et al. 2005 [33] | |
| 5-ASA in PPARγ+/− | No efficacy of 5-ASA | ||
|
| |||
| Acetic acid ischaemia |
THSG | Attenuated colon lesions | Zeng et al. 2011 [34] |
| Rosiglitazone | Protection | Nakaijma et al. 2001 [35] | |
| 15-d-PGJ2 | Reduced injury | Cuzzocrea et al. 2003 [36] | |
| NS-398 | Protection | Sato et al. 2005 [37] | |
| Glutamine | Protection | Sato et al. 2006 [38] | |
|
| |||
| Bacterial | CLA | Attenuated inflammation | Hontecillas et al. 2002 [39] |
|
| |||
| Chronic colitis | |||
|
| |||
| DSS | Triglitazone | ↓Cell proliferation | Tanaka et al. 2001 [40] |
| TNBS | Rosiglitazone | Protection | Sànchez-Hidalgoet al. 2005 [41] |
| CD4-CD45RBhi | CLA | Reduced inflammation | Bassaganya-Riera et al. 2004 [42] |
| IL-10 KO | Rosiglitazone | Slow onset colitis | Lytle et al. 2005 [43] |
| SAMP1/Yirfc | Rosiglitazone | Decreased severity | Sugawara et al. 2005 [44] |
|
| |||
| Genetic models | |||
|
| |||
| PPARγ +/− | Desreumaux et al. 2001 [30] | ||
| Ischaemia | More severe damage | Nakaijma et al. 2011 [35] | |
| Saubermann et al. 2002 [22] | |||
| DSS + PUA | Loss protective effect PUA | Hontecillas et al. 2011 [25] | |
|
| |||
| AdPPARγ | Katayama et al. 2003 [39] | ||
|
| |||
| SAMP1/yifc | Sugawara et al. 2005 [44] | ||
|
| |||
| PPARγCre+ | Bassaganya-Riera et al. 2004 [42] | ||
|
| |||
| PPARγΔMφ | DSS | Increased susceptibility | Shah et al. 2007 [45] |
|
| |||
| PPARγfifi | DSS | Accelerated colitis | Guri et al. 2010 [46] |
|
| |||
| Worsen colonic lesions | Mohapatra et al. 2010 [47] | ||
5-ASA: 5-aminosalycilic acid; 15dPGJ2: 15-deoxy-Δ12,14-prostaglandin J2; CLA: conjugated linoleic acid; PUFA: n-3 polyunsaturated fatty acids; DSS: dextransodiumsulphate; FMOC-L-leu: fluorenylmethyloxycarbonyl-L-leucine; IL-10 KO: interleukin 10 knockout mice; PPARγCre: PPARγ conditional knockout mice; TNBS: 2,4,6-trinitrobenzene sulfonic acid; PUA: punicic acid; THSG: 2,3,5,4′-tetrahydroxystilbene-2-O-beta-D-glucoside.
Taken together lessons from animal studies suggest that: (a) natural and synthetic PPARγ ligands are both effective in the treatment of acute and chronic animal models of inflammation; (b) the prophylactic effect is more pronounced than the therapeutic effect; (c) the therapeutic effect is apparently dependent by the abundance of PPARγ in the target tissue as demonstrated by the genetically modified animals. This information translated into clinical ground could suggest a major role of PPARγ agonists in maintenance rather than induction of remission in IBD patients. Moreover, with PPARγ being expressed not only in the epithelial cells but also in macrophages, T, and B cells, more investigations are needed to disclose which cell type expression of PPARγ is more crucial for the potential therapeutic effect.
5. Dietary Modulation of PPARγ
A large number of dietary nutrients are able to modulate PPARγ (see Table 2). Fatty acids and their metabolites can affect gene expression by binding to PPARγ. The effect of n-3 PUFAs is well documented; linoleic acid is the major PUFA in human diet and several derivatives like conjugated linoleic acid (CLA), nitrolinoleic acid, and gamma linoleic acid have shown activation property on PPARγ [54, 55]. Another fatty acid-derived metabolite known to be a strong PPARγ inducer is the prostaglandin 15d-PGJ2 as demonstrated in several animal models [36]. Glutamine is the preferential substrate of enterocytes and is considered essential in stress situations. In a rodent model of ischemia reperfusion, glutamine also acted as PPARγ agonist, as protective effect was abrogated by a PPARγ inhibitor [38]. Various spicy foods such as curcumin and capsaicin have been shown to activate PPARγ. The anti-inflammatory property of curcumin is also expressed by the inhibition of NF-κB, but is clearly blocked by PPARγ inhibitor [56–58]. Also ginsenosides, compounds derived by ginseng, may have opposite effects being ginsenoside 20S a strong inducer and Rh2 an inhibitor of PPARγ [59, 60]. Finally, other inducers are flavonoids, epigallocatechingallate derived from green tea, resveratrol derived from grapes and wine, butyrate, and micronutrients such as vitamin E and selenium [61–64, 72] (Table 2).
