

Abstract
OTHER ARTICLES PUBLISHED ON ANCA IN THIS ISSUE
Animal models of anti-neutrophil cytoplasmic antibody-associated vasculitis. Clinical and Experimental Immunology 2012, 169: 229–37.
Neutrophils are pivotal to host defence during infectious diseases. However, activated neutrophils may also cause undesired tissue damage. Ample examples include small-vessel inflammatory diseases (vasculitis) that are associated with anti-neutrophil cytoplasmic autoantibodies (ANCA) residing in the patients' plasma. In addition to being an important diagnostic tool, convincing evidence shows that ANCA are pathogenic. ANCA–neutrophil interactions induce important cellular responses that result in highly inflammatory necrotizing vascular damage. The interaction begins with ANCA binding to their target antigens on primed neutrophils, proceeds by recruiting transmembrane molecules to initiate intracellular signal transduction and culminates in activation of effector functions that ultimately mediate the tissue damage.
Keywords: ANCA, neutrophils, pathogenesis, signal transduction, vasculitis
OTHER ARTICLES PUBLISHED ON ANCA IN THIS ISSUE
Animal models of anti-neutrophil cytoplasmic antibody-associated vasculitis. Clinical and Experimental Immunology 2012, 169: 229–37.
Anti-neutrophil cytoplasmic autoantibodies (ANCA) and the expression of ANCA target antigens by the neutrophil
ANCA must recognize and bind their target antigens, proteinase 3 (PR3) or myeloperoxidase (MPO), in order to initiate signalling events and to subsequently activate the neutrophil. Thus, ANCA must either be internalized by the neutrophil or the antigens must be accessible on the cell surface, or both may occur. Many studies exploring the membrane expression of ANCA antigens have been performed. MPO and the vast majority of PR3 antigens reside in azurophilic granules, which can be mobilized during activation in vitro and in vivo[1,2]. In contrast to MPO, PR3 is also stored in specific granules and in secretory vesicles that are mobilized more easily [3]. Moreover, significant PR3 amounts are already expressed on the surface of resting cells with a strong increased expression after activation. Thus, there are major differences in PR3 and MPO membrane expression. Notably, and in contrast to PR3, MPO is not detected on the plasma membrane of resting neutrophils. Furthermore, the membrane MPO that increases after cell activation is small compared to PR3.
Neutrophils must be primed for subsequent ANCA-induced activation. Priming includes ANCA antigen translocation and can be achieved in vitro by various mediators, including tumour necrosis factor (TNF)-α, interleukin (IL)-1, IL-6, IL-18, N-formyl-Met-Leu-Phe (fMLF) and complement 5a (C5a) [4–7]. In-vivo priming may occur during infections that frequently precede the clinical manifestation of ANCA vasculitis. Indeed, patients with active disease show increased neutrophil ANCA antigen membrane expression [5,8,9]. A synergistic effect for increased mPR3 expression by cytokines, adhesion and anti-PR3 antibodies was demonstrated that could become relevant when neutrophils leave the circulating blood [10]. Recently, α1-anti-trypsin polymers have been described to prime the neutrophil for ANCA activation, indicating that additional priming mechanisms exist [11].
An important observation established that PR3, but not MPO, has a bimodal membrane expression pattern. mPR3low- and mPR3high-expressing neutrophils can be distinguished with a percentage of mPR3high neutrophils ranging between 0 and 100% [12]. The expression pattern is extremely stable in a given individual, does not change with activation and correlates strongly in monozygotic twins and human leucocyte antigen (HLA)-matched siblings [13,14]. The clinical significance of the mPR3 phenotype was established in independent cohorts showing that a large subset of mPR3high neutrophils is a risk factor for ANCA vasculitis. The risk factor has a negative effect on clinical patient outcomes [13,15–17]. Compared to the mPR3low cells, mPR3high neutrophils generate more superoxide and degranulate more strongly to PR3–ANCA, but not to other stimuli. This provides a potential explanation for the clinical observation on risk and outcome [18].
Because MPO and PR3 are not transmembrane molecules, elucidating how ANCA antigens are anchored in the plasma membrane is another important step in understanding how signal transduction may begin. PR3 presentation on the neutrophil membrane occurs by at least two different mechanisms. PR3 can be inserted directly into the plasma membrane, as predicted by molecular dynamics simulations using a membrane model [19]. This model suggested that PR3 associates strongly with anionic membranes, whereby basic residues mediate the orientation of PR3 at the membrane and hydrophobic amino acids mediate anchoring of the molecule. Kantari et al. mutated the basic and the hydrophobic amino acids and observed that the modified PR3 preserved its enzymatic activity. However, the mutated protein lost its plasma membrane expression in a myeloid rat basophilic leukaemic (RBL) cell model [20]. Another way of expressing PR3 on the neutrophil membrane is its presentation by a glycosylphosphatidylinositol (GPI)-linked receptor, namely CD177 (or human neutrophil antigen B1, NB1) [21,22]. Although all neutrophils contain intracellular PR3, only those cells that express NB1 protein on the neutrophil plasma membrane show high mPR3 surface expression. Studies have demonstrated further that PR3 and NB1 were not only co-expressed on the same neutrophil subset, but that both molecules co-localize and co-immunoprecipitate. Co-transfection experiments in human embryonic kidney 293 (HEK293) cells showed that NB1 was a sufficient receptor for PR3, but not for pro-PR3 [23]. Future experiments need to elucidate the control mechanisms of NB1 expression and why only a subset of neutrophils generates NB1 protein. Korkmaz et al. showed that a unique hydrophobic patch, present on human and absent from gibbon and murine PR3, enabled binding to NB1 [24]. Choi et al. performed high-throughput screening using a small molecule library and identified compounds that inhibited the interaction between NB1 and PR3 [25]. Kuhl et al. characterized conformational PR3 epitopes recognized by monoclonal anti-PR3 antibodies or PR3–ANCA from patients. These epitopes are distinct from the catalytic site and from the hydrophobic patch that allowed binding to membranes and NB1 [26]. All these studies have improved our understanding with respect to PR3 presentation on the neutrophil membrane and its accessibility for anti-PR3 antibodies. Consistent with the co-expression of NB1 and PR3 on the same cell, a larger percentage of mNB1-expressing neutrophils was a risk factor for ANCA vasculitis [27]. The role of the lacking PR3–NB1 interaction in mice could be one reason for the difficulty in generating an anti-PR3 antibody-mediated disease model, and needs further study.
