

Abstract
Keratins are the largest subgroup of intermediate filament proteins, which are an important constituent of the cellular cytoskeleton. The principally expressed keratins (K) of the intestinal epithelium are K8, K18 and K19. The specific keratin profile of a particular epithelium provides it with strength and integrity. In the colon, keratins have been shown to regulate electrolyte transport, likely by targeting ion transporters to their correct location in the colonocytes.
Keratins are highly dynamic and are subject to post-translational modifications including phosphorylation, acetylation and glycosylation. These affect the filament dynamics and hence solubility of keratins and may contribute to protection against degradation. Keratin null mice (K8−/−) develop colitis, and abnormal keratin mutations have been shown to be associated with inflammatory bowel disease (IBD). Abnormal expression of K7 and K20 has been noted in colitis-associated dysplasia and cancers. In sporadic colorectal cancers (CRCs) may be useful in predicting tumour prognosis; a low K20 expression is noted in CRCs with high microsatellite instability; and keratins have been noted as dysregulated in peri-adenomatous fields. Caspase-cleaved fragment of K18 (M30) in the serum of patients with CRC has been used as a marker of cancer load and to assess response to therapy. These data suggest an emerging importance of keratins in maintaining normal function of the gastrointestinal epithelium as well as being a marker of various colorectal diseases. This review will primarily focus on the biology of these proteins, physiological functions and alterations in IBD and CRCs.
Keratins and intermediate filaments
Intermediate filaments (IFs), along with microfilaments (MF) and microtubules (MT), constitute the key components of the cellular cytoskeleton, a scaffolding within the cytoplasm of animal cells (Fuchs & Cleveland 1998; Ku et al. 1999). The IFs are subdivided into five major types based on amino acid sequences and protein structure. Types I–IV form cytoplasmic IFs, whereas type V IF proteins, the lamins, form the nucleoskeleton (Schweizer et al. 2006; Kim & Coulombe 2007). Keratins are the largest subgroup of IF proteins and are classified as type I and type II proteins. They are composed of polymerized dimers of a type I (acidic) and type II (basic) keratin. Dimerization is facilitated by the interaction of coiled-coil domains in the protein pairs, generating coiled structures with free globular domains at the amino (N)- and carboxy(C)-termini (Omary et al. 2009).
The IF cytoskeleton radiates throughout the cell (shown in Figure 1a) and is interlinked mechanically and functionally to MF and MT cytoskeletons (Svitkina et al. 1996; Goldman et al. 1999). There is a strong similarity between the members of the class I and class II keratins (Figure 2), implying a similarity in function; however, the profile of keratin expression is specific to the particular epithelium, and the close association of IFs with desmosomes and hemidesmosomes is thought to reflect a role for IFs in providing the epithelium with strength and integrity (Herrmann & Aebi 2004).
Figure 1.

Organization and distribution of keratin 8 in colorectal cells. (Panel a) Representative images of immunocytochemical staining of keratin 8 in HCT116 cells. K8 (green) forms filaments, which radiate throughout the cell. (Panel b) Immunohistochemical staining of keratin 8 (i) and 18 (ii) staining in normal colon crypt.
Figure 2.

Similarities between members of the type I and type II keratin subfamilies. Panel a shows alignment between human type I keratins and panel b shows type II keratins. A star beneath the alignment indicates identical residue at a position, sequences accessed from Uniprot on 28.02.12 and alignment undertaken using Blast through Uniprot.
Keratins are expressed in different epithelial cells (Moll et al. 1982). The keratins that are primarily expressed in simple (single-layered) epithelia are known as simple epithelial keratins and comprise K7, K8, K18, K19, K20 and K23 (Omary et al. 2009); the principal keratins in the intestinal epithelia being K8, K18 and K19 (Ku et al. 2004). In stratified epithelia, the main keratins include K5, K14 as well as K6 and K16 (Moll et al. 1982).
Isolation and solubilization of keratins
An important feature of keratins by which they differ from other cytoskeletal filament proteins such as actin, myosin and tubulin is that they are resistant to buffers containing non-denaturing detergents and high concentrations of ions, including chaotropic salts such as potassium chloride or iodide (Franke et al. 1982; Steinert et al. 1982; Achtstaetter et al. 1986). This is critically important while attempting to study the expression of keratins because they exist in a soluble form as well as an insoluble filamentous state. Usually, 5% of the keratin pool exists in a soluble state (Chou et al. 1993), although the state of solubility is determined by various factors including cellular stress (Liao et al. 1995) and the presence of post-translational modifications (PTM) (described below). The insoluble fraction can be solubilized with high concentrations of urea (9–10 M) or guanidine hydrochloride (4–6 M) to produce soluble tetrameric or oligomeric subunits to result in solubilization of the denatured monomers (Achtstaetter et al. 1986; Ku et al. 2004).
Isolation of keratins from mammalian cell culture monolayers has usually been described using a sequential fractionation technique (Herrmann et al. 2004; Ku et al. 2004). This technique has generally been modified for human tissue samples (Herrmann et al. 2004) although this has not been studied specifically for colorectal biopsies. We have described a further modification of the technique for the isolation of IF from colorectal biopsies and their effective solubilization in 4 M guanidine hydrochloride to facilitate parallel analysis of IFs by gel-based and gel-free liquid chromatography MS/MS (LC-MS/MS) approaches (Majumdar et al. in press).
PTM of keratin
Following translation, most proteins undergo changes through PTM (Cho 2007). Such modifications modulate the function of proteins in a manner which is not directly coded for by genes (Cho 2007). Keratins are highly dynamic and reorganize in response to many stimuli particularly to cellular stress as well as during apoptosis and mitosis (Ku et al. 1999; Toivola et al. 2010). Characteristic PTMs of keratins include serine phosphorylation (Oshima 1982; Omary et al. 1998, 2006) threonine phosphorylation (Steinert 1988), tyrosine phosphorylation (Feng et al. 1999), lysine acetylation (Leech et al. 2008), serine/threonine glycosylation (Chou et al. 1992), cleavage (Makowski & Ramsby 1998; Ditzel et al. 2002) and sumoylation (Snider et al. 2011).
Phosphorylation
Phosphorylation is the most studied PTMs. To date, there are still relatively few of the know phosphorylation sites whose cognate kinase/phosphatase activities have been defined. Empirically determined modification sites of K8, K18 and K19 are summarized in Figure 3. This figure acts as a density map for PTMs, and it is apparent that the majority of modifications occur in the N- and C-terminal domains, which have lower secondary structure, than in the coiled-coil domains (marked on the figure). Serine phosphorylations have been demonstrated on Ser 431, Ser 73 and Ser 23 on K8, and Ser 33 and Ser 52 on K18 (Omary et al. 1998). These changes have been associated with the regulation of filament formation/depolymerization although movement of the K8/K18 heterodimer pairs in and out of the filament (Ku & Omary 1994; Mizuuchi et al. 2009). Different protein kinases have been shown to phosphorylate keratins. Keratin-8 residue, Ser 73, has been identified as a substrate for phosphorylation by p38 kinase and c-Jun N-terminal kinase (both of which are involved in cellular responses to stresses, cytokines and apoptosis) (Kyriakis & Avruch 1996; Ip & Davis 1998; Davis 2000) and this has likewise been shown to regulate filament organization (He et al. 2002; Ku et al. 2002). The serine residue Ser 431, on the other hand, has been shown to be phosphorylated by the kinase ERK1 (MAP kinase) mediated by stimulation of epidermal growth factor (Omary et al.1998). These changes not only affect the solubility of keratins, but hyperphosphorylation may protect K18 from degradation by caspases (Ku & Omary 2001).
Figure 3.
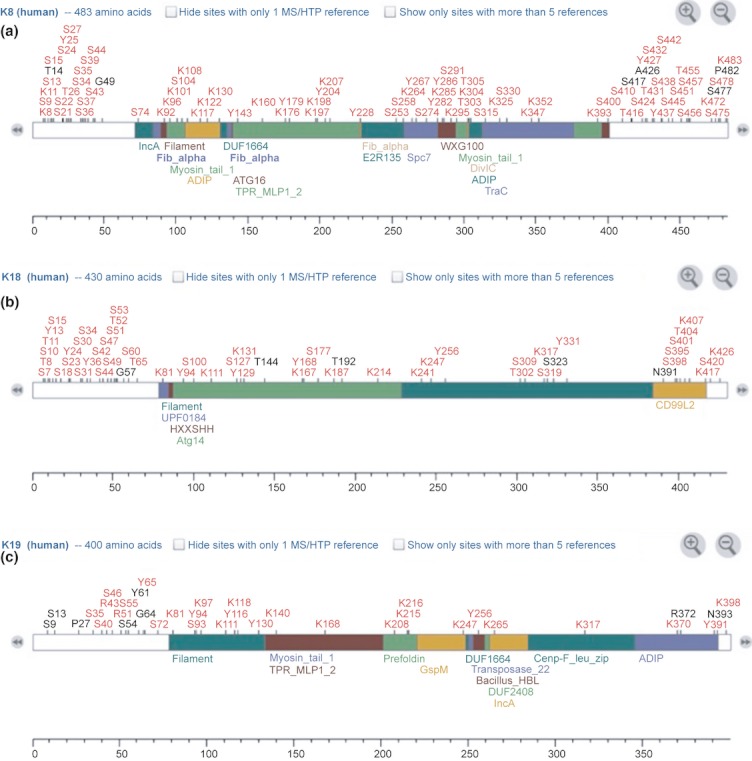
Summary of post-translational modifications of keratin 8 (Panel a), keratin 18 (Panel b) and keratin 19 (Panel c) (accessed from phosphosite.org, 21 February 2012). p, Phosphorylation; a, acetylation; m, methylation; m1, mono-methylation; m2, di-methylation; m3, tri-methylation; u, ubiquitination; S, sumoylation; n, neddylation; g, O-GlcNAc; h, palmitoylation; ad, adenylylation; sn, S-nitrosylation; ca, caspase cleavage.