Table 2.
Nutrients with demonstrated anti-inflammatory effects mediated through PPARγ.
| Nutrient | Dietary source | Models | Authors |
|---|---|---|---|
| α-linoleic acid | Green vegetables, flax | Intestinal epithelial cells | Marion-Letellier 2008 [54] |
| Docosahexaenoic | Fish | Intestinal epithelial cells | Marion-Letellier 2008 [54] |
| Eicosapentaenoic ac. | |||
| Conjugated linoleic acid | Beef, bovine milk | Intestinal epithelial cells DSS colitis | Allred et al. 2008 [55] |
| Glutamine | Beef, chicken, fish | Ischaemia reperfusion | Sato et al. 2006 [38] |
| Curcumin | Tumeric powder | TNBS colitis | Salh et al. 2003, Deguchi et al. 2007 [56]/[57] |
| Capsaicin | Cayenne pepper | Intestinal epithelial cells | Kim et al. 2004 [58] |
| Ginsenoids | Ginseng | Adypocites | Han et al. 2006, Hwang et al. 2007 [59]/[60] |
| Resveratrol | Grapes, wine, peanuts | Intestinal epithelial cells | Morikawa et al. 2007 [61] |
| Butyrate | Unabsorbed carbohydrate | Intestinal epithelial cells | Schwab et al. 2007 [62] |
| Vitamin E | Nuts, seeds, oils | Colon cancer cell lines | Campbell et al. 2003 [63] |
| Selenium | Plant foods | Macrophages | Vunta et al. 2007 [64] |
6. PPARγ and Therapy of Ulcerative Colitis
5-ASA is one of the oldest anti-inflammatory agents used for treatment of IBD, although the mechanism underlying its effects is still unknown. It is the mainstay of therapy for the majority of patients with UC for the induction of remission, maintenance, and possibly chemoprevention of colorectal cancer [73]. Recently, functional, biological, pharmacological, and chemical evidence has shown that aminosalicylates are a new functional synthetic ligand for PPARγ in colonic epithelial cells [33]. PPARγ is indeed the key receptor mediating the 5-ASA activity, by trans-repressing several key target genes such as nuclear factor κB, signal transducers, and activators of transcription.
Since in animal models treatment with PPARγ ligands has been demonstrated to attenuate inflammatory cytokines production such as IL-1β and TNF-α, it has been hypothesized the use of PPARγ ligands, like thiazolidinedione (TZD), in the therapy of UC [15]. One potential candidate is rosiglitazone, an antidiabetic drug. A first open-label pilot study in 15 patients with mild to moderate UC refractory to 5-ASA has evaluated the efficacy of the PPARγ ligand rosiglitazone (4 mg orally twice daily) (Table 3). These patients were refractory to conventional treatment, including corticosteroids and immune modulators. After 12 weeks of treatment, a striking reduction in disease activity index score was reported, with clinical and endoscopic remission in 27% and 20% of patients, respectively [68]. Liang and Quayang performed a clinical trial in China in 42 patients with mild to moderate UC [69]. Patients were allocated alternatively to the treatment of rosiglitazone 4 mg/day plus 5-ASA 2 gr or sulfasalazine 3 gr, while the control group received 5-ASA or sulfasalazine alone for 4 weeks. The remission rate was greater in the rosiglitazone group (71.4% versus 57.1%), with a significant improvement of the histologic score (P < 0.05). Moreover in the treatment group the PPARγ expression was increased compared to baseline [69].
Table 3.