We have reviewed the data describing modes of ANCA antigen expression on the neutrophil membrane and how ANCA can bind to their targets on the plasma membrane to initiate activation. Also conceivable is the possibility that ANCA internalization by the neutrophil contributes to activation. In fact, ANCA penetration into neutrophils has been observed by different investigators; however, the mechanisms and significance of this observation for the activation process are not yet understood [9,28,29]. Furthermore, reactivation of PR3 and MPO transcription has been observed and epigenetic mechanisms that control this process are beginning to be characterized [30,31]. It will be interesting to see if this process results in a protein or cellular localization distinct from those of the ‘original’ PR3 antigen.
An additional ANCA target is the lysosomal membrane glycoprotein lysosomal-associated membrane protein 2 (LAMP-2) that was implicated in pauci-immune necrotizing glomerulonephritis by Kain et al. [32,33]. LAMP-2 is a heavily glycosylated protein expressed in many cell types, including neutrophils and endothelial cells. Lysosomal membrane proteins were detected in membranes of different cellular compartments such as lysosomes, multi-vesicular bodies, the trans-Golgi and plasma membranes [34]. LAMP-2 was found mainly in granule membranes of resting neutrophils and its plasma membrane expression was increased with fMLF treatment [32]. The clinical significance of LAMP-2 as an ANCA antigen in small vessel vasculitis was challenged by the Chapel Hill group. The investigators found much lower anti-LAMP antibody titres compared with antibodies to PR3 and MPO, no correlation with vasculitis disease activity and no disease induction by passive antibody transfer into rats [35]. Kain et al. were able, very recently, to repeat their findings in different European patient cohorts [36]. The conflicting data have no obvious explanation, but may be related to methodological and population differences as discussed by Flint et al. [37]. Major findings with respect to ANCA antigens are summarized in Fig. 1.
Fig. 1.
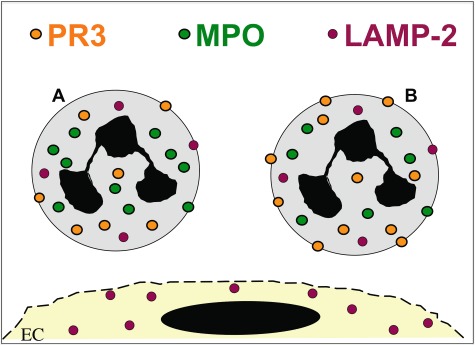
Anti-neutrophil cytoplasmic autoantibodies (ANCA) antigen expression. Myeloperoxidase (MPO) and proteinase 3 (PR3) are expressed by neutrophils and monocytes only, whereas lysosomal-associated membrane protein 2 (LAMP-2) is also found in endothelial cells. PR3 has a bimodal expression pattern on the neutrophil membrane in that a mPR3low- and mPR3high-expressing subset can be distinguished.
The ANCA antigen-binding site and the Fc part co-operate in neutrophil activation
Once ANCA have bound their neutrophil-expressed antigens, signalling and activation are initiated. Several investigators have characterized the part of the ANCA molecule that is important for neutrophil activation. Conflicting data exist, but the emerging picture is that both the antigen-binding part and the Fc part are needed. We found that ANCA Fab bind to their antigens expressed on the neutrophil, but did not trigger activation. In contrast, ANCA F(ab)2, or ANCA Fabs that were cross-linked, did activate superoxide generation. In the same study, and in contrast to these human ANCA data, F(ab)2 from a murine monoclonal antibody (mAb) had no activating capacity [38]. A PR3- and MPO-ANCA F(ab)2-induced respiratory burst was confirmed in another study [39], but not observed by other investigators [40–42]. The use of human versus murine antibodies, the strength of the activation response, assaying intra- or extracellular oxidant generation and the antigen specificity of the antibodies that were employed may, at least in part, explain some of the differences in the results. Williams et al. observed that ANCA F(ab)2 induced p21ras activation that was necessary, but not sufficient, for the respiratory burst [43]. Moreover, gene arrays showed that both ANCA F(ab)2 and ANCA immunoglobulin (Ig)G induce leucocyte gene transcription [44]. Interestingly, some of the transcribed genes were unique to intact ANCA IgG and some to the F(ab)2, whereas others were induced by both fragments. Thus, ANCA F(ab)2 bind to the neutrophil and trigger several neutrophil responses that do not depend on FcγR engagement. Few studies investigated this issue in monocytes. Weidner et al. showed that human ANCA also activated respiratory burst in monocytes and that ANCA F(ab)2 triggered a similar response compared to the complete ANCA IgG [45].
In addition to the antigen-binding fragments, the Fc part of the ANCA molecule is also important. ANCA IgG bind to FcγRIIa (CD32A) and FcγRIIIb (CD16B). FcγRIIa blockade abrogated ANCA-induced activation, whereas the role of the FcγRIIIb blockade is somewhat more controversial [38,40–42,46]. The FcγRIIa has two allelic variants with either a histidine or an arginine at amino acid position 131, resulting in a high-responder and low-responder receptor form. Neutrophils with the high-responder variant showed a stronger response to anti-PR3 and anti-MPO IgG1 mAbs in vitro[40]. This FcγRIIa also has high affinity to the IgG3 subclass, which is the dominant ANCA subclass in patients with active disease, and had the strongest capability to induce neutrophil adhesion in vitro[47,48].
Kocher et al. observed that ANCA IgG also bind to the FcγRIIIb on neutrophils that is expressed approximately 10 times higher than the FcγRIIa [46]. Distinct patterns of CD11b increase and CD62L shedding suggested that FcγIIIb is involved in ANCA-induced neutrophil activation. FcγRIIIb has two common genetic variants named NA1 and NA2, the former triggering a stronger neutrophil activation than the latter. A recent study on a large cohort of patients with granulomatosis with polyangiitis (GPA, also known as Wegener's granulomatosis) demonstrated a similar NA1 allele frequency in patients compared to controls. However, the presence of NA1 was associated with more severe renal disease [49]. The FcαR (CD89) also has genetic variants characterized by a single nucleotide polymorphism (SNP) that changes the amino acid sequence in the coding region. A serine (A) is associated with less inflammatory cytokine release and a glycine (G) with more phagocytosis and cell activation [50]. Kelley et al. studied also IgA ANCA and the SNP variants of the FcαR in their GPA patient cohorts [49]. IgA ANCA were present in 27% of the GPA patients, and were less frequent in those patients who developed end-stage renal disease and more frequent in those with upper airway manifestation. The G allele was, however, found more frequently in patients with renal disease and less frequently in those with upper airway manifestation. Neutrophils with the proinflammatory allelic FcαR variant triggered a stronger activation response to IgA ANCA in vitro. Thus, the data indicate that FcγR and FcαR genotypes influence manifestation patterns and disease severity in patients with ANCA-induced vasculitis.