Dephosphorylation of K8 in colorectal cancer (CRC) tumour cells has been shown to be associated with tumour progression (Mizuuchi et al. 2009). In this study, K8 has been demonstrated as a physiological substrate of phosphatase of regenerating liver-3 (PRL-3) a subclass of tyrosine phosphatases (Stephens et al. 2005) that play a role in cancer migration and metastasis including in CRCs (Bardelli et al. 2003; Kato et al. 2004). CRC tumour cells in metastasis and at the invasive front were shown to have high PRL-3 associated with reduced or absent K8 phosphorylation thus demonstrating a role of K8 dephosphorylation in increased tumour aggressiveness.
Acetylation
Acetylation as a form of post-translational modifier of protein functions is being increasingly recognized as being as important as phosphorylation (Kouzarides 2000). Acetylation levels are the product of the balance of lysine acetyl transferase (KAT) (previously called histone acetyl transferase) and histone deacetylases (HDAC) (Sadoul et al. 2011). It is being increasingly recognized that HDACs regulate acetylation and function of proteins other than histones (Sterner et al. 1979; L'Hernault & Rosenbaum 1985). Acetylation may contribute to the regulation of protein–protein interaction (Chen et al. 1999) and protein stabilization (Martinez-Balbas et al. 2000). Two studies have recently reported a large number of acetylation sites in K8 (Leech et al. 2008; Choudhary et al. 2009). We have demonstrated five acetylated lysine residues: lys10, lys100, lys392, lys471 and lys482 in K8 in the insoluble IFs isolated from cultured HCT116 colon cancer cell lines using a multiple reaction monitoring-initiated detection and sequencing (MIDAS) workflow (Leech et al. 2008), which uses existing knowledge to predict the potential masses of acetylated peptides, then searches for such masses and sequences them to confirm the presence; three of these five (K10, K471 and K482) occur predominantly outside the coiled-coil domains with K482 at the C-terminus. Two other acetylation sites were noted to lie near the ends of the coiled-coil domains. Additionally, an acetylation site was also noted in K18. In contrast, an analysis of the global acetylome (presumably primarily using soluble material) identified six acetylation sites within the coiled-coil domain (Choudhary et al. 2009). We have demonstrated that acetylation of K8 is associated with depolymerization of filaments, and furthermore that this occurs at a similar concentration and time to the acetylation of lysine 482 (Drake et al. 2009). It has not been possible to distinguish cause from effect but this is supportive of a potential role of acetylation in the regulation of filament stability.
Glycosylation
Glycosylation of keratins have also been studied. Using human colon adenocarcinoma cell lines (HT29), Chou et al. (1992) demonstrated dynamic glycosylation of K8 and 18 at multiple sites with a single O-linked N-acetylglucosamine (O-GlcNAc). A subsequent study identified three sites of glycosylation on K18 at Ser 29, 30 and 48 (Ku & Omary 1995). Ku et al. (2010) investigated the role of K18 glycosylation by generating mice overexpressing human K18 S30/31/49A substitution mutants – K18-Gly− mice that are not glycosylated and compared them to wild type mice. K18-Gly− mice were also noted to be susceptible to apoptosis and liver and pancreatic injury by streptozocin, thus suggesting a role for K18 glycosylation in protecting epithelial cells from injury. Similar to phosphorylation, glycosylation too has been shown to play an important role in modulating equilibrium of keratin between soluble and filamentous insoluble state (Srikanth et al. 2010). In this study, malignant HepG2 hepatoma cell lines (which have decreased O-GlcNAcylation of cellular proteins because of an increased activity of the enzyme O-GlcNAcase), when compared to hepatocyte cell lines, demonstrate decreased solubility of K8 and K18.
Sumoylation
Sumoylation, a reversible PTM by a process of addition and removal of small ubiquitin-like modifier (SUMO) polypeptides, has emerged as an important mechanism of regulating protein activity (Seeler & Dejean 2003; Geiss-Friedlander & Melchior 2007). This process creates an isopeptide bond between the C-terminal Gly residue of the modifier protein and the ε-amino group of a Lys residue of the target protein (Geiss-Friedlander & Melchior 2007). The role of Ubc9, which covalently joins SUMOs to proteins in the gastrointestinal tract, was studied using inducible knockout mouse line for the SUMO E2 enzyme Ubc9 (Ubc9fl/-/ROSA26CreERT2) (by intra-peritoneal injection of 4-hydroxytamoxifen) (Demarque et al. 2011). Decreased sumoylation, which primarily affected the small intestine, resulted in severe diarrhoea along with the depletion of the intestinal proliferative compartment. K8 as a SUMO substrate was demonstrated by the presence of SUMO-modified K8 on Western blotting; this coupled with enterocyte fragility (at the top of the villi with detachment of enterocytes from the underlying basal lamina) suggests a putative role of sumoylation in tissue fragility disorders. In addition, an in vitro study (Snider et al. 2011) has shown that keratin sumoylation is augmented during apoptosis, oxidative stress and by phosphatase inhibition. In this study, human mutations in K8 that predispose their carriers to liver disease exhibit hypersumoylation and decreased solubility, thus demonstrating an effect of sumoylation on filament dynamics.
Cleavage
Keratins may be modified by cleavage in vivo. Degradation products of keratins have been identified in cancers (Ditzel et al. 2002), and several new neo-epitopes have been identified including the widely used M30 (Leers et al. 1999). Calpain cleavage of K8 has been identified as a para-apoptotic event (Makowski & Ramsby 1998).
Keratin distribution and function in the normal colonic epithelium
We recently reported the distribution of K8 expression in the morphologically normal colon mucosa in subjects with cancer (Khan et al. 2011). K8 is predominantly expressed in the flat mucosa, with expression decreasing from the top towards the base of the crypt axis. We noted that there is considerable variation between subjects, in the extent of expression along the crypt axis (Figure 1b).
The functional roles of keratins vary according to the organ. The major function of K8/K18 in the liver is protection from mechanical and non-mechanical forms of stress (Omary et al. 2002). Cytoprotective, non-mechanical functions of keratins also include protection from apoptosis (Eriksson et al. 2009). There is increasing evidence that IFs also have a role in cell-death signalling pathways, in particular apoptosis mediated by tumour necrosis factor-alpha (TNF-α) and Fas (Paramio & Jorcano 2002). Epithelial cells lacking K8 and K18 are significantly more sensitive to TNF-mediated apoptosis (Caulin et al. 2000).
The function of keratins in the intestine is poorly understood, although the K8-null mouse phenotype suggests they may have a potential role in cell growth, differentiation or targeting of proteins to the apical compartment (Ameen et al. 2001; Toivola et al. 2004). K8-null (K8−/− and K8+/−) mice in comparison with K8+/+ mice have been shown to have abnormal colonic active ion transport (Na+ and Cl-). Similarly, the short circuit current (Isc) was significantly decreased in K8−/− and K8+/− mice compared to K8+/+ mice. Conductance of colonic tissues and paracellular transport were, however, noted to be normal (Toivola et al. 2004). These findings suggest a putative role of colonic keratins in regulating electrolyte transport, likely by targeting ion transporters in the colonocytes (Toivola et al. 2004).
Network analyses of the interactomes of the three major colorectal keratins are shown in Figure 4. It is notable that K19 does not directly or indirectly occur in the interactomes of K8 and K19 (which holds when the networks are expanded). The networks show close interaction with both MAP Kinase and p53 pathways, highlighting the importance of keratins in linking physical function into cell signalling and cell fate determination.
Figure 4.