Efficacy of rosiglitazone therapy in ulcerative colitis (*P values < 0.05).
| Authors | N° pts | Study design | Treatment | % Efficacy | ||
|---|---|---|---|---|---|---|
| Response | Remission | Mucosal healing | ||||
| Lewis et al. 2001 [68] | 15 | Open 12 weeks | 4 mg tid | — | 27 | 20 |
| Liang and Ouyang 2006 [69] | 42 | Random versus 5-ASA | 4 mg | — | 71 versus 57* | — |
| Lewis et al. 2008 [70] | 105 | 12 wks versus Plac | 4 mg tid | 44 versus 23* | 17 versus 2* | 8 versus 2 |
| Pederson and Brynskov 2010 [71] | 14 | Open versus 5-ASA | 4 mg versus 1 enema | = 5-ASA | = 5-ASA | — |
Recently, a randomized multicenter double-blind, placebo-controlled trial has been published by using rosiglitazone 4 mg orally twice daily versus placebo for 12 weeks in 105 patients with mild to moderate ulcerative colitis [70]. Disease activity was measured by Mayo score with a primary endpoint of a clinical response (≥2 points reduction) at week 12, while clinical remission, endoscopic remission, and quality of life changes were considered secondary outcomes. After 12 weeks of therapy, 23 patients (44%) treated with rosiglitazone and 12 patients (23%) treated with placebo achieved clinical response (P = 0.04). Remission was achieved in 9 patients (17%) treated with rosiglitazone and 1 patient (2%) of the placebo arm (P = 0.01). However, endoscopic remission was uncommon in either arms (8% versus 2%; P = 0.34). Clinical improvement was clearly evident already at 4 week (P = 0.049), while quality of life was significantly improved at week 8 (P = 0.01), but not at week 4 and 12. The safety profile was remarkably safe, with adverse events occurring at similar rates in both groups; in particular edema and weight gain, as expected, were more common in the rosiglitazone group. Of interest, no cases of symptomatic hypoglycemia were reported.
Pederson and Brynskov reported the use of rosiglitazone enema compared to mesalazine in fourteen patients with distal UC [71]. Rosiglitazone had a similar effect compared to mesalazine enema, with a significant reduction of Mayo score (P < 0.01). In addition rosiglitazone restored the PPARγ activity in the inflamed area which was fourfold reduced before treatment compared with noninflamed areas and controls.
Although substantial research has focused on potential anti-inflammatory effects of TZD PPARγ ligands, their mechanism of action, particularly in the colon, is not well defined. The 5-ASA compounds largely used in UC are able to bind to PPARγ [33]. In the study of Lewis et al. [70] the majority of patients were on concomitant therapy with 5-ASA. Since rosiglitazone has a higher affinity to PPARγ compared to 5-ASA, one possible explanation of the efficacy is a more powerful stimulation and anti-inflammatory property of PPARγ. Alternatively, the effect could be mediated at the mucosal level, where the PPARγ is largely expressed [74]. Of note, large clinical trials with rosiglitazone in the treatment of psoriasis, another inflammatory disease, did not demonstrate efficacy, thus suggesting a “topical” and not a systemic effect in patients with UC [75].
Being also involved in cell proliferation, apoptosis, and modulation of cytokine production with antitumorigenic effect, PPARγ is also extremely important for the basis of chemoprevention strategies against colorectal cancer. For these reasons, there is an active ongoing research to disclose and investigate safer PPARγ agonists, with topical effect and direct targeting of the colon, possibly void of metabolic and systemic effect.
7. PPARγ and Therapy of Crohn's Disease
Recent data have suggested that the role of PPARγ in IBD physiopathology is not limited to UC but might involve also CD. Based on SAMP1/YitFc animal example, developing a spontaneous ileitis due to a defect of expression of PPARγ in ileal crypts, the polymorphisms of PPARγ has been tested in CD. Sugawara et al. [44] demonstrated that two intronic SNPs exhibited a significant lower frequency in CD compared to controls. However, these findings have not been independently replicated yet. Moreover, no data are available by using PPARγ ligands in medical therapy of CD, in which 5-ASA compounds are generally believed to be of little or no efficacy [73].
8. Conclusion and Take-Home Messages
PPARγ receptors are widely and highly expressed in the colon, being a key regulator factor of bacteria-induced mucosal inflammation. Moreover, they are directly involved in the mechanism of action of mesalazine, which is largely used and effective in UC. In addition, they are involved in the process of tumor suppression, especially in the colon. Therefore, beside the potential interest in the IBD physiopathology and genetic predisposition which is still under evaluation, it is highly expected that new molecules specifically targeting the intestinal receptors and void of action in the adipose tissue and insulin action could be developed and tested. Several tens of compounds have been already synthetized, some with 30–50-fold higher affinity against PPARγ and potentially higher efficacy than 5-ASA. These compounds are not that far from clinical application with potential implication in controlling the inflammation, better handling of host-bacterial interactions, and possible chemoprevention. In addition, a better understanding of the role of microbiota on PPARγ receptors should be elucidated, since some commensal bacterial or natural ligands of foods may directly activate and increase the expression of PPARγ, thus determining a “biologic” anti-inflammatory action.