Post-translational modifications such as sialylation might be an additional mechanism to change the activating capability of ANCA. It has been shown that the PR3–ANCA sialylation ratio was significantly lower in patients with active disease correlating with the Birmingham Vasculitis Activity Score (BVAS) score. Moreover, the in-vitro respiratory burst was correlated inversely with sialylation of the PR3–ANCA IgG [51]. All these findings suggest an important interplay between the ANCA antigen-binding fragment, the Fc part with its isotype and class characteristics and post-translational ANCA modifications as well as important genetic variants in the corresponding Fcα and Fcγ receptors on the neutrophil that may determine the mechanisms and strength by which ANCA interact with the neutrophil. The bacterial enzyme endoglycosidase S resulted in hydrolysis of ANCA IgG glycans and attenuated ANCA-induced neutrophil activation necrotizing crescentic glomerulonephritis (NCGN) in an anti-MPO antibody-mediated mouse model [52].
ANCA antigens exist in a larger signalling complex allowing neutrophil activation
MPO and PR3 are not transmembrane molecules, and therefore need to co-operate with other molecules to start intracellular signal transduction. Previous data using blocking antibodies had implicated β2-integrins in ANCA-induced neutrophil activation [42]. David et al. characterized a direct interaction between PR3 and CD11b/CD18 (Mac-1) on the neutrophil membrane and suggested that PR3 modulates neutrophil adhesion by activating Mac-1 [53]. The same group described later that PR3 was present in lipid rafts together with the GPI-linked FcγRIIIb and p22phox, an essential component of nicotinamide adenine dinucleotide phosphate-oxidase (NADPH) oxidase complex [54]. An interesting finding in their study was that using phospholipase D to cleave GPI-linkers resulted in a reduction of both PR3 and FcγRIIIb, suggesting that a GPI-anchored receptor indeed mediates mPR3 presentation. As discussed above, the NB1 is also a GPI-linked protein and is a sufficient receptor for mPR3 presentation [23]. However, even knowing that PR3 exists in a complex with GPI-linked molecules could not explain how neutrophils become activated after PR3–ANCA binding, as none of these components have transmembrane domains. Precipitating CD177 from the neutrophil membrane and performing mass spectrometry, we found that several molecules co-precipitated with CD177. Among those proteins were the FcγIIIR as well as Mac-1 [55]. CD177 and Mac-1 co-localized, co-precipitated and showed direct protein interactions by plasmon-resonance analysis and when Mac-1 transfected cells interacted with immobilized NB1. We subsequently established that Mac-1 was a functionally important transmembrane component of the PR3 membrane complex, allowing subsequent PR3–ANCA-induced activation predominantly of mPR3high/NB1positive neutrophils (Fig. 2). However, we observed that degranulation and extracellular superoxide generation, but not intracellular hydrogen peroxide formation depended on the mPR3 phenotype. Interestingly, PR3–ANCA were equally potent in inducing DHR oxidation in mPR3high/NB1positive and mPR3low/NB1negative cells an observation also made by Hu et al. [27]. The underlying mechanism for this finding still needs to be elucidated.
Fig. 2.
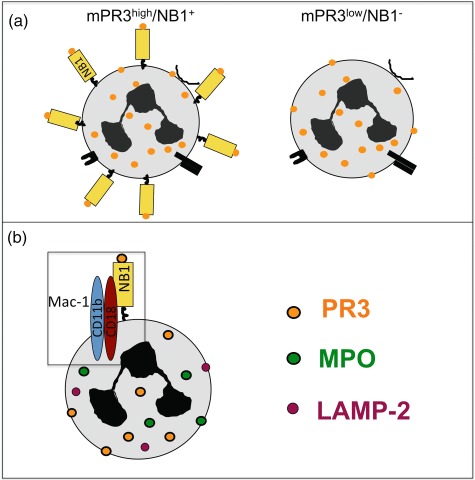
Neutrophil antigen B1 (NB1) is a receptor for proteinase 3 (PR3) presentation on the neutrophil membrane. (a) A mPR3high/NB1positive and a mPR3low/NB1negative neutrophil subset exist. (b) PR3 and NB1 are part of a larger signalling complex in the lipid rafts. The transmembrane β2-integrin Mac-1 is part of this complex and mediates PR3–anti-neutrophil cytoplasmic autoantibodies (ANCA)-induced neutrophil activation.
As mentioned, MPO membrane expression by neutrophils is somewhat scarce and much less is known as to how signalling is initiated after MPO–ANCA bind their target. Hess et al. found that large amounts of MPO can be acquired by resting neutrophils from supernatants of activated neutrophils. This acquired surface MPO allowed MPO–ANCA binding and neutrophil activation [56]. Others showed that MPO is presented by CD11b promoting neutrophil activation even in the absence and presence of anti-MPO antibodies [57,58].
ANCA activate signalling pathways and neutrophil functions that participate in the vasculitic damage
Initial studies on ANCA-induced signalling events showed that distinct intracellular signalling events mediated ANCA-induced neutrophil activation. Tyrosine kinase and protein kinase C activation by ANCA, but not by control IgG, was observed by Radford et al. [59]. Blocking both kinases using pharmacological inhibitors abrogated ANCA-induced superoxide generation. These experiments encouraged further characterization of the signal transduction cascade involved in ANCA-induced neutrophil activation. The implication was to block important key elements specifically and thereby identify novel and more specific treatment targets.
P38 mitogen-activated protein kinase (MAPK) and extracellular regulated kinase (ERK) are important during both priming and the ANCA-induced neutrophil activation. Priming increases the amount of membrane-expressed antigens, but also sparks signalling pathways that are needed for a subsequent ANCA-induced full-blown activation. Both p38 MAPK and ERK are initiated during TNF-α priming and their blockade abrogates subsequent ANCA-induced activation. However, both pathways show differential effects in that p38 MAPK, but not ERK, controls the ANCA-antigen translocation [60]. Interestingly, the 3-hydroxy-3-methylglutaryl co-enzyme A reductase inhibitors cerevastatin and simvastatin, which are known to exert actions above and beyond lipid lowering, abrogated ANCA-induced respiratory burst [61]. The effect was independent of the mevalonat pathway and involved ERK, but not p38 MAPK inhibition. Activated p38 MAPK was detected in glomerular neutrophils and intrinsic cells in biopsies from ANCA patients [62]. The importance of p38 MAPK for ANCA-induced NCGN was demonstrated recently in a disease mouse model [63] and is discussed in the adjacent review by Robson.