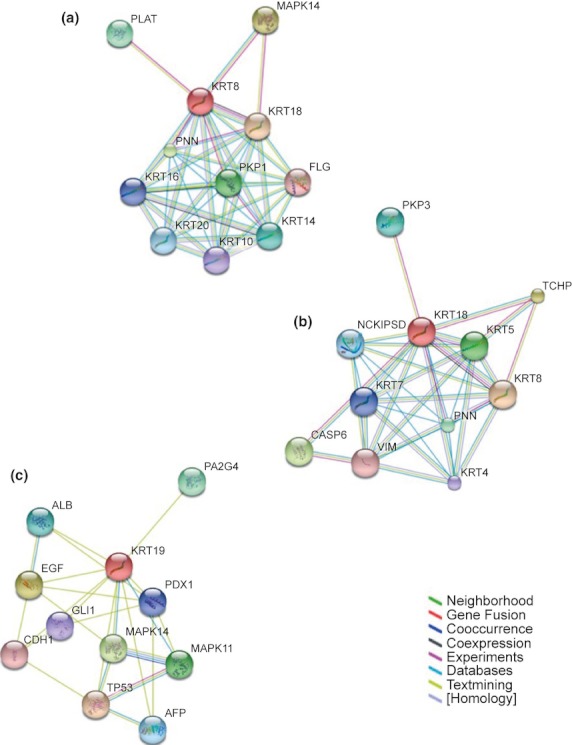
Protein–protein interaction network view summarizing the network of predicted associations for keratins 8, 18 and 19 (data accessed from STRING 9.0 (http://string-db.org/). The network nodes represent proteins, and edges demonstrate predicted functional associations between them. The colour of the node for the protein analysed is indicated in red; other nodes are coloured to indicate a direct link with the protein. Seven different coloured lines indicate the edges representing the seven types of evidence used in predicting the associations. The lines indicate the following evidences: red line – fusion; green line – neighbourhood; blue line – co-occurrence; purple line – experimental; yellow line – text-mining; light blue line – database; black line – co-expression.
Keratins and colorectal disease
Inflammatory bowel disease
Ulcerative colitis (UC) along with Crohn's disease comprises the chronic, inflammatory bowel diseases (IBD), both conditions characterized by a relapsing and remitting clinical course (Silverberg et al. 2005). The main symptoms of the conditions include diarrhoea, often bloody diarrhoea with passage of mucus. Other symptoms that can be variably present include abdominal pain, tenesmus or fever (Carter et al. 2004). The aetiology of UC remains unknown, and no single agent or distinct single mechanism has been implicated in the pathogenesis (Fiocchi 1998); rather it is believed that several factors viz. genetic susceptibility, environmental triggers and an altered immune response (Kucharzik et al. 2006; Xavier & Podolsky 2007) to luminal microbial antigens interact for the condition to become clinically apparent (Sartor 2006). Perturbations in the intestinal epithelial barrier, which provide a protective barrier against the luminal microflora, are now considered to play an important roles in the pathogenesis of the condition (Dignass et al. 2004).
Keratins and IBD
Several lines of evidence point towards an association of keratins with IBD, although this linkage is not fully clear. TNF-α is a pro-inflammatory cytokine, the levels of which are increased in blood, colonic tissue and stools of patients with UC (Murch et al. 1991, 1993; Braegger et al. 1992). K8 and K18 have been noted to co-localize with cytoplasmic domain of TNF receptor 2 (TNFR2) and moderate TNF-induced, Jun NH2-terminal kinase intracellular signalling and NFκB activation (Caulin et al. 2000). In addition, normal and malignant epithelial cells lacking in K8 and K18 were found to be sensitive to TNF-mediated cell death. It is suggested that attenuating effects on TNF function by K18 may be mediated by TNFR1-associated death domain protein (Inada et al. 2001).
Effects of interleukin 6 (IL-6) an immunoregulatory cytokine on keratin expression and its implications on the intestinal mucosal barrier function have been studied (Wang et al. 2007). Intestinal disorders including IBD are often linked to perturbations in the barrier function of the epithelial cells. It is believed that such loss of barrier function results in sensitization of the immune system to antigens previously localized to the intestinal lumen (Dignass et al. 2004; Shen & Turner 2006). Using a combination of Caco2-BBE cell line and IL-6 null mice, Wang et al. (2007) demonstrated that IL-6 significantly induces not only the expression of insoluble cytoskeletal fraction of K8 and K18, but also serine phosphorylation of K8 at phosphoserine 431 and 73, suggesting PTM of K8 in response to IL-6. IL-6 null mice also demonstrated significantly increased intestinal permeability with dextran sodium sulphate administration. Taken together, these findings demonstrate regulation of colonic expression of K8 and K18 by IL-6 as well as a potential role of K8 in mediating effects of IL-6 on the barrier function.
Two animal studies have supported a role of keratins in the pathogenesis of IBD. In one study (Baribault et al. 1994), homozygous mK8−/− FVB/N mice were shown to develop colonic hyperplasia, colitis and rectal prolapse. Histology revealed hyperplasia without any abnormalities in the crypt cells or mucin-producing goblet cells, suggesting that the inflammatory component of mK8- bowel disease may be secondary to colorectal hyperplasia. This colonic inflammation in K8-null mice may thus represent a unique model for IBD resulting from a primary epithelial rather than an immune cell defect (Baribault et al. 1994).
K8 deficient mice (K8−/−) have also been shown to develop spontaneous chronic T helper type 2 colitis (Th2 colitis) (Habtezion et al. 2005). In this study, a significant increase in T-cell receptor beta (TCRβ)-positive CD4+ T cells infiltrating the colon lamina propria in association with enhanced Th2 cytokine (IL-4, IL-5 and IL-13) production have been noted in K8−/− mice by immunohistochemistry and flow cytometry. Colonic epithelial cells in the K8−/− mice expressed MHC class II antigens; such antigen expression in K8+/+ mice was limited to the lamina propria. The colonic inflammation in the K8−/− mice was, however, prevented by administering antibiotics (vancomycin and imipenem) suggesting a possible role of luminal bacteria in triggering colitis in mice with a primary epithelial defect (Habtezion et al. 2005).
The role of keratins in the pathogenesis of colitis and the mechanisms by which K8−/− mice develop colonic inflammation and hyperplasia are poorly understood. To investigate further the colonic mucosal changes in K8−/− mice and their response to antibiotics, a genome-wide microarray was undertaken using isolated mouse colonocytes (Habtezion et al. 2011). Microarray analysis demonstrated differential regulation of several genes in K8+/+ and K8−/−-isolated mouse colonocytes, with apoptosis being the major altered pathway. These differentially altered genes in K8−/−colon were almost normalized with antibiotic pretreatment. Similar to results demonstrated with isolated colonocytes, K8−/− compared to K8+/+ colons were also noted to be resistant to the induction of apoptosis by both staurosporine and Fas. This suggests that lack of K8 confers resistance to apoptosis [unlike in hepatocytes where absence or mutation in K8 or K18 renders them susceptible to apoptosis (Omary et al. 2009)]. The authors hypothesized that difference between the hepatocytes and colonocytes probably reflects the influence of the luminal microflora on the colonocytes (Habtezion et al. 2011).
Lack of keratin filaments in the colon has been noted to cause abnormalities in transport of ions, protein mistargeting and diarrhoea even before occurrence of hyperproliferation and inflammation (Toivola et al. 2004). In this study, K8−/− mice were noted to have diarrhoea and significantly higher stool water content compared with K8+/+ mice. Colonic tissue conductance, a measure of the function of tight junctions, was not significantly different, suggesting that the diarrhoea was unrelated to an alteration of the paracellular transport or increased tight junction permeability. Colonic ion transport studies revealed marked reduction in short circuit current (ISC) with markedly decreased net Na+ absorption associated with reversal of net Cl− movement to net Cl− secretion in K8−/− mice. In comparison with wild type mice, K8-null mice distal colon revealed an altered distribution of epithelial markers H,K-ATPase and F-actin to apical/lateral locations in the enterocytes. Colonic expression of anion exchanger AE1/2 was significantly higher in K8−/− mice, whereas the Na-transporter ENaC showed an altered distribution.
Mistargeting in K8−/− colons, which normalizes after antibiotic treatment, suggests that luminal bacteria and/or their consequent inflammatory response may promote the observed protein mistargeting (Habtezion et al. 2005). The mistargeted ion transporters may be responsible in creating an environment for pathogenic bacteria to thrive, which in turn stimulate the colitis and maintenance of the mislocalized transport proteins. These studies may lend credence to the role of luminal bacteria in the pathogenesis of colonic inflammation and colitis. Although the aetiology of IBD is multifactorial, luminal microflora particularly an altered host tolerance to them is believed to play an important role (Fava & Danese 2011).
Genomic DNA from patients with UC and Crohn's disease, 50 with sporadic and 47 with familial IBD, has been studied (Owens et al. 2004). Heterozygous missense mutations in K8 were identified in five (5.2%) unrelated IBD patients in this study. Three separate KRT8 mutations (G62C) resulting in amino acid substitutions at K8 were identified: two were in unrelated UC patients with a family history of IBD and one was in a Crohn's disease patient with no family history. Patient with sporadic Crohn's disease was found to have a mutation in K8 (I63V), whereas another mutation resulting in amino acid substitution in the tail domain of K8 (K464N) was found in an UC patient with family history of IBD. Four patients were noted to have heterozygous sequence variation in KRT18 resulting to K18 (S230T) in the L12 linker region. All these changes were noted to lie within the non-helical domains, which are usually associated with milder forms of disease. The K8 (K464N) mutation was, however, not found in 194 controls, whereas the K8 (G62C) and K8 (I63V) sequences were each identified once. However, none of these changes noted were compared to identify statistical difference from controls; and morphological changes in keratins in tissue samples were not investigated.