References
- 1.Podolsky DK. Inflammatory bowel disease. New England Journal of Medicine. 2002;347(6):417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 2.Cosnes J, Gowerrousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology. 2011;140(6):1785–1794. doi: 10.1053/j.gastro.2011.01.055. [DOI] [PubMed] [Google Scholar]
- 3.Danese S, Fiocchi C. Ulcerative colitis. New England Journal of Medicine. 2011;365:1713–1725. doi: 10.1056/NEJMra1102942. [DOI] [PubMed] [Google Scholar]
- 4.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474(7351):307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson CA, Boucher G, Lees CW, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nature Genetics. 2011;43(3):246–252. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Franke A, McGovern DPB, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nature Genetics. 2010;42(12):1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho JH. Immunochip-based analysis of a large IBD case-control cohort identifies 50 novel loci, refining definitions of disease pathways. Gastroenterology. 2012;142(supplement 1):S149–S150. [Google Scholar]
- 8.Poliska S, Penyige A, Lakatos PL, et al. Association of peroxisome proliferator-activated receptor gamma polymorphisms with inflammatory bowel disease in a hungarian cohort. Inflammatory Bowel Diseases. 2012;18(3):472–479. doi: 10.1002/ibd.21798. [DOI] [PubMed] [Google Scholar]
- 9.Aoyagi Y, Nagata S, Kudo T, et al. Peroxisome proliferator-activated receptor γ 2 mutation may cause a subset of ulcerative colitis. Pediatrics International. 2010;52(5):729–734. doi: 10.1111/j.1442-200X.2010.03195.x. [DOI] [PubMed] [Google Scholar]
- 10.Shrestha UK, Karimi O, Crusius JBA, et al. Distribution of peroxisome proliferator-activated receptor-gamma polymorphisms in Chinese and Dutch patients with inflammatory bowel disease. Inflammatory Bowel Diseases. 2010;16(2):312–319. doi: 10.1002/ibd.21059. [DOI] [PubMed] [Google Scholar]
- 11.Atug O, Tahan V, Eren F, et al. Pro12Ala polymorphism in the peroxisome proliferator-activated receptor-gamma (PPARγ) gene in inflammatory bowel disease. Journal of Gastrointestinal and Liver Diseases. 2008;17(4):433–437. [PubMed] [Google Scholar]
- 12.Mwinyi J, Grete-Wenger C, Eloranta JJ, Kullak-Ublick GA. The impact of PPARγ genetic variants on IBD susceptibility and IBD disease course. PPAR Research. 2012;2012:13 pages. doi: 10.1155/2012/349469.349469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andersen V, Christensen J, Ernst A, et al. Polymorphisms in NF-κB, PXR, LXR, PPARγ and risk of infammatory bowel disease. World Journal of Gastroenterology. 2011;17(2):197–206. doi: 10.3748/wjg.v17.i2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Z-F, Yang N, Zhao G, Zhu L, Wang L-X. Association between the Pro12Ala polymorphism of peroxisome proliferator-activated receptor gamma 2 and inflammatory bowel disease: a meta-analysis. PLoS One. 2012;7(1) doi: 10.1371/journal.pone.0030551.e30551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dubuquoy L, Rousseaux C, Thuru X, et al. PPARγ as a new therapeutic target in inflammatory bowel diseases. Gut. 2006;55(9):1341–1349. doi: 10.1136/gut.2006.093484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dubuquoy L, Å Jansson E, Deeb S, et al. Impaired expression of peroxisome proliferator-activated receptor γin ulcerative colitis. Gastroenterology. 2003;124(5):1265–1276. doi: 10.1016/s0016-5085(03)00271-3. [DOI] [PubMed] [Google Scholar]
- 17.Yamamoto-Furusho JK, Peñaloza-Coronel A, Sánchez-Muñoz F, Barreto-Zuñiga R, Dominguez-Lopez A. Peroxisome proliferator-activated receptor-gamma (PPAR-γ) expression is downregulated in patients with active ulcerative colitis. Inflammatory Bowel Diseases. 2011;17(2):680–681. doi: 10.1002/ibd.21322. [DOI] [PubMed] [Google Scholar]