Several studies explored the role of phosphatidylinositol 3-kinase (PI3K) in ANCA-induced neutrophil activation. PI3K generates phosphatidylinositol-3,4,5-triphosphate (PIP3) and phosphatidylinositol-3,4-diphosphate (PIP2). Both substances recruit the serine/threonine kinase Akt. Ben-Smith et al. observed that ANCA induced PIP3, but did not activate p85/p110 PI3K. This PI3K isoform was, however, activated by simple FcγR cross-linking, again underscoring the fact that ANCA-induced activation is not merely a consequence of FcγR cross-linking and that other transmembrane molecules are required [64]. In contrast, ANCA activated the p101/110γ PI3K. Inhibition of all PI3K isoforms by LY294002 blocked ANCA-triggered superoxide generation. We confirmed the functional importance of PI3K. In addition, we investigated activation of the downstream kinase Akt by ANCA. Akt is phosphorylated by phosphoinositide-dependent kinase 1 (PDK1) and by PDK2. P38 MAPK can function as PDK2, and we showed that both the p38 MAPK and PI3K participate in Akt activation by ANCA [65]. TNF-α priming also resulted in Akt phosphorylation by both upstream kinases and promoted the association of Akt with the actin regulatory protein PAK1. ANCA patients frequently suffer from febrile infections that complicate immunosuppressive therapy. During these events neutrophils are exposed to increased temperatures. Anti-pyretics are distributed generously to fight fever, although its biological role is not so clear. We observed two interesting effects of short fever-like temperature spikes on neutrophils that could be clinically relevant in ANCA patients. Heat exposure abrogated PI3K/Akt activation and respiratory burst in primed neutrophils challenged by ANCA [66]. ANCA-induced phosphorylation of p38 MAPK and ERK was not affected. However, heat exposure prevented the increase in ANCA antigen expression in neutrophils that were treated with lipopolysaccharide (LPS) overnight [67]. This effect was mediated, at least in part, by diminishing TNF-α that was released from LPS-treated neutrophils. TNF-α required p38 MAPK to up-regulate ANCA antigen expression on the neutrophil surface, and heat accelerated p38 MAPK protein degradation in LPS-treated neutrophils. These data suggest that fever-like temperatures could modulate ANCA-mediated inflammatory responses via PI3K/Akt and p38 MAPK pathways.
The functional role of the PI3Kγ isoform was confirmed in vivo by a genetic approach in a murine bone-marrow (BM) transplantation model of anti-MPO antibody-induced NCGN [68]. NCGN occurred in mice that had received BM from wild-type, but not from PI3Kγ gene-deleted mice. Moreover, a γ isoform-specific inhibitor abrogated ANCA-induced superoxide generation, degranulation and neutrophil migration in vitro and oral treatment with this compound prevented NCGN in mice, suggesting that specific PI3Kγ inhibition could be used therapeutically (Fig. 3).
Fig. 3.
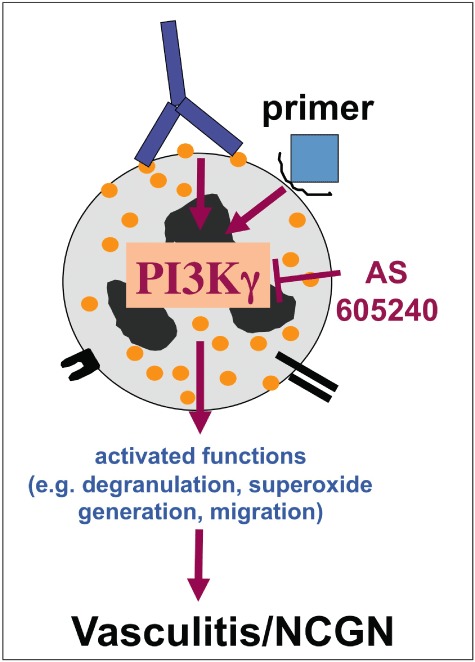
Anti-neutrophil cytoplasmic autoantibodies (ANCA)-induced neutrophil activation is controlled by intracellular signalling pathways. phosphatidylinositol 3-kinase γ (PI3Kγ) is depicted as an example for a signal molecule that was characterized as a key molecule for ANCA-induced neutrophil activation. PI3Kγ can be targeted therapeutically to prevent necrotizing crescentic glomerulonephritis (NCGN).
Several investigators have now implicated the participation of complement activation in ANCA-induced inflammation. In fact, animal studies narrowed the alternative pathway and particularly C5 as an important component in ANCA-induced NCGN [69,70]. In-vitro experiments elucidated that C5a is generated by ANCA-activated neutrophils and that this component further provides additional neutrophil priming for ANCA activation. Thus, ANCA-induced C5a would then act as an acceleration loop, further enhancing inflammation. C5a is connected to the important PI3K pathway in that the C5a receptor belongs to the G protein-coupled receptors that signal via PI3Kγ[71]. Importantly, mice lacking the C5a receptor in myeloid cells only were protected from anti-MPO antibody-induced NCGN [6]. These data imply that the C5a receptor may provide an additional treatment target in patients with ANCA vasculitis.
ANCA stimulation induces neutrophils and monocytes to produce and release cytokines [44,72–74]. Proinflammatory IL-1β may be of particular clinical interest because it is increased by ANCA, the lack of IL-1βR in renal cells protected from glomerular injury in murine anti-GBM model and an IL-1R blocker is available in the clinic [72,75,76]. Active IL-1β is generated from inactive precursor pro-IL-1β. The classical enzyme that mediates this process is caspase-1. Alternative IL-1β converting enzymes were suggested. We showed recently that active neutrophil serine proteases (NSPs) are critical for IL-1β generation in ANCA-stimulated monocytes and neutrophils. The IL-1β amount produced by monocytes was clearly higher compared to neutrophils, but neutrophils outnumber monocytes in vivo, suggesting that both cell types are possibly important. Murine monocytes and neutrophils lacking dipeptidylpeptidase I (DPPI) and therefore lacking active NSPs produced significantly less IL-1β in response to anti-MPO antibodies [77]. Preincubation of human monocytes with cell-permeable serine protease inhibitors or a caspase-1 inhibitor also diminished IL-1β generation. NSPs consist of human neutrophil elastase (HNE), PR3 and cathepsin G (CG). Exogenous PR3 rescued IL-1β generation in DPPI-deficient monocytes. DPPI- and PR3/HNE-deficient myeloid cells as well the IL-1R blocker Anakinra protected from NCGN in an anti-MPO antibody-mediated NCGN mouse model. These findings demonstrate that at least two mechanisms participate in IL-1β generation, namely caspase-1 and PR3, and that PR3 alone or in combination with HNE is important for ANCA-induced NCGN. It is conceivable that additional NSP-mediated effects participate in ANCA-mediated tissue damage. These proteases may cleave extracellular matrix proteins and injure the endothelium. Lu et al. demonstrated that ANCA-activated neutrophils released serine proteases, but not superoxide when co-cultured with EC, and that serine proteases mediated EC damage resulting in von Willebrand factor (vWF) release [78]. Serine proteases that are packaged in ANCA-induced neutrophil microparticles or in neutrophil extracellular traps (NETs) possibly also participate in endothelial damage [79,80].