Such mutations were also noted to be responsible for incompetent polymerization of keratins particularly on assembly of K8/K18 filaments in vitro and in cultured cells (Owens et al. 2004). Another German study investigated 217 patients with Crohn's disease, 131 patients with UC and 560 healthy controls (Buning et al. 2004). Heterozygous G62C mutation in K8 was detected in five (2.3%) and three (2.3%) patients with Crohn's disease and UC, respectively, whereas 9 (1.6%) control subjects were noted to carry such mutation. None of the patients or controls was homozygous for this mutation and they did not show the Y54H mutation. Patients with IBD demonstrating the heterozygous mutation did not show any phenotypic difference from those without such mutation.
A more recent study investigated the association of mutations in KRT8 and KRT19 in patients with IBD (Tao et al. 2007). In this study, 16 of the 184 patients with IBD demonstrated heterozygous variants in KRT8 (G62C, R341H); 4 of 70 unaffected/unrelated volunteers also carried the KRT8 (R341H) mutation. A novel KRT8 (R341C) mutation correlating with extensive UC was identified. KRT19 variant frequencies were not different between the groups. KRT8 variants (G62C, R341H and R341C) were tested in 682 independent nuclear families (with both parents and at least one IBD affected offspring) and 273 controls. They failed to show any significant departure from random transmission. Thus, overall, the genetic studies fail to provide any conclusive evidence of keratin mutations in the pathogenesis of IBD.
Sporadic CRC
CRC is among the most common malignancies worldwide. Single-gene abnormalities account for a minority of CRCs commonly in the form of either familial adenomatous polyposis or hereditary non-polyposis CRC; the vast majority of CRCs being of sporadic origin arising from adenomas (Hisamuddin & Yang 2004). The traditional adenoma-carcinoma sequence in the pathogenesis of CRC involves a stepwise progression of normal colonic epithelium to malignant neoplasm involving progressive sequential accumulation of molecular and genetic abnormalities in each step leading to the development of malignancy via adenomatous polyps (Arnold et al. 2005). The pathways involve either the classical chromosomal instability pathway or a microsatellite instability pathway (Lengauer et al. 1998). A small number arise from either long-standing colitis (Choi & Zelig 1994) or sessile serrated adenomas, a subset of hyperplastic-like polyps that arise mainly in the right colon of middle-aged women (Jass 2003).
The bulk of data on keratins in colorectal diseases come from studies on patients with sporadic CRCs although there have been some studies from patients with colitis-associated cancers and cancers arising from serrated adenomas.
Keratin expression in sporadic adenomas and CRC
Field change or changes in the surrounding uninvolved colonic mucosa in patients with CRCs have been described previously (Shen et al. 2005). To date, there remain relatively few reports on keratin expression profile in the normal colon and in early neoplasia, indicating an unmet need for understanding its potential roles.
Polley et al. (2006) used a two dimensional gel electrophoresis and mass spectrometry (MALDI/TOF) approach to investigate proteomic changes in CRC tissue and compared them with morphologically normal colonic mucosa of patients with CRC (i.e. cancer field) as well as colonic adenomatous polyps and colonic mucosa of controls. In this study, differential expression of seven forms of K8, four of K19 and one each of K9 and K20 was observed. In comparison with healthy mucosa (of controls, cancer and polyp patients), all four forms of K19 were under expressed in tumour tissue and one spot identified as K9 was absent. In comparison with the colonic mucosa of controls, field biopsies in cancer patients showed overexpression of seven K8 forms, one of K9 and one of K20; field mucosa in polyp patients too revealed overexpression of K8 and K9. There were multiple forms of keratins differing slightly in mass and charge although the data did not allow determination of what the novel form of keratins were. The findings of this study support the hypothesis of changes in uninvolved field mucosa both in patients with cancer and in those with polyps. Taken together with the data from K8-null mice (mK8−/− FVB/N) indicating a risk of colonic hyperplasia (Baribault et al. 1994) and data from hepatocytes implicating keratin in regulation of cell cycle and apoptosis (processes which are deranged in cancer) (Ku et al. 2007) may imply a role for alteration in keratin expression and function at the earliest stages of colorectal carcinogenesis with increased expression of K8 in the morphologically normal mucosa as the adenoma-carcinoma sequence progresses.
Similar to pathogenesis of IBD, mutations in K8 too have not yet been directly implicated in the development of CRCs. A direct link between keratin mutation and development of cancer has only been demonstrated in cutaneous basal cell cancers (BCC) (Stacey et al. 2009). In this study, a G138E substitution in the keratin 5 (KRT5) gene was noted to confer an increased risk of BCC.
Serrated adenoma-associated CRC
Serrated adenomas have now been recognized as a distinct neoplastic lesion separate from sporadic adenomas with a serrated architecture that containing areas of epithelial dysplasia (Longacre & Fenoglio-Preiser 1990). It is now known that a subset of such adenomas can undergo malignant transformation (Jass 2003). Using an immunohistochemical approach, Tatsumi et al. (2005) investigated K7 and K20 expression in a variety of colorectal lesions including serrated adenomas, hyperplastic polyps, traditional adenomas and colorectal carcinomas. Majority of serrated adenomas (71%) demonstrated a K7+/K20+ pattern; in contrast, most sporadic adenomas and adenocarcinomas showed a K−/K20+ pattern.
Colitis-associated CRC
Ulcerative colitis as a risk factor for CRC was first described by Crohn & Rosenberg (1925). Patients with long-standing UC have an increased risk of developing CRC. Although CRC in IBD constitutes 1% of all cases of CRCs, patients with IBD represent one of the highest risk groups for developing this complication (Choi & Zelig 1994).
Dysplasia (defined as unequivocal neoplasia of the epithelium confined to the basement membrane without invasion into the lamina propria) represents the premalignant phase of the condition (Riddell et al. 1983). Colitis-associated CRC is usually preceded by and co-exists with dysplastic changes elsewhere in the bowel. Neoplastic changes in colitis have been shown to be associated with genomic abnormalities throughout the whole colon, including in the non-dysplastic mucosa. This is referred to as molecular field defect or genomic field defect (Rabinovitch et al. 1999; Issa et al. 2001; O'Sullivan et al. 2002; Chen et al. 2003).
Various factors are associated with an increased risk of CRC in UC; the most important factor identified across most studies is the duration of the disease (Eaden et al. 2001). Others include anatomical extent of the disease in the colon (Ekbom et al. 1990), presence of family history of CRC (Askling et al. 2001), co-existent primary sclerosing cholangitis – a rare chronic inflammatory and fibrotic disease of the biliary tract of unknown aetiology (Soetikno et al. 2002) – and severity of inflammation in the colon (Rutter et al. 2004).
Keratins in colitis-associated CRC
Similar to sporadic CRC, K7 and K20 expression in dysplastic and neoplastic changes in UC has been investigated (Tatsumi et al. 2006; Stenling et al. 2007). Immunohistochemical staining of paraffin-embedded tissue sections has shown that in contrast to sporadic adenocarcinomas, vast majority of UC-associated neoplasm expressed K7, including those with low- and high-grade dysplasia (LGD and HGD), K20 expression was not significantly different between the two groups (Tatsumi et al. 2006). In a study from northern Sweden in subjects undergoing colonoscopic surveillance, colonic biopsies were analysed for immunohistochemical expression of K7 and K20 in different groups of patients with UC. Active disease was present in 10 patients; LGD, HGD and cancer were present in 10, 6 and 5 patients respectively. K20 expression was noted increased in the lower part of the crypts in neoplasia-associated lesions, while 2 of 5 patients with CRC, 3 of 6 patients with HGD and 7 of 10 patients with LGD were positive for K7 (Stenling et al. 2007).
The significance of such changes in keratin expression in the colorectal mucosa in patients with colitis is still not clear; it is possible that they may represent markers of neoplastic changes in patients with UC.
Keratins as tumour markers of CRC
One important characteristic of keratins and other IFs is that they remain relatively stable even after transformation to pathological states including transformation of normal cells into malignant cells. This property has been exploited with keratins being used as tumour markers (Omary et al. 2009). Keratins are not organ specific, which limits their diagnostic utility. However, their role as markers of tumour prognosis and for monitoring treatment response is relatively well established (Barak et al. 2004). Several studies have investigated keratin expression in sporadic CRC. Moll et al. (1983) found K8 and K19 to be the most abundant, while Chu et al. reported that while gastrointestinal carcinomas uniformly express K8, 18 and 19, most colorectal adenocarcinomas also express K20 (Chu et al. 2000; Chu & Weiss 2002). Unlike K8, 18 and 19, K20 expression is limited to gastric and intestinal epithelium, urothelium and Merkel cells (Moll et al. 1992). Moll et al. (1992) proposed K20 as a diagnostic marker to distinguish types of carcinoma when disease is present but the primary site is unknown, or in cases where accurate diagnosis is difficult because of poor differentiation or invasion into adjacent anatomical structures. Yun et al. (2000) sought to determine K20 specificity in detecting submicroscopic lymph node metastases by correlating its expression with mutant K-RAS expression (the K-RAS gene is mutated in 30–40% of colorectal tumours) (Sidransky et al. 1992; Pretlow et al. 1993) to show that K20 expression in lymph nodes in patients with CRC genuinely reflects metastatic disease. By using reverse transcription polymerase chain reaction (RT-PCR) assays for K20, they calculated a specificity of 97.6% in 42 patients with tumour and 41 controls (Yun et al. 2000).