- 18.Wendelsdorf K, Bassaganya-Riera J, Hontecillas R, Eubank S. Model of colonic inflammation: immune modulatory mechanisms in inflammatory Bowel disease. Journal of Theoretical Biology. 2010;264(4):1225–1239. doi: 10.1016/j.jtbi.2010.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spiegelman BM. PPAR-γ: adipogenic regulator and thiazolidinedione receptor. Diabetes. 1998;47(4):507–514. doi: 10.2337/diabetes.47.4.507. [DOI] [PubMed] [Google Scholar]
- 20.Mansén A, Guardiola-Diaz H, Rafter J, Branting C, Gustafsson JÅ. Expression of the peroxisome proliferator-activated receptor (PPAR) in the mouse colonic mucosa. Biochemical and Biophysical Research Communications. 1996;222(3):844–851. doi: 10.1006/bbrc.1996.0832. [DOI] [PubMed] [Google Scholar]
- 21.Su CG, Wen X, Bailey ST, et al. A novel therapy for colitis utilizing PPAR-γ ligands to inhibit the epithelial inflammatory response. Journal of Clinical Investigation. 1999;104(4):383–389. doi: 10.1172/JCI7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saubermann LJ, Nakajima A, Wada K, et al. Peroxisome proliferator-activated receptor gamma agonist ligands stimulate a Th2 cytokine response and prevent acute colitis. Inflammatory Bowel Diseases. 2002;8(5):330–339. doi: 10.1097/00054725-200209000-00004. [DOI] [PubMed] [Google Scholar]
- 23.Ramakers JD, Verstege MI, Thuijls G, Te Velde AA, Mensink RP, Plat J. The PPARγ agonist rosiglitazone impairs colonic inflammation in mice with experimental colitis. Journal of Clinical Immunology. 2007;27(3):275–283. doi: 10.1007/s10875-007-9074-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takagi T, Naito Y, Tomatsuri N, et al. Pioglitazone, a PPAR-γ ligand, provides protection from dextran sulfate sodium-induced colitis in mice in association with inhibition of the NF-κB-cytokine cascade. Redox Report. 2002;7(5):283–289. doi: 10.1179/135100002125000802. [DOI] [PubMed] [Google Scholar]
- 25.Hontecillas R, Horne WT, Climent M, et al. Immunoregulatory mechanisms of macrophage PPAR-γ in mice with experimental inflammatory bowel disease. Mucosal Immunology. 2011;4(3):304–313. doi: 10.1038/mi.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schaefer KL, Denevich S, Ma C, et al. Intestinal antiinflammatory effects of thiazolidenedione peroxisome proliferator-activated receptor-γ ligands on T helper type 1 chemokine regulation include nontranscriptional control mechanisms. Inflammatory Bowel Diseases. 2005;11(3):244–252. doi: 10.1097/01.mib.0000160770.94199.9b. [DOI] [PubMed] [Google Scholar]
- 27.Bassaganya-Riera J, Hontecillas R. CLA and n-3 PUFA differentially modulate clinical activity and colonic PPAR-responsive gene expression in a pig model of experimental IBD. Clinical Nutrition. 2006;25(3):454–465. doi: 10.1016/j.clnu.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 28.Bassaganya-Riera J, Viladomiu M, Pedragosa M, De Simone C, Hontecillas R. Immunoregulatory mechanisms underlying prevention of colitis-associated colorectal cancer by probiotic bacteria. PLoS One. 2012;7(4) doi: 10.1371/journal.pone.0034676.e34676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lewis SN, Brannan L, Guri AJ, et al. Dietary α-eleostearic acid ameliorates experimental inflammatory Bowel disease in mice by activating peroxisome proliferator-activated receptor-γ . Plos One. 2011;6(8) doi: 10.1371/journal.pone.0024031.e24031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Desreumaux P, Dubuquoy L, Nutten S, et al. Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome proliferator-activated receptor γ (PPARγ) heterodimer: a basis for new therapeutic strategies. Journal of Experimental Medicine. 2001;193(7):827–838. doi: 10.1084/jem.193.7.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sánchez-Hidalgo M, Martín AR, Villegas I, Alarcón de la Lastra C. Rosiglitazone, a PPARγ ligand, modulates signal transduction pathways during the development of acute TNBS-induced colitis in rats. European Journal of Pharmacology. 2007;562(3):247–258. doi: 10.1016/j.ejphar.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 32.Rocchi S, Picard F, Vamecq J, et al. A unique PPARγ ligand with potent insulin-sensitizing yet weak adipogenic activity. Molecular Cell. 2001;8(4):737–747. doi: 10.1016/s1097-2765(01)00353-7. [DOI] [PubMed] [Google Scholar]