Together, ANCA induce a variety of neutrophil responses in vitro. Some of these were shown to be significant in vivo, such as p38 MAPK, PI3Kγ, C5a and serine proteases. Others that are thought to be important await further in-vivo proof, including the role of ANCA-induced reactive oxygen generation.
Conclusion
The neutrophil is both the cell that expresses target ANCA antigens and a major effector cell in ANCA-induced small vessel vasculitis. The ANCA antigens PR3 and MPO differ substantially in their expression pattern on the neutrophil plasma membrane. ANCA bind to membrane expressed target antigens and initiate intracellular signalling events. The PR3–NB1–Mac-1 membrane complex is one example showing that larger signalling complexes with transmembrane molecules exist. Distinct signalling pathways triggered by ANCA F(ab)2 and the intact ANCA IgG molecule were identified and co-operate in neutrophil activation. Detailed characterization of the activation process will identify novel treatment targets that need to be tested in animal models and subsequently in patients.
Acknowledgments
Ralph Kettritz was supported by grants from the Deutsche Forschungsgemeinschaft and the Experimental and Clinical Research Center, a joint co-operation between the Charité Medical Faculty and the Max-Delbrück Center for Molecular Medicine Berlin-Buch.
Disclosure
Nothing to declare.
References
- 1.Sengelov H, Follin P, Kjeldsen L, Lollike K, Dahlgren C, Borregaard N. Mobilization of granules and secretory vesicles during in vivo exudation of human neutrophils. J Immunol. 1995;154:4157–65. [PubMed] [Google Scholar]
- 2.Sengelov H, Kjeldsen L, Borregaard N. Control of exocytosis in early neutrophil activation. J Immunol. 1993;150:1535–43. [PubMed] [Google Scholar]
- 3.Witko Sarsat V, Cramer EM, Hieblot C, et al. Presence of proteinase 3 in secretory vesicles: evidence of a novel, highly mobilizable intracellular pool distinct from azurophil granules. Blood. 1999;94:2487–96. [PubMed] [Google Scholar]
- 4.Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA. 1990;87:4115–19. doi: 10.1073/pnas.87.11.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charles LA, Caldas ML, Falk RJ, Terrell RS, Jennette JC. Antibodies against granule proteins activate neutrophils in vitro. J Leukoc Biol. 1991;50:539–46. doi: 10.1002/jlb.50.6.539. [DOI] [PubMed] [Google Scholar]
- 6.Schreiber A, Xiao H, Jennette JC, Schneider W, Luft FC, Kettritz R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol. 2009;20:289–98. doi: 10.1681/ASN.2008050497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hewins P, Morgan MD, Holden N, et al. IL-18 is upregulated in the kidney and primes neutrophil responsiveness in ANCA-associated vasculitis. Kidney Int. 2006;69:605–15. doi: 10.1038/sj.ki.5000167. [DOI] [PubMed] [Google Scholar]
- 8.Csernok E, Ernst M, Schmitt W, Bainton DF, Gross WL. Activated neutrophils express proteinase 3 on their plasma membrane in vitro and in vivo. Clin Exp Immunol. 1994;95:244–50. doi: 10.1111/j.1365-2249.1994.tb06518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Csernok E, Schmitt WH, Ernst M, Bainton DF, Gross WL. Membrane surface proteinase 3 expression and intracytoplasmic immunoglobulin on neutrophils from patients with ANCA-associated vasculitides. Adv Exp Med Biol. 1993;336:45–50. doi: 10.1007/978-1-4757-9182-2_5. [DOI] [PubMed] [Google Scholar]
- 10.Brachemi S, Mambole A, Fakhouri F, et al. Increased membrane expression of proteinase 3 during neutrophil adhesion in the presence of anti proteinase 3 antibodies. J Am Soc Nephrol. 2007;18:2330–9. doi: 10.1681/ASN.2006121309. [DOI] [PubMed] [Google Scholar]
- 11.Morris H, Morgan MD, Wood AM, et al. ANCA-associated vasculitis is linked to carriage of the Z allele of alpha antitrypsin and its polymers. Ann Rheum Dis. 2011;70:1851–6. doi: 10.1136/ard.2011.153569. [DOI] [PubMed] [Google Scholar]
- 12.Halbwachs-Mecarelli L, Bessou G, Lesavre P, Lopez S, Witko-Sarsat V. Bimodal distribution of proteinase 3 (PR3) surface expression reflects a constitutive heterogeneity in the polymorphonuclear neutrophil pool. FEBS Lett. 1995;374:29–33. doi: 10.1016/0014-5793(95)01073-n. [DOI] [PubMed] [Google Scholar]
- 13.Schreiber A, Busjahn A, Luft FC, Kettritz R. Membrane expression of proteinase 3 is genetically determined. J Am Soc Nephrol. 2003;14:68–75. doi: 10.1097/01.asn.0000040751.83734.d1. [DOI] [PubMed] [Google Scholar]
- 14.von Vietinghoff S, Busjahn A, Schonemann C, et al. Major histocompatibility complex HLA region largely explains the genetic variance exercised on neutrophil membrane proteinase 3 expression. J Am Soc Nephrol. 2006;17:3185–91. doi: 10.1681/ASN.2006050522. [DOI] [PubMed] [Google Scholar]
- 15.Witko-Sarsat V, Lesavre P, Lopez S, et al. A large subset of neutrophils expressing membrane proteinase 3 is a risk factor for vasculitis and rheumatoid arthritis. J Am Soc Nephrol. 1999;10:1224–33. doi: 10.1681/ASN.V1061224. [DOI] [PubMed] [Google Scholar]
- 16.Rarok AA, Stegeman CA, Limburg PC, Kallenberg CGM. Neutrophil membrane expression of proteinase 3 (PR3) is related to relapse in PR3–ANCA-associated vasculitis. J Am Soc Nephrol. 2002;13:2232–8. doi: 10.1097/01.asn.0000028642.26222.00. [DOI] [PubMed] [Google Scholar]
- 17.Schreiber A, Otto B, Ju X, et al. Membrane proteinase 3 expression in patients with Wegener's granulomatosis and in human hematopoietic stem cell-derived neutrophils. J Am Soc Nephrol. 2005;16:2216–24. doi: 10.1681/ASN.2004070609. [DOI] [PubMed] [Google Scholar]