Likewise, K7 immunostaining in the majority of colorectal adenocarcinomas tends to be weak or negative compared to other adenocarcinomas (for example, ovary, pancreas and lung), leading to its use in diagnosing the primary tumour in case of adenocarcinoma metastases (Moll et al. 1992; Chu et al. 2000). When combined with K20, for example a K7−/K20+ phenotype, a metastasis is very likely to be of colorectal origin (Moll et al. 1992; Chu et al. 2000).
Paraffin-embedded tissue sections of 196 colonic adenocarcinomas were investigated by immunohistochemistry for K7 and K20 (Bayrak et al. 2011). K7 and K20 expression was noted in 17.3% and 81.1% of patients; K7−/K20+ had the greatest proportion (65.8%) while K7+/K20+ immunophenotype was identified in 15.3%. K20 positivity was greater in low-grade than in high-grade carcinomas (85.1% vs. 47.6%) and in rectal and sigmoid carcinomas than in proximal colon carcinomas (88.2% vs. 63.2% and 88.9% vs. 63.2%, respectively). Tumours with lymph node metastasis showed increased K7 expression than non-metastatic tumours (25.3% vs. 11%).
RT-PCR assays for K20 have also been used to detect disseminated tumour cells in the peripheral and central blood of patients undergoing resection of the primary tumour (Weitz et al. 1998) in an effort to predict metastasis and add weight to the ‘no-touch isolation technique’ of surgical resection with lymphovascular ligation prior to resection of the tumour (Barnes 1952; Turnbull et al. 1967).
Keratins as prognostic markers of CRC
Tumour prognosis could be predicted using the pattern of expression of keratins. In case of CRCs, a low K20 expression is noted in CRCs with high microsatellite instability (McGregor et al. 2004). This group constitutes a distinct 15% of CRCs and has been noted to have improved survival compared with CRCs that have low microsatellite instability (McGregor et al. 2004). Similarly, a reduced expression of K8 and K20 has been noted to be associated with some CRC cell epithelial to mesenchymal transition (Knosel et al. 2006). Such changes are usually associated with more aggressive tumours and decreased survival (Knosel et al. 2006). K8 dephosphorylation by phosphatase of regenerating liver-3 (PRL-3) has also been shown to be a factor in contributing towards metastasis of CRC cells (Mizuuchi et al. 2009).
PTM and cleavage of keratins in colorectal neoplasia
Data from our group (Arasaradnam, Riley, Shaw & Corfe, unpublished) show increase in lysine acetylation in fields and in adenoma, which we have associated with depolymerization in vitro (Drake et al. 2009). These data imply that not only the level, but also the PTM (and by implication the enzymes regulating them) of keratin may be important in contributing to the development and progression of neoplasia.
Finally, majority of pathways leading to apoptosis involve caspase activation and initiation of a caspase cascade (Petak & Houghton 2001). The caspases cleave specific cellular substrates, and most type 1 keratins are thought to be substrates for caspases (Ku & Omary 2001). In the case of K18, this leads to caspase-cleaved fragments in the serum that can be analysed to assess the cancer load, cancer progression and response to chemotherapy by distinguishing between necrosis and apoptosis (Linder 2007). Ausch et al. (2009) analysed a caspase-cleaved fragment of K18 (M30) in the serum samples of patients with CRC pre- and postoperatively and found that M30 correlated significantly with tumour recurrence where it remained raised postoperatively. M30 neo-epitope in K18 on circulating tumour cells (CTCs) has been investigated to identify proportion of apoptotic and viable cells in peripheral blood of patients with epithelial cancers (Rossi et al. 2010). In this study, it was noted that a change in balance of M30-negative/positive CTC might indicate active disease.
Concluding comments
Keratins, key constituents of the IF proteins, are now established as having a significant role in maintaining epithelial integrity, in structuring and organizing cells, and in cell signalling pathways. Expression of keratins in colorectal epithelium has been shown to vary in health and in disease states. Keratins undergo PTM in response to various stimuli; the full significance of these still remains to be understood but has been implicated as having functional consequences. Likewise, the regulatory enzymes governing keratin modification are largely uncharacterized yet represent valuable potential targets in the search for new strategies to halt epithelial-mesenchymal transition. In sporadic CRCs, keratins have already been used as a tool for characterization of tumours, as well as in identifying their prognoses. Human studies in well-defined IBD patients categorized into disease type and extent or severity of disease are, however, lacking. Despite the increasing interest in keratins in both CRCs and IBD, further studies are needed to define their role in the pathogenesis of these conditions, identify expression states and solubility (including occurrence of PTM) in different disease states and to evaluate their secondary potential as diagnostic and therapeutic targets.
Declaration of interest
The authors declare no competing interest. This work was funded by the Bardhan Research and Education Trust.
References
- Achtstaetter T, Hatzfeld M, Quinlan RA, Parmelee DC, Franke WW. Separation of cytokeratin polypeptides by gel electrophoretic and chromatographic techniques and their identification by immunoblotting. Methods Enzymol. 1986;134:355–371. doi: 10.1016/0076-6879(86)34102-8. [DOI] [PubMed] [Google Scholar]
- Ameen NA, Figueroa Y, Salas PJ. Anomalous apical plasma membrane phenotype in CK8-deficient mice indicates a novel role for intermediate filaments in the polarization of simple epithelia. J. Cell Sci. 2001;114:563–575. doi: 10.1242/jcs.114.3.563. [DOI] [PubMed] [Google Scholar]
- Arnold CN, Goel A, Blum HE, Boland CR. Molecular pathogenesis of colorectal cancer: implications for molecular diagnosis. Cancer. 2005;104:2035–2047. doi: 10.1002/cncr.21462. [DOI] [PubMed] [Google Scholar]
- Askling J, Dickman PW, Karlen P, et al. Family history as a risk factor for colorectal cancer in inflammatory bowel disease. Gastroenterology. 2001;120:1356–1362. doi: 10.1053/gast.2001.24052. [DOI] [PubMed] [Google Scholar]
- Ausch C, Buxhofer-Ausch V, Olszewski U, et al. Circulating cytokeratin 18 fragment m65-a potential marker of malignancy in colorectal cancer patients. J. Gastrointest. Surg. 2009;13:2020–2026. doi: 10.1007/s11605-009-0992-6. [DOI] [PubMed] [Google Scholar]
- Barak V, Goike H, Panaretakis KW, Einarsson R. Clinical utility of cytokeratins as tumor markers. Clin. Biochem. 2004;37:529–540. doi: 10.1016/j.clinbiochem.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Bardelli A, Saha S, Sager JA, et al. PRL-3 expression in metastatic cancers. Clin. Cancer Res. 2003;9:5607–5615. [PubMed] [Google Scholar]
- Baribault H, Penner J, Iozzo RV, Wilson-Heiner M. Colorectal hyperplasia and inflammation in keratin 8-deficient FVB/N mice. Genes Dev. 1994;8:2964–2973. doi: 10.1101/gad.8.24.2964. [DOI] [PubMed] [Google Scholar]
- Barnes JP. Physiologic resection of the right colon. Surg. Gynecol. Obstet. 1952;94:722–726. [PubMed] [Google Scholar]
- Bayrak R, Yenidunya S, Haltas H. Cytokeratin 7 and cytokeratin 20 expression in colorectal adenocarcinomas. Pathol. Res. Pract. 2011;207:156–160. doi: 10.1016/j.prp.2010.12.005. [DOI] [PubMed] [Google Scholar]
- Braegger CP, Nicholls S, Murch SH, Stephens S, MacDonald TT. Tumour necrosis factor alpha in stool as a marker of intestinal inflammation. Lancet. 1992;339:89–91. doi: 10.1016/0140-6736(92)90999-j. [DOI] [PubMed] [Google Scholar]
- Buning C, Halangk J, Dignass A, et al. Keratin 8 Y54H and G62C mutations are not associated with inflammatory bowel disease. Dig. Liver Dis. 2004;36:388–391. doi: 10.1016/j.dld.2004.01.020. [DOI] [PubMed] [Google Scholar]