- 33.Rousseaux C, Lefebvre B, Dubuquoy L, et al. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator-activated receptor-γ . Journal of Experimental Medicine. 2005;201(8):1205–1215. doi: 10.1084/jem.20041948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zeng C, Xiao JH, Chang MJ, Wang JL. Beneficial effects of THSG on acetic acid-induced experimental colitis: involvement of upregulation of PPAR-γ and inhibition of the NF-κB inflammatory pathway. Molecules. 2011;16:8552–8568. doi: 10.3390/molecules16108552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakajima A, Wada K, Miki H, et al. Endogenous PPARγ mediates anti-inflammatory activity in murine ischemia-reperfusion injury. Gastroenterology. 2001;120(2):460–469. doi: 10.1053/gast.2001.21191. [DOI] [PubMed] [Google Scholar]
- 36.Cuzzocrea S, Pisano B, Dugo L, et al. Rosiglitazone and 15-deoxy-Δ12,14-prostaglandin J 2, ligands of the peroxisome proliferator-activated receptor-γ (PPAR-γ), reduce ischaemia/reperfusion injury of the gut. British Journal of Pharmacology. 2003;140(2):366–376. doi: 10.1038/sj.bjp.0705419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sato N, Kozar RA, Zou L, et al. Peroxisome proliferator-activated receptor γ mediates protection against cyclooxygenase-2-induced gut dysfunction in a rodent model of mesenteric ischemia/reperfusion. Shock. 2005;24(5):462–469. doi: 10.1097/01.shk.0000183483.76972.ae. [DOI] [PubMed] [Google Scholar]
- 38.Sato N, Moore FA, Kone BC, et al. Differential induction of PPAR-γ by luminal glutamine and iNOS by luminal arginine in the rodent postischemic small bowel. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2006;290(4):G616–G623. doi: 10.1152/ajpgi.00248.2005. [DOI] [PubMed] [Google Scholar]
- 39.Hontecillas R, Wannemeulher MJ, Zimmerman DR, et al. Nutritional regulation of porcine bacterial-induced colitis by conjugated linoleic acid. Journal of Nutrition. 2002;132(7):2019–2027. doi: 10.1093/jn/132.7.2019. [DOI] [PubMed] [Google Scholar]
- 40.Tanaka T, Kohno H, Yoshitani SI, et al. Ligands for peroxisome proliferator-activated receptors α and γ inhibit chemically induced colitis and formation of aberrant crypt foci in rats. Cancer Research. 2001;61(6):2424–2428. [PubMed] [Google Scholar]
- 41.Sánchez-Hidalgo M, Martín AR, Villegas I, Alarcón De La Lastra C. Rosiglitazone, an agonist of peroxisome proliferator-activated receptor gamma, reduces chronic colonic inflammation in rats. Biochemical Pharmacology. 2005;69(12):1733–1744. doi: 10.1016/j.bcp.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 42.Bassaganya-Riera J, Reynolds K, Martino-Catt S, et al. Activation of PPAR γ and δ by conjugated linoleic acid mediates protection from experimental inflammatory bowel disease. Gastroenterology. 2004;127(3):777–791. doi: 10.1053/j.gastro.2004.06.049. [DOI] [PubMed] [Google Scholar]
- 43.Lytle C, Tod TJ, Vo KT, Lee JW, Atkinson RD, Straus DS. The peroxisome proliferator-activated receptor γ ligand rosiglitazone delays the onset of inflammatory bowel disease in mice with interleukin 10 deficiency. Inflammatory Bowel Diseases. 2005;11(3):231–243. doi: 10.1097/01.mib.0000160805.46235.eb. [DOI] [PubMed] [Google Scholar]
- 44.Sugawara K, Olson TS, Moskaluk CA, et al. Linkage to peroxisome proliferator-activated receptor-γ in SAMP1/YitFc mice and in human Crohn’s disease. Gastroenterology. 2005;128(2):351–360. doi: 10.1053/j.gastro.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 45.Shah YM, Morimura K, Gonzalez FJ. Expression of peroxisome proliferator-activated receptor-γ in macrophage suppresses experimentally induced colitis. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2007;292(2):G657–G666. doi: 10.1152/ajpgi.00381.