- 18.Schreiber A, Luft FC, Kettritz R. Membrane proteinase 3 expression and ANCA-induced neutrophil activation. Kidney Int. 2004;65:2172–83. doi: 10.1111/j.1523-1755.2004.00640.x. [DOI] [PubMed] [Google Scholar]
- 19.Hajjar E, Mihajlovic M, Witko-Sarsat V, Lazaridis T, Reuter N. Computational prediction of the binding site of proteinase 3 to the plasma membrane. Proteins. 2008;71:1655–69. doi: 10.1002/prot.21853. [DOI] [PubMed] [Google Scholar]
- 20.Kantari C, Millet A, Gabillet J, et al. Molecular analysis of the membrane insertion domain of proteinase 3, the Wegener's autoantigen, in RBL cells: implication for its pathogenic activity. J Leukoc Biol. 2011;90:941–50. doi: 10.1189/jlb.1210695. [DOI] [PubMed] [Google Scholar]
- 21.Bauer S, Abdgawad M, Gunnarsson L, Segelmark M, Tapper H, Hellmark T. Proteinase 3 and CD177 are expressed on the plasma membrane of the same subset of neutrophils. J Leukoc Biol. 2007;81:458–64. doi: 10.1189/jlb.0806514. [DOI] [PubMed] [Google Scholar]
- 22.von Vietinghoff S, Tunnemann G, Eulenberg C, et al. NB1 mediates surface expression of the ANCA antigen proteinase 3 on human neutrophils. Blood. 2007;109:4487–93. doi: 10.1182/blood-2006-10-055327. [DOI] [PubMed] [Google Scholar]
- 23.von Vietinghoff S, Eulenberg C, Wellner M, Luft FC, Kettritz R. Neutrophil surface presentation of the anti-neutrophil cytoplasmic antibody-antigen proteinase 3 depends on N-terminal processing. Clin Exp Immunol. 2008;152:508–16. doi: 10.1111/j.1365-2249.2008.03663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korkmaz B, Kuhl A, Bayat B, Santoso S, Jenne DE. A hydrophobic patch on proteinase 3, the target of autoantibodies in Wegener granulomatosis, mediates membrane binding via NB1 receptors. J Biol Chem. 2008;283:35976–82. doi: 10.1074/jbc.M806754200. [DOI] [PubMed] [Google Scholar]
- 25.Choi M, Eulenberg C, Rolle S, von Kries JP, Luft FC, Kettritz R. The use of small molecule high-throughput screening to identify inhibitors of the proteinase 3-NB1 interaction. Clin Exp Immunol. 2010;161:389–96. doi: 10.1111/j.1365-2249.2010.04174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuhl A, Korkmaz B, Utecht B, et al. Mapping of conformational epitopes on human proteinase 3, the autoantigen of Wegener's granulomatosis. J Immunol. 2010;185:387–99. doi: 10.4049/jimmunol.0903887. [DOI] [PubMed] [Google Scholar]
- 27.Hu N, Westra J, Huitema MG, et al. Coexpression of CD177 and membrane proteinase 3 on neutrophils in antineutrophil cytoplasmic autoantibody-associated systemic vasculitis: anti-proteinase 3-mediated neutrophil activation is independent of the role of CD177-expressing neutrophils. Arthritis Rheum. 2009;60:1548–57. doi: 10.1002/art.24442. [DOI] [PubMed] [Google Scholar]
- 28.van der Woude FJ, Rasmussen N, Lobatto S, et al. Autoantibodies against neutrophils and monocytes: tool for diagnosis and marker for disease activity in Wegener's granulomatosis. Lancet. 1985;i:425–9. doi: 10.1016/s0140-6736(85)91147-x. [DOI] [PubMed] [Google Scholar]
- 29.Deutsch M, Guejes L, Zurgil N, et al. Antineutrophil cytoplasmic autoantibodies penetrate into human polymorphonuclear leukocytes and modify their apoptosis. Clin Exp Rheumatol. 2004;22:S35–40. [PubMed] [Google Scholar]
- 30.Yang JJ, Pendergraft WF, Alcorta DA, et al. Circumvention of normal constraints on granule protein gene expression in peripheral blood neutrophils and monocytes of patients with antineutrophil cytoplasmic autoantibody-associated glomerulonephritis. J Am Soc Nephrol. 2004;15:2103–14. doi: 10.1097/01.ASN.0000135058.46193.72. [DOI] [PubMed] [Google Scholar]
- 31.Ciavatta DJ, Yang J, Preston GA, et al. Epigenetic basis for aberrant upregulation of autoantigen genes in humans with ANCA vasculitis. J Clin Invest. 2010;120:3209–19. doi: 10.1172/JCI40034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kain R, Matsui K, Exner M, et al. A novel class of autoantigens of anti-neutrophil cytoplasmic antibodies in necrotizing and crescentic glomerulonephritis: the lysosomal membrane glycoprotein h-lamp-2 in neutrophil granulocytes and a related membrane protein in glomerular endothelial cells. J Exp Med. 1995;181:585–97. doi: 10.1084/jem.181.2.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kain R, Exner M, Brandes R, et al. Molecular mimicry in pauci-immune focal necrotizing glomerulonephritis. Nat Med. 2008;14:1088–96. doi: 10.1038/nm.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carlsson SR, Roth J, Piller F, Fukuda M. Isolation and characterization of human lysosomal membrane glycoproteins, h-lamp-1 and h-lamp-2. Major sialoglycoproteins carrying polylactosaminoglycan. J Biol Chem. 1988;263:18911–19. [PubMed] [Google Scholar]
- 35.Roth AJ, Brown MC, Smith RN, et al. Anti-LAMP-2 antibodies are not prevalent in patients with antineutrophil cytoplasmic autoantibody glomerulonephritis. J Am Soc Nephrol. 2012;23:545–55. doi: 10.1681/ASN.2011030273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kain R, Tadema H, McKinney EF, et al. High prevalence of autoantibodies to hLAMP-2 in anti-neutrophil cytoplasmic antibody-associated vasculitis. J Am Soc Nephrol. 2012;23:556–66. doi: 10.1681/ASN.2011090920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Flint SM, Savage CO. Anti-LAMP-2 autoantibodies in ANCA-associated pauci-immune glomerulonephritis. J Am Soc Nephrol. 2012;23:378–80. doi: 10.1681/ASN.2012010065. [DOI] [PubMed] [Google Scholar]
- 38.Kettritz R, Jennette JC, Falk RJ. Crosslinking of ANCA-antigens stimulates superoxide release by human neutrophils. J Am Soc Nephrol. 1997;8:386–94. doi: 10.1681/ASN.V83386. [DOI] [PubMed] [Google Scholar]