- Carter MJ, Lobo AJ, Travis SP. Guidelines for the management of inflammatory bowel disease in adults. Gut. 2004;53(Suppl 5):V1–V16. doi: 10.1136/gut.2004.043372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulin C, Ware CF, Magin TM, Oshima RG. Keratin-dependent, epithelial resistance to tumor necrosis factor-induced apoptosis. J. Cell Biol. 2000;149:17–22. doi: 10.1083/jcb.149.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lin RJ, Xie W, Wilpitz D, Evans RM. Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell. 1999;98:675–686. doi: 10.1016/s0092-8674(00)80054-9. [DOI] [PubMed] [Google Scholar]
- Chen R, Rabinovitch PS, Crispin DA, et al. DNA fingerprinting abnormalities can distinguish ulcerative colitis patients with dysplasia and cancer from those who are dysplasia/cancer-free. Am. J. Pathol. 2003;162:665–672. doi: 10.1016/S0002-9440(10)63860-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho WC. Proteomics technologies and challenges. Genomics Proteomics Bioinformatics. 2007;5:77–85. doi: 10.1016/S1672-0229(07)60018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi PM, Zelig MP. Similarity of colorectal cancer in Crohn's disease and ulcerative colitis: implications for carcinogenesis and prevention. Gut. 1994;35:950–954. doi: 10.1136/gut.35.7.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou CF, Smith AJ, Omary MB. Characterization and dynamics of O-linked glycosylation of human cytokeratin 8 and 18. J. Biol. Chem. 1992;267:3901–3906. [PubMed] [Google Scholar]
- Chou CF, Riopel CL, Rott LS, Omary MB. A significant soluble keratin fraction in ‘simple’ epithelial cells. Lack of an apparent phosphorylation and glycosylation role in keratin solubility. J. Cell Sci. 1993;105(Pt 2):433–444. doi: 10.1242/jcs.105.2.433. [DOI] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Chu PG, Weiss LM. Keratin expression in human tissues and neoplasms. Histopathology. 2002;40:403–439. doi: 10.1046/j.1365-2559.2002.01387.x. [DOI] [PubMed] [Google Scholar]
- Chu P, Wu E, Weiss LM. Cytokeratin 7 and cytokeratin 20 expression in epithelial neoplasms: a survey of 435 cases. Mod. Pathol. 2000;13:962–972. doi: 10.1038/modpathol.3880175. [DOI] [PubMed] [Google Scholar]
- Crohn UB, Rosenberg H. The sigmoidoscopic picture of chronic ulcerative colitis (non-specific) Am. J. Med. Sci. 1925;170:220–228. [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Demarque MD, Nacerddine K, Neyret-Kahn H, et al. Sumoylation by Ubc9 regulates the stem cell compartment and structure and function of the intestinal epithelium in mice. Gastroenterology. 2011;140:286–296. doi: 10.1053/j.gastro.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Dignass AU, Baumgart DC, Sturm A. Review article: the aetiopathogenesis of inflammatory bowel disease–immunology and repair mechanisms. Aliment. Pharmacol. Ther. 2004;20(Suppl 4):9–17. doi: 10.1111/j.1365-2036.2004.02047.x. [DOI] [PubMed] [Google Scholar]
- Ditzel HJ, Strik MC, Larsen MK, et al. Cancer-associated cleavage of cytokeratin 8/18 heterotypic complexes exposes a neoepitope in human adenocarcinomas. J. Biol. Chem. 2002;277:21712–21722. doi: 10.1074/jbc.M202140200. [DOI] [PubMed] [Google Scholar]
- Drake PJ, Griffiths GJ, Shaw L, Benson RP, Corfe BM. Application of high-content analysis to the study of post-translational modifications of the cytoskeleton. J. Proteome Res. 2009;8:28–34. doi: 10.1021/pr8006396. [DOI] [PubMed] [Google Scholar]
- Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48:526–535. doi: 10.1136/gut.48.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekbom A, Helmick C, Zack M, Adami HO. Ulcerative colitis and colorectal cancer. A population-based study. N. Engl. J. Med. 1990;323:1228–1233. doi: 10.1056/NEJM199011013231802. [DOI] [PubMed] [Google Scholar]
- Eriksson JE, Dechat T, Grin B, et al. Introducing intermediate filaments: from discovery to disease. J. Clin. Invest. 2009;119:1763–1771. doi: 10.1172/JCI38339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fava F, Danese S. Intestinal microbiota in inflammatory bowel disease: friend of foe? World J. Gastroenterol. 2011;17:557–566. doi: 10.3748/wjg.v17.i5.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Zhou X, Liao J, Omary MB. Pervanadate-mediated tyrosine phosphorylation of keratins 8 and 19 via a p38 mitogen-activated protein kinase-dependent pathway. J. Cell Sci. 1999;112(Pt 13):2081–2090. doi: 10.1242/jcs.112.13.2081. [DOI] [PubMed] [Google Scholar]
- Fiocchi C. Inflammatory bowel disease: etiology and pathogenesis. Gastroenterology. 1998;115:182–205. doi: 10.1016/s0016-5085(98)70381-6. [DOI] [PubMed] [Google Scholar]
- Franke WW, Schmid E, Schiller DL, et al. Differentiation-related patterns of expression of proteins of intermediate-size filaments in tissues and cultured cells. Cold Spring Harb. Symp. Quant. Biol. 1982;46(Pt 1):431–453. doi: 10.1101/sqb.1982.046.01.041. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Cleveland DW. A structural scaffolding of intermediate filaments in health and disease. Science. 1998;279:514–519. doi: 10.1126/science.279.5350.514. [DOI] [PubMed] [Google Scholar]
- Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol. 2007;8:947–956. doi: 10.1038/nrm2293. [DOI] [PubMed] [Google Scholar]
- Goldman RD, Chou YH, Prahlad V, Yoon M. Intermediate filaments: dynamic processes regulating their assembly, motility, and interactions with other cytoskeletal systems. FASEB J. 1999;13(Suppl 2):S261–S265. doi: 10.1096/fasebj.13.9002.s261. [DOI] [PubMed] [Google Scholar]
- Habtezion A, Toivola DM, Butcher EC, Omary MB. Keratin-8-deficient mice develop chronic spontaneous Th2 colitis amenable to antibiotic treatment. J. Cell Sci. 2005;118:1971–1980. doi: 10.1242/jcs.02316. [DOI] [PubMed] [Google Scholar]
- Habtezion A, Toivola DM, Asghar MN, et al. Absence of keratin 8 confers a paradoxical microflora-dependent resistance to apoptosis in the colon. Proc. Natl Acad. Sci. U.S.A. 2011;108:1445–1450. doi: 10.1073/pnas.1010833108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He T, Stepulak A, Holmstrom TH, Omary MB, Eriksson JE. The intermediate filament protein keratin 8 is a novel cytoplasmic substrate for c-Jun N-terminal kinase. J. Biol. Chem. 2002;277:10767–10774. doi: 10.1074/jbc.M111436200. [DOI] [PubMed] [Google Scholar]
- Herrmann H, Aebi U. Intermediate filaments: molecular structure, assembly mechanism, and integration into functionally distinct intracellular Scaffolds. Annu. Rev. Biochem. 2004;73:749–789. doi: 10.1146/annurev.biochem.73.011303.073823. [DOI] [PubMed] [Google Scholar]
- Herrmann H, Kreplak L, Aebi U. Isolation, characterization, and in vitro assembly of intermediate filaments. Methods Cell Biol. 2004;78:3–24. doi: 10.1016/s0091-679x(04)78001-2. [DOI] [PubMed] [Google Scholar]
- Hisamuddin IM, Yang VW. Genetics of colorectal cancer. MedGenMed. 2004;6:13. [PMC free article] [PubMed] [Google Scholar]
- Inada H, Izawa I, Nishizawa M, et al. Keratin attenuates tumor necrosis factor-induced cytotoxicity through association with TRADD. J. Cell Biol. 2001;155:415–426. doi: 10.1083/jcb.200103078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip YT, Davis RJ. Signal transduction by the c-Jun N-terminal kinase (JNK)–from inflammation to development. Curr. Opin. Cell Biol. 1998;10:205–219. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- Issa JP, Ahuja N, Toyota M, Bronner MP, Brentnall TA. Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res. 2001;61:3573–3577. [PubMed] [Google Scholar]
- Jass JR. Hyperplastic-like polyps as precursors of microsatellite-unstable colorectal cancer. Am. J. Clin. Pathol. 2003;119:773–775. doi: 10.1309/UYN7-0N9W-2DVN-9ART. [DOI] [PubMed] [Google Scholar]
- Kato H, Semba S, Miskad UA, Seo Y, Kasuga M, Yokozaki H. High expression of PRL-3 promotes cancer cell motility and liver metastasis in human colorectal cancer: a predictive molecular marker of metachronous liver and lung metastases. Clin. Cancer Res. 2004;10:7318–7328. doi: 10.1158/1078-0432.CCR-04-0485. [DOI] [PubMed] [Google Scholar]