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guri AJ, Mohapatra SK, Horne WT, Hontecillas R, Bassaganya-Riera J. The Role of T cell PPAR γ in mice with experimental inflammatory bowel disease. BMC Gastroenterology. 2010;10, article no. 60 doi: 10.1186/1471-230X-10-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mohapatra SK, Guri AJ, Climent M, et al. Immunoregulatory actions of epithelial cell PPAR γ at the colonic mucosa of mice with experimental inflammatory bowel disease. PLoS One. 2010;5(4) doi: 10.1371/journal.pone.0010215.e10215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Auwerx J, Baulieu E, Beato M, et al. A unified nomenclature system for the nuclear receptor superfamily. Cell. 1999;97(2):161–163. doi: 10.1016/s0092-8674(00)80726-6. [DOI] [PubMed] [Google Scholar]
- 49.Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs: from orphan receptors to drug discovery. Journal of Medicinal Chemistry. 2000;43(4):527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- 50.Kliewer SA, Umesono K, Noonan DJ, Heyman RA, Evans RM. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature. 1992;358(6389):771–774. doi: 10.1038/358771a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamazaki K, Shimizu M, Okuno M, et al. Synergistic effects of RXRα and PPARγ ligands to inhibit growth in human colon cancer cells—phosphorylated RXRα is a critical target for colon cancer management. Gut. 2007;56(11):1557–1563. doi: 10.1136/gut.2007.129858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dubuquoy L, Dharancy S, Nutten S, Pettersson S, Auwerx J, Desreumaux P. Role of peroxisome proliferator-activated receptor γ and retinoid X receptor heterodimer in hepatogastroenterological diseases. Lancet. 2002;360(9343):1410–1418. doi: 10.1016/S0140-6736(02)11395-X. [DOI] [PubMed] [Google Scholar]
- 53.Spiegelman BM. PPARγ in monocytes: less pain, any gain? Cell. 1998;93(2):153–155. doi: 10.1016/s0092-8674(00)81567-6. [DOI] [PubMed] [Google Scholar]
- 54.Marion-Letellier R, Butler M, Déchelotte P, Playford RJ, Ghosh S. Comparison of cytokine modulation by natural peroxisome proliferator-activated receptor γ ligands with synthetic ligands in intestinal-like Caco-2 cells and human dendritic cells—potential for dietary modulation of peroxisome proliferator-activated receptor γ in intestinal inflammation. American Journal of Clinical Nutrition. 2008;87(4):939–948. doi: 10.1093/ajcn/87.4.939. [DOI] [PubMed] [Google Scholar]
- 55.Allred CD, Talbert DR, Southard RC, Wang X, Kilgore MW. PPARγ1 as a molecular target of eicosapentaenoic acid in human colon cancer (HT-29) cells. Journal of Nutrition. 2008;138(2):250–256. doi: 10.1093/jn/138.2.250. [DOI] [PubMed] [Google Scholar]
- 56.Salh B, Assi K, Templeman V, et al. Curcumin attenuates DNB-induced murine colitis. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2003;285(1):G235–G243. doi: 10.1152/ajpgi.00449.2002. [DOI] [PubMed] [Google Scholar]
- 57.Deguchi Y, Andoh A, Inatomi O, et al. Curcumin prevents the development of dextran sulfate sodium (DSS)-induced experimental colitis. Digestive Diseases and Sciences. 2007;52(11):2993–2998. doi: 10.1007/s10620-006-9138-9. [DOI] [PubMed] [Google Scholar]
- 58.Kim CS, Park WH, Park JY, et al. Capsaicin, a spicy component of hot pepper, induces apoptosis by activation of the peroxisome proliferator-activated receptor γ in HT-29 human colon cancer cells. Journal of Medicinal Food. 2004;7(3):267–273. doi: 10.1089/jmf.2004.7.267. [DOI] [PubMed] [Google Scholar]
- 59.Han KL, Jung MH, Sohn JH, Hwang JK. Ginsenoside 20(S)-protopanaxatriol (PPT) activates peroxisome proliferator-activated receptor γ (PPARγ) in 3T3-L1 adipocytes. Biological and Pharmaceutical Bulletin. 2006;29(1):110–113. doi: 10.1248/bpb.29.110. [DOI] [PubMed] [Google Scholar]
- 60.Hwang JT, Kim SH, Lee MS, et al. Anti-obesity effects of ginsenoside Rh2 are associated with the activation of AMPK signaling pathway in 3T3-L1 adipocyte. Biochemical and Biophysical Research Communications. 2007;364(4):1002–1008. doi: 10.1016/j.bbrc.2007.10.125. [DOI] [PubMed] [Google Scholar]