- 39.Keogan MT, Esnault VL, Green AJ, Lockwood CM, Brown DL. Activation of normal neutrophils by anti-neutrophil cytoplasm antibodies. Clin Exp Immunol. 1992;90:228–34. doi: 10.1111/j.1365-2249.1992.tb07934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Porges AJ, Redecha PB, Kimberly WT, Csernok E, Gross WL, Kimberly RP. Anti-neutrophil cytoplasmic antibodies engage and activate human neutrophils via Fc gamma RIIa. J Immunol. 1994;153:1271–80. [PubMed] [Google Scholar]
- 41.Mulder AH, Heeringa P, Brouwer E, Limburg PC, Kallenberg CG. Activation of granulocytes by anti-neutrophil cytoplasmic antibodies (ANCA): a Fc gamma RII-dependent process. Clin Exp Immunol. 1994;98:270–8. doi: 10.1111/j.1365-2249.1994.tb06137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reumaux D, Vossebeld PJ, Roos D, Verhoeven AJ. Effect of tumor necrosis factor-induced integrin activation on Fc gamma receptor II-mediated signal transduction: relevance for activation of neutrophils by anti-proteinase 3 or anti-myeloperoxidase antibodies. Blood. 1995;86:3189–95. [PubMed] [Google Scholar]
- 43.Williams JM, Ben-Smith A, Hewins P, et al. Activation of the G(i) heterotrimeric G protein by ANCA IgG F(ab')2 fragments is necessary but not sufficient to stimulate the recruitment of those downstream mediators used by intact ANCA IgG. J Am Soc Nephrol. 2003;14:661–9. doi: 10.1097/01.asn.0000050223.34749.f4. [DOI] [PubMed] [Google Scholar]
- 44.Yang JJ, Preston GA, Alcorta DA, et al. Expression profile of leukocyte genes activated by anti-neutrophil cytoplasmic autoantibodies (ANCA) Kidney Int. 2002;62:1638–49. doi: 10.1046/j.1523-1755.2002.00619.x. [DOI] [PubMed] [Google Scholar]
- 45.Weidner S, Neupert W, Goppelt-Struebe M, Rupprecht HD. Antineutrophil cytoplasmic antibodies induce human monocytes to produce oxygen radicals in vitro. Arthritis Rheum. 2001;44:1698–706. doi: 10.1002/1529-0131(200107)44:7<1698::AID-ART294>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 46.Kocher M, Edberg JC, Fleit HB, Kimberly RP. Antineutrophil cytoplasmic antibodies preferentially engage Fc gammaRIIIb on human neutrophils. J Immunol. 1998;161:6909–14. [PubMed] [Google Scholar]
- 47.Pankhurst T, Nash G, Williams J, Colman R, Hussain A, Savage C. Immunoglobulin subclass determines ability of immunoglobulin (Ig)G to capture and activate neutrophils presented as normal human IgG or disease-associated anti-neutrophil cytoplasm antibody (ANCA)-IgG. Clin Exp Immunol. 2011;164:218–26. doi: 10.1111/j.1365-2249.2011.04367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jayne DR, Weetman AP, Lockwood CM. IgG subclass distribution of autoantibodies to neutrophil cytoplasmic antigens in systemic vasculitis. Clin Exp Immunol. 1991;84:476–81. [PMC free article] [PubMed] [Google Scholar]
- 49.Kelley JM, Monach PA, Ji C, et al. IgA and IgG antineutrophil cytoplasmic antibody engagement of Fc receptor genetic variants influences granulomatosis with polyangiitis. Proc Natl Acad Sci USA. 2011;108:20736–41. doi: 10.1073/pnas.1109227109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu J, Ji C, Xie F, et al. FcalphaRI (CD89) alleles determine the proinflammatory potential of serum IgA. J Immunol. 2007;178:3973–82. doi: 10.4049/jimmunol.178.6.3973. [DOI] [PubMed] [Google Scholar]
- 51.Espy C, Morelle W, Kavian N, et al. Sialylation levels of anti-proteinase 3 antibodies are associated with the activity of granulomatosis with polyangiitis (Wegener's) Arthritis Rheum. 2011;63:2105–15. doi: 10.1002/art.30362. [DOI] [PubMed] [Google Scholar]
- 52.van Timmeren MM, van der Veen BS, Stegeman CA, et al. IgG glycan hydrolysis attenuates ANCA-mediated glomerulonephritis. J Am Soc Nephrol. 2010;21:1103–14. doi: 10.1681/ASN.2009090984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.David A, Kacher Y, Specks U, Aviram I. Interaction of proteinase 3 with CD11b/CD18 (beta2 integrin) on the cell membrane of human neutrophils. J Leukoc Biol. 2003;74:551–7. doi: 10.1189/jlb.1202624. [DOI] [PubMed] [Google Scholar]
- 54.David A, Fridlich R, Aviram I. The presence of membrane Proteinase 3 in neutrophil lipid rafts and its colocalization with FcgammaRIIIb and cytochrome b558. Exp Cell Res. 2005;308:156–65. doi: 10.1016/j.yexcr.2005.03.034. [DOI] [PubMed] [Google Scholar]
- 55.Jerke U, Rolle S, Dittmar G, et al. Complement receptor Mac-1 is an adaptor for NB1 (CD177)-mediated PR3–ANCA neutrophil activation. J Biol Chem. 2011;286:7070–81. doi: 10.1074/jbc.M110.171256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hess C, Sadallah S, Schifferli JA. Induction of neutrophil responsiveness to myeloperoxidase antibodies by their exposure to supernatant of degranulated autologous neutrophils. Blood. 2000;96:2822–7. [PubMed] [Google Scholar]
- 57.Johansson MW, Patarroyo M, Oberg F, Siegbahn A, Nilsson K. Myeloperoxidase mediates cell adhesion via the alpha M beta 2 integrin (Mac-1, CD11b/CD18) J Cell Sci. 1997;110((Pt 9)):1133–9. doi: 10.1242/jcs.110.9.1133. [DOI] [PubMed] [Google Scholar]
- 58.Lau D, Mollnau H, Eiserich JP, et al. Myeloperoxidase mediates neutrophil activation by association with CD11b/CD18 integrins. Proc Natl Acad Sci USA. 2005;102:431–6. doi: 10.1073/pnas.0405193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Radford DJ, Lord JM, Savage COS. The activation of the neutrophil respiratory burst by anti-neutrophil cytoplasm autoantibody (ANCA) from patients with systemic vasculitis requires tyrosine kinases and protein kinase C activation. Clin Exp Immunol. 1999;118:171–9. doi: 10.1046/j.1365-2249.1999.01043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kettritz R, Schreiber A, Luft FC, Haller H. Role of mitogen-activated protein kinases in activation of human neutrophils by antineutrophil cytoplasmic antibodies. J Am Soc Nephrol. 2001;12:37–46. doi: 10.1681/ASN.V12137. [DOI] [PubMed] [Google Scholar]