- Khan AQ, Bury JP, Brown SR, Riley SA, Corfe BM. Keratin 8 expression in colon cancer associates with low faecal butyrate levels. BMC Gastroenterol. 2011;11:2. doi: 10.1186/1471-230X-11-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Coulombe PA. Intermediate filament scaffolds fulfill mechanical, organizational, and signaling functions in the cytoplasm. Genes Dev. 2007;21:1581–1597. doi: 10.1101/gad.1552107. [DOI] [PubMed] [Google Scholar]
- Knosel T, Emde V, Schluns K, Schlag PM, Dietel M, Petersen I. Cytokeratin profiles identify diagnostic signatures in colorectal cancer using multiplex analysis of tissue microarrays. Cell. Oncol. 2006;28:167–175. doi: 10.1155/2006/354295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 2000;19:1176–1179. doi: 10.1093/emboj/19.6.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku NO, Omary MB. Identification of the major physiologic phosphorylation site of human keratin 18: potential kinases and a role in filament reorganization. J. Cell Biol. 1994;127:161–171. doi: 10.1083/jcb.127.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku NO, Omary MB. Identification and mutational analysis of the glycosylation sites of human keratin 18. J. Biol. Chem. 1995;270:11820–11827. doi: 10.1074/jbc.270.20.11820. [DOI] [PubMed] [Google Scholar]
- Ku NO, Omary MB. Effect of mutation and phosphorylation of type I keratins on their caspase-mediated degradation. J. Biol. Chem. 2001;276:26792–26798. doi: 10.1074/jbc.M103315200. [DOI] [PubMed] [Google Scholar]
- Ku NO, Zhou X, Toivola DM, Omary MB. The cytoskeleton of digestive epithelia in health and disease. Am. J. Physiol. 1999;277:G1108–G1137. doi: 10.1152/ajpgi.1999.277.6.G1108. [DOI] [PubMed] [Google Scholar]
- Ku NO, Azhar S, Omary MB. Keratin 8 phosphorylation by p38 kinase regulates cellular keratin filament reorganization: modulation by a keratin 1-like disease causing mutation. J. Biol. Chem. 2002;277:10775–10782. doi: 10.1074/jbc.M107623200. [DOI] [PubMed] [Google Scholar]
- Ku NO, Toivola DM, Zhou Q, Tao GZ, Zhong B, Omary MB. Studying simple epithelial keratins in cells and tissues. Methods Cell Biol. 2004;78:489–517. doi: 10.1016/s0091-679x(04)78017-6. [DOI] [PubMed] [Google Scholar]
- Ku NO, Strnad P, Zhong BH, Tao GZ, Omary MB. Keratins let liver live: mutations predispose to liver disease and crosslinking generates Mallory-Denk bodies. Hepatology. 2007;46:1639–1649. doi: 10.1002/hep.21976. [DOI] [PubMed] [Google Scholar]
- Ku NO, Toivola DM, Strnad P, Omary MB. Cytoskeletal keratin glycosylation protects epithelial tissue from injury. Nat. Cell Biol. 2010;12:876–885. doi: 10.1038/ncb2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucharzik T, Maaser C, Lugering A, et al. Recent understanding of IBD pathogenesis: implications for future therapies. Inflamm. Bowel Dis. 2006;12:1068–1083. doi: 10.1097/01.mib.0000235827.21778.d5. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Sounding the alarm: protein kinase cascades activated by stress and inflammation. J. Biol. Chem. 1996;271:24313–24316. doi: 10.1074/jbc.271.40.24313. [DOI] [PubMed] [Google Scholar]
- Leech SH, Evans CA, Shaw L, et al. Proteomic analyses of intermediate filaments reveals cytokeratin8 is highly acetylated–implications for colorectal epithelial homeostasis. Proteomics. 2008;8:279–288. doi: 10.1002/pmic.200700404. [DOI] [PubMed] [Google Scholar]
- Leers MP, Kolgen W, Bjorklund V, et al. Immunocytochemical detection and mapping of a cytokeratin 18 neo-epitope exposed during early apoptosis. J. Pathol. 1999;187:567–572. doi: 10.1002/(SICI)1096-9896(199904)187:5<567::AID-PATH288>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- L'Hernault SW, Rosenbaum JL. Chlamydomonas alpha-tubulin is posttranslationally modified by acetylation on the epsilon-amino group of a lysine. Biochemistry. 1985;24:473–478. doi: 10.1021/bi00323a034. [DOI] [PubMed] [Google Scholar]
- Liao J, Lowthert LA, Ghori N, Omary MB. The 70-kDa heat shock proteins associate with glandular intermediate filaments in an ATP-dependent manner. J. Biol. Chem. 1995;270:915–922. doi: 10.1074/jbc.270.2.915. [DOI] [PubMed] [Google Scholar]
- Linder S. Cytokeratin markers come of age. Tumour Biol. 2007;28:189–195. doi: 10.1159/000107582. [DOI] [PubMed] [Google Scholar]
- Longacre TA, Fenoglio-Preiser CM. Mixed hyperplastic adenomatous polyps/serrated adenomas. A distinct form of colorectal neoplasia. Am. J. Surg. Pathol. 1990;14:524–537. doi: 10.1097/00000478-199006000-00003. [DOI] [PubMed] [Google Scholar]
- Majumdar D, Rosser R, Havard S, Lobo A, Evans CA, Corfe BM. An integrated workflow for fractionation, extraction and solubilisation of intermediate filaments from colorectal biopsies for proteomic analysis. Electrophoresis. doi: 10.1002/elps.201100662. in press. [DOI] [PubMed] [Google Scholar]
- Makowski GS, Ramsby ML. Degradation of cytokeratin intermediate filaments by calcium-activated proteases (calpains) in vitro: implications for formation of Mallory bodies. Res. Commun. Mol. Pathol. Pharmacol. 1998;101:211–223. [PubMed] [Google Scholar]
- Martinez-Balbas MA, Bauer UM, Nielsen SJ, Brehm A, Kouzarides T. Regulation of E2F1 activity by acetylation. EMBO J. 2000;19:662–671. doi: 10.1093/emboj/19.4.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGregor DK, Wu TT, Rashid A, Luthra R, Hamilton SR. Reduced expression of cytokeratin 20 in colorectal carcinomas with high levels of microsatellite instability. Am. J. Surg. Pathol. 2004;28:712–718. doi: 10.1097/01.pas.0000126757.58474.12. [DOI] [PubMed] [Google Scholar]
- Mizuuchi E, Semba S, Kodama Y, Yokozaki H. Down-modulation of keratin 8 phosphorylation levels by PRL-3 contributes to colorectal carcinoma progression. Int. J. Cancer. 2009;124:1802–1810. doi: 10.1002/ijc.24111. [DOI] [PubMed] [Google Scholar]
- Moll R, Franke WW, Schiller DL, Geiger B, Krepler R. The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell. 1982;31:11–24. doi: 10.1016/0092-8674(82)90400-7. [DOI] [PubMed] [Google Scholar]
- Moll R, Krepler R, Franke WW. Complex cytokeratin polypeptide patterns observed in certain human carcinomas. Differentiation. 1983;23:256–269. doi: 10.1111/j.1432-0436.1982.tb01291.x. [DOI] [PubMed] [Google Scholar]
- Moll R, Lowe A, Laufer J, Franke WW. Cytokeratin 20 in human carcinomas. A new histodiagnostic marker detected by monoclonal antibodies. Am. J. Pathol. 1992;140:427–447. [PMC free article] [PubMed] [Google Scholar]
- Murch SH, Lamkin VA, Savage MO, Walker-Smith JA, MacDonald TT. Serum concentrations of tumour necrosis factor alpha in childhood chronic inflammatory bowel disease. Gut. 1991;32:913–917. doi: 10.1136/gut.32.8.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murch SH, Braegger CP, Walker-Smith JA, MacDonald TT. Location of tumour necrosis factor alpha by immunohistochemistry in chronic inflammatory bowel disease. Gut. 1993;34:1705–1709. doi: 10.1136/gut.34.12.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omary MB, Ku NO, Liao J, Price D. Keratin modifications and solubility properties in epithelial cells and in vitro. Subcell. Biochem. 1998;31:105–140. [PubMed] [Google Scholar]
- Omary MB, Ku NO, Toivola DM. Keratins: guardians of the liver. Hepatology. 2002;35:251–257. doi: 10.1053/jhep.2002.31165. [DOI] [PubMed] [Google Scholar]
- Omary MB, Ku NO, Tao GZ, Toivola DM, Liao J. “Heads and tails” of intermediate filament phosphorylation: multiple sites and functional insights. Trends Biochem. Sci. 2006;31:383–394. doi: 10.1016/j.tibs.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Omary MB, Ku NO, Strnad P, Hanada S. Toward unraveling the complexity of simple epithelial keratins in human disease. J. Clin. Invest. 2009;119:1794–1805. doi: 10.1172/JCI37762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima RG. Developmental expression of murine extra-embryonic endodermal cytoskeletal proteins. J. Biol. Chem. 1982;257:3414–3421. [PubMed] [Google Scholar]
- O'Sullivan JN, Bronner MP, Brentnall TA, et al. Chromosomal instability in ulcerative colitis is related to telomere shortening. Nat. Genet. 2002;32:280–284. doi: 10.1038/ng989. [DOI] [PubMed] [Google Scholar]
- Owens DW, Wilson NJ, Hill AJ, et al. Human keratin 8 mutations that disturb filament assembly observed in inflammatory bowel disease patients. J. Cell Sci. 2004;117:1989–1999. doi: 10.1242/jcs.01043. [DOI] [PubMed] [Google Scholar]