- 61.Morikawa K, Ikeda C, Nonaka M, et al. Epigallocatechin gallate-induced apoptosis does not affect adipocyte conversion of preadipocytes. Cell Biology International. 2007;31(11):1379–1387. doi: 10.1016/j.cellbi.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 62.Schwab M, Reynders V, Loitsch S, Steinhilber D, Stein J, Schröder O. Involvement of different nuclear hormone receptors in butyrate-mediated inhibition of inducible NFκB signalling. Molecular Immunology. 2007;44(15):3625–3632. doi: 10.1016/j.molimm.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 63.Campbell SE, Stone WL, Whaley SG, Qui M, Krishnan K. Gamma tocopherol upregulates peroxisome proliferator activated receptor (PPAR) gamma expression in SW 480 human colon cancer cell lines. BMC Cancer. 2003;3, article no. 25 doi: 10.1186/1471-2407-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vunta H, Davis F, Palempalli UD, et al. The anti-inflammatory effects of selenium are mediated through 15-deoxy-Δ12,14-prostaglandin J2 in macrophages. Journal of Biological Chemistry. 2007;282(25):17964–17973. doi: 10.1074/jbc.M703075200. [DOI] [PubMed] [Google Scholar]
- 65.Bassaganya-Riera J, Hontecillas R. Dietary conjugated linoleic acid and n-3 polyunsaturated fatty acids in inflammatory Bowel disease. Current Opinion in Clinical Nutrition and Metabolic Care. 2010;13(5):569–573. doi: 10.1097/MCO.0b013e32833b648e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Katayama K, Wada K, Nakajima A, et al. A novel PPARγ gene therapy to control inflammation associated with inflammatory bowel disease in a murine model. Gastroenterology. 2003;124(5):1315–1324. doi: 10.1016/s0016-5085(03)00262-2. [DOI] [PubMed] [Google Scholar]
- 67.Marion-Letellier R, Déchelotte P, Lacucci M, Ghosh S. Dietary modulation of peroxisome proliferator-activated receptor gamma. Gut. 2009;58(4):586–593. doi: 10.1136/gut.2008.162859. [DOI] [PubMed] [Google Scholar]
- 68.Lewis JD, Lichtenstein GR, Stein RB, et al. An open-label trial of the PPARγ ligand rosiglitazone for active ulcerative colitis. American Journal of Gastroenterology. 2001;96(12):3323–3328. doi: 10.1111/j.1572-0241.2001.05333.x. [DOI] [PubMed] [Google Scholar]
- 69.Liang HL, Ouyang Q. A clinical trial of rosiglitazone and 5-aminosalicylate combination for ulcerative colitis. Zhonghua Nei ke Za Zhi. 2006;45(7):548–551. [PubMed] [Google Scholar]
- 70.Lewis JD, Lichtenstein GR, Deren JJ, et al. Rosiglitazone for active ulcerative colitis: a randomized placebo-controlled trial. Gastroenterology. 2008;134(3):688–695. doi: 10.1053/j.gastro.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pedersen G, Brynskov J. Topical rosiglitazone treatment improves ulcerative colitis by restoring peroxisome proliferator-activated receptor-γ activity. American Journal of Gastroenterology. 2010;105(7):1595–1603. doi: 10.1038/ajg.2009.749. [DOI] [PubMed] [Google Scholar]
- 72.Ulrich S, Loitsch SM, Rau O, et al. Peroxisome proliferator-activated receptor γ as a molecular target of resveratrol-induced modulation of polyamine metabolism. Cancer Research. 2006;66(14):7348–7354. doi: 10.1158/0008-5472.CAN-05-2777. [DOI] [PubMed] [Google Scholar]
- 73.Talley NJ, Abreu MT, Achkar JP, et al. An evidence-based systematic review on medical therapies for inflammatory bowel disease. American Journal of Gastroenterology. 2011;106(supplement 1):S2–S25. doi: 10.1038/ajg.2011.58. [DOI] [PubMed] [Google Scholar]
- 74.Fajas L, Auboeuf D, Raspé E, et al. The organization, promoter analysis, and expression of the human PPARγ gene. Journal of Biological Chemistry. 1997;272(30):18779–18789. doi: 10.1074/jbc.272.30.18779. [DOI] [PubMed] [Google Scholar]
- 75.Ellis CN, Barker JN, Haig AE, Parker CA, Daly S, Jayawardene DA. Placebo response in two long-term randomized psoriasis studies that were negative for rosiglitazone. American Journal of Clinical Dermatology. 2007;8(2):93–102. doi: 10.2165/00128071-200708020-00005. [DOI] [PubMed] [Google Scholar]