- 61.Choi M, Rolle S, Rane M, Haller H, Luft FC, Kettritz R. Extracellular signal-regulated kinase inhibition by statins inhibits neutrophil activation by ANCA. Kidney Int. 2003;63:96–106. doi: 10.1046/j.1523-1755.2003.00718.x. [DOI] [PubMed] [Google Scholar]
- 62.Polzer K, Soleiman A, Baum W, et al. Selective p38MAPK isoform expression and activation in antineutrophil cytoplasmatic antibody-associated crescentic glomerulonephritis: role of p38MAPKalpha. Ann Rheum Dis. 2008;67:602–8. doi: 10.1136/ard.2007.077263. [DOI] [PubMed] [Google Scholar]
- 63.van der Veen BS, Chen M, Muller R, et al. Effects of p38 mitogen-activated protein kinase inhibition on anti-neutrophil cytoplasmic autoantibody pathogenicity in vitro and in vivo. Ann Rheum Dis. 2011;70:356–65. doi: 10.1136/ard.2010.129106. [DOI] [PubMed] [Google Scholar]
- 64.Ben-Smith A, Dove SK, Martin A, Wakelam MJ, Savage CO. Antineutrophil cytoplasm autoantibodies from patients with systemic vasculitis activate neutrophils through distinct signaling cascades: comparison with conventional Fcgamma receptor ligation. Blood. 2001;98:1448–55. doi: 10.1182/blood.v98.5.1448. [DOI] [PubMed] [Google Scholar]
- 65.Kettritz R, Choi M, Butt W, et al. Phosphatidylinositol 3-kinase controls antineutrophil cytoplasmic antibodies-induced respiratory burst in human neutrophils. J Am Soc Nephrol. 2002;13:1740–9. doi: 10.1097/01.asn.0000019411.36000.06. [DOI] [PubMed] [Google Scholar]
- 66.von Vietinghoff S, Choi M, Rolle S, Luft FC, Kettritz R. Febrile temperatures control antineutrophil cytoplasmic autoantibody-induced neutrophil activation via inhibition of phosphatidylinositol 3-kinase/Akt. Arthritis Rheum. 2007;56:3149–58. doi: 10.1002/art.22832. [DOI] [PubMed] [Google Scholar]
- 67.Kettritz R, Choi M, Salanova B, Wellner M, Rolle S, Luft FC. Fever-like temperatures affect neutrophil NF-kappaB signaling, apoptosis, and ANCA-antigen expression. J Am Soc Nephrol. 2006;17:1345–53. doi: 10.1681/ASN.2005090948. [DOI] [PubMed] [Google Scholar]
- 68.Schreiber A, Rolle S, Peripelittchenko L, et al. Phosphoinositol 3-kinase-gamma mediates antineutrophil cytoplasmic autoantibody-induced glomerulonephritis. Kidney Int. 2010;77:118–28. doi: 10.1038/ki.2009.420. [DOI] [PubMed] [Google Scholar]
- 69.Xiao H, Schreiber A, Heeringa P, Falk RJ, Jennette JC. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am J Pathol. 2007;170:52–64. doi: 10.2353/ajpath.2007.060573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huugen D, van Esch A, Xiao H, et al. Inhibition of complement factor C5 protects against anti-myeloperoxidase antibody-mediated glomerulonephritis in mice. Kidney Int. 2007;71:646–54. doi: 10.1038/sj.ki.5002103. [DOI] [PubMed] [Google Scholar]
- 71.Ohashi PS, Woodgett JR. Modulating autoimmunity: pick your PI3 kinase. Nat Med. 2005;11:924–5. doi: 10.1038/nm0905-924. [DOI] [PubMed] [Google Scholar]
- 72.Brooks CJ, King WJ, Radford DJ, Adu D, McGrath M, Savage CO. IL-1 beta production by human polymorphonuclear leucocytes stimulated by anti-neutrophil cytoplasmic autoantibodies: relevance to systemic vasculitis. Clin Exp Immunol. 1996;106:273–9. doi: 10.1046/j.1365-2249.1996.d01-835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cockwell P, Brooks CJ, Adu D, Savage COS. Interleukin-8: a pathogenetic role in antineutrophil cytoplasmic autoantibody-associated glomerulonephritis. Kidney Int. 1999;55:852–63. doi: 10.1046/j.1523-1755.1999.055003852.x. [DOI] [PubMed] [Google Scholar]
- 74.Hruskova Z, Rihova Z, Mareckova H, et al. Intracellular cytokine production in ANCA-associated vasculitis: low levels of interleukin-10 in remission are associated with a higher relapse rate in the long-term follow-up. Arch Med Res. 2009;40:276–84. doi: 10.1016/j.arcmed.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 75.Noronha IL, Kruger C, Andrassy K, Ritz E, Waldherr R. In situ production of TNF-alpha, IL-1 beta and IL-2R in ANCA-positive glomerulonephritis. Kidney Int. 1993;43:682–92. doi: 10.1038/ki.1993.98. [DOI] [PubMed] [Google Scholar]
- 76.Timoshanko JR, Kitching AR, Iwakura Y, Holdsworth SR, Tipping PG. Leukocyte-derived interleukin-1beta interacts with renal interleukin-1 receptor I to promote renal tumor necrosis factor and glomerular injury in murine crescentic glomerulonephritis. Am J Pathol. 2004;164:1967–77. doi: 10.1016/s0002-9440(10)63757-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schreiber A, Pham CT, Hu Y, Schneider W, Luft FC, Kettritz R. Neutrophil serine proteases promote IL-1beta generation and injury in necrotizing crescentic glomerulonephritis. J Am Soc Nephrol. 2012;23:470–82. doi: 10.1681/ASN.2010080892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lu X, Garfield A, Rainger GE, Savage CO, Nash GB. Mediation of endothelial cell damage by serine proteases, but not superoxide, released from antineutrophil cytoplasmic antibody-stimulated neutrophils. Arthritis Rheum. 2006;54:1619–28. doi: 10.1002/art.21773. [DOI] [PubMed] [Google Scholar]
- 79.Hong Y, Eleftheriou D, Hussain AA, et al. Anti-neutrophil cytoplasmic antibodies stimulate release of neutrophil microparticles. J Am Soc Nephrol. 2012;23:49–62. doi: 10.1681/ASN.2011030298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kessenbrock K, Krumbholz M, Schonermarck U, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 2009;15:623–5. doi: 10.1038/nm.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]