- Paramio JM, Jorcano JL. Beyond structure: do intermediate filaments modulate cell signalling? BioEssays. 2002;24:836–844. doi: 10.1002/bies.10140. [DOI] [PubMed] [Google Scholar]
- Petak I, Houghton JA. Shared pathways: death receptors and cytotoxic drugs in cancer therapy. Pathol. Oncol. Res. 2001;7:95–106. doi: 10.1007/BF03032574. [DOI] [PubMed] [Google Scholar]
- Polley AC, Mulholland F, Pin C, et al. Proteomic analysis reveals field-wide changes in protein expression in the morphologically normal mucosa of patients with colorectal neoplasia. Cancer Res. 2006;66:6553–6562. doi: 10.1158/0008-5472.CAN-06-0534. [DOI] [PubMed] [Google Scholar]
- Pretlow TP, Brasitus TA, Fulton NC, Cheyer C, Kaplan EL. K-ras mutations in putative preneoplastic lesions in human colon. J. Natl Cancer Inst. 1993;85:2004–2007. doi: 10.1093/jnci/85.24.2004. [DOI] [PubMed] [Google Scholar]
- Rabinovitch PS, Dziadon S, Brentnall TA, et al. Pancolonic chromosomal instability precedes dysplasia and cancer in ulcerative colitis. Cancer Res. 1999;59:5148–5153. [PubMed] [Google Scholar]
- Riddell RH, Goldman H, Ransohoff DF, et al. Dysplasia in inflammatory bowel disease: standardized classification with provisional clinical applications. Hum. Pathol. 1983;14:931–968. doi: 10.1016/s0046-8177(83)80175-0. [DOI] [PubMed] [Google Scholar]
- Rossi E, Basso U, Celadin R, et al. M30 neoepitope expression in epithelial cancer: quantification of apoptosis in circulating tumor cells by CellSearch analysis. Clin. Cancer Res. 2010;16:5233–5243. doi: 10.1158/1078-0432.CCR-10-1449. [DOI] [PubMed] [Google Scholar]
- Rutter M, Saunders B, Wilkinson K, et al. Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology. 2004;126:451–459. doi: 10.1053/j.gastro.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Sadoul K, Wang J, Diagouraga B, Khochbin S. The tale of protein lysine acetylation in the cytoplasm. J. Biomed. Biotechnol. 2011;2011:970382. doi: 10.1155/2011/970382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartor RB. Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006;3:390–407. doi: 10.1038/ncpgasthep0528. [DOI] [PubMed] [Google Scholar]
- Schweizer J, Bowden PE, Coulombe PA, et al. New consensus nomenclature for mammalian keratins. J. Cell Biol. 2006;174:169–174. doi: 10.1083/jcb.200603161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeler JS, Dejean A. Nuclear and unclear functions of SUMO. Nat. Rev. Mol. Cell Biol. 2003;4:690–699. doi: 10.1038/nrm1200. [DOI] [PubMed] [Google Scholar]
- Shen L, Turner JR. Role of epithelial cells in initiation and propagation of intestinal inflammation. Eliminating the static: tight junction dynamics exposed. Am. J. Physiol. Gastrointest. Liver Physiol. 2006;290:G577–G582. doi: 10.1152/ajpgi.00439.2005. [DOI] [PubMed] [Google Scholar]
- Shen L, Kondo Y, Rosner GL, et al. MGMT promoter methylation and field defect in sporadic colorectal cancer. J. Natl Cancer Inst. 2005;97:1330–1338. doi: 10.1093/jnci/dji275. [DOI] [PubMed] [Google Scholar]
- Sidransky D, Tokino T, Hamilton SR, et al. Identification of ras oncogene mutations in the stool of patients with curable colorectal tumors. Science. 1992;256:102–105. doi: 10.1126/science.1566048. [DOI] [PubMed] [Google Scholar]
- Silverberg MS, Satsangi J, Ahmad T, et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can. J. Gastroenterol. 2005;19(Suppl A):5–36. doi: 10.1155/2005/269076. [DOI] [PubMed] [Google Scholar]
- Snider NT, Weerasinghe SV, Iniguez-Lluhi JA, Herrmann H, Omary MB. Keratin hypersumoylation alters filament dynamics and is a marker for human liver disease and keratin mutation. J. Biol. Chem. 2011;286:2273–2284. doi: 10.1074/jbc.M110.171314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soetikno RM, Lin OS, Heidenreich PA, Young HS, Blackstone MO. Increased risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis: a meta-analysis. Gastrointest. Endosc. 2002;56:48–54. doi: 10.1067/mge.2002.125367. [DOI] [PubMed] [Google Scholar]
- Srikanth B, Vaidya MM, Kalraiya RD. O-GlcNAcylation determines the solubility, filament organization, and stability of keratins 8 and 18. J. Biol. Chem. 2010;285:34062–34071. doi: 10.1074/jbc.M109.098996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey SN, Sulem P, Masson G, et al. New common variants affecting susceptibility to basal cell carcinoma. Nat. Genet. 2009;41:909–914. doi: 10.1038/ng.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinert PM. The dynamic phosphorylation of the human intermediate filament keratin 1 chain. J. Biol. Chem. 1988;263:3333–13339. [PubMed] [Google Scholar]
- Steinert P, Zackroff R, Aynardi-Whitman M, Goldman RD. Isolation and characterization of intermediate filaments. Methods Cell Biol. 1982;24:399–419. doi: 10.1016/s0091-679x(08)60667-6. [DOI] [PubMed] [Google Scholar]
- Stenling R, Lindberg J, Rutegard J, Palmqvist R. Altered expression of CK7 and CK20 in preneoplastic and neoplastic lesions in ulcerative colitis. APMIS. 2007;115:1219–1226. doi: 10.1111/j.1600-0643.2007.00664.x. [DOI] [PubMed] [Google Scholar]
- Stephens BJ, Han H, Gokhale V, Von Hoff DD. PRL phosphatases as potential molecular targets in cancer. Mol. Cancer Ther. 2005;4:1653–1661. doi: 10.1158/1535-7163.MCT-05-0248. [DOI] [PubMed] [Google Scholar]
- Sterner R, Vidali G, Allfrey VG. Studies of acetylation and deacetylation in high mobility group proteins. Identification of the sites of acetylation in HMG-1. J. Biol. Chem. 1979;254:11577–11583. [PubMed] [Google Scholar]
- Svitkina TM, Verkhovsky AB, Borisy GG. Plectin sidearms mediate interaction of intermediate filaments with microtubules and other components of the cytoskeleton. J. Cell Biol. 1996;135:991–1007. doi: 10.1083/jcb.135.4.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao GZ, Strnad P, Zhou Q, et al. Analysis of keratin polypeptides 8 and 19 variants in inflammatory bowel disease. Clin. Gastroenterol. Hepatol. 2007;5:857–864. doi: 10.1016/j.cgh.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Tatsumi N, Mukaisho K, Mitsufuji S, et al. Expression of cytokeratins 7 and 20 in serrated adenoma and related diseases. Dig. Dis. Sci. 2005;50:1741–1746. doi: 10.1007/s10620-005-2928-7. [DOI] [PubMed] [Google Scholar]
- Tatsumi N, Kushima R, Vieth M, et al. Cytokeratin 7/20 and mucin core protein expression in ulcerative colitis-associated colorectal neoplasms. Virchows Arch. 2006;448:756–762. doi: 10.1007/s00428-006-0188-3. [DOI] [PubMed] [Google Scholar]
- Toivola DM, Krishnan S, Binder HJ, Singh SK, Omary MB. Keratins modulate colonocyte electrolyte transport via protein mistargeting. J. Cell Biol. 2004;164:911–921. doi: 10.1083/jcb.200308103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toivola DM, Strnad P, Habtezion A, Omary MB. Intermediate filaments take the heat as stress proteins. Trends Cell Biol. 2010;20:79–91. doi: 10.1016/j.tcb.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull RB, Jr, Kyle K, Watson FR, Spratt J. Cancer of the colon: the influence of the no-touch isolation technic on survival rates. Ann. Surg. 1967;166:420–427. doi: 10.1097/00000658-196709000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Srinivasan S, Theiss AL, Merlin D, Sitaraman SV. Interleukin-6 induces keratin expression in intestinal epithelial cells: potential role of keratin-8 in interleukin-6-induced barrier function alterations. J. Biol. Chem. 2007;282:8219–8227. doi: 10.1074/jbc.M604068200. [DOI] [PubMed] [Google Scholar]
- Weitz J, Kienle P, Magener A, et al. [Detection of isolated disseminated tumor cells of colorectal carcinomas in lymph nodes] Langenbecks Arch. Chir. Suppl. Kongressbd. 1998;115:319–322. [PubMed] [Google Scholar]
- Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- Yun K, Merrie AE, Gunn J, Phillips LV, McCall JL. Keratin 20 is a specific marker of submicroscopic lymph node metastases in colorectal cancer: validation by K-RAS mutations. J. Pathol. 2000;191:21–26. doi: 10.1002/(SICI)1096-9896(200005)191:1<21::AID-PATH581>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]